Abstract
Novel substituted 5,7-diaryl-2,3-dihydro-1,4-diazepines and 4,6-diaryl-2-aminopyrimidines were synthesized and tested for their antiproliferative activity. Title compounds were obtained by cyclocondensation of a substituted flavone with ethylenediamine and guanidine respectively. The cytotoxicity in vitro against various human leukemic cancer cell lines viz., Jurkat, HL60, MOLT3, NCEB-1, K562 was determined.
Introduction
The present work is in conjunction with our ongoing programme Citation1-3 on the syntheses and biological studies of various diaryl substituted heterocyclic systems. There is an overwhelming evidence indicating that various heterocyclic cores attached to a diaryl system possess diverse pharmacological activities viz., COX II inhibition [Citation4], allosteric modulation of GABAA receptor [Citation5] and estrogen receptor [Citation6], adenosine receptor antagonism [Citation7], selective p38α inhibition [Citation8], cannabinoid receptor antagonism [Citation9], antimitotic activity [Citation10] (combretastatin analogs), antiplatelet activity [Citation11] and anti-HIV-1 activity [Citation12] (TMC-125).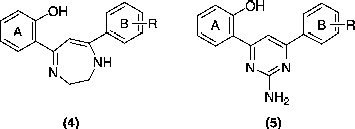
Recent reports indicate the multifarious activities [Citation13] of various benzodiazepines and diazepines condensed with different heterocyclic systems. In light of the reports available for diaryl heterocyclic systems, we envisaged the synthesis of diaryldiazepines and diaryl pyrimidines from a common intermediate. Here, we report some 5,7-diaryl-1,4-diazepine (4) and 4,6-diaryl-2-aminopyrimidine (5) compounds as antiproliferative agents.
Materials and methods
Chemistry
Melting points were determined on a Toshniwal melting point apparatus and are uncorrected. IR (cm− 1) spectra in KBr pellets were performed on a Shimadzu 8300 instrument and 1H NMR spectra were recorded on a Brüker spectrometer (300 or 400 MHz) using d6-DMSO or CDCl3 as solvent and tetramethylsilane as an internal standard. Chemical shift data are reported in parts per million (δ in ppm) where s, bs, and m designate singlet, broad singlet, and multiplet respectively. Elemental analyses were recorded on a Perkin-Elmer PE 2400 CHNS analyzer. Mass spectra were recorded on a JEOL SX 102/DA-6000 mass spectrometer. Thin-layer chromatography (TLC) was performed on precoated Silica gel Merck plates. Compounds were visualized by illuminating with UV light (254 nm) or exposure to iodine vapors. Solvents were purified using standard methods.
Flavones 3 were prepared according to literature methods [Citation14].
General method for the preparation of 5,7-diaryl-2,3-dihydro-1,4-diazepine derivatives
A mixture of the appropriate flavone 3a–k in ethylenediamine (20 mL) was refluxed for 2 h on an oil bath. The cooled mixture was poured into ice-water and the precipitate was filtered. The crude product was recrystallized from methanol.
5-(2-Hydroxyphenyl)-7-phenyl-2,3-dihydro-1H-1,4-diazepine (4a)
Mp 208-210°C. IR (KBr), νmax(cm− 1): 3231(N–H), 3000 (OH), 1605 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 3.65 and 3.95 (4H, bs, diazepine-C2 & C3-(CH2)2), 5.71(1H, s, diazepine-C6-H), 6.45–7.75 (9H, m, ArH), 8.3 (1H, bs, NH). ES-MS (m/z): 264 (M+, 100%). Anal. Calcd for C17H16N2O (264.32): C, 77.25; H, 6.10; N, 10.60. Found: C, 77.18; H, 6.24; N, 10.52%.
5-(2-Hydroxyphenyl)-7-(4-methylphenyl)-2,3-dihydro-1H-1,4-diazepine (4b)
Mp 235–237°C. IR (KBr), νmax(cm− 1): 3176(N–H), 2916 (OH), 1595 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 2.39 (3H, s, CH3), 3.72 and 3.88 (4H, bs, diazepine-C2 & C3-(CH2)2), 5.8 (1H, s, diazepine-C6-H), 6.53–7.62 (8H, m, ArH), 8.25 (1H, bs, NH). ES-MS (m/z): 279 (M++1, 100%). Anal. Calcd for C18H18N2O (278.35): C, 77.67; H, 6.52; N, 10.06. Found: C, 77.74; H, 6.50; N, 9.88%.
5-(2-Hydroxyphenyl)-7-(3-methylphenyl)-2,3-dihydro-1H-1,4-diazepine (4c)
Mp 206–208°C. IR (KBr), νmax(cm− 1): 3232 (N–H), 3000 (OH), 1608 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 2.45 (3H, s, CH3), 3.65 and 3.9 (4H, bs, diazepine-C2 & C3-(CH2)2), 5.7 (1H, s, diazepine-C6-H), 6.48–7.65 (8H, m, ArH), 8.27 (1H, bs, NH). ES-MS (m/z): 279 (M++1, 100%). Anal. Calcd for C18H18N2O (278.35): C, 77.67; H, 6.52; N, 10.06. Found: C, 77.70; H, 6.61; N, 9.99%.
5-(2-Hydroxyphenyl)-7-(4-methoxyphenyl)-2,3-dihydro-1H-1,4-diazepine (4d)
Mp 228–230°C. IR (KBr), νmax(cm− 1): 3221(N–H), 2922 (OH), 1604 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 3.63 (2H, s, diazepine-C3-H), 3.88 (5H, bs, OCH3 & diazepine-C2-H), 5.7 (1H, s, diazepine-C6-H), 6.45–7.65 (8H, m, ArH), 8.25 (1H, bs, NH). ES-MS (m/z): 295 (M++1, 100%). Anal. Calcd for C18H18N2O2 (294.35): C, 73.45; H, 6.16; N, 9.52. Found: C, 73.49; H, 6.13; N, 9.64%.
5-(2-Hydroxyphenyl)-7-(3-methoxyphenyl)-2,3-dihydro-1H-1,4-diazepine (4e)
Mp 185–188°C. IR (KBr), νmax(cm− 1): 3217 (N–H), 2985 (OH), 1600 (C = N) 1230 & 1048 (OCH3). 1H-NMR δ (400 MHz, DMSO-d6): 3.63 (2H, s, diazepine-C3-H), 3.9 (5H, bs, OCH3 & diazepine-C2-H), 5.7 (1H, s, diazepine-C6-H), 6.45–7.66 (8H, m, ArH), 8.3 (1H, bs, NH). ES-MS (m/z): 295 (M++1, 100%). Anal. Calcd for C18H18N2O2 (294.35): C, 73.45; H, 6.16; N, 9.52. Found: C, 73.56; H, 6.19; N, 9.49%.
5-(2-Hydroxyphenyl)-7-(4-chlorophenyl)-2,3-dihydro-1H-1,4-diazepine (4f)
Mp 243–245°C. IR (KBr), νmax(cm− 1): 3231(N–H), 3000 (OH), 1598 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 3.75 and 3.85 (4H, bs, diazepine-C2 & C3-(CH2)2), 5.74 (1H, s, diazepine-C6-H), 6.61–7.74 (8H, m, ArH), 8.3 (1H, bs, NH). ES-MS (m/z): 298 (M+, 100%). Anal. Calcd for C17H15ClN2O (298.77): C, 68.34; H, 5.06; N, 9.38. Found: C, 68.48; H, 5.04; N, 9.35%.
5-(2-Hydroxyphenyl)-7-(3-chlorophenyl)-2,3-dihydro-1H-1,4-diazepine (4g)
Mp 208–210°C. IR (KBr), νmax(cm− 1): 3200(N–H), 2916 (OH), 1610 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 3.62 and 3.92 (4H, bs, diazepine-C2 & C3-(CH2)2), 5.70 (1H, s, diazepine-C6-H), 6.5-7.7 (8H, m, ArH), 8.28 (1H, bs, NH). ES-MS (m/z): 298 (M+, 100%). Anal. Calcd for C17H15ClN2O (298.77): C, 68.34; H, 5.06; N, 9.38. Found: C, 68.41; H, 4.97; N, 9.50%.
5-(2-Hydroxyphenyl)-7-(2-chlorophenyl)-2,3-dihydro-1H-1,4-diazepine (4h)
Mp 210–212°C. IR (KBr), νmax(cm− 1): 3232(N–H), 2916 (OH), 1598 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 3.63 and 4.0 (4H, bs, diazepine-C2 & C3-(CH2)2), 5.37 (1H, s, diazepine-C6-H), 6.49–7.64 (8H, m, ArH), 8.4 (1H, bs, NH). ES-MS (m/z): 298 (M+, 100%). Anal. Calcd for C17H15ClN2O (298.77): C, 68.34; H, 5.06; N, 9.38. Found: C, 68.30; H, 5.08; N, 9.45%.
5-(2-Hydroxyphenyl)-7-(4-fluorophenyl)-2,3-dihydro-1H-1,4-diazepine (4i)
Mp 234–236°C. IR (KBr), νmax(cm− 1): 3203(N–H), 2916 (OH), 1604 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 3.65 and 3.9 (4H, bs, diazepine-C2 & C3-(CH2)2), 5.68 (1H, s, diazepine-C6-H), 6.5-7.8 (8H, m, ArH), 8.37 (1H, bs, NH). ES-MS (m/z): 282 (M+, 100%). Anal. Calcd for C17H15FN2O (282.31): C, 72.32; H, 5.36; N, 9.92. Found: C, 72.31; H, 5.29; N, 10.01%.
5-(2-Hydroxyphenyl)-7-(2-furyl)-2,3-dihydro-1H-1,4-diazepine (4j)
Mp 168–170°C. IR (KBr), νmax(cm− 1): 3206(N–H), 3000 (OH), 1608 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 3.65 and 3.9 (4H, bs, diazepine-C2 & C3-(CH2)2), 6.1 (1H, s, diazepine-C6-H), 6.53–7.96 (7H, m, ArH), 8.28 (1H, bs, NH). ES-MS (m/z): 254 (M+, 100%). Anal. Calcd for C15H14N2O2 (254.28): C, 70.85; H, 5.55; N, 11.02. Found: C, 70.96; H, 5.48; N, 10.98%.
5-(2-Hydroxyphenyl)-7-(2-thienyl)-2,3-dihydro-1H-1,4-diazepine (4k)
Mp 232–235°C. IR (KBr), νmax(cm− 1): 3203(N–H), 2980 (OH), 1595 (C = N). 1H-NMR δ (400 MHz, DMSO-d6): 3.6 and 3.9 (4H, bs, diazepine-C2 & C3-(CH2)2), 5.92 (1H, s, diazepine-C6-H), 6.58-7.8 (7H, m, ArH), 8.28 (1H, bs, NH). ES-MS (m/z): 270 (M+, 100%). Anal. Calcd for C15H14N2OS (270.35): C, 66.64; H, 5.22; N, 10.36. Found: C, 66.71; H, 5.20; N, 10.47%.
General method for the preparation of 4,6-diaryl-2-amino pyrimidine derivatives
A mixture of the flavone 3a–k (0.0019 mol), guanidine hydrochloride (0.01 mol) and potassium hydroxide (1.0 g) was refluxed in methanol (30 mL) for 4–6 h. After the completion of the reaction, the mixture was poured on to crushed ice containing acetic acid. The yellow solid obtained was filtered, washed with water and recrystallized from methanol to give 5a–k.
4-(2-Hydroxyphenyl)-6-phenyl-2-aminopyrimidine (5a)
Mp 174–76°C. IR (KBr), νmax(cm− 1): 3508, 3354 (NH2), 3200 (OH), 1625 (C = N). 1H-NMR δ (400 MHz, CDCl3): 5.37 (2H, s, NH2), 7.54 (1H, s, pyrimidine-C5-H), 6.9–8.0 (9H, m, ArH), 14.1 (1H, br, OH). ES-MS (m/z): 264 (M++1, 100%). Anal. Calcd for C16H13N3O (263.29): C, 72.99; H, 4.98; N, 15.96. Found: C, 72.85; H, 5.02; N, 16.0%.
4-(2-Hydroxyphenyl)-6-(4-methylphenyl)-2-aminopyrimidine (5b)
Mp 191–92°C. IR (KBr), νmax(cm− 1): 3500, 3330 (NH2), 3197 (OH), 1631(C = N). 1H-NMR δ (400 MHz, CDCl3): 2.43 (3H, s, CH3), 5.7 (2H, s, NH2), 7.54 (1H, s, pyrimidine-C5-H), 6.93–8.0 (8H, m, ArH), 13.5 (1H, br, OH). ES-MS (m/z): 278 (M++1, 100%). Anal. Calcd for C17H15N3O (277.32): C, 73.63; H, 5.45; N, 15.45. Found: C, 73.46; H, 5.54; N, 15.52%.
4-(2-Hydroxyphenyl)-6-(3-methylphenyl)-2-aminopyrimidine (5c)
Mp 124–26°C. IR (KBr), νmax(cm− 1): 3490, 3394 (NH2), 3200 (OH), 1639 (C = N). 1H-NMR δ (400 MHz, CDCl3): 2.46 (3H, s, CH3), 5.35 (2H, s, NH2), 7.54 (1H, s, pyrimidine-C5-H), 6.92–7.88 (8H, m, ArH), 14.2 (1H, br, OH). ES-MS (m/z): 278 (M++1, 100%). Anal. Calcd for C17H15N3O (277.32): C, 73.63; H, 5.45; N, 15.45. Found: C, 73.52; H, 5.38; N, 15.40%.
4-(2-Hydroxyphenyl)-6-(4??-methoxyphenyl)-2-aminopyrimidine (5d)
Mp 161–63°C. IR (KBr), νmax(cm− 1): 3492, 3327 (NH2), 3201 (OH), 1639 (C = N), 1249, 1025 (OCH3). 1H-NMR δ (400 MHz, CDCl3): 3.88 (3H, s, OCH3), 5.4 (2H, s, NH2), 7.50 (1H, s, pyrimidine-C5-H), 6.7-8.2 (8H, m, ArH), 14.3 (1H, br, OH). ES-MS (m/z): 294 (M++1, 100%). Anal. Calcd for C17H15N3O2 (293.32): C, 69.61; H, 5.15; N, 14.33. Found: C, 69.74; H, 5.08; N, 14.38%.
4-(2-Hydroxyphenyl)-6-(3-methoxyphenyl)-2-aminopyrimidine (5e)
Mp 203–06°C. IR (KBr), νmax(cm− 1): 3400, 3313 (NH2), 3176 (OH), 1647 (C = N), 1236, 1031 (OCH3). 1H-NMR δ (400 MHz, CDCl3): 3.92 (3H, s, OCH3), 6.0 (2H, s, NH2), 7.52 (1H, s, pyrimidine-C5-H), 6.9–7.8 (8H, m, ArH), 14.2 (1H, br, OH). ES-MS (m/z): 294 (M++1, 100%). Anal. Calcd for C17H15N3O2 (293.32): C, 69.61; H, 5.15; N, 14.33. Found: C, 69.68; H, 5.14; N, 14.30%.
4-(2-Hydroxyphenyl)-6-(4-chlorophenyl)-2-aminopyrimidine (5f)
Mp 239–41°C. IR (KBr), νmax(cm− 1): 3502, 3343 (NH2), 3217 (OH), 1641 (C = N). 1H-NMR δ (400 MHz, CDCl3): 6.2 (2H, s, NH2), 7.35 (1H, s, pyrimidine-C5-H), 6.9–8.0 (8H, m, ArH), 13.5 (1H, br, OH). ES-MS (m/z): 298 (M++1, 100%). Anal. Calcd for C16H12ClN3O (297.74): C, 64.54; H, 4.06; N, 14.11. Found: C, 64.72; H, 4.17; N, 13.98%.
4-(2-Hydroxyphenyl)-6-(3-chlorophenyl)-2-aminopyrimidine (5g)
Mp 183–85°C. IR (KBr), νmax(cm− 1): 3502, 3346 (NH2), 3203 (OH), 1616 (C = N). 1H-NMR δ (400 MHz, CDCl3): 5.24 (2H, s, NH2), 7.52 (1H, s, pyrimidine-C5-H), 6.9–8.0 (8H, m, ArH), 13.5 (1H, br, OH). ES-MS (m/z): 298 (M++1, 100%). Anal. Calcd for C16H12ClN3O (297.74): C, 64.54; H, 4.06; N, 14.11. Found: C, 64.65; H, 4.02; N, 14.14%.
4-(2-Hydroxyphenyl)-6-(2-chlorophenyl)-2-aminopyrimidine (5h)
Mp 190–192°C. IR (KBr), νmax(cm− 1): 3423, 3315 (NH2), 3187 (OH), 1629 (C = N). 1H-NMR δ (400 MHz, CDCl3): 5.68 (2H, s, NH2), 7.43 (1H, s, pyrimidine-C5-H), 6.76–7.8 (8H, m, ArH), 14.3 (1H, br, OH). ES-MS (m/z): 298 (M++1, 100%). Anal. Calcd for C16H12ClN3O (297.74): C, 64.54; H, 4.06; N, 14.11. Found: C, 64.73; H, 3.86; N, 14.00%.
4-(2-Hydroxyphenyl)-6-(4-fluorophenyl)-2-aminopyrimidine (5i)
Mp 221–23°C. IR (KBr), νmax(cm− 1): 3490, 3321 (NH2), 3201 (OH), 1647 (C = N). 1H-NMR δ (400 MHz, CDCl3): 5.5 (2H, s, NH2), 7.51 (1H, s, pyrimidine-C5-H), 6.9–8.1 (8H, m, ArH), 14.1 (1H, br, OH). ES-MS (m/z): 282 (M++1, 100%). Anal. Calcd for C16H12FN3O (281.28): C, 68.32; H, 4.30; N, 14.94. Found: C, 68.46; H, 4.52; N, 14.72%.
4-(2-Hydroxyphenyl)-6-(2-furyl)-2-aminopyrimidine (5j)
Mp 211–13°C. IR (KBr), νmax(cm− 1): 3417, 3285 (NH2), 3163 (OH), 1635 (C = N). 1H-NMR δ (400 MHz, CDCl3): 5.3 (2H, s, NH2), 7.52 (1H, s, pyrimidine-C5-H), 6.59–7.87 (7H, m, ArH), 14.3 (1H, br, OH). ES-MS (m/z): 254 (M++1, 100%). Anal. Calcd for C14H11N3O2 (253.26): C, 66.40; H, 4.38; N, 16.59. Found: C, 66.24; H, 4.56; N, 16.66%.
4-(2-Hydroxyphenyl)-6-(2-thienyl)-2-aminopyrimidine (5k)
Mp 182–84°C. IR (KBr), νmax(cm− 1): 3502, 3438 (NH2), 3342 (OH), 1635 (C = N). 1H-NMR δ (400 MHz, CDCl3): 5.27 (2H, s, NH2), 7.44 (1H, s, pyrimidine-C5-H), 6.9–7.8 (7H, m, ArH), 13.5 (1H, br, OH). ES-MS (m/z): 270 (M++1, 100%). Anal. Calcd for C14H11N3OS (269.32): C, 62.43; H, 4.12; N, 15.60. Found: C, 62.48; H, 4.01; N, 15.54%.
In vitro cytotoxicity assay
The results of the cytotoxic potencies of the synthesized compounds tested in vitro against five leukemic cell lines NCEB-1, HL60-DS, Jurkat E6-1, K562 and Molt-3 are summarized in . The synthesized compounds were dissolved in DMSO (5 μL of each compound at a 10 μM final concentration) and dispensed into an assay plate using acustom built low volume 384-well head tool. The assay plates were then loaded with 45 μL of cells and allowed to incubate with the compounds for 48 h at 37°C. Then, 5 μL of alamar blue reagent was added to the assay plate and incubated for 24 h at 37°C. Alamar blue reduction [Citation15] was measured on a CCD-based optical imaging reader.
Table I. Antiproliferative activity IC50 (μM) values of 5,7-diaryldiazepines 4a−k and 4,6-diarylpyrimidines 5a−k.
For initial screening, 1% DMSO was utilized as the high control to represent maximum reduction of alamar blue from cellular metabolism. The cytotoxic agent staurosporine was utilized at 50 μM to represent minimal reduction of alamar blue as a result of total cellular killing. These controls are used to calculate Z′ as a test of the functionality of the assay and to determine its range, robustness and reliability. For these cell lines, the controls gave a Z′ factor of 0.598 or better and a good signal to noise ratio indicating a broad dynamic range making the study reliable in our initial screening.
Results and discussion
Chemistry
The diaryldiazepine heterocyclic derivatives presented in this paper were prepared according to the route described in Scheme . The 1,3-diketones required for the work were obtained from the esters, by the base-catalyzed Baker-Venkataraman transformation [Citation16]. Condensation of 2-hydroxyacetophenones with various substituted benzoic acids in dry pyridine and POCl3 furnished the esters (1a–k). In the IR spectra, the 1,3-diketones showed absorption bands for C = O in the range 1615–1625 cm− 1. Furthermore, characteristic absorption bands of C = O of ketone and ester in the region 1681 and 1740 cm− 1 seen in (1a–k), are absent in the IR spectrum of the 1,3-diketones (2a–k). The traditional approach [Citation17] to the synthesis of 2,3-dihydro-1,4-diazepines is based on the reaction of 1,3-diketones with ethylenediamine in acidic medium. However, a competitive reaction also occurs giving flavones as bi products. To circumvent this problem we transformed the 1,3-diketones to the flavones (3a–k) which were then reacted with ethylenediamine to afford the required diazepines. This approach is by analogy with the report [Citation18] where substituted flavones undergo cyclocondensation reaction with amines in basic medium to form diazepines. The formation of the flavones [Citation14] 3a–k was confirmed by the appearance of C = O absorption bands at 1640–1660 cm− 1.
Scheme 1 Synthetic route to the diazepines (4a–k).
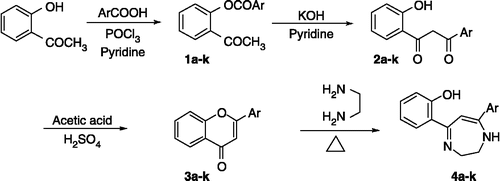
Substituted diazepines 4a–k resulted when the corresponding flavones were condensed with ethylenediamine which gave intensely yellow or orange colored diaryldiazepines in fairly good yield. The structures of the 5,7-diaryldiazepines are consistent with the IR and NMR spectra. The ring expansion was confirmed by the appearance of C = N stretching at 1595–1610 cm− 1 and NH stretching at 3200 cm− 1 in the IR spectrum. The –CH2–CH2– protons of the diazepine ring appeared as two broad singlets at δ 3.7 and 3.8, the N–H protons appeared as a broad singlet (δ 7.0–9.0) and the H-6 proton showed a sharp singlet at δ 5.8–6.0. The aromatic protons appeared as a multiplet. The hydroxyl proton could not be traced in the NMR spectra over the range 1–15 ppm. The mass spectra revealed the molecular ion as the base peak for most of the compounds.
Treatment of the flavones 3a–k with a slight excess of guanidine hydrochloride in alkaline medium afforded 4,6-diaryl-2-aminopyrimidines 5a–k (Scheme ). Compounds 5a–k showed a characteristic peak at 3200 cm− 1 for O–H str, asymmetric and symmetric stretching N–H bands at 3500 and 3350 cm− 1 respectively in the IR spectrum. In the 1H NMR spectra, hydroxyl protons were seen as a broad peak at δ 14–15, NH2 protons δ 5.3–5.5 and the pyrimidinyl proton (C5–H) appeared as a sharp singlet at δ 7.3–7.5. The aromatic protons appeared as a multiplet at δ 6.9–8.0.
Scheme 2 Synthetic route to the pyrimidines (5a–k).
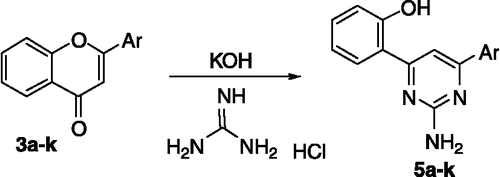
Cytotoxicity
Initial evaluation of 4a–k and 5a–k were carried out at the Memorial Sloan Kettering Cancer Center (MSKCC), New York (USA). The cytotoxic effects of 5,7-diaryl-1,4-diazepine and 4,6-diarylpyrimidine derivatives were tested using Alamar blue assay. Alamar blue (Resazurin) is commonly employed as an indicator of cell number and viability, since it is reduced to a (Resorufin) pink fluorescent dye in the medium by cell activity (possibly by oxygen consumption through metabolism). Alamar Blue is nontoxic to cells and does not necessitate killing of cells to obtain measurements, as is the case with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT).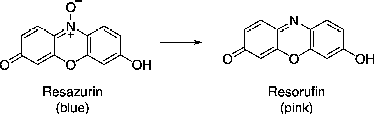
The various human leukemic cancer cell lines Jurkat, HL60, Molt3, NCEB-1 and K562 were incubated with various concentrations of the 5,7-diaryldiazepines and 4,6-diarylpyrimidines for 72 hours (48 h plus 24 h with dye). The resulting IC50 values for the compounds are summarized in . Compound 4a showed significant cytotoxic activity with IC50 values of 9.12 and 10.59 μM against K562 and Jurkat cell lines, respectively. Introduction of a methyl group at the meta position of the diaryldiazepine nucleus as in compound 4c, demonstrated high cytotoxic activity against the K562 and Jurkat cell lines (70 & 77% cell death, respectively, after 72 hours (48 h plus 24 h with dye) incubation time at 10 μM) with IC50 values of 4.55 and 8.47 μM respectively. Compound 4e, in which the methyl group was replaced with a greater electron donating group, a methoxy group, was also found to be cytotoxic but significantly less so than compound 4c. Results indicate that among the halo-substituted diaryldiazepines, compound 4 h, bearing a chloro substituent at the ortho position showed significant activity with an IC50 value of 7.66 μM against Jurkat cell line.
The other diaryl series, compounds 5a–k, in which the diazepine nucleus was replaced with a pyrimidine heterocyclic ring were all devoid of cell killing activity with the exception of compound 5c. Compound 5c showed considerable cytotoxic activity with an IC50 value of 11.95 μM against K562 cell line. The results indicate that some of the diazepines show moderate antiproliferative activity.
In conclusion, 4c and 4e are the most potent towards the K562 cell line, 4 h is potent towards the Jurkat cell line and 5c is quite good against all five types of cell lines.
Acknowledgements
The authors are grateful to the Indian Council of Medical Research (ICMR), New Delhi, India, for the award of a Senior Research Fellowship to R.R. The authors wish to acknowledge the AICTE, New Delhi for granting the RPS Project to RG. The authors also kindly acknowledge CDRI, Lucknow, India, for providing the NMR, Mass and Elemental data for the compounds. The authors also acknowledge the financial support from Mr. William H. Goodwin, and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the William Randolph Hearst Fund in Experimental Therapeutics, and the MSKCC Experimental Therapeutics Center.
References
- Ramajayam R, Giridhar R, Yadav MR. Synthesis of novel substituted diaryl-1,4-diazepines, Khim Geterotsikl Soedin 2006; 1042.
- Yadav MR, Shirude ST, Parmar A, Balaraman R, Giridhar R. Synthesis and anti-inflammatory activity of 2,3-diaryl-4(3H)-quinazolinones, Khim Geterotsikl Soedin 2006; 1198.
- Ramajayam R, Giridhar R, Yadav MR, de Clercq E. Identification of novel non-nucleoside reverse transcriptase inhibitors using fragment-based lead generation. Med Chem Res. (In press).
- Almansa C, de Arriba AF, Cavalcanti FL, Gomez LA, Miralles A, Merlos M, Rafanell JG, Forn J. Synthesis and SAR of a new series of COX-2-selective inhibitors: Pyrazolo[1,5-a]pyrimidines. J Med Chem 2001; 44: 350
- Asproni B, Talani G, Busonero F, Pau A, Sanna S, Cerri R, Mascia MP, Sanna E, Biggio G. Synthesis, structure-activity relationships at the GABAA receptor in rat brain, and differential electrophysiological profile at the recombinant human GABAA receptor of a series of substituted 1,2-diphenylimidazoles. J Med Chem 2005; 48: 2638
- von Rauch M, Busch S, Gust R. Investigations on the effects of basic side chains on the hormonal profile of (4R,5S)/(4S,5R)-4,5-Bis(4-hydroxyphenyl)-2-imidazolines. J Med Chem 2005; 48: 466
- Goblyos A, de Vries H, Brussee J, IJzerman AP. Synthesis and biological evaluation of a new series of 2,3,5-substituted [1,2,4]-thiadiazoles as modulators of adenosine A1 receptors and their molecular mechanism of action. J Med Chem 2005; 48: 1145
- Gill AL, Frederickson M, Cleasby A, Woodhead SJ, Carr MG, Woodhead AJ, Walker MT, Congreve MS, Devine LA, Tisi D, Reilly MO, Seavers LCA, Davis DJ, Curry J, Anthony R, Padova A, Murray CW, Carr RAE, Jhoti H. Identification of novel p38 MAP kinase inhibitors using fragment-based lead generation. J Med Chem 2005; 48: 414
- Lange JHM, Coolen HKAC, van Stuivenberg HH, Dijksman JAR, Herremans AHJ, Ronken E, Keizer HG, Tipker K, McCreary AC, Veerman W, Wals HC, Stork B, Verveer PC, den Hartog AP, de Jong NMJ, Adolfs TJP, Hoogendoorn J, Kruse CG. Synthesis, biological properties, and molecular modeling investigations of novel 3,4-diarylpyrazolines as potent and selective CB1 cannabinoid receptor antagonists. J Med Chem 2004; 47: 627
- Tron GC, Pagliai F, Grosso ED, Genazzani AA, Sorba G. Synthesis and cytotoxic evaluation of combretafurazans. J Med Chem 2005; 48: 3260
- Meanwell NA, Rosenfeld MJ, Kim Wright JJ, Brassard CL, Buchanan J0, Federici ME, Fleming JS, Gamberdella M, Hartl KS, Zavoico GB, Seiler SM. Nonprostanoid prostacyclin mimetics. 4. Derivatives of 2-[3-[2-(4,5-diphenyl-2-oxazolyl)ethyl]phenoxy]acetic acid substituted.alpha. to the oxazole ring. J Med Chem 1993; 36: 3871
- Guillemont J, Pasquier E, Palandjian P, Vernier D, Gaurrand S, Lewi PJ, Heeres J, de Jonge MR, Koymans LMH, Daeyaert FFD, Vinkers MH, Arnold E, Das K, Pauwels R, Andries K, de Bethune MP, Bettens E, Hertogs K, Wigerinck P, Timmerman P, Janssen PAJ. Synthesis of novel diarylpyrimidine analogues and their antiviral activity against human immunodeficiency virus type 1. J Med Chem 2005; 48: 2072
- Nawrocka W, Sztuba B, Opolski A, Wietrzyk J, Kowalska MW, Glowiak T. Synthesis and antiproliferative activity in vitro of novel 1,5-benzodiazepines, Part II. Arch Pharm Pharm Med Chem 2001; 334: 3, and references cited therein.
- Thakar KA, Muley PR. Indian J Chem 1975; 14: 226
- O'Brien J, Wilson I, Orton T, Pognan F. Investigation of the alamar blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem 2000; 267: 5421
- Borkhade KT, Marathey MG. Indian J Chem 1970; 8: 796
- Lloyd D, McDougall RH, Marshall DR. Part V- 2,3-Dihydro-1H-1,4-diazepines. J Chem Soc 1966; 4: 780
- Ghosh C, Tewari NJ. Heterocyclic systems. 8. condensation reactions of 4-oxo-4H-[1]benzopyran-3-carbonitrile. J Org Chem 1980; 45: 1964