Abstract
Nine potential non-symmetrical xylene-bridged AChE reactivators were synthesized using modifications of currently known synthetic pathways. Their potency to reactivate AChE inhibited by the nerve agent tabun and the insecticide paraoxon together with nine symmetrical xylene-bridged compounds, was tested in vitro. Seven compounds were promising against paraoxon-inhibited AChE. Two compounds were found to be more potent against tabun-inhibited AChE than obidoxime at a concentration applicable in vivo.
Introduction
Organophosphorus compounds (OP) are commonly known as nerve agents (soman, sarin, tabun, VX, etc.), pesticides (chlorpyrifos, paraoxon, diazinon, etc.), compounds used for industrial purposes (tributylphosphate) or as potential therapeutics (metrifonate) Citation1-4. Their chemical structure is derived from phosphonic and phosphoric acid or their thio-analogues respectively Citation1-2. They are able to irreversibly phosphonylate or phosphorylate the serine hydroxyl in the active site of acetylcholinesterase (AChE, EC 3.1.1.7) inhibiting the cleavage of the neurotransmitter acetylcholine which is essential to terminate cholinergic transmission (Scheme ) Citation1-2. The accumulated acetylcholine causes cholinergic overstimulation and subsequent cholinergic crisis which normally causes serious malfunction of the breathing centre in the medulla oblongata followed by death [Citation2]. In the last decade, the risk of intoxication by these compounds has rapidly increased with growing agricultural production and with the threat of terrorist attacks Citation5-6.
Scheme 1 AChE inhibited by nerve agent tabun.

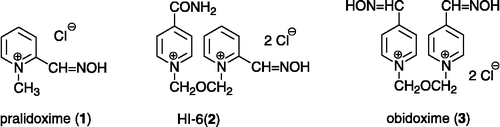
Anticholinergic drugs such as atropine are used to counteract the effects of OP-inhibited enzyme at peripheral cholinergic receptors [Citation7]. However, they are not able to restore natural enzyme activity. For this reason, AChE reactivators are used to cleave the covalent bond OP-enzyme by a reactive nucleophilic group and to restore the activity of AChE (Scheme ) [Citation2]. The commonly used reactivators are monoquaternary or bisquaternary compounds carrying the hydroxyiminomethyl (oxime) group as nucleophilic agent, e.g. pralidoxime – “the golden standard of AChE reactivators” (1, 2-hydroxyiminomethyl-1-methylpyridinium chloride), oxime HI-6 (2, 1-(2-hydroxyiminomethylpyridinium)-3-(4-carbamoylpyridinium)-2-oxapropane dichloride), obidoxime (3, Toxogonine®, 1,3-bis(4-hydroxyiminomethylpyridinium)-2-oxapropane dichloride) () Citation8-11. Nevertheless, every type of OP needs a specific structure for the AChE reactivator and there is not a broad spectrum reactivator after more than fifty years of investigations Citation12-15. Therefore, the development and selection of new effective reactivators of AChE-like antidotes of OP are very important.
Scheme 2 Oxime induced reactivation of tabun-inhibited AChE.
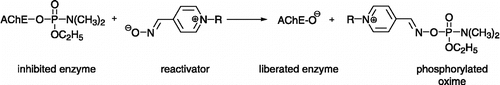
Figure 1 Examples of oxime reactivators currently used against OP intoxications.
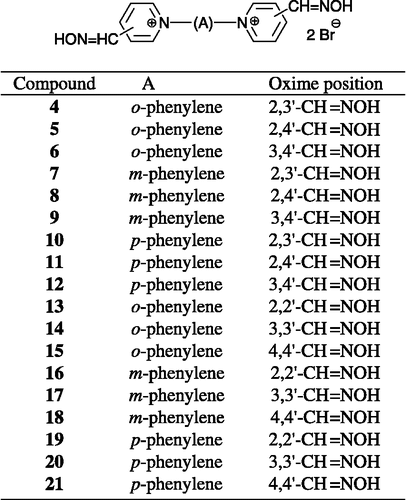
In this work, eighteen compounds (4–21) are described (Scheme ). Our research was focused on finding more rigid bisquaternary structures than the commonly used reactivators possess (2, 3). In order to fix the conformation of the molecule, we decided to use a xylene linker connecting two pyridinium rings bearing oxime moieties non-symmetrically (nine novel compounds, 4–12), in contrast with symmetrical ones (nine compounds prepared previously, 13–21) [Citation16]. The idea and design of an aromatic linker was used by comparison with inhibitors of AChE with a triazole linker prepared using click chemistry methods Citation17-18. Moreover, each position of oxime on the pyridinium ring is able to reactivate another type of inhibitor. While position four is more suitable for tabun or pesticide-inhibited AChE, position two is effective against sarin, soman or VX-inhibited AChE Citation19-21. Position three has usually low efficacy due to the dissociation constant at the pH of human blood [Citation19]. Therefore, the combination of various positions should extend the reactivation to a broader spectrum of OP inhibitors. Both symmetrical and non-symmetrical compounds were tested in vitro on tabun (GA) and paraoxon-inhibited AChE.
Scheme 3 Potential oxime reactivators tested against tabun- and paraoxon-inhibited AChE.
Materials and methods
Chemistry
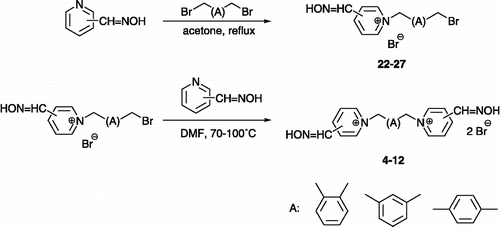
The compounds were synthesized using conventional methods Citation15-16. Initially, the hydroxyiminomethylpyridine was mixed in acetone with 5 equivalent excess of the corresponding dibromoxylene to afford the monoquaternary compounds (Scheme ). The monoquaternary salt was easily purified by the recrystallization from acetonitrile, whereas the bi-product is almost insoluble. Secondly, a different hydroxyiminomethylpyridine was added in DMF to obtain the non-symmetrical bisquaternary salt, usually in satisfactory yield (27-91%).
Scheme 4 Two step synthesis of non-symmetrical bisquaternary compounds.
Preparation of quaternary salts
(A) Preparation of monoquaternary salts – A solution of the hydroxyiminomethylpyridine (1.0 g, 8.2 mmol) and dibromoxylene (10.8 g, 40.9 mmol) in acetone (100 mL) was stirred at reflux. The reaction mixture was then cooled to room temperature; the crystalline crude product was collected by filtration, washed with acetone (3 × 20 mL) and recrystallized from acetonitrile (MeCN). (B) Preparation of bisquaternary salts – A solution of the monoquaternary salt (0.50 g, 1.3 mmol) and hydroxyiminomethylpyridine (0.30 g, 2.4 mmol) in DMF (10 mL) was stirred at 70–100°C. The reaction mixture was then cooled to room temperature and portioned with acetone (50 ml); the crystalline crude product was collected by filtration, washed with acetone (3 × 20 mL) and recrystalized from MeCN.
2,3′-bis(hydroxyiminomethyl)-1,1′-(1,2-phenylenedimethyl)-bispyridinium dibromide (4)
Prepared by method B via 22. The reaction mixture was stirred at 70°C and stopped after 10 h. Yield 0.48 g (73%), TLC Rf 0.15, m.p. 215–216 °C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.37 (s, 1H, PyrH), 9.18 (d, 1H, J = 6.0 Hz, PyrH), 9.06 (d, 1H, J = 6.0 Hz, PyrH), 8.84 (d, 1H, J = 8.1 Hz, PyrH), 8.76-8.64 (m, 2H, PyrH + -CH = NOH), 8.57 (d, 1H, J = 8.1 Hz, PyrH), 8.44 (s, 1H, -CH = NOH), 8.31-8.17 (m, 2H, PyrH), 7.56-7.27 (m, 3H, ArH), 6.61 (d, 1H, J = 7.4 Hz, ArH), 6.36 (s, 2H, -CH2-), 6.22 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 147.77, 147.76, 147.73, 147.70, 146.34, 146.28, 143.30, 142.73, 141.28, 133.80, 133.78, 132.66, 131.40, 130.22, 128.59, 128.53, 126.27, 125.96, 60.41, 57.94. Analysis: calculated 47.27% C, 3.97% H, 11.02% N; found 47.07% C, 4.18% H, 11.04% N. ESI-MS: m/z 347.1 [M2 + -H+] (calculated for [C20H20N4O22 + -H+] 347.16).
2,4′-bis(hydroxyiminomethyl)-1,1′-(1,2-phenylenedimethyl)-bispyridinium dibromide (5)
Prepared by method B via 23. The reaction mixture was stirred at 70°C and stopped after 10 h. Yield 0.50 g (76%), TLC Rf 0.15, m.p. 205–206°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.15 (d, 2H, J = 6.3 Hz, PyrH), 9.05 (d, 1H, J = 6.3 Hz, PyrH), 8.76-8.64 (m, 2H, PyrH + -CH = NOH), 8.60-8.48 (m, 2H, PyrH + -CH = NOH), 8.33 (d, 2H, J = 6.3 Hz, PyrH), 8.26-8.19 (m, 1H, PyrH), 7.52-7.35 (m, 2H, ArH), 7.31 (d, 1H, J = 7.4 Hz, ArH), 6.61 (d, 1H, J = 7.4 Hz, ArH), 6.35 (s, 2H, -CH2-), 6.16 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 149.00, 147.71, 146.26, 146.13, 145.42, 145.10, 141.32, 132.52, 131.72, 130.10, 129.70, 129.34, 128.15, 126.26, 126.04, 124.42, 59.68, 57.93. Analysis: calculated 47.27% C, 3.97% H, 11.02% N; found 46.53% C, 4.17% H, 10.79% N. ESI-MS: m/z 347.1 [M2 + -H+] (calculated for [C20H20N4O22 + -H]+347.16).
3,4′-bis(hydroxyiminomethyl)-1,1′-(1,2-phenylenedimethyl)-bispyridinium dibromide (6)
Prepared by method B via 23. The reaction mixture was stirred at 100°C and stopped after 2 h. Yield 0.57 g (86%), TLC Rf 0.15, m.p. 224–226°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.31 (s, 1H, PyrH), 9.14-9.04 (m, 3H, PyrH), 8.81 (d, 1H, J = 8.1 Hz, PyrH), 8.49 (s, 1H, -CH = NOH), 8.40 (s, 1H, -CH = NOH), 8.33-8.19 (m, 3H, PyrH), 7.57-7.47 (dd, 2H, J = 3.3 Hz, ArH), 7.34-7.23 (m, 2H, ArH), 6.19 (d, 4H, J = 13.5 Hz, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 149.01, 145.34, 145.05, 144.73, 143.23, 142.63, 142.18, 133.76, 132.81, 132.42, 130.14, 130.05, 129.81, 129.63, 128.55, 124.36, 60.38, 59.66. Analysis: calculated 47.27% C, 3.97% H, 11.02% N; found 46.07% C, 4.27% H, 10.93% N. ESI-MS: m/z 347.1 [M2 + -H+] (calculated for [C20H20N4O22 + -H+] 347.16).
2,3′-bis(hydroxyiminomethyl)-1,1′-(1,3-phenylenedimethyl)-bispyridinium dibromide (7)
Prepared by method B via 24. The reaction mixture (MeCN-30mL/EtOH-10mL/DMF-8mL) was stirred at 70°C and stopped after 26 h. Yield 0.36 g (27%), TLC Rf 0.15, m.p. 199–201°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.47 (s, 1H, PyrH), 9.30 (d, 1H, J = 6.0 Hz, PyrH), 9.19 (d, 1H, J = 6.0 Hz, PyrH), 8.77 (d, 1H, J = 8.1 Hz, PyrH), 8.71-8.61 (m, 2H, PyrH + -CH = NOH), 8.48-8.35 (m, 2H, PyrH + -CH = NOH), 8.26-8.16 (m, 2H, PyrH), 7.64-7.45 (m, 3H, ArH), 7.29 (d, 1H, J = 7.4 Hz, ArH), 6.15 (s, 2H, -CH2-), 5.93 (s, 2H, -CH2-). 13C NMR spectrum (75 MHz, DMSO d6): δ (ppm) 147.09, 146.56, 146.02, 146.00, 144.34, 143.19, 142.40, 141.97, 141.35, 134.93, 134.89, 133.71, 130.03, 129.27, 128.46, 128.13, 127.92, 126.11, 62.78, 59.92. Analysis: calculated 47.27% C, 3.97% H, 11.02% N; found 47.10% C, 4.08% H, 10.98% N. ESI-MS: m/z 347.1 [M2 + -H+] (calculated for [C20H20N4O22 + -H+] 347.16).
2,4′-bis(hydroxyiminomethyl)-1,1′-(1,3-phenylenedimethyl)-bispyridinium dibromide (8)
Prepared by method B via 25. The reaction mixture was stirred at 70°C and stopped after 8.5 h. Yield 0.43 g (65%), TLC Rf 0.15, m.p. 208–210°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.30 (d, 1H, J = 6.0 Hz, PyrH), 9.19 (d, 2H, J = 6.0 Hz, PyrH), 8.71-8.61 (m, 2H, PyrH + -CH = NOH), 8.48-8.39 (m, 2H, PyrH + -CH = NOH), 8.30-8.18 (m, 3H, PyrH), 7.60-7.45 (m, 3H, ArH), 7.29 (d, 1H, J = 7.4 Hz, ArH), 6.15 (s, 2H, -CH2-), 5.88 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 148.75, 147.09, 146.56, 146.00, 145.04, 144.97, 141.38, 135.15, 134.88, 130.04, 129.18, 128.07, 127.93, 127.80, 126.14, 124.32, 62.05, 59.94. Analysis: calculated 47.27% C, 3.97% H, 11.02% N; found 47.06% C, 4.21% H, 11.17% N. ESI-MS: m/z 347.1 [M2 + -H+] (calculated for [C20H20N4O22 + -H+] 347.16).
3,4′-bis(hydroxyiminomethyl)-1,1′-(1,3-phenylenedimethyl)-bispyridinium dibromide (9)
Prepared by method B via 25. The reaction mixture was stirred at 70°C and stopped after 5 h. Yield 0.32 g (48%), TLC Rf 0.15, m.p. 200–202°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.51 (s, 1H, PyrH), 9.31-9.19 (m, 3H, PyrH), 8.78 (d, 1H, J = 8.1 Hz, PyrH), 8.46 (s, 1H, -CH = NOH), 8.41 (s, 1H, -CH = NOH), 8.31-8.16 (m, 3H, PyrH), 7.85 (s, 1H, ArH), 7.65-7.47 (m, 3H, ArH), 5.95 (d, 4H, J = 14.4 Hz, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 147.10, 146.46, 145.98, 144.42, 144.41, 143.20, 142.39, 142.02, 141.34, 134.99, 134.67, 133.74, 129.57, 128.50, 128.04, 126.07, 62.65, 59.70. Analysis: calculated 47.27% C, 3.97% H, 11.02% N; found 46.45% C, 4.07% H, 10.79% N. ESI-MS: m/z 347.1 [M2 + -H+] (calculated for [C20H20N4O22 + -H+] 347.16).
2,3′-bis(hydroxyiminomethyl)-1,1′-(1,4-phenylenedimethyl)-bispyridinium dibromide (10)
Prepared by method B via 26. The reaction mixture was stirred at 100°C and stopped after 8 h. Yield 0.49 g (74%), TLC Rf 0.15, m.p. 228–230°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.48 (s, 1H, PyrH), 9.26 (dd, 2H, J = 6.2 Hz, PyrH), 8.80-8.70 (m, 2H, PyrH + -CH = NOH), 8.70-8.60 (m, 1H, PyrH), 8.43 (d, 1H, J = 8.2 Hz, PyrH), 8.36 (s, 1H, -CH = NOH), 8.26-8.16 (m, 2H, PyrH), 7.63 (d, 2H, J = 7.8 Hz, ArH), 7.35 (d, 2H, J = 7.8 Hz, ArH), 6.16 (s, 2H, -CH2-), 5.95 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 147.10, 146.46, 145.98, 144.42, 144.41, 143.20, 142.39, 142.02, 141.34, 134.99, 134.67, 133.74, 129.57, 128.50, 128.04, 126.07, 62.65, 59.70. Analysis: calculated 47.27% C, 3.97% H, 11.02% N; found 47.22% C, 4.13% H, 10.86% N. ESI-MS: m/z 347.1 [M2 + -H+] (calculated for [C20H20N4O22 + -H+] 347.16).
2,4′-bis(hydroxyiminomethyl)-1,1′-(1,4-phenylenedimethyl)-bispyridinium dibromide (11)
Prepared by method B via 27. The reaction mixture was stirred at 100°C and stopped after 8 h. Yield 0.48 g (73%), TLC Rf 0.15, m.p. 239–241°C. 1H NMR spectrum (300 MHz, DMSO d6): δ (ppm) 9.29 (d, 1H, J = 6.0 Hz, PyrH), 9.21 (d, 2H, J = 6.0 Hz, PyrH), 8.73 (s, 1H, -CH = NOH), 8.70-8.60 (m, 1H, PyrH), 8.49-8.40 (m, 2H, PyrH + -CH = NOH), 8.32-8.17 (m, 3H, PyrH), 7.61 (d, 2H, J = 7.3 Hz, ArH), 7.35 (d, 2H, J = 7.3 Hz, ArH), 6.16 (s, 2H, -CH2-), 5.89 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 148.75, 147.09, 146.46, 145.98, 145.02, 141.35, 134.92, 129.48, 128.06, 127.91, 126.07, 124.36, 61.91, 59.71. Analysis: calculated 47.27% C, 3.97% H, 11.02% N; found 46.98% C, 4.33% H, 10.76% N. ESI-MS: m/z 347.1 [M2 + -H+] (calculated for [C20H20N4O22 + -H+] 347.16).
3,4′-bis(hydroxyiminomethyl)-1,1′-(1,4-phenylenedimethyl)-bispyridinium dibromide (12)
Prepared by method B via 27. The reaction mixture was stirred at 100°C and stopped after 2 h. Yield 0.60 g (91%), TLC Rf 0.15, m.p. 264–265°C. 1H NMR spectrum (300 MHz, DMSO d6): δ (ppm) 9.48 (s, 1H, PyrH), 9.29-9.17 (m, 3H, PyrH), 8.75 (d, 1H, J = 8.1 Hz, PyrH), 8.43 (s, 1H, -CH = NOH), 8.36 (s, 1H, -CH = NOH), 8.26 (d, 2H, J = 6.3 Hz, PyrH), 8.23-8.16 (m, 1H, PyrH), 7.72-7.61 (m, 4H, ArH), 5.93 (d, 4H, J = 15.8 Hz, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 148.75, 145.03, 144.43, 143.20, 142.36, 142.01, 135.37, 135.07, 133.73, 129.66, 129.55, 128.50, 124.36, 62.70, 61.95. Analysis: calculated 47.27% C, 3.97% H, 11.02% N; found 47.12% C, 4.38% H, 10.91% N. ESI-MS: m/z 174.1 [M2 + ] (calculated for [C10H10N2O2 + ] 174.08).
1-(2-bromomethylbenzyl)-3-hydroxyiminomethylpyridinium bromide (22)
Prepared by method A. The reaction mixture was stopped after 2 h. Yield 2.87 g (91%), TLC Rf 0.60, m.p. 189–190°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.30 (s, 1H, PyrH), 9.07 (d, 1H, J = 6.0 Hz, PyrH), 8.78 (d, 1H, J = 8.0 Hz, PyrH), 8.39 (s, 1H, -CH = NOH), 8.27-8.17 (m, 1H, PyrH), 7.60 (d, 1H, J = 7.0 Hz, ArH), 7.55-7.41 (m, 2H, ArH), 7.37 (d, 1H, J = 7.0 Hz, ArH), 6.12 (s, 2H, -CH2-), 4.96 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 144.68, 143.25, 142.48, 142.18, 137.19, 133.58, 131.93, 131.60, 130.62, 130.11, 129.68, 128.38, 60.31, 31.70. Analysis: calculated 43.55% C, 3.65% H, 7.26% N; found 43.67% C, 3.91% H, 7.32% N. ESI-MS: m/z 305.0 [M+] (calculated for [C14H14BrN2O+] 305.03).
1-(2-bromomethylbenzyl)-4-hydroxyiminomethylpyridinium bromide (23)
Prepared by method A. The reaction mixture was stopped after 2 h. Yield 3.10 g (98%), TLC Rf 0.60, m.p. 204–206°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.06 (d, 2H, J = 6.0 Hz, PyrH), 8.46 (s, 1H, -CH = NOH), 8.28 (d, 2H, J = 6.0 Hz, PyrH), 7.60 (d, 1H, J = 6.6 Hz, ArH), 7.53-7.38 (m, 2H, ArH), 7.31 (d, 1H, J = 6.6 Hz, ArH), 6.06 (s, 2H, -CH2-), 4.95 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 148.87, 145.33, 145.06, 137.07, 132.26, 131.55, 130.34, 129.95, 129.64, 124.23, 59.55, 31.68. Analysis: calculated 43.55% C, 3.65% H, 7.26% N; found 43.46% C, 4.01% H, 7.25% N. ESI-MS: m/z 305.0 [M+] (calculated for [C14H14BrN2O+] 305.03).
1-(3-bromomethylbenzyl)-3-hydroxyiminomethylpyridinium bromide (24)
Prepared by method A. The reaction mixture was stopped after 2 h. Yield 2.80 g (89%), TLC Rf 0.60, m.p. 210–211°C. 1H NMR spectrum (300 MHz, DMSO d6): δ (ppm) 9.45 (s, 1H, PyrH), 9.22 (d, 1H, J = 6.0 Hz, PyrH), 8.75 (d, 1H, J = 8.0 Hz, PyrH), 8.38 (s, 1H, -CH = NOH), 8.20 (dd, 1H, J = 6.5 Hz), 7.59-7.31 (m, 4H, ArH), 5.93 (s, 2H, -CH2-), 4.49 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 144.38, 143.68, 143.28, 142.24, 141.99, 133.80, 133.70, 128.95, 128.50, 127.38, 127.23, 126.73, 63.44, 62.35. Analysis: calculated 43.55% C, 3.65% H, 7.26% N; found 43.61% C, 3.99% H, 7.27% N. ESI-MS: m/z 305.0 [M+] (calculated for [C14H14BrN2O+] 305.03).
1-(3-bromomethylbenzyl)-4-hydroxyiminomethylpyridinium bromide (25)
Prepared by method A. The reaction mixture was stopped after 2 h. Yield 3.10 g (98%), TLC Rf 0.60, m.p. 157–158°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.18 (d, 2H, J = 6.0 Hz, PyrH), 8.43 (s, 1H, -CH = NOH), 8.27 (d, 2H, J = 6.0 Hz, PyrH), 7.61 (s, 1H, ArH), 7.53-7.38 (m, 3H, ArH), 5.87 (s, 2H, -CH2-), 4.69 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 148.75, 145.03, 139.04, 134.73, 130.13, 129.55, 129.32, 128.60, 124.38, 62.24, 33.63. Analysis: calculated 43.55% C, 3.65% H, 7.26% N; found 43.57% C, 3.91% H, 7.31% N. ESI-MS: m/z 305.0 [M+] (calculated for [C14H14BrN2O+] 305.03).
1-(4-bromomethylbenzyl)-3-hydroxyiminomethylpyridinium bromide (26)
Prepared by method A. The reaction mixture was stopped after 2 h. Yield 2.90 g (92%), TLC Rf 0.60, m.p. 189–190°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.44 (s, 1H, PyrH), 9.21 (d, 1H, J = 8.0 Hz, PyrH), 8.75 (d, 1H, J = 8.0 Hz, PyrH), 8.37 (s, 1H, -CH = NOH), 8.24-8.14 (m, 1H, PyrH), 7.61-7.48 (m, 4H, ArH), 5.93 (s, 2H, -CH2-), 4.71 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 144.42, 143.24, 142.33, 141.96, 139.24, 133.99, 133.74, 129.98, 129.18, 128.49, 62.92, 33.54. Analysis: calculated 43.55% C, 3.65% H, 7.26% N; found 43.53% C, 3.95% H, 7.25% N. ESI-MS: m/z 305.0 [M+] (calculated for [C14H14BrN2O+] 305.03).
1-(4-bromomethylbenzyl)-4-hydroxyiminomethylpyridinium bromide (27)
Prepared by method A. The reaction mixture was stopped after 2 h. Yield 3.05 g (97%), TLC Rf 0.60, m.p. 198–201°C. 1H NMR (300 MHz, DMSO d6): δ (ppm) 9.18 (d, 2H, J = 6.0 Hz, PyrH), 8.43 (s, 1H, -CH = NOH), 8.26 (d, 2H, J = 6.0 Hz, PyrH), 7.52 (d, 4H, J = 1.4 Hz, ArH), 5.86 (s, 2H, -CH2-), 4.71 (s, 2H, -CH2-). 13C NMR (75 MHz, DMSO d6): δ (ppm) 148.73, 145.03, 145.00, 139.16, 134.24, 129.98, 129.04, 124.36, 62.20, 33.54. Analysis: calculated 43.55% C, 3.65% H, 7.26% N; found 43.61% C, 3.94% H, 7.27% N. ESI-MS: m/z 305.0 [M+] (calculated for [C14H14BrN2O+] 305.03).
Biochemistry
In vitro testing of synthesized oximes involved a standard collection of experimental procedures. The 10% rat brain homogenate was used as a source of AChE. The brain homogenate (0.5 mL) was mixed with 20 μL of an isopropanol solution of GA (O-ethyl-N,N-dimethylphosphoramidocyanidate, obtained from the Military facility Brno, 95% purity) or paraoxon (O,O-diethyl-O-(4-nitrophenyl)phosphate, analytical standard 99.2% from Sigma-Aldrich) and distilled water (0.5 mL) to achieved 95% inhibition of AChE. The mixture was incubated at 25°C for 30 min. 2.5 mL of a solution of sodium chloride (3 M) was added to the mixture and adjusted to a volume of 23 mL with distilled water. Finally, 2 mL of a solution of acetylcholine iodide (0.02 M) was added. The enzyme activity (analyzed by potentiometric titration of decomposed acetylcholine iodide) was measured at pH 7.6 and temperature 25°C on an autotitrator RTS 822 (Radiometer, Denmark). The same procedure was repeated with enzyme further subjected to 10 min incubation with an aqueous solution of reactivator (0.2 mL - 10− 3 M or 10− 5 M), which replaced 0.2 mL of water. Activities of intact AChE (a0), inhibited AChE (ai) and reactivated AChE (ar) were deduced from the rate of consumption of NaOH solution (0.01 M) with time. The percentage reactivation (%) was calculated from the measured data according to the formula:
The whole method is described in detail in the work of Kuca and Cabal [Citation22]. Pralidoxime, HI-6 and obidoxime of HPLC purity, previously synthesized in our laboratory, were used as references. The obtained data are summarized in .
Table I. Reactivation potencies of tested oximes (%, mean value of three independent determinations) – time of inhibition – 30 min; time of reactivation by AChE reactivators – 10 min; pH 8; temperature 25°C.
Results and discussion
The reactivation potency of the tested compounds depends not only on the structure of the OP inhibitor Citation2Citation14-16 but also on the reactivator's structure. Moreover, a reactivator suitable for in vivo experiments should exceed 10% reactivation ability in vitro [Citation2]. Therefore, it is extraordinarily difficult to reactivate AChE inhibited by the nerve agent GA Citation23-25. The lone electron pair located on the amidic group makes nucleophilic attack almost impossible [Citation23]. In addition, some conformational changes occur in the cavity of the GA-inhibited enzyme [Citation25]. As can be seen from , obidoxime (3) is able to satisfactorily reactivate GA-inhibited AChE at 10− 3 M. However, the concentration 10− 3 M is not attainable with in vivo experiments [Citation26]. A concentration 10− 5 M is more appropriate and attainable for human use [Citation2]. Only two compounds with a xylene bridge (16, 19) have reactivation ability against GA at a concentration 10− 5 M. None of the non-symmetrical compounds show sufficient reactivation ability against GA-inhibited AChE.
On the other hand, pesticides are known as weaker inhibitors of AChE compared to nerve agents [Citation2,Citation27]. In the past, the design of AChE reactivators was focused on preparation of potent compounds against nerve agents. Formerly, it was determined that commonly used reactivators are not suitable for pesticide intoxications Citation28-29. In contrast to this, all reference compounds showed satisfactory ability in reactivation of paraoxon-inhibited AChE at concentration 10− 3 M, especially obidoxime (3). None of eighteen new compounds was able to exceed obidoxime in potency and only one (15) gave a result comparable with that of reference substances at 10− 3 M. At the 10− 5 M, the situation was changed. Of the reference substances, only obidoxime showed ability to reactivate paraoxon-inhibited AChE. Moreover, seven xylene-linked compounds (7-8, 10-11, 16, 18-19) exceeded the potency of obidoxime at 10− 5 M. In addition, there was the finding of this interesting phenomenon that these compounds showed higher reactivation ability at lower concentration (e.g. 7-8, 10-11, 16, 18-21). This is probably caused by coincident reactivation and inhibition of the enzyme by the reactivator itself as was described earlier Citation15-16.
Consequently, we can recommend the structural factors appropriate for reactivation of paraoxon-inhibited AChE by novel compounds [Citation14]. The oxime functional group breaks down the bond OP inhibitor-enzyme and is essential for activity of the reactivator Citation30-31. The results confirm that the position of hydroxyiminomethyl groups influences the reactivation potency Citation30-33. In this case, compounds bearing oxime groups in positions 3-4 (6, 9, 12) and 3-3 (14, 17, 20) were almost ineffective, compounds with oxime groups in positions 2-3 (7, 10), 2-4 (8, 11), 2-2 (16, 19) and 4-4 (15, 18, 21) showed satisfactory reactivation ability against paraoxon-inhibited AChE. Another important factor for reactivation potency is the structure of the connecting chain as was discussed previously Citation14-16. For the xylene-linked compounds, the less sterically limited connecting bridge (m- and pphenylene; 7-8, 10-11, 16, 18-19) seemed to be better than the o-phenylene chain (4-5, 13-15) for paraoxon-inhibited AChE. Some cation-π interaction probably occurs in the cavity of enzyme but more investigations are required to confirm this hypothesis. No less, the quaternary nitrogen is also important for the good affinity of reactivator to the enzyme Citation34-35. Additionally, the previously obtained data for the pesticide chlorpyrifos confirm our results for paraoxon-inhibited AChE [Citation16].
In conclusion, nine novel non-symmetrical reactivators with xylene linker were prepared in satisfactory yield and purity. Ability of these nine compounds together with nine symmetrical ones to reactivate GA and paraoxon-inhibited AChE was measured in vitro. Seven compounds were found to be promising against paraoxon-inhibited AChE at physiological concentration. Only two symmetrical reactivators exceed the potency of obidoxime against GA. Although the non-symmetrical compounds were less potent than the symmetrical ones, they confirm the dominance of the sterically less m- and p-xylene connecting chain in reactivation of paraoxon-inhibited AChE.
Acknowledgements
The authors express their appreciation to Mrs M. Hrabinova for her technical assistance. The work was supported by a grant from the Grant Agency of Charles University No. 302/2005/B-CH/FaF and by a grant from the Ministry of Defence of Czech Republic No. FVZ0000501.
References
- Marrs TC. Organophosphate poisoning. Pharmacol Therapeut 1993; 58: 51–66
- Bajgar J. Organophosphates/nerve agent poisoning: Mechanism of action, diagnosis, prophylaxis, and treatment. Adv Clin Chem 2004; 38: 151–216
- Marklund A, Andersson B, Haglund P. Organophosphorus flame retardants and plasticizers in air from various indoor environments. J Environ Monit 2005; 7: 814–819
- Ringman JM, Cummings JL. Metrifonate (Trichlorfon): A review of the pharmacology, pharmacokinetics and clinical experience with a new acetylcholinesterase inhibitor for Alzheimer's disease. Opin Invest Drugs 1999; 8: 463–471
- Satoh T, Hosokawa M. Organophosphates and their impact on the global environment. Neurotoxicol 2000; 21: 223–227
- Krivoy A, Layish I, Rotman E, Goldberg A, Yehezkelli Y. OP or not OP: the medical challenge at the chemical terrorism scene. Prehospital Disaster Med 2005; 20: 155–158
- Kassa J. The influence of anticholinergic drug and oxime selection on the effectiveness of antidotal treatment against tabun-induced poisoning in mice. Acta Medica 2002; 45: 75–78
- Petroianu GA, Hasan MY, Arafat K, Nurulain SM, Schmitt A. Weak inhibitors protect cholinesterases from strong inhibitors (paraoxon): In vitro effect of tiapride. J Appl Toxicol 2005; 25: 562–567
- Rousseaux CG, Dua AK. Pharmacology of HI-6, an H-series oxime. Can J Physiol Pharmacol 1989; 67: 1183–1189
- Kassa J, Cabal J, Bajgar J, Szinicz L. The choice: HI-6,. pralidoxime or obidoxime against nerve agents?. ASA Newslett 1997; 97: 16–18
- Sevelova L, Kuca K, Krejcova-Kunesova G. Antidotal treatment of GF-agent intoxication in mice with bispyridinium oximes. Toxicol 2005; 207: 1–6
- Kuca K, Kassa J. A comparison of the ability of new bispyridinium oxime 1-(4-hydroxyiminomethylpyridinium)-4-(4-carbamoylpyridinium)butane dibromide and currently used oximes to reactivate nerve agent-inhibited rat brain acetylcholinesterase by in vivo methods. J Enz Inhib Med Chem 2003; 18: 529–535
- Kuca K, Patocka J. Reactivation of cyclosarin-inhibited rat brain acetylcholinesterase by pyridinium-oximes. J Enz Inhib Med Chem 2004; 19: 39–43
- Kuca K, Jun D, Musilek K. Mini-Rev Med Chem 2006; 6: 269
- Musilek K, Kuca K, Jun D, Dohnal V, Dolezal M. Synthesis of the novel series of bispyridinium compounds bearing (E)-but-2-ene linker and evaluation of their reactivation activity against chlorpyrifos-inhibited acetylcholinesterase. Bioorg Med Chem Lett 2006; 16: 622–627
- Musilek K, Kuca K, Jun D, Dohnal V, Dolezal M. Synthesis of a novel series of bispyridinium compounds bearing a xylene linker and evaluation of their reactivation activity against chlorpyrifos-inhibited acetylcholinesterase. J Enz Inhib Med Chem 2005; 20: 409–415
- Manetsch R, Krasinski A, Radic Z, Raushel J, Taylor P, Sharpless KB, Kolb HC. In situ click chemistry: Enzyme inhibitors made to their own specifications. J Am Chem Soc 2004; 126: 12809–12818
- Radic Z, Manetsch R, Krasinski A, Raushel J, Yamauchi J, Garcia C, Kolb H, Sharpless KB, Taylor P. Molecular basis of interactions of cholinesterases with tight binding inhibitors. Chem Biol Interact 2005; 157–158: 133–141
- Patocka J, Cabal J, Kuca K, Jun D. Oxime reactivation of acetylcholinesterase inhibited by toxic phosporus ester: In vitro kinetics and thermodynamics. J Appl Biomed 2005; 3: 91–99
- Kuca K, Cabal J, Kassa J. A comparison of the potency of newly developed oximes (K005, K027, K033, K048) and currently used oximes (pralidoxime, obidoxime, HI-6) to reactivate sarin-inhibited rat brain acetylcholinesterase by in vitro methods. J Toxicol Env Health A 2005; 68: 677–686
- Kuca K, Cabal J, Jun D, Kassa J, Bartosova L, Kunesova G. In vitro reactivation potency of some acetylcholinesterase reactivators against sarin- and cyclosarin-induced inhibitions. J Appl Toxicol 2005; 25: 296–300
- Kuca K, Cabal J. Evaluation of newly synthesized reactivators of the brain cholinesterase inhibited by sarin nerve agent. Toxicol Mech Method 2005; 15: 247–252
- Bajgar J, Patocka J. In vitro inhibition of soluble brain acetylcholinesterase by organophosphates of the O-ethyl-S-(dialkylaminoethyl)-methylphosphonothiolate type. Coll Czech Chem Commun 1977; 42: 770–776
- Cabal J, Kuca K, Kassa J. Specification of the structure of oximes able to reactivate tabun-inhibited acetylcholinesterase. Basic Clin Pharmacol Toxicol 2004; 95: 81–86
- Ekstrom F, Akfur C, Tunemalm AK, Lundberg S. Biochemistry 2006; 45: 74–81
- Tattersall JE. Ion channel blockade by oximes and recovery of diaphragm muscle from soman poisoning in vitro. Br J Pharmacol 1993; 108: 1006–1015
- Kim TH, Kuca K, Jun D, Jung YS. Design and synthesis of new bis-pyridinium oxime reactivators for acetylcholinesterase inhibited by organophosphorous nerve agents. Bioorg Med Chem Lett 2005; 15: 2914–2917
- Thiermann H, Szinicz L, Eyer F, Worek F, Eyer P, Felgenhauer N, Zilker T. Modern strategies in therapy of organophosphate poisoning. Toxicol Lett 1999; 107: 233–239
- Worek F, Thiermann H, Szinicz L, Eyer P. Kinetic analysis of interactions between human acetylcholinesterase, structurally different organophosphorus compounds and oximes. Biochem Pharmacol 2004; 68: 2237–2248
- Kuca K, Bielavsky J, Cabal J, Kassa J. Synthesis of a new reactivator of tabun-inhibited acetylcholinesterase. Bioorg Med Chem Lett 2003; 13: 3545–3547
- Pang YP, Kollmeyer TM, Hong F, Lee JC, Hammond PI, Haugabouk SP, Brimijoin S. Rational design of alkylene-linked bis-pyridiniumaldoximes as improved acetylcholinesterase reactivators. Chem Biol 2003; 10: 491–502
- Chennamaneni SR, Vobalaboina V, Garlapati A. Quaternary salts of 4,3′ and 4,4′ bis-pyridinium monooximes: Synthesis and biological activity. Bioorg Med Chem Lett 2005; 15: 3076–3080
- Srinivas Rao C, Venkateswarlu V, Achaiah G. Quaternary salts of 4,3′ and 4,4′ bis-pyridinium monooximes. Part 2: Synthesis and biological activity. Bioorg Med Chem Lett 2006; 2134–2138
- Kenley RA, Howd RA, Mosher CW, Winterle JS. Nonquaternary cholinesterase reactivators. Dialkylaminoalkyl thioesters of alpha-ketothiohydroximic acids as reactivators of diisopropyl phosphorofluoridate inhibited acetylcholinesterase. J Med Chem 1981; 24: 1124–1133
- Kenley RA, Bedford CD, Howd RA, Jackson SE. Reactivation of ethyl methylphosphonylated eel acetylcholinesterase in vitro by 2PAM, H16, and a series of nonquaternary alpha-ketothiohydroximates. Biochem Pharmacol 1985; 34: 3606–3608