Abstract
Albumin is generally regarded as an inert protein with no enzyme activity. However, albumin has esterase activity as well as aryl acylamidase activity. A new acetanilide substrate, o-nitrotrifluoroacetanilide (o-NTFNAC), which is more reactive than the classical o-nitroacetanilide, made it possible to determine the catalytic parameters for hydrolysis by fatty-acid free human serum albumin. Owing to the low enzymatic activity of albumin, kinetic studies were performed at high albumin concentration (0.075 mM). The albumin behavior with this substrate was Michaelis-Menten like. Kinetic analysis was performed according to the formalism used for catalysis at high enzyme concentration. This approach provided values for the turnover and dissociation constant of the albumin-substrate complex: kcat = 0.13 ± 0.02 min − 1 and Ks = 0.67 ± 0.04 mM. MALDI-TOF experiments showed that unlike the ester substrate p-nitrophenyl acetate, o-NTFNAC does not form a stable adduct (acetylated enzyme). Kinetic analysis and MALDI-TOF experiments demonstrated that hydrolysis of o-NTFNAC by albumin is fully rate-limited by the acylation step (kcat = k2). Though the aryl acylamidase activity of albumin is low (kcat/Ks = 195 M− 1min− 1), because of its high concentration in human plasma (0.6–1 mM), albumin may participate in hydrolysis of aryl acylamides through second-order kinetics. This suggests that albumin may have a role in the metabolism of endogenous and exogenous aromatic amides, including drugs and xenobiotics.
Introduction
Albumin is capable of binding numerous compounds and displays several enzymatic activities [Citation1], including esterase activity with p-nitrophenyl acetate and other aryl-esters, including drugs and prodrugs Citation2-4. The esteratic site of human albumin was found to be the binding site for several drugs, organophosphates and fatty acids. The topology of this active site was probed by site-directed mutagenesis [Citation5] and through the determination of the x-ray structure of albumin complexes [Citation6]. Recently, an aryl acylamidase (AAA, EC. 3.5.1.13) activity of albumin was demonstrated with o-nitroacetanilide as the substrate [Citation7]. The organophosphate diisopropylfluorophosphate progressively inhibited the AAA activity of albumin. This study provided evidence that the AAA active site is identical to the esteratic site. However, the activity of albumin with o-nitroacetanilide was so slow that no catalytic parameters of hydrolysis were determined. The present study was performed using a more reactive acetanilide, o-nitrotrifluoroacetanilide (o-NTFNAC) under conditions of catalysis at high enzyme concentration. The hydrolysis was Michaelis-Menten like which allowed determination of catalytic parameters. MALDI-TOF experiments showed that unlike the ester substrate p-NPA, o-NTFNAC does not form a stable adduct (acetylated enzyme) on Tyr 411. This indicated that the rate-determining step of hydrolysis of this substrate is acylation.
Materials and methods
Chemicals
Essentially fatty acid-free human serum albumin (FAF-HSA)>96% pure and ∼0.005% fatty acid, was from Sigma Chemical Co (Saint Quentin Fallavier, France). No further purification of FAF-HSA was carried out. o-NTFNAC was synthesized by Dr. R.S. McDonald (Mount Saint Vincent University, Halifax) as described [Citation8]. Butyrylthiocholine iodide was from Sigma France. Pepsin from porcine gastric mucosa (3690 units/mg) was purchased from Sigma (St. Louis, MO; P6887). A 1 mg/mL solution was prepared in 10 mM HCl and stored at − 80°C. A stock solution of 0.1 M p-nitrophenyl acetate (p-NPA, Sigma, St. Louis, MO; N8130) was prepared in acetonitrile. Alpha-cyano-4-hydroxycinnamic acid (Fluka 70990) was recrystallized from ethanol. All other chemicals were of biochemical grade.
Gel electrophoresis of FAF-HSA
Non-denaturing polyacrylamide gel (T = 6%) electrophoresis on slab gels was carried out under standard conditions [Citation9]. Loaded samples were: fresh human plasma (3 and 6 μL) and 2.5 to 20 μL of FAF-HSA (0.75 mM) in 60 mM Tris/HCl buffer, pH 8.0. The cholinesterase activity was revealed on gels using the method of Karnovsky and Roots [Citation11] with butyrylthiocholine iodide (1 mM) as the substrate. Non-denaturing gels were also stained for AAA activity using 10 mM o-nitroacetanilide as substrate and N-(naphthyl)ethylenediamine dihydrochloride (Sigma) to develop the color [Citation12].
Measurement of the AAA activity of albumin with o-NTFNAC
The AAA activity of FAF-HSA was determined with o-NTFNAC as the substrate in 60 mM Tris/HCl buffer, pH 8.0 at 25°C [Citation10]. The substrate concentration varied from 0.005 to 3 mM. Kinetic analysis was performed at high concentration of FAF-HSA ([E] = 0.075 mM). Stock solution of albumin (0.75 mM) was prepared by weighing the lyophilized FAF-HSA powder. Stock solutions of o-NTFNAC were 10 or 60 mM in 66% aqueous acetonitrile. The final acetonitrile concentration in the assay was kept constant at 3.5%. Release of the chromogenic product (o-nitroaniline) was continuously monitored for 5 min by spectrophotometry at 430 nm (ε = 3954 M− 1cm− 1). The time courses of substrate hydrolysis were linear over more than 60 min. The catalytic parameters were determined by non-linear fitting of the rate equation, v versus [S], using the Sigma plot 4 program (Jandel Sci., San Raphael, CA, USA).
Commercial preparations of human serum albumin that were not fatty acid free displayed lower AAA activity and were not used. Fatty acids block AAA activity because they compete with amides for binding to the same domain.
Measurement of AAA activity of human paraoxonase
Human paraoxonase (PON1) purified to homogeneity [Citation13] was tested for AAA activity with o-NTFNAC as described above. Because the concentration of PON1 in human plasma is about 1000 fold less than that of albumin, activity assays with PON1 were carried out with an enzyme concentration less than 0.1 μM.
Maldi-tof
Sample Preparation For Maldi-tof
FAF-HSA at a concentration of 5 mg/mL (0.075 mM) in TrisCl pH 8.0 buffer was treated with 5 mM o-NTFNAC or with 5 mM p-nitrophenyl acetate at 25°C. The control albumin in a volume of 1 mL was treated with 0.05 mL of acetonitrile. A 40 μL aliquot of albumin was removed, at intervals, between 10 min to 8 h after addition of substrate, and the pH was immediately adjusted to pH 2.1 by the addition of 8 μL of 1% TFA. Albumin was digested with 1 μL of 1 mg/mL pepsin for 2 h at 37°C. The reaction was stopped by diluting 10 μL of the digest into 600 μL of 50% acetonitrile, 0.1% TFA. The concentration of the diluted albumin was 1 pmol/μL. The diluted digests were applied in 1 μL aliquots to the MALDI-TOF target plate and overlaid with 1 μL of a saturated solution of alpha-cyano-4-hydroxycinnamic acid matrix.
Maldi-tof Spectrometry
Mass spectra were acquired with the Voyager DE-PRO MALDI-TOF mass spectrometer (Applied Biosystems, MDS Sciex, Foster City, CA) in linear positive ion mode, 20,000 volts accelerating voltage, 94% grid voltage, 0.1% guide wire, 350 ns extraction delay time, automated laser intensity adjustment from 1000 to 1600. The instrument was calibrated with a peptide calibration mixture from New England Biolabs. Mass accuracy for each standard was within 0.05% of the corresponding average molecular weight. Spectra were acquired in automatic mode, by examining signals from random spots on a target. The signals from the first 10 spots that met the acceptance criteria were summed into one final profile mass spectrum The acceptance criteria were signal intensities between 1000 and 55000 counts, with signal to noise ratios of 10 or greater, and minimum resolution of 50. The final spectrum was the average of 1000 shots.
The MS-Digest program from the UCSF Mass Spectrometry Facility was used to calculate the masses of the peptic peptides expected from digests of human serum albumin (accession number gi:28592).
Results
Absence of contaminating enzymes in FAF-HSA
The potential presence of human plasma butyrylcholinesterase (BuChE) as an estero-amidase contaminant of the HSA preparation was checked by gel electrophoresis. Indeed, BuChE was found to be a possible contaminant of human albumin preparations [Citation8] mostly because its monomer (C1) forms a covalent conjugate with albumin (C2 component in the multiple molecular form pattern of human plasma BuChE) [Citation9]. In addition, BuChE displays a high AAA activity with o-NTFNAC [Citation10]. However, in contrast to HSA, the FAF-HSA preparation was found to be free of BuChE based on highly sensitive cholinesterase activity detection on polyacrylamide gel.
Specific AAA activity gel staining [Citation12] detected BuChE tetramers (G4 form) in plasma but did not detect a band of AAA activity for albumin. At least 250 μL of human plasma had to be loaded per lane before an AAA band for BuChE was seen. The absence of an AAA band for albumin reflects the low catalytic activity of albumin toward o-nitroacetanilide.
Human plasma contains only three esterases: albumin, BuChE and paraoxonase [Citation14,Citation15]. To rule out the possibility that the AAA activity of HSA is due to contamination by paraoxonase, the AAA activity of highly purified human PON1 was tested. It was found that PON1 had no AAA activity toward o-NTFNAC.
Hydrolysis Of O-ntfnac By Faf-hsa
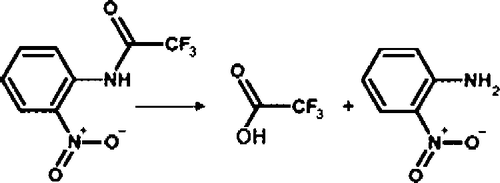
The hydrolysis of o-NTFNAC by FAF-HSA, to give the chromogenic product o-nitroaniline and trifluoroacetate, is illustrated in . Because there was neither substrate activation, substrate inhibition, nor product inhibition, the hydrolysis reaction can be depicted by a simple two-step enzyme hydrolysis mechanism (Scheme ).
Figure 1 Hydrolysis of o-NTFNAC to produce trifluoroacetic acid and o-nitroaniline.
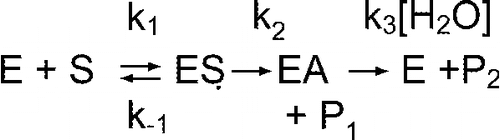
Ks = k− 1/k1 is the dissociation constant of the albumin-substrate complex, k2 the rate constant of acylation, and k3 the rate constant of deacylation. P1 is the arylamine hydrolysis product (o-nitroaniline) and P2 the carboxylic acid product (trifluoroacetic acid). The turnover number is kcat = k2k3/(k2+k3).
Because of the low affinity and slow reactivity of albumin with acetanilide substrates, assays were performed at high albumin concentration, [E] = 0.075 mM. shows the reaction velocity versus the substrate concentration. The AAA activity of FAF-HSA displays a Michaelis-Menten like behavior. That is, the reaction follows simple saturation kinetics and shows no substrate activation, no substrate inhibition, and no co-operativity.
Figure 2 Plot of velocity versus [S] for hydrolysis of o-NTFNAC by FAF-HSA ([E] = 0.075 mM) in 60 mM Tris/HCl buffer, pH 8.0 at 25°C. The continuous line is the fit to Equation (2). The velocity is expressed as the change in optical density (ΔOD) at λ = 430 nm per min.
![Figure 2 Plot of velocity versus [S] for hydrolysis of o-NTFNAC by FAF-HSA ([E] = 0.075 mM) in 60 mM Tris/HCl buffer, pH 8.0 at 25°C. The continuous line is the fit to Equation (2). The velocity is expressed as the change in optical density (ΔOD) at λ = 430 nm per min.](/cms/asset/ca8a0ee6-1fc2-4411-8da3-cc7cb4a3f75d/ienz_a_238282_f0003_b.gif)
Determination of the catalytic parameters of albumin-catalyzed hydrolysis of o-NTFNAC was possible using a formalism that applies to catalysis at high enzyme concentration [Citation16,Citation17]. This formalism was used rather than the standard Michaelis-Menten equation because the derivation of the Michaelis-Menten equation assumes that the enzyme concentration, [E]0, is far less than the total substrate concentration, [S]0, so that depletion of free substrate by binding to the enzyme is negligible under the assay conditions. These conditions did not apply to the assays in this study. Errors in estimates of kinetic parameters kcat and Km become important when [E]0 approaches Km.
The kinetic behavior at high enzyme concentrations has been theoretically investigated Citation17-19. The method of Cha [Citation16] requires that several approximations be made to take into account the depletion of substrate. When this method was applied to data in this study, it led to intolerable errors in values of kcat and Km. Alternatively, Schnell and coworkers demonstrated that when [E]0>>[S]0, S is in thermodynamic equilibrium with ES (d[S]/dt ≈ 0), and a reverse quasi-steady-state (rQSS) approximation could be made [Citation18,Citation20,Citation21]. The rQSS approximation led to a more dependable velocity equation for enzyme reactions at high [E]0:
In Equation (1), Ks is the dissociation constant of ES. However, errors can be introduced when using this equation because under rQSS conditions, [S] = [S]0-[ES], therefore [S] can be significantly different from the initial free substrate concentration, [S0]. Thus, we used the general rate equation that is valid at high concentration when [E]0 is of the same order as [S]0 and k2 ≈ k− 1 [Citation16,Citation17,Citation22]:
Objections to the use of Equation (2) based on the failure to account for formation of product in the substrate mass balance [Citation17] do not apply to the current application because velocity data were obtained from the initial rate, at which time product concentrations were very small relative to the initial substrate concentration and therefore did not affect the substrate mass balance.
Data from were fitted to Equation (2) and provided Ks = 0.67 ± 0.04 mM and kcat = 0.13 ± 0.02 min− 1. This gives a bimolecular constant, kcat/Ks, of 195 ± 30 M− 1min− 1. Note that fitting the data to Equation (1) gave Ks = 0.76 ± 0.04 mM. That value corresponds to Ks+[E]0, which is the apparent value of Ks that is obtained at low [S]0, taking into account the depletion of free substrate by binding to the enzyme [Citation16].
The rate constant for the spontaneous hydrolysis of o-NTFNAC, k0, in 60 mM Tris buffer pH 8.0, i.e., [OH− ] = 10− 6M, at 25°C was determined by monitoring the release of product P1, o-nitroaniline, over 5 min, using different concentrations of o-NTFNAC ranging from 0.05 to 2 mM. A plot of d[P1]/dt = k0[S] provided the value k0 = 0.0027 min− 1 at pH 8.0. Thus the ratio of kcat/k0 = 48 ± 7.
Maldi-tof
A procedure was developed for demonstrating the existence of an acylated albumin adduct during the hydrolysis of p-NPA by albumin. This technique was applied to the acetanilide substrate o-NTFNAC. The method requires a stable acylated adduct that can be captured as a peptide fragment after digestion of the protein by pepsin. The size of the peptide before and after reaction with substrate is measured in the mass spectrometer. The method also requires knowledge of the active site residue that covalently binds the acyl group of the substrate during catalysis. The active site of human albumin for esterase activity is Tyr 411 [Citation5,Citation22,Citation24].
lists the sequence of the Tyrosine 411 peptic peptides of human albumin and their masses before and after covalent attachment of the acyl group from p-NPA and from o-NTFNAC. Tyr 411 loses a hydrogen when it covalently binds these substrates, thus explaining why the added masses are 42 and 96 rather than 43 and 97. Two peptides are shown because in our experiments there was always one missed cleavage.
Table I. Expected sizes of Tyr 411 containing peptides after digestion of human albumin (accession# gi:28592) with pepsin, before and after reaction with substrates p-NPA or o-NTFNAC.
Panel A of shows the masses of peptic peptides from untreated albumin. Two Tyr 411 containing peptides had masses of 1718 and 1831 amu. Panel B shows that the reaction of albumin with p-NPA produced two new peptides with masses of 1760 and 1873. The acyl group of p-NPA is covalently attached to Tyr 411 in peptides 1760 and 1873. This result confirms the identity of Tyr 411 as the active site for albumin esterase activity. 100% of the Tyr 411 residues of albumin are occupied as shown by the complete absence of peptides with masses of 1718 and 1831 in panel B. All time points from 10 min to 8 h gave the same result as in panel B. It is concluded that the rate of formation of the acylated albumin adduct is fast (k2), and that the slow step in hydrolysis of the p-NPA ester is deacylation (k3). This provides direct evidence that deacylation is the rate-limiting step (k3 < k2) in albumin-catalyzed hydrolysis of p-NPA, confirming results obtained from burst kinetics [Citation2].
Figure 3 MALDI-TOF mass spectra showing the acyl-albumin adduct on Tyr 411. Panel A is untreated control albumin digested with pepsin. The Tyr 411 containing peptides at 1718 and 1831 amu are indicated. Panel B shows the acylated albumin peptides at 1760 and 1873 amu derived from the reaction of albumin with p-NPA. Panel C shows that the reaction of albumin with o-NTFNAC yields the same peptides as untreated control. The absence of acylated peptides for the reaction with o-NTFNAC suggests that deacylation is rapid with this substrate and that the rate determining step is the formation of the acyl-albumin adduct.
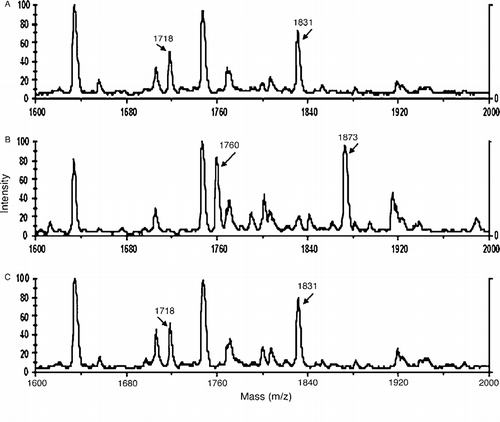
The reaction of o-NTFNAC with albumin rapidly produced a yellow color (o-nitroaniline), demonstrating that the acetanilide substrate was being hydrolyzed. However, Panel C in shows no acylated albumin adduct. At all time points tested, the Tyr 411 peptides had masses of 1718 and 1831 amu, indicating that Tyr 411 was not acylated. This suggests that the deacylation step is rapid for the trifluoroacetate adduct. The slow step in the turnover of o-NTFNAC is the formation of the acylated intermediate (k2) rather than deacylation (k3).
The possibility was considered that the active site for hydrolysis of o-NTFNAC might be a residue other than Tyr 411. However, comparison of all the peptides obtained from untreated control albumin and o-NTFNAC-treated albumin revealed no evidence for a modified peptide. Support for the conclusion that the active site for hydrolysis of o-NTFNAC is Tyr 411 is the finding that hydrolysis was blocked by pretreatment of albumin with soman, an organophosphorus compound that makes a covalent adduct with Tyr 411 [Citation25]. We conclude that the rate-determining step in hydrolysis of o-NTFNAC by albumin is formation of the acyl-albumin adduct, and that deacylation is rapid.
Discussion
Catalytic mechanism
The present study confirms previous observations that albumin has an intrinsic catalytic activity and extends the known substrates Citation1-4 to include the aryl acylamide, N-(o-nitrophenyl)trifluoroacetamide. Although the esterase/AAA activity of albumin is low, its enzymatic activity is characterized by: a) dependence on the presence of Tyr411 [Citation5], b) inhibition by covalent phosphorylation of Tyr411 [Citation5,Citation25], and c) saturable kinetics, displaying a Michaelis-Menten like behavior.
Means and Wu have shown that a stable acyl-albumin adduct is formed by reaction of 1.2 molar equivalents of p-NPA with human serum albumin. The adduct is stable to dialysis against pH 4.2 buffer for several days [Citation23]. The acylated residue was inferred to be Tyr 411. The conclusions of Means and Wu were confirmed by our MALDI-TOF experiments, which proved conclusively that Tyr 411 is the active site residue for the esterase activity of human serum albumin. Since the acetanilide hydrolase activity of albumin also requires Tyr 411 [Citation7], it was anticipated that an acyl-albumin adduct would be found in the reaction of o-NTFNAC with albumin. However, o-NTFNAC did not yield a stable acyl-peptide. This strongly suggests that the rate-determining step in the hydrolysis of o-NTFNAC by albumin is formation of the acyl-albumin adduct, and that deacylation is rapid and not rate-limiting in the overall catalytic mechanism. Thus, k2 = kcat.
Comparison of our kinetic data for hydrolysis of o-NTFNAC to previous data for hydrolysis of p-NPA by albumin [Citation4,Citation23] corroborates the above conclusion. The catalytic efficiency of albumin for hydrolysis o-NTFNAC (kcat/Ks = 195 M− 1min− 1 at pH = 8.0) is more than two orders of magnitude lower than for hydrolysis of p-NPA (kcat/Ks = 24200 M− 1min− 1 at pH = 7.4[Citation4]). The lower reactivity of o-NTFNAC can be attributed to a decrease in its acylation rate. The decreased acylation rate is due in part to resonance stabilization of amide bonds, which makes the energy needed to break the amide bond much higher than that needed to break an ester bond. In addition, the ortho-nitro substitution on the aromatic ring in the amide substrate (o-NTFNAC) has unfavorable effect on the acylation reaction compared to the para-nitro substitution in the ester substrate (p-NPA). For instance, kcat/Ks for hydrolysis of p-NPA is 13-fold higher than for o-NPA [Citation4]. Thus, the energy needed to acylate albumin should be higher for o-NTFNAC than for p-NPA, making k2(o-NTFNAC) < k2(p-NPA). This is in agreement with the value, k2 = 12 min− 1, reported by Means and Wu for acylation of human albumin by pNPA [Citation23]. Conversely, the electron-withdrawing effect of the three fluorine atoms in the trifluoroacetyl intermediate makes the carbonyl group more susceptible to hydrolysis of the acyl bond on Tyr411 relative to the acetyl-adduct from p-NPA. The differences in the molecular behaviors of the acetyl- and trifluoroacetyl-albumin moieties are reflected in the MALDI-TOF experiments (acetyl-albumin adduct detected, no trifluoroacetyl-albumin adduct observed, ), and in the kinetic parameters. Thus, k3(o-NTFNAC)>k3(p-NPA). Decreasing k2 and increasing k3 should cause a decrease in the accumulation of the acyl-intermediate, which is what is observed.
Perspectives
Enzymes that selectively hydrolyze aryl acylamides (i.e., serine carboxylesterase-amidases) have been found in various tissues, mostly brain and liver. They are involved in deacetylation of endogenous aryl acylamides (e.g melatonin) and exogenous aromatic amides (e.g. drugs and xenobiotics) [Citation26]. Thus there is a clear need for aryl acylamidase activity in vivo. The activity of HSA toward aromatic amides (o-nitroanilide [Citation7] and o-NTFNAC) coupled with the large concentration of albumin in serum raises the question of the physiological and pharmaco-toxicological relevance of the AAA activity of albumin.
Exogenous aromatic amides pass through the blood to reach their biological targets. It may be reasonably assumed that their concentrations in plasma, [C], are always far less than 67 μM, the value of Ks we determined for o-NTFNAC. Therefore, if albumin were involved in their degradation, the reaction would be pseudo-first order:
where kobs, the pseudo-first order rate constant, is:
Taking [HSA] as 0.6 mM and assuming kcat/Ks ≈ 200 M− 1min− 1 for hydrolysis of most aryl acylamide compounds by albumin (similar to the value found for o-NTFNAC), Equation (4) yields a value for kobs = 0.12 min− 1. Despite the high concentration of albumin in plasma this kobs is rather low. However, there are examples in the literature of enzymes that are considered to have an important function despite similarly poor kinetic constants. For example, the heme-regulated phosphodiesterase has a rate constant of 0.15 min− 1 with cAMP and a dissociation constant of 0.3 mM for nitric oxide [Citation27].
It is noteworthy that the arylacyl-amide bond cleaved by albumin is similar to that in pain-relieving drugs such as paracetamol and phenacetin. Binding of paracetamol to albumin both in plasma and in pure HSA solution was reported [Citation28]. This makes paracetamol and other arylacylamide-containing drugs potential substrates for albumin. In conclusion, albumin may be involved in deacetylation of endogenous and drug amides.
The AAA activity of albumin also raises the question of the contribution of this activity to the AAA activity of brain, liver and other organs. A part of the AAA activity in these organs has been ascribed to cholinesterases [Citation29]. However, measurements of the AAA activity in embryonic organ homogenates of double knockout mice for acetylcholinesterase and butyrylcholinesterase showed high levels of activity. This AAA activity may be ascribed to albumin (unpublished). Because it is impossible to completely eliminate albumin from blood vessels and from the lymphatic system in perfused isolated organs, albumin contributes to the AAA activity detected in various tissues.
| Abbreviations | ||
| AAA: | = | aryl acylamidase |
| FAF-HSA: | = | fatty acid-free human serum albumin |
| HSA: | = | human serum albumin |
| o-NTFNAC: | = | N-(o-nitrophenyl)trifluoroacetamide |
| p-NPA: | = | p-nitrophenylacetate |
Acknowledgements
This work was supported by Edgewood Biological Chemical Center, contract W911S-04-C-0019 (to OL), and DGA 03co010-05/PEA 01 08 7 and EMA/LR 06 to PM. Mass spectra were obtained with support from the Protein Structure Core Facility at the University of Nebraska Medical Center.
References
- Kragh-Hansen U, Tuam Giam Chuang V, Otagiri M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol Pharm Bull 2002; 25: 695–704
- Means GE, Bender ML. Acetylation of human serum albumin by p-nitrophenyl acetate. Biochemistry. 1975; 14: 4989–4994
- Salvi A, Carrupt PA, Mayer JM, Testa B. Esterase-like activity of human serum albumin toward prodrug esters of nicotinic acid. Drug Metab Dispos 1997; 25: 395–398
- Sakurai Y, Ma SF, Watanabe H, Yamaotsu N, Hirono S, Kurono Y, Kragh-Hansen U, Otagiri M. Esterase-like activity of serum albumin: Characterization of its structural chemistry using p-nitrophenyl esters as substrates. Pharm Res 2004; 21: 285–292
- Watanabe H, Tanase S, Nakajou K, Maruyama T, Kragh-hansen U, Otagiri M. Role of Arg-410 and Tyr-411 in human serum albumin for ligand binding and esterase-like activity. Biochem J 2000; 349: 813–819
- Ghuman J, Zunszain PA, Petitpas I, Bhattachrya AA, Otagiri M, Curry S. Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol 2005; 353: 38–52
- Manoharam I, Boopathy R. Diisopropylfluorophosphate-sensitive aryl acylamidase activity of fatty acid free human serum albumin. Arch Biochem Biophys 2006; 452: 186–188
- Chapuis N, Bruhlmann C, Reist M, Carrupt PA, Mayer JM, Testa B. The esterase-like activity of serum albumin may be due to cholinesterase contamination. Pharm Res 2001; 18: 1435–1439
- Masson P. A naturally-occuring molecular form of human plasma cholinesterase is an albumin conjugate. Biochim Biophys Acta 1989; 988: 258–268
- Darvesh S, McDonald RS, Darvesh KV, Mataija D, Mothana S, Cook H, Carneiro KM, Richard N, Walsh R, Martin E. On the active site for hydrolysis of aryl amides and choline esters by human cholinesterases. Bioorg Med Chem 2006; 14: 4586–4599
- Karnovsky MJ, Roots L. A “direct-coloring” thiocholine method for cholinesterases. J Histochem Cytochem 1964; 12: 219–220
- Jaganathan L, Boopathy R. A direct method to visualise the aryl acylamidase activity on cholinesterases in polyacrylamide gels. BMC Biochem 2000; 1: 3, http://www.biomedcentral.com/1471-2091/1/3
- Renault F, Chabrière E, Andrieu JP, Dublet B, Masson P, Rochu D. Tandem purification of two HDH-associated partner proteins in human plasma, paraoxonase (PON1) and phosphate binding protein (HPBP) using hydroxyapatite chromatography. J Chromagr B 2006; 836: 15–21
- Li B, Sedlacek M, Manoharan I, Boopathy R, Duysen EG, Masson P, Lockridge O. Butyrylcholinesterase, paraoxonase, and albumin esterase, but no carboxylesterase, are present in human plasma. Biochem Pharmacol 2005; 70: 1673–1684
- Satoh T, Hosokawa M. Structure, function and regulation of carboxylesterases. Chem Biol Interact 2006; 162: 195–211
- Cha S. Kinetic behavior at high enzyme concentrations. J Biol Chem 1970; 245: 4814–4818
- Schnell S, Maini PK. A century of enzyme kinetics: reliability of the KM and Vmax estimates. Comments on Theor Biol 2003; 8: 169–187
- Schnell S, Maini PK. Enzyme kinetics at high concentration. Bull Math Biol 2000; 62: 483–499
- Tzafriri AR. Michaelis-Menten kinetics at high enzyme concentrations. Bull Math Biol 2003; 65: 1111–1129
- Schnell S, Maini PK. Enzyme kinetics far from the standard quasi-steady-state and equilibrium approximations. Math Comput Moldel 2002; 35: 137–144
- Schnell S, Mendoza C. The condition for pseudo-first-order kinetics in enzymatic reactions is independent of the initial enzyme concentration. Biophys Chem 2004; 107: 165–174
- Toti P, Petri A, Pelaia V, Osman AM, Paolini M, Bauer C. A linearization method for low catalytic activity enzyme kinetic analysis. Biophys J 2005; 114: 245–251
- Means GE, Wu HL. The reactive tyrosine residue of human serum albumin: Characterization of its reaction with diisopropylfluorophosphate. Arch Biochem Biophys 1979; 194: 526–530
- Schopfer LM, Champion MM, Tamblyn N, Thompson CM, Lockridge O. Characteristic mass spectral fragments of the organophosphorus agent FP-biotin and FP-biotinylated peptides from trypsin and bovine albumin (Tyr410). Anal Biochem 2005; 345: 122–132
- Li B, Froment MT, Nachon F, Verdier L, Debouzy JC, Brasme B, Gillon E, Schopfer LM, Lockridge O, Masson P. Binding and hydrolysis of soman by human serum albumin, (to be submitted).
- Satoh T. Role of carboxylesterases in xenobiotic metabolism. Rev Biochem Toxicol 1987; 8: 155–181
- Sasakura Y, Yoshimura-Suzuki T, Kurokawa H, Shimizu T. Structure-function relationships of EcDOS, a heme-regulated phosphodiesterase from Escherichia coli. Acc Chem Res 2005; 39: 37–43
- Milligan TP, Morris HC, Hammond PM, Price CP. Studies on paracetamol binding to proteins. Ann Clin Biol 1994; 31: 492–496
- George ST, Balasubramanian AS. The aryl acylamidases and their relationship to cholinesterases in human serum, erythrocyte and liver. Eur J Biochem 1981; 121: 177–186