Abstract
The kinetic and thermodynamic effects of aspirin and diclofenac on the activity of adenosine deaminase (ADA) were studied in 50 mM phosphate buffer pH = 7.5 at 27 and 37°C, using UV-Vis spectrophotometry and isothermal titration calorimetry (ITC). Aspirin exhibits competitive inhibition at 27 and 37°C and the inhibition constants are 42.8 and 96.8 μM respectively, using spectrophotometry. Diclofenac shows competitive behavior at 27°C and uncompetitive at 37°C with inhibition constants of 56.4 and 30.0 μM, at respectively. The binding constant and enthalpy of binding, at 27°C are 45 μM, − 64.5 kJ/mol and 61 μM, − 34.5 kJ/mol for aspirin and diclofenac. Thermodynamic data revealed that the binding process for these ADA inhibitors is enthalpy driven. QSAR studies by principal component analysis implemented in SPSS show that the large, polar, planar, and aromatic nucleoside and small, aromatic and polar non-nucleoside molecules have lower inhibition constants.
Introduction
Adenosine deaminase (E.C.3.5.4.4) is a monomeric protein (34.5 kDa), which catalyzes the deamination of adenosine and 2′-deoxyadenosine nucleosides to their respective inosine derivative nucleosides and ammonia with a rate enhancement of 2 × 1012 relative to the non-enzymatic reaction [Citation1]. This catalysis requires a Zn2?+ cofactor [Citation2].
The enzyme is present virtually in all human tissues, but the highest levels are found in the lymphoid system such as lymph nodes, spleen, and thymus Citation3-5. Higher levels of ADA in the alimentary tract and decidual cells of the developing fetal-maternal interface put ADA among those enzymes performing unique roles related to the growth rate of cells, embryo implantation, and other undetermined functions [Citation6,Citation7]. ADA is widely distributed in the brain, and one important function of this enzyme is probably associated with regulation of the extracellular level of adenosine and 2′-deoxyadenosine in contact with cerebral blood vessels. The inhibition of adenosine deaminase in the brain would allow for an accumulation of adenosine, which would produce vasodilatation and increase in cerebral blood flow [Citation8].
In recent years, adenosine has come to be considered as an important factor in the attenuation of inflammation [Citation9,Citation10] since it has been reported that the concentration of adenosine is increased in inflammatory lesions [Citation11,Citation12]. Furthermore, it is also believed that ecto-ADA has an extra enzymatic function via binding with CD26 on the surface of activated lymphocytes [Citation13] and could perpetuate chronic inflammation by metabolizing adenosine released at inflamed sites that are toxic for lymphocytes [Citation14]. Therefore, it is considered that an ADA inhibitor may prevent adenosine released specifically at inflamed sites from metabolism by ecto-ADA and would have great potential as an anti-inflammatory drug with few side effects.
A number of ADA inhibitors have been reported in the literature. However, almost all are purine nucleoside or alkyladenine analogues, for example, (+)-erythro-9- (2-hydroxy-3-nonyl) adenine ((+)-EHNA) (ground-state inhibitor)[Citation15], pentostatin (transition-state inhibitor)[Citation16], and various derivatives [Citation17]. As a result, they have many problems, such as poor pharmacokinetics [Citation18,Citation19] and/or several toxicities [Citation20,Citation21]. Because of these problems, pentostatin is the only ADA inhibitor in clinical use; however, it is limited to the treatment of adult patients with hairy cell leukemia and is only available via intravenous administration [Citation20,Citation21]. ADA inhibitors with reduced toxicity and oral bioavailability would be expected to not only improve the treatment of leukemia but also have potential use as anti-inflammatory drugs.
Non-steroid anti-inflammatory drugs (NSAIDs) such as aspirin and diclofenac are major drugs against inflammation and pain. Aspirin uncouples oxidative phosphorylation leading to ATP catabolism, and pharmacologically relevant concentrations of sodium salicylate diminish intracellular ATP concentrations in vitro, thereby releasing micromolar amounts of adenosine, an autacoids with potent anti-inflammatory properties into extracellular fluids. Aspirin is an anti-inflammatory pain killer (NSAID), which is extensively used, worldwide, for pain relief, to reduce inflammation and pyrexin by affecting the prostaglandins, and to reduce the risk of heart attacks and strokes. It is widely used to prevent heart attacks and strokes [Citation22].
Diclofenac is also a commonly used non-steroidal anti-inflammatory drug. It acts by reducing hormones that cause inflammation and pain and stiffness caused by many conditions, such as osteoarthritis, rheumatoid arthritis, abdominal cramps associated with menstruation, and ankylosing spondylitis [Citation23].
The effect of inosine [Citation24], caffeine [Citation25], theophyline [Citation26], acetaminophen [Citation27] and theobromine [Citation28] as inhibitors on ADA activity has been studied by calorimetery and spectroscopy. The enthalpy, equilibrium and inhibition constants for binding were obtained [Citation29,Citation30].
Understanding the interaction of ADA with its inhibitors and substrates at the molecular level will be important in the development of the next generation of pharmaceutical agents that acts as inhibitors or substrates. X-ray crystallography [Citation31,Citation32] and molecular dynamics [Citation33] were analyzed at the molecular level for the ADA-inhibitor complex. Binding sites for some of inhibitors and the effect of their structure on the enzyme were investigated.
It is well known that, drug design is dependent on the structure of the drug, inter- and intra-molecular interactions between drug and receptor. X-ray crystallography, NMR and computational methods give the most information about the three-dimensional structure and molecular interactions but these methods are relatively expensive.
On the other hand, performing QSAR analysis for several series of drugs, enzyme and biological active compounds is now well appreciated. It is less expensive and a simple method relative to the above cited methods. Kinetic parameters were used for the QSAR analysis and as such, we found some theoretical descriptors which correlated the binding affinity of ADA towards several adenine nucleosides as inhibitors. The kinetic parameters of adenosine deaminase such as Km and KI were determined in the absence and presence of adenine derivatives as well as the QSAR of these derivatives was studied. QSAR analysis revealed that the binding affinity of the adenine nucleoside upon interaction with ADA depends on the molecular volume, dipole moment of the molecule, electric charge, and the highest positive charge for the related molecules [Citation34].
In line with that proposed research, we investigated the effect of aspirin and diclofenac on ADA using spectrophotometry and isothermal titration calorimetry. Thermodynamic parameters such as enthalpy, entropy, Gibbs free energy and the inhibition constant (KI) were obtained. Using other data sets from literature, correlation between KI for 20 inhibitors and 25 descriptors were investigated by multiple linear regression (MLR) and principal component analysis (PCA).
Materials and methods
Materials
Adenosine deaminase (type IV, from calf intestinal mucosa), were obtained from Roche. Aspirin and diclofenac from Sigma, adenosine and other related chemicals, of the highest grade, were obtained from Merck. The solutions were prepared in doubly distilled water.
Methods
Enzyme assay
Enzymatic activities were assayed by UV-Vis spectrophotometry with a GBC 916 spectrophotometer, by following the decrease in absorbance at 265 nm resulting from the conversion of adenosine to inosine based on the Kaplan method [Citation35]. This method uses the change in the absorbance coefficient of adenosine (ϵ = 8400 M− 1cm− 1) on conversion to inosine by the catalytic activity of the enzyme. The concentration of the enzyme in the assay mixture (50 mM sodium phosphate buffer, pH 7.5) was 0.94 nM with a final volume of 2 ml. Activities were measured using at least seven different concentrations of adenosine and the assays were performed at least in triplicate. The adenosine concentration range used was between 0.25–2.5 times that of the Km. Care was taken to use experimental conditions where the enzyme reaction was linear during the first minutes of the reaction.
Isothermal titration calorimetry (ITC)
Isothermal titration microcalorimetric experiments were performed with a 4-channel commercial microcalorimetric system, Thermal activity monitor 2277 (Thermometric, Sweden). The inhibitor solution (1.5 mM) was injected, using a Hamilton syringe, into a stirred calorimetric titration vessel, which contained 2 mL of enzyme solution. The injection of 50 μL inhibitor solution into the perfusion vessel was repeated 20 times. The heat of dilution of the drug solution was measured as described above except that the buffer solution was injected into the protein solution in the sample cell. The enthalpies of dilution for the drug and protein solutions were subtracted from the enthalpy of the ADA-aspirin interaction as previously reported Citation24-28. The molecular weight of ADA was taken to be 34 500 [Citation36].
Computational methods
A Data set was made for the literature selected compounds Citation24-33 and this work. QSAR analysis was performed on the cited compounds with regarding to the following steps 1) entry of the molecular structures into adequate software to perform the structural optimization, 2) generation of the molecular descriptors, 3) statistical analysis through the multiple linear regression (MLR) and principal component analysis, 4) defining of the model validation.
In the first step, the molecular structure of different nucleoside and non-nucleoside inhibitors was constructed and the three-dimensional structure optimized by the PM3 semi-empirical method in the Hyperchem–7.0 medium. In the second step, quantum mechanical descriptors were generated using the Hyperchem-7.0 and Dragon-3.0 programs [Citation37,Citation38]. Dragon calculates 1497 descriptors in 18 classes such as topological, geometrical, empirical, constitutional, charge and so on. The third step, was variable selection which was performed by the MLR method based on the construction of a linear mathematical model with regard to the observed inhibition constants. The final linear equation was formed by stepwise selection of terms. Some of descriptors may be correlated to each other. For the prevention of repeating descriptors and classifying them, we have used principal component analysis (PCA) [Citation39]. PCA involves a mathematical procedure that transforms a number of (possibly) correlated variables into a (smaller) number of uncorrelated variables called principal components (PCs). Objectives of principal component analysis are to, 1) Discover or reduce the dimensionality of the data set. 2) Identify new meaningful underlying variables.
This method reduces the descriptors into a few factors. Each factor describes a property same as geometrical, topological, electronic and lipophilic properties. These factors can be considered as a new more overall descriptor that includes previous descriptors. We also performed MLR analysis on these factors and obtained a general equation between biological activity and common properties or factors.
In the fourth step, model validation was defined through randomly dividing the data-set into two groups, a training set and a prediction set was used for model generation and a prediction set for evaluation of the model. Generally, the good linear agreement between the predicted and experimental values can be used as an adequate parameter for testing the validity of the model. It should be noted that, in our statistical analysis the total number of selected compounds was 20. In the literature, there are many QSAR reports where a smaller set of compounds is utilized for statistical studies Citation40-42. So, it can be deduced that here, an acceptable accuracy existed for the accomplished statistical work. In QSAR analysis it has been commonly accepted that the data set should contain five times as many compounds as the descriptors. The reason for this is that too few compounds relative to the number of descriptors will give a falsely high correlation. This aspect has also been considered in our QSAR study.
Results and discussion
shows a double reciprocal Lineweaver-Burk plots for ADA at different fixed concentrations of aspirin and diclofenac, in phosphate buffer, pH = 7.5 at 27°C and 37°C. The Km values of the enzyme obtained were 39.0 and 61.8 μM at 27 and 37°C. The values of Vmax are unchanged by aspirin at 27 and 37°C, and diclofenac at 27°C but the apparent Michaelis constant (K'm or Km,app) values are increased, confirming the competitive inhibition of these drugs on ADA at 27°C. On the other hand, at 37°C diclofenac shows uncompetitive inhibition because Vmax has changed and Km remains unchanged (Figure 1b). The values of K'm at any fixed concentration of aspirin and diclofenac were obtained from Figure 1 and then plotted against concentrations of the inhibitor (in the inset of Figure, secondary plot) to obtain the inhibition constant (KI). KI values at 27 and 37°C were 42.8 and 96.8 μM for aspirin, and 56.4 and 30.0 μM, for diclofenac, respectively. The figures for aspirin at 37°C and diclofenac at 27°C were similar to aspirin at 27°C (data not shown).
Figure 1 Double reciprocal Lineweaver–Burk plots for kinetics of 0.94 nM ADA at pH = 7.5 in the presence of 0.0 (▪), 7.5? (▪), 15.0 (▴), 22.5 (▵) and 30.0 μM (() of (a) aspirin at 27°C and (b) diclofenac at 37°C. Inset of each figure (secondary plots): apparent Michaelis constant, K'm (Km,app) versus concentration of inhibitor, [I].
![Figure 1 Double reciprocal Lineweaver–Burk plots for kinetics of 0.94 nM ADA at pH = 7.5 in the presence of 0.0 (▪), 7.5? (▪), 15.0 (▴), 22.5 (▵) and 30.0 μM (() of (a) aspirin at 27°C and (b) diclofenac at 37°C. Inset of each figure (secondary plots): apparent Michaelis constant, K'm (Km,app) versus concentration of inhibitor, [I].](/cms/asset/c8c197a4-c134-4fe4-b944-16b9bba7a54c/ienz_a_222827_f0001_b.gif)
By titration of a solution containing an enzyme (E) with a solution of inhibitor (I), the equilibrium reaction moves toward an increasing concentration of the EI complex. The heat of the reaction depends on the concentration of the EI complex. Thus, the reaction under consideration can be written as follows [Citation29,Citation43]:
Where
where qi is the sum of heat evolutions following the i-th titration step, Vi is the volume of the reaction solution and ΔHo is the enthalpy of binding. Ai, Bi and Ci can be calculated in each injection; thus, Equation (2) contains two unknowns, KI and ΔHo. A series of reasonable values for KI is inserted into Equation (2), corresponding values for ΔHo are calculated and a graph of ΔHo vs. KI is constructed. Curves of all titration steps will intersect at one point, which represents the true value for ΔHo and KI.
The plots of ΔHo vs. KI, according to Equation (2), for the first 10 injections are shown in . The intersection of the curves gives ΔHo and KI which are − 64.5 kJ/mol, 45.0 μM, − 34.5 kJ/mol and 61.0 μM, for aspirin and diclofenac, respectively.
Figure 2 ΔHo versus KI for first 10 injections, according to Equation (2) for (a) aspirin (b) diclofenac. The coordinates of intersection point of curves give true values for ΔHo and KI.
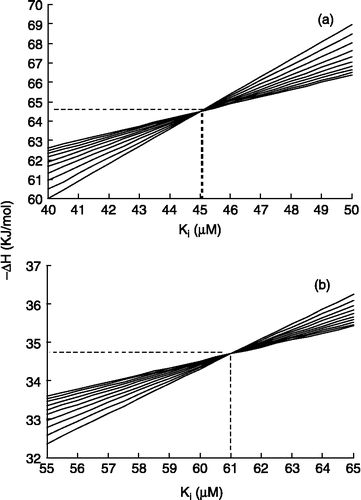
Good conformity of the dissociation binding constant (KI) obtained from thermodynamic and kinetic studies were observed. In addition, calorimetric measurements showed that the interaction between ADA and the two drugs was an exothermic process.
The standard Gibbs free energy change of binding for aspirin and diclofenac to ADA can be calculated using the inverse of the KI value as the association binding constants (K = 1/KI). Molar Gibbs free energy (ΔGo) and entropy (ΔSo) of binding can be obtained by the following equations:
The molar ΔGo and ΔSo of binding for aspirin were − 25.0 kJ/mol and − 131.7 J/(mol K) and for diclofenac were − 24.2 kJ/mol and − 32.0 J/(mol.K), respectively. Therefore, the binding process for these inhibitors on ADA is spontaneous enthalpy driven. The Binding constant of aspirin is higher than that of diclofenac.
To gain insight into the structure-activity relationship for the inhibitors, QSAR analysis has been performed through ascertaining the several suitable descriptors followed by defining of any linear correlation between the cited descriptors and the parameters of inhibition. Thus, a number of molecular descriptors have been primarily determined for a set of competitive inhibitors including nucleoside and non-nucleoside inhibitors. Fortunately, in the literature, Citation24-33 there are several inhibitors, where their kinetic parameters were reported during their interaction with ADA under environmental conditions that are similar those of our studies. In the previous work we reported only the QSAR of nucleoside inhibitors [Citation34]. However, in the present study, we considered QSAR and factor analysis on nucleoside (Scheme , compounds 1–11) and non-nucleoside inhibitors (Scheme 1b compounds 12–20).
Scheme 1 Molecular structure of a) nucleoside and b) non-nucleoside inhibitors.
The majority of these compounds had been experimentally studied previously. Only aspirin and diclofenac (compounds 19, 20) were studied here by us. shows the abbreviation and definition of the 25 descriptors.
Table I. List of descriptors used in this work.
The related values obtained from Hyperchem and Dragon were tabulated in. These descriptors belong to different properties such as geometric, topologic, electronic, hydrophobic and aromatic properties. In the first step correlation between logKI and these descriptors was investigated (final column of ); a negative sign shows a negative correlation.
Table II. Values of descriptors was calculated by Hyperchem-7.0 and Dragon-3.0.
Table II. continued.
Then the best descriptors were selected on the basis of the correlation obtained from stepwise selection in SPSS. Whenever the number of entered descriptors increases the correlation coefficient is enhanced. Thus, different models are observed on the basis of the number of entered descriptors. There are 20 compounds and so the number of descriptors is 1/5 number of compounds, i.e. 4 descriptors. Therefore, a model having 4 descriptors was selected. A Correlation between logKI and selected descriptors was obtained for 20 compounds by MLR methods as follow:
As we see, the logKI decreases with hydrophobicity (MLOGP), total dipole moment (MUTOT) and folding degree index (FDI) and increases with dipole moment in the Y-direction (MUY). On the other hand, aspirin has a lower value of MUY and MLOGP and a higher value of MUTOT relative to diclofenac. This means that aspirin is less hydrophobic and more polar relative to diclofenac.
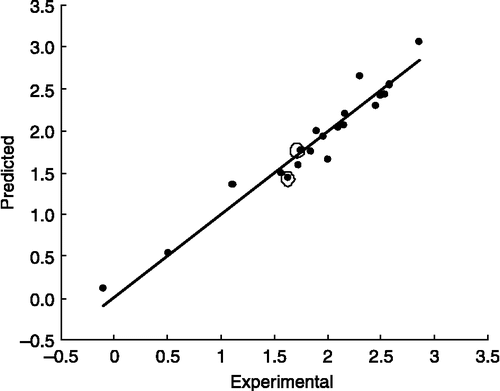
Predicted values by this equation were tabulated in [See column 3, logKI(pred1)]. shows a correlation between experimental and predicted data. The values for aspirin and diclofenac are encircled.
Figure 3 The linear correlation between the experimental values of logKI and the related values of the prediction set by MLR analysis on descriptors.
The correlation data () shows that there are some descriptors such as volume, surface area, and polarizability, refractivity, molecular weight, which have similar properties. They describe the size of molecule. Also geometrical descriptors such as L/BW, RGYR, SPH, ASP and FDI have similar properties. Therefore, it seems we can classify and reduce similar descriptors to a family or group.
So, in the next step, principal component analysis (PCA) was used for reduction and classification of descriptors. This method reduces the descriptors to a few factors or principal components (PCs). These factors include those descriptors which have similar effects and properties. In the current study, 25 descriptors were reduced into three factors which are tabulated in .
Table III. Rotated component matrix for studied descriptors.
Factor 1 includes volume, molecular weight, accessible and polar surface area, refractivity, polarizability, aromaticity, total dipole moment and the Z-direction dipole moment. The majority of them describe the size, thus we refer to them as the size factor. In previous work [Citation34] we also showed that volume decreases the logKI while here we obtained several descriptors which show a size property.
Factor 2 includes L/BW, RGYR, SPH, ASP and FDI which belong to geometrical and HOMA and AROM which belong to aromaticity indices [Citation39]. Aromaticity descriptors (HOMA, HOMT, RCI, AROM, ARR) distribute between three factors, because aromaticity depends on size, electronic properties and molecular shape. This factor is related to planarity or is two-dimensional such as aromaticity descriptors (HOMA, AROM, L/BW) or non-planarity three-dimensional (RGYR, SPH, ASP and FDI) descriptors. Therefore we refer to factor 2 as a shape factor.
Factor 3 includes MLOGP, HY, MUX, MUY, ARR and HE. MLOGP, HY and HE are related to hydrophobicity and electronic properties. Dipole moment in the X (MUX), Y (MUY) direction and ARR belong to electronic properties and describe hydrophobicity or electronic descriptors. We refer to this factor as the electronic factor. It is difficult to clearly define the boundary of these factors but each factor majority describes an overall property. Dipole moment depends on electronic charges and distance between them. Lengthy, polar molecule have a higher dipole moment. If we consider that the surface of the molecule locates in XY, then Z is perpendicular to the surface of the molecule. Increasing the MUX and MUY is related to enhancing the surface and planarity. On the other hand, more conjugated planar molecules have higher aromaticity. Increasing the MUZ reduces the two-dimensional shape (planarity) and increases the three-dimensional shape (spherosity or cylindrical). , show that increasing the MUTOT, MUZ, MUX, bond conjugation and aromaticity decreases logKI and so increases the binding constant. Thus lengthy, planar, aromatic and polar molecule has a lower logKI and higher tendency to bind to ADA. These data are compatible with other reports where more aromatic and large inhibitors have a higher binding constant or lower inhibition constant Citation31-33. Therefore, using these finding we can design a desirable drug for other proposals.
On the other hand, we can obtain the dependence of logKI on these factors by MLR. The resulting equation is as follow:
The equation shows that logKI decreases with the size and electronic or hydrophobicity factor and increases with the shape factor. Calculated data are tabulated in [logKI(Pred2)]. The correlation coefficient, standard error and mean effect are tabulated in . The effect of each descriptor appears in the beta factor (mean effect).
Table IV. Specification of the multiple linear regression model for factors.
The predicted value for aspirin is higher than that of diclofenac by this method. For improving it, we performed PCA analysis separately for the two categories. In addition the resulting equation from MLR of PCs for non-nucleosides is as follows:
and for nucleosides is:
The coefficients, their standard errors and mean effects for the three groups are tabulated in . The predicted values for non-nucleoside specially for aspirin and diclofenac, are shown in [log KI(Pred3)].
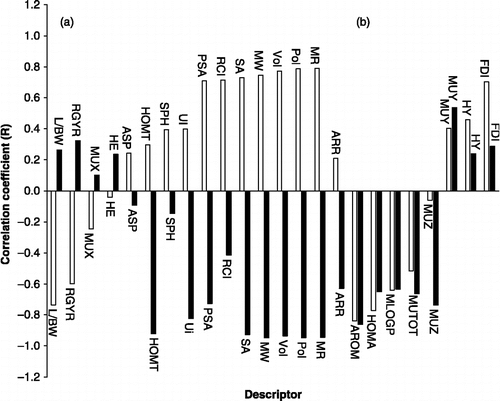
It is observed that all of the factors for nucleosides are negative but in non-nucleosides only the electronic factor is negative. The mean effects show that, the size in nucleosides and shape in non-nucleosides are more effective than the others. This finding related to correlation coefficient between logKI and descriptors. shows the differences between the correlation coefficients of the two categories. It is interesting to note that the size and shape descriptors have a reverse correlation in the two groups (region A). It may be due to the differences in inhibitor structure, binding site and type of interaction between inhibitors and ADA. It can help us to prediction an inhibitor's behavior. We also found that diclofenac has a higher size factor and higher logKI relative to aspirin which is compatible with non-nucleoside behavior.
Figure 4 Correlation coefficients logKI and descriptors for nucleoside (black) and non-nucleoside (white) bar line.
Thus we can gain an insight into structure-activity relationships and drug design with favorable properties by these methods, which are complementary methods for other studies.
Conclusion
Aspirin and diclofenac are anti-inflammatory drugs and inhibit the activity of ADA. They show different effects at different temperatures. The binding process for these ADA inhibitors is enthalpy driven. Diclofenac is a larger and more aromatic molecule compatible to its higher inhibition constant. Molecular structure and statistical analysis show that polar, aromatic, ribbon shape and planar molecules have higher tendency for binding to the enzyme and a lower inhibition constant. Analysis of the correlation between logKI and factors of two categories shows that nucleosides have a negative correlation with size, shape and electronic parameters while non-nucleosides have a positive correlation with size and shape and negative with electronic. In nucleosides, size is more effective than shape, while in non-nucleosides it is the reverse. All the data show a proper prediction of logKI for aspirin and diclofenac.
Acknowledgements
The financial support of the Damghan University of Basic Science and Research Council of the University of Tehran are gratefully acknowledged.
References
- Frick L, Wolfenden R, Smal E, Baker DC. Transition-state stabilization by adenosine deaminase: Structural studies of its inhibitory complex with deoxycoformycin. Biochemistry 1986; 8: 1616–1621
- Wilson DK, Rudolph FB, Quiocho FA. Atomic structure of adenosine deaminase complexed with a transition-state analog: Understanding catalysis and immunodeficiency mutations. Science 1991; 252: 1278–1284
- Chechik BE, Schreder WP, Minowada J. An immunomorphological study of adenosine deaminase. distribution in human thymus tissue, normal lymphocytes, and hematopoietic cell lines. J Immunol 1981; 126: 1003–1007
- Herschfeld MS, Mitchell BS. The metabolic and molecular basis of inherited disease, CR Scriver, AL Beaudet, WS Sly, D Valle. McGraw-Hill, New York 1995; 1725–1768
- Herschfield MS. Adenosine deaminase deficiency: clinical expression, molecular basis and therapy. Semin Hematol 1998; 35: 291–298
- Gan TE, Dadonna PE, Mitchell BS. Genetic expression of adenosine deaminase in human lymphoid malignancies. Blood 1987; 69: 1376–1380
- Hong L, Mulholland J, Chinsky JM, Knudsen TB, Kellems KE, Glasser SR:. Developmental expression of adenosine deaminase during decidualization in the rat uterus. Biol Reprod 1991; 44: 83–93
- Phillis JW, Wu PH. The role of adenosine and its nucleotides in central synaptic transmission. Biol Reprod Prog Neurobiol 1981; 16: 187–239
- Cronstein BN. Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol 1994; 76: 5–13
- Ohta A, Sitkovsky M. Critical Role of G protein-coupled adenosine Receptors in Down Regulation of Inflammation and Prevention of Tissue Damage In Vivo Nature 2001; 414: 916–920
- Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischemia Trends. Pharmacol Sci 1992; 13: 439–445
- Marquardt DL, Gruber HE, Wasserman SI. Adenosine release from stimulated mast cells. Proc Natl Acad Sci 1984; 81: 6192–6196, U.S.A.
- Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C. Direct association of adenosine deaminase with aT cell activation antigen, CD26. Science 1993; 261: 466–469
- Franco R, Valenzuela A, Lluis C, Blanco J. Enzymatic and extraenzymatic role of ecto-adenosine deaminase in lymphocytes. Immunol Rev 1998; 161: 27–42
- Baker DC, Hanvey JC, Hawkins LD, Murphy J. Identification of the bioactive enantiomer of. erythro-3-(adenine-9-yl)-2-nonanol, (EHNA), a semi-tight binding inhibitor of. adenosine deaminase. Biochem Pharmacol 1981; 30: 1159–1160
- Agerwal RP, Spector T, Parks RE. Inhibition of adenosine deaminases by various inhibitors Biochem. Pharmacol 1977; 26: 359–367
- Cristalli G, Costanzi S, Lambertucci C, Lupidi G, Vittori S, Volpini R, Camaioni E. Adenosine deaminase: functional implications and different classes of inhibitors Med. Res Rev 2001; 21: 105–128
- McConnell WR, Furner RL, Hill DL. Pharmokinetics of 2’-deoxycoformycin in normal and L1210 leukemic mice drug metab. Dispos 1979; 7: 11–13
- McConnell WR, El-Dareer SM, Hill DL. Metabolism and disposition of erythro-9-(2-hydroxy-3-nonyl)[14C]adenine in the rhesus monkey drug metab. Dispos 1980; 8: 5–7
- Brogden RN, Sorkin EM. Pentostatin: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in lymphoproliferative disorders. Drugs 1993; 46: 652–677
- Rafel M, Cervantes F, Beltran JM, Zuazu J, Nieto LH, Rayon C, Talavera JG, Montserrat E. Deoxycoformycin in the treatment of patients with hairy cell leukemia. Cancer 2000; 88: 352–357
- Warner TD, Giuliano F, Vojnovici I, Bukasa A, Mitchell JA. Vane JR. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: A full in vitro analysis. Proc Natl Acad Sci USA 1999; 96: 7563–7568
- Cronstein BN. Molecular and cellular basis of inflammation, CN Serhan, PA Ward. Humana, Totowa, NJ 1998; 259–274
- Saboury AA, Divsalar A, Ataie G, Moosavi-Movahedi AA, Housaindokht MR, Hakimelahi GH. A product inhibition study on adenosine deaminase by spectroscopy and calorimetry. J Biochem Mol Biol 2002; 35: 302–305
- Saboury AA, Divsalar A, Ataie G, Amanlou M, Moosavi-Movahedi AA. Inhibition study of adenosine deaminase by caffeine using spectroscopy and isothermal titration calorimetry. Acta Biochemica Polinica 2003; 50: 849–855
- Saboury AA, Bagheri S, Ataie G, Amanlou M, Moosavi-Movahedi AA, Hakimelahi GH, Cristalli G, S. Binding properties of adenosine deaminase interacted with theophylline. Chem Pharm Bull 2004; 52: 1179–1182
- Ataie G, Safarian S, Divsalar A, Saboury AA, Moosavi-Movahedi AA, Ranjbar B, Cristalli G, Mardanian S. Kinetic and structural analysis of adenosine deaminase upon interaction with acetaminophen. J Enz Inhib Med Chem 2004; 19: 71–78
- Saboury AA, Bagheri S, Ataie G, Moosavi-Movahedi AA, Hakimelahi GH, Cristalli G, Mardanian SA. Thermodynamic and kinetic study on the inhibition of adenosine deaminase by theobromine Asian. J Chem 2005; 17: 233–239
- Saboury AA. A simple method for determination of binding isotherm by isothermal titration calorimetry and its application to the interaction between Cu2+ and myelin basic protein. J Thermal Anal Cal 2004; 77: 997–1004
- Ataie G, Moosavi-Movahedi AA, Saboury AA, Hakimelahi GH, Hwu JR, Tsay SC. Enthalpy and enzyme activity of modified histidine residues of adenosine deaminase and diethyl pyrocarbonate complexes Int. J Biol Macromol 2000; 27: 29–33
- Terasaka T, Kinoshita T, Kuno M, Nakanishi I. A highly potent non-nucleoside adenosine deaminase inhibitor: Efficient drug discovery by intentional lead hybridization. J Am Chem Soc 2004; 126: 34–35
- Terasaka T, Okumura H, Tsuji K, Kato T, Nakanishi I, Kinoshita T, Kato Y, Kuno M, Seki N, Naoe Y, Inoue T, Tanaka K, Nakamura K. Structure-based design and synthesis of non-nucleoside, potent and orally bioavailable adenosine deaminase inhibitors. J Med Chem 2004; 47: 2728–2731
- Marron TJ, Straatsma TP, Briggs JM, Wilson DK, Quicho FA, McCammon JA. Theoretical study of inhibition of adenosine deaminase by 8R-Coformycin and 8R-Deoxycoformycin. J Med Chem 1996; 39: 277–284
- Moosavi-Movahedi AA, Safarian S, Hakimelahi GH, Ataei G, Ajloo D, Panjehpour S, Riahi S, Mousavi MF, Mardanyan S. QSAR analysis for ADA upon interaction with a series of adenine derivatives as inhibitors nucleosides. Nucleotides & Nucleic acids. 2004; 3: 613–624
- Kaplan NO. Meth Enzymol. Academic Press, New York 1955; Vol. 2: 473–480
- Brady TG, O'sullivan W. The effect of pH, urea and guanidine on adenosine deaminase isolated from bovine mucosa Biochim. Biophys Acta 1967; 132: 127–137
- Todeschini R, Consonni V, Mauri A, Pavan M, Dragon-3.0
- Todeschini R, Consonni V. Various molecular descriptors Handbook of Molecular descriptors. Wiley-VCH, Weinheim, Germany 2000.
- Gibson S, McGuire R, Rees DC. Principal components describing biological activities and molecular diversity of heterocyclic aromatic ring fragments. J Med Chem 1996; 39: 4065–4072
- Gupta SP. Quantitative structure-activity relationship studies on local anesthetics. Chem Rev 1991; 91: 1109–1118
- Hadjipavlou-Litina D, Hansch C. Quantitative structure-activity relationship of the benzodiazepines. Chem Rev 1994; 94: 1493–1505
- Zhang L, Gao H, Hansch C, Salassie CD. Molecular orbital parameters and comparative QSAR in the analysis of phenol toxicity to leukemia cells. J Chem Soc Perkin Trans 1998; 2: 2553–2556
- Ghadermarzi AA, Saboury AA, Moosavi-Movahedi AA. A microcalorimetry and spectroscopy study on the interaction of catalase with cyanide ion. Polish J Chem 1998; 72: 2024–2029