Abstract
The kinetics of microperoxidase-11 (MP-11) in the oxidation reaction of guaiacol (AH) by hydrogen peroxide was studied, taking into account the inactivation of enzyme during reaction by its suicide substrate, H2O2. Concentrations of substrates were so selected that: 1) the reaction was first-order in relation to benign substrate, AH and 2) high ratio of suicide substrate to the benign substrate, [H2O2]>>[AH]. Validation and reliability of the obtained kinetic equations were evaluated in various nonlinear and linear forms. Fitting of experimental data into the obtained integrated equation showed a close match between the kinetic model and the experimental results. Indeed, a similar mechanism to horseradish peroxidase was found for the suicide-peroxide inactivation of MP-11. Kinetic parameters of inactivation including the intact activity of MP-11, αi, and the apparent inactivation rate constant, ki, were obtained as 0.282 ± 0.006 min− 1 and 0.497 ± 0.013 min− 1 at [H2O2] = 1.0 mM, 27°C, phosphate buffer 5.0 mM, pH = 7.0. Results showed that inactivation of microperoxidase as a peroxidase model enzyme can occur even at low concentrations of hydrogen peroxide (0.4 mM).
Introduction
Understanding the structure/function relationship of hemoproteins needs investigations on the interactions between the heme and the peptide or apoprotein matrix. For this purpose, hemoprotein models, which can simplify the complex relationships, can be used Citation1-3. Hemoenzyme models such as microperoxidases are able to reproduce the coordination properties and/or the catalytic function of hemoproteins Citation4-7.
Microperoxidase as a heme peptide with a molecular weight of only 1900 has been found useful as an ultra-structural tracer [Citation4]. Heme-based mini-enzymes or microperoxidases form a new generation of biocatalysts and hemoenzyme models [Citation5,Citation6]. Capability of these biocatalysts to act as peroxidase Citation2Citation6-11, cytochrome C and nitric reductase [Citation3] has made them interesting models for studying the chemistry of native peroxidases [Citation6]. Microperoxidases have peroxidatic activity in a magnitude of about 1/4 that of HRP and 150 times that of cytochrome C [Citation4].
Microperoxidase-11 (MP-11) is a product of the proteolytic degradation of horseheart cytochrome C [Citation11,Citation12] and consists of a heme prosthetic group (Iron(III) protoporphyrin IX) covalently attached to a short polypeptide chain (11 amino acid residues) through two thioether bonds. The advantage of using MP-11 over simple heme (e.g. hemin) is that MP-11 shows better water solubility, lower tendency to aggregate/dimerize, and a spatial configuration, determined by the polypeptide chain, that resembles that in true hemoproteins [Citation13]. The microperoxidase family consists of four members (MP-6, MP-8, MP-9 and MP-11) [Citation14]. The smallest member with a short chain of five residues was also known in 1999 [Citation14]. These heme-based mini-enzymes are able to mimic peroxidase oxidation reactions [Citation5].
Some of such studies, which include hydroperoxo-iron heme intermediate (compound I) were reported and reviewed, previously [Citation6,Citation9]. Also it has been reported that in the presence of a radical scavenger like ascorbate, dealkylation reactions catalyzed by microperoxidase proceeds through a non radical-type mechanism [Citation9,Citation10].
Another important structural feature of MP-11 is that, similar to peroxidases [Citation13], the heme group in MP-11 is exposed directly to the solvent, allowing easy access even for bulky substrates Citation15-17. Recently, MP-11 was used for oxidation of pyocyanin [Citation13] and anthracycline [Citation15]. Also, MP-11 can catalyze oxidation reactions in organic media [Citation16]. Effects of the microenvironment on the function of MP-11 have been investigated [Citation18].
Hemoproteins have been the subject of extensive mechanistic Citation17-23, kinetic Citation24-27 and thermodynamic Citation28-31 studies Citation32-35. In the presence of high concentrations of hydrogen peroxide, all hemoproteins including peroxidases inactivate [Citation1,Citation36,Citation37] through a pathway referred to as “suicide-peroxide inactivation”. Many aspects of this process have not yet been fully elucidated [Citation1]. Microperoxidases may be also inactivated by high concentration of hydrogen peroxide, even more easily than the peroxidases because the heme group is not protected by the polypeptide chain.
The kinetic behaviour of the suicide substrates of enzymes has been analyzed [Citation38,Citation39] for significant [Citation40,Citation41] and negligible [Citation42,Citation43] substrate depletion. These kinetic analyses have been applied to enzymes such as tyrosinase [Citation44], horseradish peroxidase [Citation45], Coprinus cinereus peroxidase[Citation46], thyroid and lactoperoxidase [Citation47,Citation48].
The kinetic model for suicide-peroxide inactivation of catalase (as a monosubstrate hemoenzyme) [Citation49] and horseradish peroxidase (as a bisubstrate hemoenzyme) has been introduced previously [Citation37]. In the latter model, the inactivation process can be easily monitored through the time-course (progress curve) of the oxidation reaction of an aromatic hydrogen donor substrate [Citation37]. The main feature of this study was presentation of the kinetic equations and indicating the validity and reliability of the kinetic model. Since MP-11 is a peroxidase model enzyme, we examined the same kinetic model for peroxide-inactivation of microperoxidase. The present work involves a comparative study on the suicide inactivation of microperoxidase-11 and horseradish peroxidase. Validation of the kinetic model is evaluated and the comparative strength of inactivation as well as the kinetic parameters including the intact activity (αi), initial activity (αo) and the apparent rate constant of inactivation of MP-11 (ki) were determined.
Experimental
Materials
MP-11 (sodium salt) and horseradish peroxidase (type II) were obtained from Sigma. Guaiacol (ortho-methoxyphenol), Na2HPO4.2H2O and hydrogen peroxide 30% (V/V) were obtained from Merck. Phosphate buffer 5.0 mM, as the solvent system was prepared in CO2-free deionized water (Barnstead NanoPure D4742 deionizer, Electrical Resistance = 18.3 MΩ).
Methods
Absorbance changes and electronic spectra were recorded using a Varian spectrophotometer model Cary 50 equipped with fiber optic dip probe accessory and a xenon light source. The fiber optic probe provides the data recording at the time of mixing in the reaction vessel. Using this accessory, no cuvette is required. The ionic strength and pH of the solutions were kept constant by using a 5.0 mM phosphate buffer. Temperature of the reaction solution in the jacketed laboratory reactor (4 mL) was adjusted to 25°C ( ± 0.1) using a Lauda oil circulating thermobath equipped with an external temperature sensor. Specific activity of MP-11 was determined spectrophotometrically in a 1-min reaction time course using guaiacol as hydrogen donor [Citation50]. The specific activity of MP-11 was equal to 52 U/mg. The same spectrophotometer in the kinetic mode was used for recording the progress curves of the inactivation process. MP-11 concentration was determined at pH = 7.0 using an extinction coefficient of 176 mM− 1.cm− 1 at 395 nm [Citation16].
Concentration of hydrogen peroxide was estimated by measuring absorbance of the solution using ε240 = 43.6 cm− 1.M− 1 [Citation14,Citation51]. In all calculations, a molecular weight of 1861 was used for MP-11. Suicide-peroxide inactivation of MP-11 was monitored at 470 nm (λmax for the product of the catalytic reaction) by difference spectrophotometry. The details of the procedure of measuring the product concentration, the rate of reaction, and determination of the progress curves have been described previously [Citation37]. Progress curves were determined by following absorbance of the reaction mixture at 470 nm in an appropriate time course of about 30 min. After starting the reaction, 30 s was considered as the dead time for the reaction to reach the stationary state. The unreacted part of AH was obtained from the recorded absorbance data and suitable relations [Citation37].
Each experiment was repeated three times and standard deviations were estimated for the obtained results.
“End-point procedure” experiments were carried out in order to determine the maximum elevation of absorbance (A∞) and the value of intact activity (αi). Thus, the reaction mixture was incubated for a prolonged time (about 2 h) according to the “end-point procedure” which is described in detail elsewhere [Citation37]. In the end-point method by adding a fresh and excess quantity of MP-11 solution to the incubated mixture, values of A∞, the unreacted parts of AH ([AH]∞) and also the intact activity of the enzyme (αi), could be estimated. Intact activity (αi) is defined as the enzyme activity before it is exposed or reacted with the substrate.
Results and discussion
An important structural feature of MP-11 is that, similar to most peroxidases, the fifth coordination position of iron in the heme is occupied by the imidazole group of histidine present in the polypeptide chain (His.18). The sixth position is occupied by a water molecule [Citation6] and this weak ligand can be easily replaced by the peroxide substrate [Citation52]. The chemical structure of MP-11 is shown below:
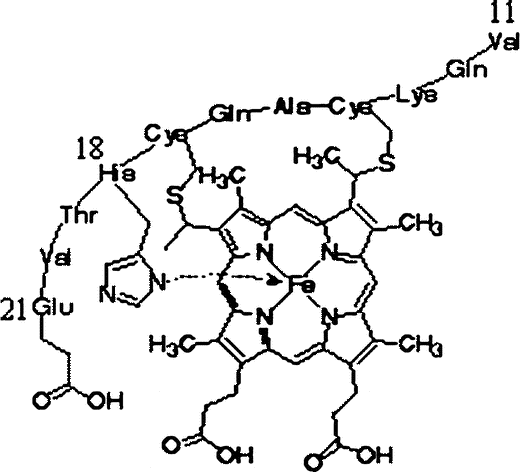
In the catalytic cycle, displacement of the water molecule by hydrogen peroxide and the subsequent formation of compound I can easily occurr. In comparison to hemoproteins, the heme periphery of MP-11 is exposed directly to the solvent, allowing easy access even for bulky substrates. From the kinetic point of view, formation of compound I is the rate-limiting step [Citation10]. Indeed, in the presence of H2O2, microperoxidases form a reactive intermediate (an analog of peroxidase compound I which is responsible for the catalytic activity [Citation13,Citation52]. Thus, in accordance with the spectroscopic data it is believed that microperoxidase and HRP have a similar kinetic mechanism [Citation6].
There are two major catalytic pathways for the kinetics of peroxidase: 1) a two-electron transfer as the catalase pathway and 2) a one-electron transfer as the compound III pathway, which is a minor pathway compared with that of the catalase reaction [Citation1,Citation53].
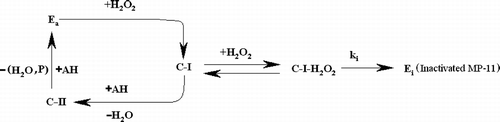
It has been shown that in the presence of high concentrations of peroxide, in addition to the two above described major catalytic pathways, peroxidase can also enter into an inactivation pathway namely “suicide-substrate inactivation” Citation1Citation37Citation54-57. The inactivation process starts by the reaction of excess H2O2 with compound-I which has a central role in the suicide-substrate inactivation pathway. The product of the inactivation process (P-670) is a catalytically inactive form of peroxidase (Ei in ) Citation37Citation58-60. This inactive form indicates a λmax in its electronic spectra at 670 nm (see ). P-670 has two characteristics: 1) It has a low molar absorptivity that would involve the use of high enzyme concentrations and 2) P-670 is unstable and may convert into the other inactive forms. Hence, following the inactivation of peroxidase and peroxidase models is not recommended through the quantitative monitoring of P-670.
Among the MP active forms (including MP, C-I and C-II), Compound I as a high valent intermediate and strong oxidant shows the maximum capacity and potential to oxidize aromatic substrates.
According to Scheme I, two major reactions could be attributed to the reaction system:
Reaction a is the inactivation process and b (oxidation of guaiacol by peroxide) is the catalyzed reaction which is also used to monitor the inactivation process.
Ea, Ei, AH, and P denote the whole forms of active enzyme, inactivated enzyme, hydrogen donor (reductant-substrate) and the catalytic reaction product (tetraguaiacol), respectively. Parameters of ki and α denote the apparent rate constant of inactivation (reaction a) and the apparent catalytic rate constant of the oxidation reaction (reaction b), respectively. In the catalytic reaction cycle, the active enzyme species are free active enzyme (Ea), C-I and C-II. The rate constants of formation of C-I, C-II and Ea are extremely large compared with the ki value. Accordingly, the whole active enzymes are supposed to be in form of C-I, which may undergo inactivation by the peroxide (see ). The following conditions are recommended for the simultaneous advancement of the two reactions (a and b):
Scheme I The catalytic reaction cycle and the inactivation pathway for MP-11.
a) Higher concentration of hydrogen peroxide with respect to the concentration of hydrogen donor (AH)
b) Low concentration of hydrogen donor (“benign substrate”, a substrate that does not damage the enzyme); so the donor substrate does not saturate the enzyme (according to the initial linear part of the Michaelis-Menten plot).
The reaction b is essentially first-order in relation to the hydrogen donor and an expected linear dependence on the initial velocity of reaction b and lower range of AH concentration was observed (data not shown) [Citation37]. As a result, the rate of reaction a seems to be proportional to the active enzyme concentration. Using a steady-state approximation, it is possible to derive the differential and integrated kinetic equations. In principle, by monitoring AH concentration in the catalytic reaction cycle, one can estimate the extent of decrease in the level of active forms of the enzyme (due to the suicide inactivation process).
Combination of the rate equations of the two above reactions i.e. consumption of substrate and inactivation of enzyme, can be written as:
where αo can be defined as the value of α at the start time of measurements (t = 0). The remaining amount of guaiacol (ortho-methoxy phenol, [AH]t) was determined using the following relationships:
where A∞ and At are the absorbance of the colored product (at 470 nm) and concentration of guaiacol at time t and t = ∞ (end of the catalytic reaction), respectively. [AH]t is concentration of guaiacol (AH) at time t.
The present model involves the advantage of monitoring of the inactivation reaction with no need to measure the concentrations of enzyme species (Ea, C-I, C-II, Ei). In fact, the overall concentration of active enzyme species, α, (in the form of C-I) defines the first order rate law at each time (α = αo e(–ki .t)). Instead of activity measurements, αo can be obtained by fitting the experimental data into the overall integrated kinetic equation (Equation (4)) of the process. Indeed, the rate of the consumption of reductant substrate (AH) and the overall rate of conversion of active enzyme (Ea) to the inactivated form (Ei) enables us to obtain the differential kinetic equation as well as the integrated kinetic relation (according to our previous model) [Citation37,Citation49,Citation61]:
where [AH]o is the molar concentration of AH at t = 0. Equation (4) is the relationship between the remaining concentration of the aromatic substrate and time, whilst the decrease in enzyme activity during the simultaneous inactivation reaction has been taken into account. Equation (4) can be used for the determination of αo and ki in a non-linear regression manner by fitting the experimental data into the equation. Common computer software such as Excel Solver was used for this purpose. indicates the obtained values of the kinetic parameters for suicide inactivation of MP-11.
Table I. ki and αi values for HRP and MP-11 were obtained for a time course of 10 min of the progress curves data at 27°C, pH=7.0, phosphate buffer 5.0 mM.
On the other hand, at adequately low concentrations of H2O2 (about 0.1–0.2 mM) the normal catalytic cycle proceeds without the suicidal inactivation effect of hydrogen peroxide. shows the various types of inactivation progress curves at different concentrations of hydrogen peroxide. Progress curves were obtained as described in the Methods. Details of the experiments are described in the legend of . The excellent coincidence between experimental data (points) and the fitted curve (calculated) based on Equation (4) was observed as shown in . On the other hand, illustrates the accuracy of the kinetic model and the Equation (4) for the suicide-peroxide inactivation of microperoxidase-11.
Figure 1 Progress curves for suicide inactivation of MP-11 by hydrogen peroxide, as the variation of guaiacol concentration versus reaction time, according to Equation (4). Inactivation rate constant, ki and the initial activity of MP-11, αo, were estimated by successive approximation and non-linear fitting of the experimental data into Equation (4). Solid lines indicate the calculated curve data using the obtained ki and αo parameters in Equation (4).Values of the kinetic parameters are shown in . Each parameter was systematically adjusted to produce best fit curves that gave a minimum value of the sum-of-squares of residuals (SSR, as the differences between observed and calculated values of progress curves, i.e. SSR = Σ(yobs.–ycalc.)2). An Excel Solver program was used for this purpose. A detailed description of the suicide-peroxide inactivation model is given in references [Citation33] and [Citation37]. Reactions were started by adding hydrogen peroxide to the mixture of AH and MP-11 (1.0 μM) having an initial activity of 0.3 min− 1, time course of about 30 min at 27°C, phosphate buffer 5.0 mM, pH = 7.0. a) [AH] = 200 μM, [H2O2] = 1.0 mM, ki = 0.487 min− 1 b) [AH] = 100 μM, [H2O2] = 1.0 mM, ki = 0.491 min− 1, c) [AH] = 100 μM, [H2O2] = 0.40 mM, ki = 0.120 min− 1 d) The hypothetical curve with ki = 0 (using value of ki = 10− 8 for solving Equation (4)), i.e. without suicidal effects of peroxide. Here [AH]o and αo were considered to be the same as state (c). AH concentration was followed from the absorbance of the reaction mixture at 470 nm using Equations (2) and (3). In order to reach the stationary state of the reaction, a dead time of 30 seconds was considered for processing the progress curves data.
![Figure 1 Progress curves for suicide inactivation of MP-11 by hydrogen peroxide, as the variation of guaiacol concentration versus reaction time, according to Equation (4). Inactivation rate constant, ki and the initial activity of MP-11, αo, were estimated by successive approximation and non-linear fitting of the experimental data into Equation (4). Solid lines indicate the calculated curve data using the obtained ki and αo parameters in Equation (4).Values of the kinetic parameters are shown in Table I. Each parameter was systematically adjusted to produce best fit curves that gave a minimum value of the sum-of-squares of residuals (SSR, as the differences between observed and calculated values of progress curves, i.e. SSR = Σ(yobs.–ycalc.)2). An Excel Solver program was used for this purpose. A detailed description of the suicide-peroxide inactivation model is given in references [Citation33] and [Citation37]. Reactions were started by adding hydrogen peroxide to the mixture of AH and MP-11 (1.0 μM) having an initial activity of 0.3 min− 1, time course of about 30 min at 27°C, phosphate buffer 5.0 mM, pH = 7.0. a) [AH] = 200 μM, [H2O2] = 1.0 mM, ki = 0.487 min− 1 b) [AH] = 100 μM, [H2O2] = 1.0 mM, ki = 0.491 min− 1, c) [AH] = 100 μM, [H2O2] = 0.40 mM, ki = 0.120 min− 1 d) The hypothetical curve with ki = 0 (using value of ki = 10− 8 for solving Equation (4)), i.e. without suicidal effects of peroxide. Here [AH]o and αo were considered to be the same as state (c). AH concentration was followed from the absorbance of the reaction mixture at 470 nm using Equations (2) and (3). In order to reach the stationary state of the reaction, a dead time of 30 seconds was considered for processing the progress curves data.](/cms/asset/c4f0dcae-54dc-4f27-bb6b-2ebde7f964c4/ienz_a_226977_f0002_b.gif)
Generally, there is a delay from the start of the reaction to the start of measurements (t = 0) for homogenizing the mixture and allowing the reaction to reach the steady state condition. Certainly, some of the enzyme activity may be lost in such a delay period. αo is the activity of enzyme at the end of this delay time. Therefore, a correction is necessary for calculation of αi as the intact value of activity just at the start of reaction. The intact activity defines by a modified form of Equation (1) {αi = αo × e–ki .τ} in which τ is the delay time of measurement. The inactivation rate constant, ki, must be independent of changes in the initial value of either enzyme or AH concentration. and shows such independency of ki from both of the above variables. depicts the independency of αo versus changes in the concentration of reductant substrate ([AH]).
Figure 2 a) Variation of ki with initial concentration of guaiacol, AH. ki values were obtained (27°C, phosphate buffer 5.0 mM, pH = 7.0) from the related progress curves and Equation (4) as described in legend of Figure 1. as a function of activity (concentration) of MP-11, αo. Values of ki and αo were obtained (27°C, phosphate buffer 5.0 mM, pH = 7.0, [H2O2] = 1.0 mM) from the related progress curves and Equation (4) as described in the text and in the legend of Figure 1. c) αo as a function of concentration of hydrogen donor substrate, [AH]. Values of αo were obtained (27°C, phosphate buffer 5.0 mM, pH = 7.0, [H2O2] = 1.0 mM) from the related progress curves and Equation (4) as described in the text and in the legend of Figure 1.
![Figure 2 a) Variation of ki with initial concentration of guaiacol, AH. ki values were obtained (27°C, phosphate buffer 5.0 mM, pH = 7.0) from the related progress curves and Equation (4) as described in legend of Figure 1. as a function of activity (concentration) of MP-11, αo. Values of ki and αo were obtained (27°C, phosphate buffer 5.0 mM, pH = 7.0, [H2O2] = 1.0 mM) from the related progress curves and Equation (4) as described in the text and in the legend of Figure 1. c) αo as a function of concentration of hydrogen donor substrate, [AH]. Values of αo were obtained (27°C, phosphate buffer 5.0 mM, pH = 7.0, [H2O2] = 1.0 mM) from the related progress curves and Equation (4) as described in the text and in the legend of Figure 1.](/cms/asset/b5ea697e-610a-4171-9c64-4a23a583ffeb/ienz_a_226977_f0003_b.gif)
Determination of ki and αi is also possible from the “end-point” approach, using linear plots. Based on the end-point procedure, the relation between the remaining concentration of substrate at the end of reaction (t∞) assigned by [AH]∞ and the other variables can be predicted from Equation (4) by setting t = ∞, as below:
where [AH]i is the initial concentration of AH. According to Equation (5) a linear plot of [AH]∞ versus [AH]i represents a slope of e(–αi /ki). shows a linear relation between [AH]∞ versus [AH]i. Also, shows the linear plot of ln[AH]∞ versus ln[AH]i with a calculated slope of unity and Y-intercept of –αi/ki, according to Equation (6). Furthermore, on the basis of Equation (7) a plot of ln([AH]i/[AH]∞) versus initial activity of MP-11 would be a line with the slope of 1/ki. This linear plot is shown in . Each of the above plots i.e. ,b,c were obtained from independent experiments. Good agreements between the predicted and the experimental values for the slopes and intercepts of the lines (according to the related equation) indicate the validity and reliability of the kinetic model. The related slopes and intercepts are shown in the legend of the Figures. Equations (5-7) demonstrate that 1) variation of initial concentration of aromatic substrate ([AH]i) must result in a proportional change in its final concentration ([AH]∞), when the other parameters remain unchanged, 2) variation of the intact activity parameter (αi) must lead to the expected changes in the ratio of ([AH]i/[AH]∞) according to Equation (7).
Figure 3 Linear plots based on the end-point procedure () illustrating dependence of initial concentration of AH ([AHi]) on the remaining (unreacted) concentration of AH ([AH∞]). Reactions were carried out at constant activity of MP-11. a) Linear plot based on Equation (5). A mixture of MP-11 (1.0 μM) and various initial concentrations of guaiacol ([AHi]) were incubated in the presence of [H2O2] = 1.0 mM at 27°C for 2 h (t = ∞). Reactions were carried out at constant activity of MP-11 (0.3 min− 1). Slope = Exp(–αi/ki) = 0.976 and αi/ki = 0.0243, Y-intercept = 0.009. b) Logarithmic plot based on Equation (6). A mixture of MP-11 (1.0 μM) and various initial concentrations of guaiacol ([AHi]) were incubated in the presence of [H2O2] = 1.0 mM at 27°C for 2 h (t = ∞). Reactions were carried out at constant activity of MP-11 (0.3 min− 1). Slope = 1.003, Y-intercept = –αi/ki = − 0.0256 and αi/ki = 0.0256 c): Linear plot according to Equation (7) for the variation of ln([AHi]/[AH∞]) with the initial activity of MP-11, αi. The mixture of AH and MP-11 (1.0 μM) were incubated in the presence of 1.0 mM H2O2, phosphate buffer 5.0 mM at 27°C for 2 h (t = ∞). Slope = 1/ki = 2.023 and ki = 0.494 min− 1., Y-intercept = 0.0.
![Figure 3 Linear plots based on the end-point procedure () illustrating dependence of initial concentration of AH ([AHi]) on the remaining (unreacted) concentration of AH ([AH∞]). Reactions were carried out at constant activity of MP-11. a) Linear plot based on Equation (5). A mixture of MP-11 (1.0 μM) and various initial concentrations of guaiacol ([AHi]) were incubated in the presence of [H2O2] = 1.0 mM at 27°C for 2 h (t = ∞). Reactions were carried out at constant activity of MP-11 (0.3 min− 1). Slope = Exp(–αi/ki) = 0.976 and αi/ki = 0.0243, Y-intercept = 0.009. b) Logarithmic plot based on Equation (6). A mixture of MP-11 (1.0 μM) and various initial concentrations of guaiacol ([AHi]) were incubated in the presence of [H2O2] = 1.0 mM at 27°C for 2 h (t = ∞). Reactions were carried out at constant activity of MP-11 (0.3 min− 1). Slope = 1.003, Y-intercept = –αi/ki = − 0.0256 and αi/ki = 0.0256 c): Linear plot according to Equation (7) for the variation of ln([AHi]/[AH∞]) with the initial activity of MP-11, αi. The mixture of AH and MP-11 (1.0 μM) were incubated in the presence of 1.0 mM H2O2, phosphate buffer 5.0 mM at 27°C for 2 h (t = ∞). Slope = 1/ki = 2.023 and ki = 0.494 min− 1., Y-intercept = 0.0.](/cms/asset/acd1b23f-6199-4a16-9e3d-07b827a6df48/ienz_a_226977_f0004_b.gif)
On the other side, effect of variation of H2O2 concentration on ki is shown in which indicates that the rate of inactivation is proportional to the concentration of suicide substrate, a factor which drives the free MP-11 to a substantially reactive intermediate, compound I and then crashes the latter as a catalytically inactive derivative, P-670. According to , a linear relation in the form of ki = 0.445 × [H2O2] can be easily used to predict and estimate the apparent rate constant of inactivation of MP-11 by hydrogen peroxide. shows that the intact activity (αi) of MP-11 is not influenced by the suicide substrate, H2O2. This means that the rate limiting step of the catalytic reaction cycle is not the H2O2 consuming step.
Figure 4 a) Dependence of ki on concentration of suicide-substrate, [H2O2]. [AH] = 0.4 mM, [MP-11] = 1.0 μM, pH = 7.0, phosphate buffer 5.0 mM and temperature of 27°C. b) Independency of αi on [H2O2]. [AH] = 0.4 mM, pH = 7.0, phosphate buffer 5.0 mM and temperature of 27°C.
![Figure 4 a) Dependence of ki on concentration of suicide-substrate, [H2O2]. [AH] = 0.4 mM, [MP-11] = 1.0 μM, pH = 7.0, phosphate buffer 5.0 mM and temperature of 27°C. b) Independency of αi on [H2O2]. [AH] = 0.4 mM, pH = 7.0, phosphate buffer 5.0 mM and temperature of 27°C.](/cms/asset/874b8e47-2c87-442c-b6f0-ed55e7ef3d9b/ienz_a_226977_f0005_b.gif)
shows the electronic spectra for the native and inactivated MP-11 (in the presence of 3.0 mM H2O2) after 1 minute of incubation. The inactivated form shows a maximum absorption at 670 nm which is the same as for inactivated HRP. Also during the inactivation, due to the conversion of the active heme into the inactive verdoheme, the Soret band absorbance is expected to be decreased as shown in .
All of the above mentioned findings show that MP-11 and HRP have similar a suicide inactivation mechanism regarding both the inactivated product (see ) and kinetics of suicide-peroxide inactivation. On the other hand, similar to HRP, MP-11 also has two substrates: one of them is AH, the “benign substrate” and the other is H2O2, the damaging substrate. The benign substrate which does not damage the enzyme, is only used for monitoring the progress of the reaction.
Figure 5 Electronic spectra of native and inactivated microperoxidase 11 (150 μM) at pH = 7.0, phosphate buffer 5.0 mM. Solid line, Native MP-11 (in the absence of hydrogen peroxide). Dashed line, produced inactivated MP-11 (or P-670) after 5 min incubation with [H2O2] = 3.0 mM.
![Figure 5 Electronic spectra of native and inactivated microperoxidase 11 (150 μM) at pH = 7.0, phosphate buffer 5.0 mM. Solid line, Native MP-11 (in the absence of hydrogen peroxide). Dashed line, produced inactivated MP-11 (or P-670) after 5 min incubation with [H2O2] = 3.0 mM.](/cms/asset/4983bdf7-84cd-4a8f-8896-46fa1f780c2f/ienz_a_226977_f0006_b.gif)
Comparison of the inactivation parameters for HRP and MP-11, in addition to providing an estimation of kinetic parameters for the microperoxidase family also gives the extent of inactivation for each biocatalyst. Suicide-peroxide inactivation parameters for MP-11 and horseradish peroxidase are shown in . Comparing the ki values (see ) shows that the rate of inactivation of MP-11 is about 10 times faster than HRP. It must be noted that the minimum concentration for detection of inactivation of HRP was about 2 mM while the same value for MP-11 is about 0.4 mM of H2O2. This result was expected because of the availability of the heme group to the suicide substrate (H2O2). Furthermore, in the presence of 3.0 mM of hydrogen peroxide, MP-11 inactivates much more rapidly with a rate constant of ki = 1.30 min− 1 which shows a 2.65 times (1.30/0.491) increase in the strength of inactivation in relation to [H2O2] = 1.0 mM (see ). In the peroxidase, the heme group is sandwiched and protected between two helices and only the edge of the pyrol (IV) of heme is accessible to the solvent or substrate.
For HRP the extent of inactivation is negligible below [H2O2] < 0.5 mM and/or [H2O2]/[AH]∼1.3 ([AH] = 0.4 mM, ki = 0.009 min− 1), while for the ratio of [H2O2]/[AH]∼25 peroxidase inactivates completely ([H2O2] = 10 mM, [AH] = 0.4 mM, ki = 0.18 min− 1) within a 10 minute reaction cycle [Citation37].
According to , the inactivation extent for MP-11 at [H2O2] = 3.0 mM becomes about 2.65 times greater in relation to 1.0 mM H2O2. On the other side, in comparison with HRP the extent of inactivation of MP-11 at [H2O2] = 3.0 mM is about 28.3 times (1.30/0.046) faster (see ). This problem needs to be solved for the application of MP-11 in systems containing hydrogen peroxide. This is the subject of our future research on protecting the active site of MP-11 against the destroying effect of hydrogen peroxide, by using efficient water soluble polymers and osmolytes.
Conclusion
Suicide-peroxide inactivation of microperoxidase takes place in a manner and mechanism similar to horseradish peroxidase, but at much more rapid rates. Inactivation of MP-11 as a peroxidase model enzyme can occur even at low concentrations of hydrogen peroxide (0.4 mM). A complete and fast inactivation of MP-11 takes place within about 20 minutes at [H2O2]∼3 mM. This clearly indicates a significant need to proceed with when such enzyme models are used for clinical, biochemical or chemical purposes. Furthermore, introducing the means and solutions for protection and preventing such severe inactivation effects is of prior and great importance, and is currently in progress in our laboratory.
| Abbreviations | ||
| MP: | = | Microperoxidase |
| HRP: | = | Horseradish Peroxidase |
| AH: | = | Hydrogen Donor |
| A: | = | Absorbance |
| C-I: | = | Compound I |
| C-II: | = | Compound II |
Acknowledgements
The financial support of the research council of Islamic Azad University, research council of Tehran University and Iran National Science Foundation are gratefully acknowledged.
References
- Valderrama B, Ayala M, Vazquez-Duhalt R. Chem Biol 2002; 9: 555–565
- Ricoux R, Lecomte S, Policar C, Boucher JL, Mahy JP. J Inorg Biochem 2005; 99: 1165–1173
- Suruga K, Murakami K, Taniyama Y, Hama T, Chida H, Satoh T, Yamada S, Hakamata W, Kawachi R, Isogai Y, Nishio T, Oku T. Biochem Biophys Res Commun 2004; 315(4)815–822
- Feder N. J Cell Biol 1971; 51: 339–343
- Boersma MG, Primus JL, Koerts J, Veeger C, Rietjens IMCM. J Biochem 2000; 267: 6673–6678
- Veeger C. J Inorg Biochem 2002; 91: 35–45
- Osman AM, Boeren S, Veeger C, Rietjens IMCM. Chem Biol Int 1997; 104(2–3)147–164
- Willner I, Arad G, Katz E. Bioelectrochem Bioenergetics 1998; 44(2)209–214
- Ichinose H, Wariishi H, Tanaka H. Enz Microb Technol 2002; 30(3)334–339
- Baldwin DA, Marques HM, Pratt JM. J Inorg Biochem 1987; 30(3)203–217
- Okazaki S, Nagasawa S, Goto M, Furusaki S, Wariishi H, Tanaka H. Biochem Eng J 2002; 12: 237–241
- Laszlo JA, Compton DL. J Mol Cat B 2002; 18: 109–120
- Chuang WJ, Chang YD, Jeng WY. J Inorg Biochem 1999; 75: 93–97
- Reszka KJ, Malley Y, Mccormick ML, Denning GM, Britigan BE. Free Rad Biol Med 2004; 36(11)1448–1459
- Reszka KJ, Mccormick ML, Britigan BE. Free Rad Biol Med 2003; 35(1)78–93
- Oyadomari M, Kabuto M, Wariishi H, Tanaka H. Biochem Eng J 2003; 15: 159–164
- Aitken SM, Ouellet M, Percival MD, English AM. Biochem J 2003; 375: 613–621
- Kadnikova EN, Kostic NM. J Org Chem 2003; 68(7)2600–2608
- Dunford HB, Stillman JS. Coord Chem Rev 1976; 19: 187–251
- Hosler JP. Biochim Biophys Acta 2004; 1655: 332–339
- Rodriguez-Lopez JN, Lowe DJ, Hernandez-Ruiz J, Hiner AN, Garcia-Canovas F, Thorneley RN. J Am Chem Soc 2001; 123(48)11838–11847
- Adak S, Mazumdar A, Banerjee RK. J Biol Chem 1997; 272(17)11049–11056
- Huang Q, Huang Q, Pinto RA, Griebenow K, Schweitzer-Stenner R, Weber WJ. J Am Chem Soc 2005; 127(5)1431–1437
- Candeias LP, Folkes LK, Wardman P. Biochemistry 1997; 36: 7081–7085
- Diederix REM, Fittipaldi M, Worrall JAR, Huber M, Ubbink M, Canters GW. Inorg Chem 2003; 42(22)7249–7257
- Rodriguez-Lopez JN, Hernandez-Ruiz J, Garcia-Canovas F, Thorneley RNF, Acosta M, Arnao MB. J Biol Chem 1997; 272(9)5469–5476
- Hiner ANP, Rodrigeuz-Lopez JN, Arnao MB, Raven EL, Garcia-Canoves F, Acosta M. Biochem J 2000; 348: 321–328
- Nazari K, Moosavi-Movahedi AA, Coll Surf B. Coll Surf B 2000; 18: 63–70
- Battistuzzi G, Borsari M, Ranieri A, Sola M. J Am Chem Soc 2002; 124(1)5315–5324
- Moosavi-Movahedi AA, Nazari K. Int J Biol Macromol 1995; 17: 43–47
- Nazari K, Saboury AA, Moosavi-Movahedi AA. Thermochim Acta 1997; 302: 131–135
- Prasad S, Maiti NC, Mazumdar S, Mitra S. Biochim Biophys Acta 2002; 1596: 63–75
- Nazari K, Mahmoudi A, Shahrooz M, Khodafarin R, Moosavi-Movahedi AA. J Enz Inhib Med Chem 2005; 20(3)285–292
- Filizola M, Loew GH. J Am Chem Soc 2000; 122: 18–25
- Doerge DR, Chang HC. J Chromatog B 2002; 777: 269–279
- Bratton MR, Pressler MA, Hosler JP. Biochemistry 1999; 38: 16236–16245
- Moosavi-Movahedi AA, Nazari K, Ghadermarzi M. Ital J Biochem 1999; 48(1)9–17
- Abeles RH. Basic Life Sci 1983; 25: 287–305
- Walsh C. Horiz Biochem Biophys 1977; 3: 36–81
- Waley SG. Biochem J 1980; 185(3)771–773
- Waley SG. Biochem J 1985; 227(3)843–849
- Garcia-Canovas F, Tudela J, Varon R, Vazquez AM. J Enzy Inhib 1989; 3(2)81–90
- Garcia-Canovas F, Tudela J, Martinez MC, Varon R, Garcia-Carmona F, Lozano JA. Biochim Biophys Acta 1987; 912(3)417–423
- Tudela J, Garcia-Canovas F, Varon R, Jimenez M, Garcia-Carmona F, Lozano JA. Biophys Chem 1988; 30(3)303–310
- Arnao MB, Acosta M, del Rio JA, Varon R, Garcia-Canovas F. Biochim Biophys Acta 1990; 1041(1)43–47
- Chang HC, Holland RD, Bumpus JA, Churchwell MI, Doerge DR. Chem Biol Interact 1999; 123(3)197–217
- Divi RL, Doerge DR. Biochemistry 1994; 33(32)9668–9674
- Doerge DR, Decker CJ, Takazawa RS. Biochemistry 1993; 32(1)58–65
- Ghadermarzi M, Moosavi-Movahedi AA. Ital J Biochem 1997; 46(4)197–205
- Maehly AC. Meth Enzymol 1965; 2: 801–813
- George P. Biochem J 1953; 54: 267–271
- Casella L, Gioia LD, Silvestri GF, Monzani E, Redaelli C, Roncone R, Santagostini L. J Inorg Biochem 2000; 79: 31–40
- Azevedo AM, Martins VC, Prazeres DMF, Vojinovie V, Cabral JMS, Fonseca LP. Biotechnol Ann Rev 2003; 9: 1387–2656
- Nakajima R, Yamazaki I. J Biol Chem 1987; 262: 2576–2581
- Mazmudar A, Adak S, Chatterjee R, Banerjee K. Biochem J 1997; 324: 713–719
- Noble RW, Gibson QH. J Biol Chem 1970; 245: 2409–2413
- Yousefi R, Saboury AA, Ghadermarzi M, Moosavi-Movahedi AA. Bull Korean Chem Soc 2000; 21(6)567–570
- Ator MA, Shantha K, Ortiz de Montellano PR. J Biol Chem 1987; 262: 14954–14960
- Yamazaki I, Nakajima R. Molecular and physiological aspects of plant peroxidases, H Greppin, C Penel, T Gaspar. University of Geneva, Geneva, Switzerland 1992; 71–84
- Hayashi Y, Yamazaki I. J Biol Chem 1979; 254: 9101–9106
- Ghadermarzi M, Moosavi-Movahedi AA, Ghadermarzi M. Biochim Biophys Acta 1999; 1431: 30–36