Abstract
This study is part of a long term project designed to explore the hypothesis that stimulation of cancer cells followed by treatment with one or more cytotoxic agents may create greater damage to tumours than to the corresponding normal tissues. The aim of the present investigation was to discover various compounds which stimulate a protein tyrosine kinase, namely fyn kinase. The N-acyl-3,5-bis(arylidene)-4-piperidones and related analogues activated this enzyme using concentrations of 25 μM while representative molecules achieved this result at 0.1 μM. Molecular modelling suggested that the compounds interact transiently with the ATP binding site of fyn kinase thereby enhancing the catalytic phosphorylation of proteins. In the future, candidate antineoplastic agents will be designed which incorporate the structural features of these enzyme stimulators with the goal of their being formed in vitro and in vivo prior to the release of cytotoxins.
Introduction
A number of different strategies have been employed in the design of novel antineoplastic agents. One of these approaches entitled sequential cytotoxicity, which originated from this laboratory, is the concept that successive chemical insults may be more detrimental to malignant cells than the corresponding normal tissues. The rationale behind this theory as well as various clusters of compounds prepared to examine this hypothesis have been described[Citation1,Citation2]. In this scenario, the first interaction with cellular constituents leads to chemosensitization to further drug-induced toxicity preferentially in the tumours compared to related non-cancerous cells. A development of this concept which may result in preferential chemosensitization in malignant cells by sequential interactions with cellular constituents is as follows. In this approach, the initial response is stimulation of one or more enzymes associated with cellular proliferation, leading to increased rate of growth thereby causing the tumour to be more susceptible to an antineoplastic agent than non-malignant cells.
This hypothesis of chemosensitivity by enzyme stimulation is based on the observation that certain tumour cells divide more rapidly than the corresponding normal cells and are therefore more susceptible to anticancer drugs. For example, the antineoplastic alkylating agents cause most damage to rapidly proliferating cells[Citation3]. Chemosensitivity would be predicted to be enhanced in those cases where enzyme stimulation and subsequently growth rates are greater in malignant rather than non-neoplastic cells.
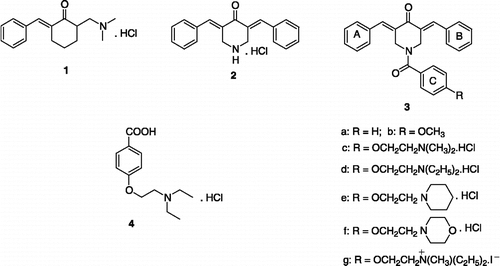
In order to evaluate this latter hypothesis, namely that selective toxicity to malignant cells may arise by chemosensitization due to enzyme stimulation, the choice of an appropriate enzyme and the discovery of various compounds which stimulate this enzyme were addressed initially and these considerations form the substance of this report. The regulation of cellular proliferation is controlled to a large extent by protein kinases which phosphorylate proteins[Citation4]. In particular, enhanced activity of protein tyrosine kinase (PTK) enzymes, which catalyze the transfer of the phosphoryl group of adenosine triphosphate (ATP) to the tyrosine residues of the target proteins[Citation5], have been noted in certain cancers[Citation6]. The PTK chosen was fyn kinase since a previous study from this laboratory showed that the Mannich base 1 stimulated this enzyme[Citation7]. However murine toxicity was noted with 1[Citation7] and other acyclic Mannich bases (3-aminoketones)[Citation8,Citation9], suggesting that pursuit of this concept should proceed using less toxic analogues. On the other hand 2, which is a 4-piperidone (a cyclic 3-aminoketone) and contains arylmethylene groups, as well as related N-acyl analogues, are well tolerated in mice[Citation10,Citation11]. An initial exploration revealed that 2 stimulated fyn kinase vide infra. While this result was encouraging, the percentage stimulation was only modest. Thus molecular modification of 2 was planned with a view of forming compounds having additional capacities to interact with polar and nonpolar groups of the enzyme. The attachment of an aroyl group on the piperidyl nitrogen atom of 2 leading to 3a should allow van der Waals bonding between ring C and amino acids containing aryl groups. In addition, the presence of various functional groups attached to ring C having differing polar and nonpolar groups may enhance interactions with complementary portions on the enzyme. Such considerations led to the decision to prepare 3b-g as candidate stimulators of fyn kinase. The structures of 1-3 as well as the related acid 4 vide infra are presented in figure 1.
In summary, the initial objective of this study was to determine novel compounds which would stimulate fyn kinase. In the future such structures would be incorporated into more complex molecules whereby stimulation of fyn kinase would take place initially and a cytotoxic species would be released subsequently.
Figure 1 Structures of compounds 1-4.
Materials and methods
Synthesis of compounds 2-4
The synthesis of 2[Citation12] and 4[Citation13] have been reported previously. Condensation of the free base of 2 with the acid chloride of 4 led to the formation of 3d. The related compounds 3a-c,e,f were prepared in a similar fashion from the free base of 2 using the appropriate acid chloride while quaternization of 3d with methyl iodide produced 3g. The syntheses of the compounds in series 3 will be presented elsewhere.
Fyn kinase assay
The fyn kinase expressed plasmid was a gift from Dr C. J. Pallen, University of British Columbia, Canada. The expression and purification of fyn kinase (pGEX-KG-fyn) was accomplished using a reported methodology[Citation14]. In brief, E. coli BL21 with the fyn kinase was grown in LB medium at 37°C to the stationery phase. The protein was purified and mixed with glycerol (20% w/v) and stored at − 80°C until required for use. The kinase assay was undertaken at 37°C using a literature procedure[Citation15]. In brief, the reaction mixture contained fyn kinase and a synthetic peptide which had been prepared by the Alberta Peptide Institute. This peptide (KVEK1GEGTYGVVKK) corresponded in sequence to residues 6–20 of cdc 2. After 30 min., the reaction was terminated by spotting onto phosphocellulose filter paper (Whatman P81) and washed as described previously[Citation16]. Radioactivity was measured using a Beckman Ready Safe Liquid Scintillation mixture in a Beckman Liquid Scintillation Counter.
Molecular modelling
Molecular model building, dynamic simulations, energy minimizations and docking for fyn kinase and the enzyme complexes with 2, 3c and 3d were carried out using Insight II modelling software (Accelrys Inc., San Diego, CA) on a Silicon Graphics Octane 2 workstation[Citation17]. The secondary structural predictions and homology alignments were performed using Homology and Jpred. The initial alignment was undertaken with five structures of which two to three were used to employ a model. In total six initial models were created which was followed by a structural check of each molecule using Accelrys Homology ProStat Struct_Check of bond lengths and bond angles with a normalized standard deviation cutoff of 3. Three of these molecules were carried forward and used in the docking simulations. Src structures were used as the starting structural template. Molecular models for fyn kinase were constructed using non-complexed human c-src (PDB # 1FMK). Structural refinements to the fyn kinase 413-426 loop region which was undefined in PDB # 1FMK (c-src 410-423) was performed using the structure of csrc 410-423 from PDB #2SRC (c-src in complex with adenylimidodiphosphate (AMP-PNP). AMP-PNP is a nonhydrolysable analogue of ATP in which the oxygen atom connecting the β and γ phosphate groups has been replaced by a nitrogen atom. Docking of the various compounds to fyn kinase were performed using the coordinates AMP-PNP (from PDB # 2SRC) and that of a selective tyrosine kinase inhibitor (from PDB # 1QFC) as a starting reference point. All docked structures were subjected to dynamic simulations and energy minimizations to obtain the final structures. The model validation data as well as the methods for dynamics and docking were carried out using the Accelrys Homology and Docking and Discovery 3 programmes.
Results
The evaluation of concentrations of 25, 50 and 100 μM of 2, 3b-g and 4 towards fyn kinase are presented in . The effect of lower concentrations of 2, 3a,b and 4 towards this enzyme are summarized in . Molecular modelling of the interactions of three representative compounds 2, 3c and 3d as the free bases with fyn kinase utilized various Insight II modules. The interaction of 3c with fyn kinase is indicated in .
Table I. Stimulation of fyn kinase by 2, 3b-g and 4.
Table II. Effects of low concentrations of 2, 3a,b and 4 on fyn kinase.
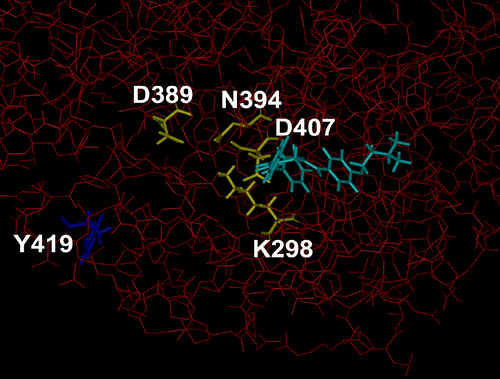
Figure 2 Structure of part of fyn kinase in complex with 3c. The heavy atoms of fyn 60-536 are coloured red and displayed as thin sticks. The residues in fyn that participate in coordinating the magnesium ion and the α- and β-phosphates of ATP (fyn K298, N394, and D407) and the proton acceptor site (fyn D389) are coloured yellow and displayed as thick sticks. The autocatalytic phosphorylation site in fyn, Y419, is coloured dark blue. The docked compound 3c is coloured light-blue. The docked compounds do not directly interact with the proton acceptor site (fyn D389) nor the residues coordinating the phosphate-magnesium ion (side chain atoms of fyn K298, N394, and D407), rather they interact with the residues coordinating the adenosine moiety of ATP, and include backbone atoms of K298 andD407. Thus, the presence of the compounds may alter the orientation of the side chains of K298 and D407 in fyn and thereby effect ATP binding. (Please see colour online)
Discussion
The concentration of 1 which causes a 50% increase in the stimulation of fyn kinase is 18.2 μM[Citation7]. Consequently 2 and 3b-g were examined initially at approximately the same concentration, namely 25 μM; solubility problems prevented the use of this concentration of 3a. The results are summarized in and the following observations were noted. First, all of these compounds stimulate the enzyme. Second, conversion of 2 into 3b-g increases the percentage stimulation approximately twofold (3b,f), threefold (3d,e) and sevenfold (3c,g). This enhancement of activity may be attributed to the interactions of the N-acyl groups with this enzyme. In order to evaluate this possibility, compound 4, which contains most of the structural features of the N-acyl group of 3d, was examined and as the data in reveal, it has a markedly stimulant effect on fyn kinase.
The concentrations of 2, 3b-g and 4 were increased to 50 and 100 μM in order to determine whether the magnitude of stimulation correlated with the quantity of the compound employed. The data in reveal that the activity of 2 was the same at all three concentrations. However in the case of 3b-g and 4, the activity of the enzyme was lower at 100 μM than 25 μM. It is possible that as the concentration rises, an enzyme-inhibitory effect coexists with the stimulation process.
The effects of lower concentrations of four representative compounds 2, 3a,b and 4 on the activity of fyn kinase was undertaken and the results are presented in . At concentrations of 0.1 μM and greater, 2, 3a,b and 4 caused stimulation of the enzyme. In the case of 3a, stimulation occurred at even lower concentrations and, in particular, the increase in activity of 56% at 0.1 nM is noteworthy. On the other hand, concentrations below 0.1 μM of 2, 3b and 4 caused some inhibition of the enzyme. For these compounds, it is possible that there are two mechanisms of inhibition, namely one binding site which is saturated by the ligand at low concentrations while another site requires substantially higher concentrations such as 100 μM in order to inhibit the enzyme.
The conclusion to be drawn from the data summarized in Tables and is that concentrations of 0.1-100 μM of 2, 3b-g and 4 stimulate fyn kinase. A number of cytotoxic drugs such as melphalan and 5-fluorouracil as well as the thiol-alkylater helenalin exert their antineoplastic effects in this concentration range[Citation18]. Stimulation of the enzyme is demonstrated consistently by 3a at even lower concentrations. Thus the structural features of these compounds may be incorporated into future candidate drug molecules in order to evaluate this sequential stimulation-cytotoxicity concept.
Finally, molecular modelling was employed in order to obtain an insight into the possible manner whereby fyn kinase was stimulated by various ligands. This enzyme is composed of a SH2, SH3 and a kinase domain. While the structures of the SH2 and SH3 domains have been solved[Citation19], the structure of the kinase domain has not been described. However fyn kinase and src share a 77.1% identity in 454 overlapping amino acids (jpred alignment) and high resolution structures of src are available[Citation20]. Hence src structures were used as the starting structural template and developed subsequently as described in the experimental section of this report vide supra. In the case of fyn kinase, the initial reaction involves phosphorylation of the tyrosine residue Y419 by ATP. The resultant negative charge on the phosphate portion of this acylated amino acid induces a conformational charge resulting in the proper three dimensional presentation of four amino acids D389, D407, K298 and N394 which comprise the active kinase binding site (site 1). Subsequently the tyrosine portion of certain proteins is phosphorylated at site 1. indicates various amino acid portions of fyn kinase. Compounds 2, 3c and 3d were docked onto the enzyme and bound at site 1 in a similar manner as illustrated for 3c in . Thus stimulation of fyn kinase is likely due to a transient interaction of the 4-piperidones at site 1 causing the enzyme to adopt a conformation which allows phosphorylation of various proteins to occur. A number of residues which surround site 1 and interact with the compounds are hydrophobic in nature and include V284, M286, A296, Y343 and L396. Thus, the introduction of lipophilic groups onto the arylidene aryl rings in the compounds in series 2-4 may enhance van der Waals bonding with these hydrophobic amino acids and enhance enzyme activity.
Conclusions
This study has led to the identification of a series of N-acyl-3,5-bis(phenylmethylene)-4-piperidones which stimulate fyn kinase. The modelling studies led to a concept of how this activation of the enzyme is achieved. The way in which the compounds affect the rate of phosphorylation of proteins will be investigated in order to evaluate the molecular modelling study. In addition, two directions of structural modification of series 3 and 4 will be pursued. First, the attachment of representative molecules to a masked cytotoxin via a labile linker will be undertaken. Ideally the liberation of the enzyme stimulator will occur initially followed by the release of a cytotoxic agent. Secondly, the preparation of a number of analogues of series 3 and 4 will be designed in an attempt to increase the activation of fyn kinase.
Acknowledgements
The authors thank the Canadian Institutes of Health Research for operating grants to R. K. Sharma and J. R. Dimmock while appreciation is extended to the Canadian Foundation for Innovation who awarded a grant to T. A. Haas. Ms B. McCullough is thanked for typing various drafts of this paper.
References
- Dimmock JR, Sidhu KK, Chen M, Reid RS, Allen TM, Kao GY, Truitt GA. Evaluation of some Mannich bases of cycloalkanones and related compounds for cytotoxic activity. Eur J Med Chem 1993; 28: 313–322
- Dimmock JR, Kandepu NM, Nazarali AJ, Motaganahalli NL, Kowalchuk TP, Pugazhenthi U, Prisciak JS, Quail JW, Allen TM, LeClerc R, Santos CL, De Clercq E, Balzarini J. Sequential cytotoxicity: A theory evaluated using novel 2-[4-(3-aryl-2-propenoyloxy)phenylmethylene]cyclohexanones and related compounds. J Med Chem 2000; 43: 3933–3940
- Remers WA. Antineoplastic agents. Wilson and Gisvold's textbook of organic medicinal and pharmaceutical chemistry10th ed., JN Delgado, WA Remers. Lippincott-Raven, Philadelphia 1998; 353
- Aronov AM, Murcko MA. Toward a pharmacophore for kinase frequent hitters. J Med Chem 2004; 47: 5616–5619
- Clark DD, Peterson BR. Analysis of protein tyrosine kinase inhibitors in recombinant yeast lacking the ERG6 gene. Chem Bio Chem 2003; 4: 101–107
- Levitzki A, Gazit A. Tyrosine kinase inhibition: An approach to drug development. Science 1995; 267: 1782–1788
- Dimmock JR, Chamankah M, Das U, Zello GA, Quail JW, Yang J, Nienaber KH, Sharma RK, Selvakumar P, Balzarini J, De Clercq E, Stables JP. Cytotoxic and topographical properties of 6-arylidene-2-dimethylaminomethylcyclohexanone hydrochlorides and related compounds. J Enz Inhib Med Chem 2004; 19: 1–10
- Dimmock JR, Patil SA, Shyam K. Evaluation of some Mannich bases of 1-aryl-1-ethanones and related ketones for anticonvulsant activities. Pharmazie 1991; 46: 538–539
- Dimmock JR, Taylor WG. Evaluation of nuclear-substituted styryl ketones and related compounds for antitumor and cytotoxic properties. J Pharm Sci 1975; 64: 241–249
- Dimmock JR, Jha A, Zello GA, Sharma RK, Shrivastav A, Selvakumar P, Allen TM, Santos CL, Balzarini J, De Clercq E, Manavathu EK, Stables JP. 3,5-Bis(phenylmethylene)-1-(N-arylmaleamoyl)-4-piperidones: A novel group of cytotoxic agents. J Enz Inhib Med Chem 2003; 18: 325–332
- Dimmock JR, Jha A, Zello GA, Quail JW, Oloo EO, Nienaber KH, Kowalczyk ES, Allen TM, Santos CL, De Clercq E, Balzarini J, Manavathu EK, Stables JP. Cytotoxic N-[4-(3-aryl-3-oxo-1-propenyl)phenylcarbonyl]-3,5-bis(phenylmethylene)-4-piperidones and related compounds. Eur J Med Chem 2002; 37: 961–972
- Dimmock JR, Padmanilayam MP, Puthucode RN, Nazarali AJ, Motaganahalli NL, Zello GA, Quail JW, Oloo EO, Kraatz H-B, Prisciak JS, Allen TM, Santos CL, Balzarini J, De Clercq E, Manavathu EK. A conformational and structure-activity relationship study of cytotoxic 3,5-bis-arylidene-4-piperidones and related N-acryloyl analogues. J Med Chem 2001; 44: 586–593
- Jones CD, Jevnikar AJ, Pike AJ, Peters MK, Black LJ, Thompson AR, Falcone JF, Clemens JA. Antiestrogens.2. Structure-activity studies in a series of 3-aroyl-2-arylbenzo[b] thiophene derivatives leading to [6-hydroxy-2-(4-hydroxyphenyl)benzo[b] thien-3-yl]-[4-[2-(1-piperidinyl)ethoxy]phenyl]methanone hydrochloride (LY 156758), a remarkably effective estrogen antagonist with only minimal intrinsic estrogenicity. J Med Chem 1984; 27: 1057–1066
- Bhanderi V, Kim KL, Pallen CJ. Physical and functional interactions between receptor-like protein-tyrosine phosphatase and p59fyn. J Biol Chem 1998; 273: 8691–8698
- Cheng HC, Nishio H, Hatase O, Ralph S, Wang JH. A synthetic peptide derived from p34cdc2 is a specific and efficient substrate of src-family tyrosine kinases. J Biol Chem 1992; 267: 9248–9256
- Sharma RK. Assay and purification of calmodulin-dependent protein kinase. Calcium signaling protocols: Methods in molecular biology, DG Lambert. Humana Press, Totowa, New Jersey 2006; Vol 312: 305–324
- Haas TA, Plow EF. Development of a structural model for the cytoplasmic domain of an integrin. Protein Eng 1997; 10: 1395–1405
- Dimmock JR, Kumar P, Nazarali AJ, Motaganahalli NL, Kowalchuk TP, Beazely MA, Quail JW, Oloo EO, Allen TM, Szydlowski J, De Clercq E, Balzarini J. Cytotoxic 2,6-bis(arylidene)cyclohexanones and related compounds. Eur J Med Chem 2000; 35: 967–977
- Arold ST, Ulmer TS, Mulhern TD, Werner JM, Ladbury JE, Campbell ID, Noble ME. The role of src homology 3-Src homology 2 interface in the regulation of Src kinases. J Biol Chem 2001; 276: 17199–17205
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase 6-Src. Nature 1997; 385: 595–602