Abstract
Cathepsins have been found to have important physiological roles. The implication of cathepsin L in various types of cancers is well established. In a search for selective cathepsin L inhibitors as anticancer agents, a series of 2-cyanoprrolidine peptidomimetics, carrying a nitrile group as warhead, were designed. Two series of compounds, one with a benzyl moiety and a second with an isobutyl moiety at P2 position of the enzyme were synthesized. The synthesized compounds were evaluated for inhibitory activity against human cathepsin L and cathepsin B. Although, none of the compounds showed promising inhibitory activity, (E)N-{(S)1-[(S)2-cyano-1-pyrrolidinecarbonyl]-3-methylbutyl}-2,3-diphenylacrylamide (24) with an isobutyl moiety at P2 was found to show selectivity as a cathepsin L inhibitor (Ki 5.3 μM for cathepsin L and Ki > 100 μM for cathepsin B). This compound could act as a new lead for the further development of improved inhibitors within this inhibitor type.
Introduction
The lysosomal compartment of the cell is responsible for the controlled recycling of cellular organelles and macromolecules[Citation1]. Both heterophagic and autophagic cargos find their final destiny in lysosomes, where they are broken down by numerous hydrolases. The recent completion of the human genome sequencing has revealed the encoding of eleven cysteine proteases of the papain superfamily[Citation2]. Proteases of the cathepsin family are among the best studied lysosomal hydrolases[Citation3]. These enzymes cleave a plethora of protein substrates often with overlapping specificities leading to the notion that there may be redundancy in their function. However, recent specific targeting in transgenic mice revealed that cathepsins have very distinctive roles in fulfilling specific cell functions[Citation4]. Cathepsins have been found to have important physiological roles in bone resorption and remodeling[Citation5,Citation6], thyroid hormone liberation[Citation7] and immune response processes[Citation8]. They have also been implicated in a number of degenerative processes that include osteoporosis, rheumatoid arthritis[Citation9], emphysema and muscular dystrophy[Citation10]. There is a strong evidence for the participation of cathepsins in cancer where, they have been shown to act intracellularly[Citation11] and extracellularly in tumor invasion and metastasis[Citation12,Citation13]. It is recognized that many cancer cells secrete cathepsin L to degrade the components of extracellular matrices and basement membranes thus, promoting tumor invasion and metastasis. It has been demonstrated that approximately 10-fold higher mature cathepsin L activity was observed extracellularly in a medium of human fibrosarcoma (HT1080) cells compared with their intracellular activity[Citation14]. Recently, it has been demonstrated that this protease acts as central mediator of the invasive capacity of S-adenosylmethionine decarboxylase-transformed malignant fibroblasts, probably by proteolytic degradation of extracellular matrix constituents[Citation15]. Its involvement in the metastasis of human melanoma cells has also been reported[Citation16]. Therefore, cathepsin L is considered to be an attractive target for the development of anticancer agents.
Development of a potent and selective cathepsin inhibitor for a particular pathological condition poses a great challenge to the researchers due to varied and vital roles played by different cysteine proteases in human physiology[Citation17]. The difficulty in designing selective cathepsin inhibitors results from the similarity in their substrate recognition as well as their common proteolytic mechanism[Citation18]. Many chemotypes have been devised as inhibitors of cystein proteases[Citation19]. A majority of the reported inhibitors[Citation20] are either peptidic or peptidomimetic compounds in which the hydrolyzable amide is replaced by an electrophilic moiety. Till recently, most of the reported cysteine protease inhibitors have been irreversible inhibitors like fluromethyl ketones[Citation21], vinylsulfones[Citation22] or epoxysuccinates[Citation23]. Numerous warheads[Citation24] have been utilized in designing of reversible inhibitors of cysteine proteases including peptidic aldehydes[Citation25], nitriles[Citation26], cyclopropanones[Citation27], diamino ketones[Citation28] and α-ketoamides[Citation29]. There has also been a report on the use of non-covalent amides as cathepsin K inhibitors[Citation30].
Nitriles have been reported to be class selective protease inhibitors which strongly inhibit cysteine proteases. Peptide nitriles bind to the active site (thiol moiety) of the enzyme forming a reversible covalent thioimidate intermediate as indicated by the NMR studies (Scheme )Citation31-33. However, reports on nitriles as inhibitors of serine proteases are also available in the literature[Citation34,Citation35].
Scheme 1 Reversible formation of thioimidates from peptide nitriles and cysteine proteases.
Endogenous inhibitors of cathepsins include high molecular weight propeptide segments and cystatins[Citation36]. Design of small size molecules as inhibitors typically involves di- or tri-peptides with an electrophilic ‘warhead’ covalently attached to the peptide chain. While the electrophilic warhead binds reversibly or irreversibly with the enzyme to cause its inhibition, hydrogen bonding and hydrophobic interactions of the peptide backbone and attached side chains to the enzyme determine its selectivity towards a specific enzyme. Nonpeptidic N-cyanopyrrolidines[Citation37] have been reported earlier as potent inhibitors of various cathepsins. Introduction of peptidic chains in such scaffolds has led to the development of compounds with major differences in inhibitory potencies and selectivities towards four different cathepsins (B, K, L and S)[Citation38]. In order to investigate whether additional potency and selectivity could be achieved by the structural modifications of such compounds, we have synthesized some dipeptides having 2-cyanopyrrolidine as electrophilic warhead. As compounds with benzyl or branched alkyl chains[Citation39,Citation40] are reported to be suitable at P2 positions we have retained these functions in the synthesized compounds. Carboxamides like morpholinecarboxamide[Citation41,Citation42], naphthalene-1-carboxamide[Citation43] and diphenylacetamide[Citation39] have been used in literature at P3 position for various cathepsin inhibitors. α,β-Unsaturated groups were used at P3 to introduce additional electrophilic functionality at this site in the compounds reported herein. To the best of our knowledge this is the first report on 2-cyanopyrrolidine peptidomimetics as selective cathepsin L inhibitors.
Materials and methods
Chemistry
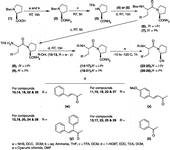
The 2-cyanopyrrolidine derivatives were prepared following the general method outlined in Scheme . Boc-proline (1), Boc-leucine (4) and Boc-phenylalanine (5) were prepared following the reported procedure[Citation44]. Cinnamic acid (10, R = w), p-methoxycinnamic acid (11, R = x) and α-phenylcinnamic acid (12, R = y) were also prepared by the reported procedures[Citation45]. All the synthesized compounds were characterized by their spectral and elemental data. Melting points (m.p.) were determined using a Veego make melting point apparatus (silicon oil type) and are uncorrected. FT-IR was recorded on Shimadzu–8300 model using KBr pelletes/neat samples. 1H-NMR spectra were recorded with Bruker spectrometer (300/400 MHz), in deuterated chloroform (CDCl3), unless specified. Chemical shifts are reported in parts per million (ppm, δ units) using TMS as an internal standard. Coupling constants (J) are reported in units of hertz (Hz). Mass spectral data were obtained on a QTRAP Applied Biosystem SCIEX spectrometer. Elemental analyses were performed on Perkin-Elmer/Carlo-Erba elemental analyzer.
Scheme 2 Synthesis of 2-cyanepyrrolidines.
H-Pro-NH2 TFA salt (3)
To an ice-cooled mixture of Boc-proline (1) (5 g, 0.023 mole) and N-hydroxysuccinimide (NHS) (2.67 g, 0.023 mole) in dichloromethane (DCM) (80 mL), N,N′-dicyclohexylcarbodiimide (DCC) (4.78 g, 0.023 mole) was added and the reaction mixture was stirred at RT for 16 h. The reaction mixture was filtered to remove N,N′-dicyclohexylurea (DCU) and the filtrate was concentrated to obtain an oily residue, which was dissolved in tetrahydrofuran (THF) (70 mL), and treated with aq ammonia (7 mL, 30%) at 0–5°C. The reaction mixture was stirred for 5 h at RT and concentrated under vacuum to afford a sticky mass which was diluted with water (50 mL) and extracted with chloroform (5 × 50 mL). The organic layer was combined and washed with brine (2 × 50 mL). After drying, the solvent was removed under vacuum and the product was crystallized from ethyl acetate to give the amide (2), (4 g, 80%); m. p. 108–10°C; IR (KBr, cm− 1): 3381, 3201, 1706, 1684, 1364 and 1164. A cold solution of compound (2) (4.28 g) in DCM (40 mL) was treated with trifluoroacetic acid (TFA) (8 mL) and stirred at RT for 90 min. It was concentrated under reduced pressure to get a yellow sticky mass (3) in quantitative yield. The crude product was used for the next step without further purification.
Boc-Leu-Pro-NH2 (6):
To a cold solution of Boc-Leu-OH (4), (5 g, 0.02 mole) in DCM (90 mL), 1-hydroxybezotriazole (1-HOBT) (2.7 g, 0.02 mole) and 1-(3-dimethylaminopropyl)-3-ethylcabodiimide hydrochloride (EDC) (3.84 g, 0.02 mole) were added. The reaction mixture was stirred for 45 min at 10–15°C. A solution of (3) (4.28 g, 0.02 mole) in DCM (20 mL) was added into the above solution followed by the addition of triethylamine (TEA) (7 mL). This reaction mixture was stirred at RT for 15 h, washed with aq. citric acid (0.5 M, 2 × 50 mL), aq. sodium bicarbonate (5%, 2 × 50 mL) and brine (2 × 50 mL), sequentially. The organic layer was dried, concentrated and purified by column chromatography using ethyl acetate in n-hexane (30%) as eluent to afford (6); Yield 4.5 g (64%); m. p. 76–78°C; IR (KBr, cm− 1): 3381, 3201, 1706, 1684, 1647, 1437, 1366 and 1170.
Boc-Phe-Pro-NH2 (7)
Compound (7) was prepared by reacting Boc-Phe-OH (5) (5.3 g, 0.02 mole) with (3) (4.28 g, 0.02 mole) as described above for compound (6). Yield 3.8 g (53%); m. p. 88–90°C; IR (KBr, cm− 1): 3370, 3221, 1700, 1679, 1642, 1437 and 1350.
General method of synthesis for compounds (14–17)
Compound 6 (3 g, 9.1 mmole) was dissolved in DCM (40 mL) and the solution was cooled to 5–10°C. TFA (5 mL) was added slowly into the above solution and the reaction mixture was stirred at RT for 90 min. The reaction mixture was concentrated to get the TFA salt (8) as yellow oil, which was dried in desiccator and dissolved in DCM (10 mL).
Cinnamic acid (10; R = w) (1.34 g, 9.1 mmol) was dissolved in DCM (40 mL) and the solution was cooled to 10–15°C. EDC (1.4 g, 9.1 mmole) and 1-HOBT (1.2 g, 9.1 mmole) were added to the above solution and the reaction mixture was stirred further for 45 min at 10–15°C. The TFA salt (8) (2.7 g, 9.1 mmol) prepared as described above was added to the above solution followed by the addition of TEA (7 mL, 50.3 mmol) at 10–15°C. The resulting mixture was stirred at RT for 15 h, washed with aq. citric acid (0.5 M, 2 × 50 mL), aq. sodium bicarbonate (5%, 2 × 50 mL) and brine (2 × 50 mL) in the given sequence. The organic layer was dried and concentrated under reduced pressure to afford compound (14) which was purified by column chromatography using silica as adsorbent and n-hexane in ethylacetate (20–30%) as eluent. The compound (14) was used as such for further reactions.
Compounds (15–17) were prepared in analogy to compound 14 starting from 4-methoxy cinnamic acid (11; R = x), α-phenyl cinnamic acid (12; R = y) and benzoic acid (13; R = z), respectively.
General method of synthesis for compounds (18–21)
Compound (7) (3.2 g, 0.01 mole) was treated with TFA under similar conditions as described for compound (8) to afford its TFA salt (9). This was treated with acids (10–13, 0.01 mole) under standard peptide coupling conditions as described for (14–17) to afford compounds (18–21). These compounds (18–21) were also used as such for the next step.
General method of synthesis for compounds (22–29)
A solution of amide (14–21) (0.0024 mole) in dry dimethyl formamide (DMF) (7 mL) was cooled to − 10°C. Cyanuric chloride (0.57 g, 0.0031 mole) was added to this solution in portions over a period of 10 min and the reaction mixture was stirred at the same temperature for 1 h. The reaction mixture was poured over crushed ice and extracted with ethyl acetate (3 × 20 mL). Combined organic layer was washed with brine (2 × 30 mL), dried and concentrated. The crude compound was purified by column chromatography using ethyl acetate in n-hexane (30%) to afford the desired products (22–29) which were crystallized from ethyl acetate and hexane.
(E)N-{(S)1-[(S)2-Cyano-1-pyrrolidinecarbonyl]-3-methylbutyl}-3-phenylacrylamide (22; R = w)
Yield 25%; m.p. 98-100°C; IR (KBr, cm− 1): 3280, 2246, 1652, 1628, 1537 and 1423; 1H-NMR: 0.98–1.00 (d, 3H, J = 3.9 Hz), 1.02–1.04 (d, 3H, J = 3.9 Hz), 1.38–1.78 (m, 3H), 2.21–2.33 (m, 4H), 3.68–3.71 (m, 1H), 3.86–3.94 (m, 1H), 4.75–4.79 (m, 1H), 4.83–4.92 (m, 1H), 6.45–6.51 (d, 1H), 7.24–7.42 (m, 5H), 7.62–7.67 (d, 1H); C20H25N3O2: Requires C, 70.77; H, 7.42; N, 12.38. Found C, 70.80; H, 7.38; N, 12.43%.
(E)N-{(S)1-[(S)2-Cyano-1-pyrrolidinecarbonyl]-3-methylbutyl}-3-(4-methoxyphenyl)acrylamide (23; R = x)
Yield 27%; m.p. 75–78°C; IR (KBr, cm− 1): 3272, 2245, 1659, 1625, 1602, 1512, 1423 and 1173; 1H-NMR: 0.97–0.99 (d, 3H, J = 4 Hz), 1.01–1.03 (d, 3H, J = 4 Hz), 1.26–1.81 (m, 3H), 2.18–2.31 (m, 4H), 3.66–3.71 (m, 1H), 3.83 (s, 3H), 3.85–3.93 (m, 1H), 4.78–4.80 (m, 1H), 4.86–4.94 (m, 1H), 6.26–6.31 (d, 1H, J = 15.6 Hz), 6.38–6.41 (d, 1H, J = 8.46 Hz), 6.86–6.89 (d, 2H, J = 8.7 Hz), 7.39–7.43 (d, 2H, J = 8.7 Hz) and 7.51–7.56 (d, 1H, J = 15.6 Hz); C21H27N3O3: Requires C, 68.27; H, 7.36; N, 11.37. Found C, 67.88; H, 7.10; N, 10.94%.
(E)N-{(S)1-[(S)2-Cyano-1-pyrrolidinecarbonyl]-3-methylbutyl}-2,3-diphenylacrylamide (24; R = y)
Yield 40%; m.p. 160–162°C; IR (KBr, cm− 1): 3422, 2232, 1666, 1652, 1620, 1507, 1423 and 1192; 1H-NMR: 0.90–0.92 (d, 3H), 0.97–0.98 (d, 3H), 1.25–1.48 (m, 1H), 1.52–1.65 (m, 2H), 2.19–3.00 (m, 4H), 3.45–3.68 (m, 1H), 3.84–3.92 (m, 1H), 4.75–4.78 (m, 1H), 4.81–4.88 (m, 1H), 6.00–6.04 (d, 1H), 6.96–6.99 (m, 2H), 7.10–7.20 (m, 3H), 7.24–7.27 (m, 2H), 7.43–7.49 (m, 3H) and 7.80 (s, 1H); C26H29N3O2: Requires C, 75.15; H, 7.03; N, 10.11. Found C, 75.70; H, 7.25; N, 10.65%.
N-{(S)1-[(S)2-Cyano-1-pyrrolidinecarbonyl]-3-methylbutyl}benzamide (25; R = z)
Yield 40%; m.p. 143–144°C; IR (KBr, cm− 1): 3268, 2245, 1673, 1635, 1528, 1490 and 1293; 1H-NMR (200 MHz): 0.98–1.00 (d, 3H, J = 4 Hz), 1.03–1.05 (d, 3H, J = 4 Hz), 1.61–1.65 (m, 1H), 1.70–1.81 (m, 2H), 2.1–2.32 (m, 4H), 3.68–3.74 (m, 1H), 3.85– 3.92 (m, 1H), 4.77–4.81 (m, 1H), 4.94–5.02 (m, 1H), 6.75–6.78 (d, 1H), 7.49 (m, 3H) and 7.78 (d, 2H); Mass (m/z): 314 (M + H)+; C18H23N2O2: Requires C, 72.21; H, 7.74; N, 9.36. Found C, 72.29; H, 7.80; N, 9.32%.
N-[(S)1-Benzyl-2-((S)2-cyano-1-pyrrolidinyl)-2-oxoethyl]-3-phenylacrylamide (26; R = w)
Yield 32%; m.p. 113–115°C; IR (KBr, cm− 1): 3272, 2245, 1659, 1625, 1602, 1512, 1423 and 1173; 1H-NMR: 2.15–2.30 (m, 4H), 3.05–3.09 (m, 2H), 3.68–3.70 (m, 1H), 3.82–3.85 (m, 1H), 4.68–4.72 (m, 1H), 4.92–5.02 (m, 1H), 6.38–6.50 (d, 1H), 7.08–7.21 (m, 5H), 7.25–7.50 (m, 10H), 7.58–7.70 (d, 1H); C23H23N3O2: Requires C, 73.97; H, 6.21; N, 11.25. Found: C, 73.83; H, 6.30; N, 11.03%.
N-[(S)1-Benzyl-2-((S)2-cyano-1-pyrrolidinyl)-2-oxoethyl]-3-(4-methoxyphenyl) acrylamide (27; R = x)
Yield 30%; m.p. 93–95°C; IR (KBr, cm− 1): 3270, 2242, 1659, 1620, 1602 and 1163; 1H-NMR: 1.90–2.72 (m, 4H), 2.97–3.05 (m, 2H), 3.41–3.47 (m, 1H), 3.72 (s, 3H), 3.75–3.80 (m, 1H), 4.64–4.67 (m, 1H), 4.85–4.92 (m, 1H), 6.30–6.36 (d, 1H), 6.40–6.43 (d, 1H), 6.92–7.41 (m, 9H), 7.48–7.56 (d, 1H); C24H25N3O3: Requires: C, 71.44; H, 6.25; N, 10.41. Found: C, 71.38; H, 6.22; N, 10.34%.
N-[(S)1-Benzyl-2-((S)2-cyano-1-pyrrolidinyl)-2-oxoethyl]-2,3-diphenylacrylamide (28; R = y)
Yield 43%; m.p. 171–172°C; IR (KBr, cm− 1): 3372, 2232, 1659, 1625, 1602, 1512, 1423 and 1173; 1H-NMR: 1.86–2.18 (m, 4H), 3.05–3.10 (m, 2H), 3.53–3.82 (m, 2H), 4.68–4.75 (m, 1H), 4.95–5.02 (m, 1H), 6.72–6.75 (d, 1H), 7.12–7.50 (m, 15H), 7.67 (s, 1H); C29H28N3O2: Requires: C, 77.31; H, 6.26; N, 9.33. Found: C, 77.24; H, 6.33; N, 9.40%.
N-[(S)1-Benzyl-2-((S)2-cyano-1-pyrrolidinyl)-2-oxoethyl]benzamide (29; R = z)
Yield 37%; m.p. 163–165°C; IR (KBr, cm− 1): 3272, 2242, 1670, 1625, 1602, 1512, 1423 and 1173; 1H-NMR: 1.95–2.20 (m, 4H), 3.01–3.07 (m, 2H), 3.30–3.67 (m, 2H), 4.68–4.75 (m, 1H), 4.88–4.97 (m, 1H), 6.53–6.56 (d, 1H), 7.20–7.58 (m, 8H), 7.70–7.76 (m, 2H); C21H21N3O2: Requires: C, 72.6; H, 6.09; N, 12.09. Found: C, 73.08; H, 6.13, N, 11.91%.
Cathepsin inhibiting activity
The substrate used for both cathepsin B and cathepsin L was Cbz-Phe-Arg-MCA. The substrate and the reversible inhibitor E-64 were purchased from Bachem (King of Prussia, PA) and Peptides International (Louisville, KY), respectively. Human cathepsin B and L were expressed and purified as described previously[Citation46,Citation47]. For the assay of cathepsin B 15 pM of cathepsin B and 20 uM of substrate was used whereas for cathepsin L assay 9 pM of cathepsin L, and 1 uM of substrate was used. Fluorescence was monitored on a Varian Gemini spectrofluorometer with the excitation and emission wavelengths at 380 and 440 nm, respectively. Cathepsin B and L stored at 4°C in inhibited form by S-methyl methanethiosulfonate (MMTS) (0.1 mM), was preactivated by incubation with dithiothreitol (DTT) (2 mM) in the same buffer as the reaction mixture. Concentration of the active enzyme was determined by titration with E-64. All kinetic measurements were carried out at 23°C at pH 6.0 in the presence of sodium phosphate (50 mM), EDTA (5 mM), DTT (2 mM), NaCl (0.2 M), and DMSO (5%). The KM values for Cbz-Phe-Arg-MCA were determined to be 100 uM with cathepsin B, and 4.5 uM with cathepsin L. The Ki values were obtained by using the rearranged Michaelis Menten Equation (Equations 1 & 2) from a graph of 1/v versus [I] by measuring the initial rate of substrate hydrolysis (v) in the presence of different concentrations of inhibitor [I] and at substrate concentration kept well below KM (Usually, substrate concentration [S] at least 10 times lower than Km is used but, a 5-fold lower ratio is also acceptable).
If then the equation becomes:
Therefore, in a graph of 1/v vs [I],
slope = and intercept =
The Ki value would be calculated as follows:
Ki = Intercept/slope
The results of the study are summarized in .
Table I. In vitro activity of compounds (22–29) against cathepsin L and B.
Results and discussion
Chemistry
The syntheses of the title cyanopyrrolidine derivatives were accomplished in accordance with the sequence of reactions depicted in Scheme . Different carboxylic acids (10–13) were coupled to the TFA salt (8) (which was obtained from intermediate (6) after Boc-cleavage) leading to the formation of amides (14–17). Cinnamic acid (10; R = w) was activated with 1-HOBT and EDC in DCM and reacted with the TFA salt (8) to obtain the diamide (14; R = w). Its IR spectrum showed characteristic signals at 3381 and 3258 (NH2 stretching), 3176 (NH), 1686 (CONH2), 1666 (amide C = O stretching) and 1632 cm− 1 (C = O of the conjugated system).
Compounds (15–17) were synthesized in the same manner as described above for compound (14). For compounds 15 (R = x), 16 (R = y) and 17 (R = z) p-methoxycinnamic acid (11; R = x), α-phenylcinnamic acid (12; R = y) and benzoic acid (13; R = z) were used instead of cinnamic acid (10; R = w). These compounds were characterized on the basis of their IR data. On similar lines, the phenylalanine analogs (18–21) were prepared by reacting compound (9) with all the four above mentioned carboxylic acids (10–13). These amides were also characterized on the basis of their IR data. The amides (14–21) were converted into their respective nitriles by dehydration using cyanuric chloride[Citation48]. The nitriles (22–29) were purified by column chromatography and crystallized. The IR spectra of the compounds (22–29) showed peaks at about 3268 for NH, 2245 for CN, and 1673 and 1635 cm− 1 (for the C = O of two amides). The proton NMR spectrum of the compound (23) is explained in detail. The vinylic protons appeared as two doublets at δ 7.51–7.56 (13-CH; J = 15.6 Hz) and 6.26–6.31
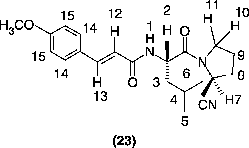
(12-CH; J = 15.6 Hz). Similarly, four aromatic protons appeared as two doublets at δ 7.39–7.43 (2H, 14-CH; J = 8.7 Hz) and 6.86–6.89 (2H, 15-CH; J = 8.7 Hz). Signal for three protons of methoxy group appeared at δ 3.83 as a singlet. The NH (1-H) proton got split into a doublet due to the adjoining proton (2-H) at δ 6.38–6.41 (J = 8.46 Hz). Methine proton (7-H) appeared as multiplet at δ 4.78–4.80. Methylene protons (C8 and C9) offered a multiplet at δ 2.18–2.31. The two protons (10-H and 11-H) appeared at different positions. One of them appeared at δ 3.66–3.71 (m, 10-CH) and the other at a more down field value, δ 3.85–3.93 (m, 11-H). This could be due to the fixed conformation of the pyrrolidine where carbonyl group is showing its anisotropic effect. The proton (2-H) sandwiched between the carbonyl and the NH appeared at 4.86–4.94 (m, 2-CH). The methylene (3-CH2) and methine (4-CH) protons appeared at 1.26–1.81 (m, 3H). Surprisingly, both the methyl protons of the isopropyl group (5-CH3 and 6-CH3) appeared as doublets at two positions; δ 1.01–1.03 (d, 3H; 5-CH3) and 0.97–0.99 (d, 3H; 6-CH3). This could be due to some steric constraint offered by the adjacent nitrile in the free rotation of the iso propyl methyl protons.
Biological activity
The synthesized compounds were screened against human cathepsin L and B following the reported method22 (). Phenylalanine derivatives (26–29) did not inhibit cathepsin B and showed only a weak affinity towards cathepsin L. Leucine derivatives (22–25) were also inactive as cathepsin B inhibitors but showed improved potency towards cathepsin L. Among the compounds (22–25) α–phenylcinnamic derivative (24) showed the highest affinity for the enzyme cathepsin L. Looking at the activity profiles of these compounds it seems that a bulky moiety at P3 should be able to provide more potent inhibitors. In compounds (22) and (23) the hydrophobic phenyl groups were tethered too far from the P2-P3 amide linkage to fit optimally at S3 pocket of the enzyme which is reflected in their lower degree of binding affinity for the enzyme as compared to compound (25), which has a phenyl group attached directly to the amidic linkage. Presence of α-phenyl group in compound (24) probably provides the required bulk for the better fitting in the active site at S3. There seems to be no involvement of α,β-unsaturated functionality in the binding to the S3 site in the enzyme. Although none of the synthesized compounds (22–29) showed promising activity against cathepsin L they may provide leads for obtaining potent and selective cathepsin L inhibitors in future.
Acknowledgements
Mr. A. K. Shinde gratefully acknowledges financial support from CSIR, New Delhi, India.
References
- de Duve C. Lysosomes revisited. Eur J Biochem 1983; 137: 391–397
- Brömme D, Kaleta J. Thiol-depedent cathepsins: Pathophysiological implications and recent advances in inhibitor design. J Curr Pharm Des 2002; 8: 1639–1658
- Fehrenbacher N, Jaattela M. Lysosomes as targets for cancer therapy. Can Res 2005; 65: 2993–2995
- Guicciardi ME, Miyoshi H, Bronk SF, Gores GJ. Cathepsin B knockout mice are resistant to tumor necrosis factor-alpha-mediated hepatocyte apoptosis and liver injury: Implications for therapeutic applications. Am J Pathol 2001; 159: 2045–2054
- Kakegawa H, Nikawa T, Tagami K, Kamioka H, Sumitani K, Kawata T, Drobnic-Kosorok M, Lenarcic B, Turk V, Katunuma N. Participation of cathepsin L on bone resorption. FEBS Lett 1993; 321: 247–250
- Drake FH, Dodds RA, James IE, Connor JR, Debouck C, Richardson S, Lee-Rykaczewski E, Coleman L, Rieman D, Barthlow R, Hastings G, Gowen M. Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J Biol Chem 1996; 271: 12511–12516
- Brix K, Lemansky P, Herzog V. Evidence for extracellularly acting cathepsins mediating thyroid hormone liberation in thyroid epithelial cells. Endocrinology 1996; 137: 1963–1974
- Chapman HA. Endosomal proteolysis and MHC class II function. Curr Opin Immunol 1998; 10: 93–102
- Esser RE, Angelo RA, Murphey MD, Watts LM, Thornburg LP, Palmer JT, Talhouk JW, Smith RE. Cysteine proteinase inhibitors decrease articular cartilage and bone destruction in chronic inflammatory arthritis. Arthritis Rheum 1994; 37: 236–247
- Katunuma N, Kominami E. Abnormal expression of lysosomal cysteine proteinases in muscle wasting diseases. Rev Physiol Biochem Pharmacol 1987; 108: 1–20
- Navab R, Chevet E, Authier F, Di Guglielmo GM, Bergeron JJM, Brodt P. Inhibition of endosomal insulin-like growth factor-I processing by cysteine proteinase inhibitors blocks receptor-mediated functions. J Biol Chem 2001; 276: 1364–1369
- Sheahan K, Shuja S, Murnane MJ. Cysteine protease activities and tumor development in human colorectal carcinoma. Cancer Res 1989; 49: 3809–3814
- Elliot E, Sloane BF. The cysteine protease cathepsin B in cancer. Perspect Drug Disc Des 1996; 6: 12–32
- Hoshimoto Y, Kondo C, Kojima T, Magata H, Moriyama A, Hayakawa T, Katunuma M. Significance of 32-kDa cathepsin L secreted from cancer cells. Cancer Biother. Radiopharm 2006; 21: 217–224
- Ravanko K, Järvinen K, Helin J, Kalkkinen N, Hölttä E. Cysteine cathepsins are central contributors of invasion by cultured adenosylmethionine decarboxylase-transformed rodent fibroblasts. Cancer Res 2004; 64: 8831–8838
- Rousselet N, Mills L, Jean D, Tellez C, Bar-Eli M, Frade R. Inhibition of umorigenicity and metastasis of human melanoma cells by anti-cathepsin L single chain variable fragment. Cancer Res 2004; 64: 146–151
- Löser R, Schilling K, Dimmig E, Gütschow M. Interaction of papain-like cysteine proteases with dipeptide-derived nitriles. J Med Chem 2005; 48: 7688–7707
- Otto HH, Schirmeister T. Cysteine proteases and their inhibitors. Chem Rev 1997; 97: 133–171
- Yamashita DS, Dodds RA. Cathepsin K and the design of inhibitors of cathepsin K. Curr Pharm Des 2000; 6: 1–24
- Donmienne L, Giovanni A, David PF. Protease inhibitors: current status and future prospects. J Med Chem 2000; 43: 305–341
- Rosenthal PJ, Wollish WS, Palmer JT, Rasnick D. J Clin Invest 1991; 88: 1467–1472
- Palmer JT, Rasnick D, Klaus JL, Bromme D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J Med Chem 1995; 38: 3193–3196
- Hanada K, Tamai M, Yamagishi M, Ohmura S, Sawada J, Tanaka I. Isolation and characterization of E-64, a new thiol protease inhibitor. Agric Biol Chem 1978; 42: 523–528
- Tavares FX, Boncek V, Deaton DN, Hassell AM, Long ST, Miller AB, Payne AA, Miller LR, Shewchuk LM, Wells-Knecht K, Willard DH, Jr., Wright LL, Zhou HQ. Design of potent, selective, and orally bioavailable inhibitors of cysteine protease cathepsin K. J Med Chem 2004; 47: 588–599
- Yasuma T, Oi S, Choh N, Nomura T, Furuyama N, Nishimura A, Fujisawa Y, Sohda T. Synthesis of peptide aldehyde derivatives as selective inhibitors of human cathepsin L and their inhibitory effect on bone resorption. J Med Chem 1998; 41: 4301–4308
- Liang TC, Abeles RH. Inhibition of papain by nitriles: Mechanistic studies using NMR and kinetic measurements. Arch Biochem Biophys 1987; 252: 626–634
- Ando R, Sakaki T, Morinaka Y, Takahashi C, Tamao Y, Yoshii N, Katayama S, Saito K, Tokuyama H, Isaka M, Nakamura E. Cyclopropenone containing cysteine protease inhibitors: Synthesis and enzyme inhibitory activities. Bioorg Med Chem 1999; 7: 571–579
- Marquis RW, Ru Y, LoCastro SM, Zeng J, Yamashita DS, Oh H,-J, Erhard KF, Davis LD, Tomaszek TA, Tew D, Salyers K, Proksch J, Ward K, Smith B, Levy M, Cummings MD, Haltiwanger RC, Trescher G, Wang B, Hemling ME, Quinn CJ, Cheng H,-Y, Lin F, Smith WW, Janson CA, Zhao B, McQueney MS, D'Alessio K, Lee C,-P, Marzulli A, Dodds RA, Blake S, Hwang S,-M, James IE, Gress CJ, Bradley BR, Lark MW, Gowen M, Veber DF. Azepanone-based inhibitors of human and rat cathepsin K. J Med Chem 2001; 44: 1380–1395
- Harbeson SL, Abelleira SM, Akiyama A, Barrett C, III R, arroll RM, Straub JA, Tkacz JN, Wu C, Musso GF. Stereospecific synthesis of peptidyl α-keto amides as inhibitors of calpain. J Med Chem 1994; 37: 2918–2929
- Altmann E, Renaud J, Green J, Farley D, Cutting B, Jahnke W. Arylaminoethyl amides as novel noncovalent cathepsin K inhibitors. J Med Chem 2002; 45: 2352–2354
- Moon JB, Coleman RS, Hanzlik R-P. Reversible covalent inhibition of papain by a peptide nitrile. 13C NMR evidence for a thioimidate ester adduct. J Am Chem Soc 1986; 108: 1350–1351
- Brisson J-R, Carey PR, Storer AC. Benzoylamidoacetonitrile is bound as a thioimidate in the active site of papain. J Biol Chem 1986; 261: 9087–9089
- Ward YD, Thomson DS, Frye LL, Cywin CL, Morwick T, Emmanuel MJ, Zindell R, McNeil D, Bekkali Y, Giradot M, Hrapchak M, DeTuri M, Crane K, White D, Pav S, Wang Y, Hao M,-H, Grygon CA, Labadia ME, Freeman DM, Davidson W, Hopkins JL, Brown ML, Spero DM. Design and synthesis of dipeptide nitriles as reversible and potent cathepsin S inhibitors. J Med Chem. 2002; 45: 5471–5482
- Peters JU. 11 Years of cyanopyrrolidines as DPP - IV inhibitors. Curr Top Med Chem 2007; 7: 579–595
- Fukushima H, Hiratate A, Takahashi M, Saito M, Munetomo E, Kitano K, Saito H, Takaoka Y, Yamamoto K. Synthesis and structure-activity relationships of potent 3- or 4-substituted-2-cyanopyrrolidine dipeptidyl peptidase IV inhibitors. Bioorg Med Chem 2004; 12: 6053–6061
- Calkins CC, Sloane BF. Mammalian cysteine protease inhibitors: Biochemical properties and possible roles in tumor progression. Biol Chem Hoppe Seyler 1995; 376: 71–80
- Falgueyret JP, Oballa RM, Okamoto O, Wesolowski G, Aubin Y, Rydzewski RM, Prasit P, Riendeau D, Rodan SB, Percival MD. Novel, nonpeptidic cyanamides as potent and reversible inhibitors of human cathepsins K and L. J Med Chem 2001; 44: 94–104
- Rydzewski RM, Bryant C, Oballa R, Wesolowski G, Rodan SB, Bass KE, Wong DH. Peptidic-1-cyanopyrrolidines: Synthesis and SAR of a series of potent, selective cathepsin inhibitors. Bioorg Med Chem 2002; 10: 3277–3284
- Greenspan PD, Clark KL, Tommasi RA, Cowen SD, McQuire LD, Farley DL, van Duzer JH, Goldberg RL, Zhou H, Fitt JJ, Coppa DE, Fang Z, Macchia W, Zhu L, Capparelli MP, Goldstein R, Wigg AM, Doughty JR, Bohacek RS, Knap AK. Identification of dipetidyl nitriles as potent and selective inhibitors of cathepsin B through structure-based drug design. J Med Chem 2001; 44: 4524–4534
- Picken PP, Guthrie DJ, Walker B. Inhibition of bovine cathepsin B by amino acid-derived nitriles. Biochem Soc Trans 1990; 18: 316–416
- Ward YD, Thomson DS, Frye LL, Cywin CL, Morwick T, Emmanuel MJ, Zindell R, McNeil D, Bekkali Y, Girardot M, Hrapchak M, DeTuri M, Crane K, White D, Pav S, Wang Y, Hao M, Grygon CA, Labadia ME, Freeman DM, Davidson W, Hopkins JL, Brown ML, Spero DM. Design and synthesis of dipeptide nitriles as reversible and potent cathepsin S inhibitors. J Med Chem 2002; 45: 5471–5482
- Bekkali Y, Thomson DS, Betageri R, Emmanuel MJ, Hao M, Hickey E, Liu W, Patel U, Ward YD, Young ERR, Nelson R, Kukulka A, Brown ML, Crane K, White D, Freeman DM, Labadia ME, Wildeson J, Spero DM. Identification of a novel class of succinyl-nitrile-based Cathepsin S inhibitors. Bioorg Med Chem Lett 2007; 17: 2465–2469
- Marquis RW, James I, Zeng J, Trout REL, Thompson S, Rahman A, Yamashita DS, Xie R, Ru Y, Gress CJ, Blake S, Lark MA, Hwang S, Tomaszek T, Offen P, Head MS, Cummings MD, Veber DF. Azepanone-based inhibitors of human cathepsin L. J Med Chem 2005; 48: 6870–6878
- Organic Synthesis Collective Volume-VII., Freeman JP, Ed., John Willey and Sons Inc., New York, p 70-5.
- Vogel's Textbook of Practical Organic Chemistry. BS Furniss, AJ Hannaford, V Rogers, PGW Smith, AR Tatchell. ELBS publication. 1984; 801–803
- Nägler DK, Storer AC, Portaro FCV, Carmona E, Juliano L, Ménard R. Major increase in endopeptidase activity of human cathepsin B upon removal of occluding loop contacts. Biochemistry 1997; 36: 12608–12615
- Carmona E, Dufour E, Plouffe C, Takebe S, Manson P, Mort JS, Menard R. Potency and selectivity of the cathepsin L propeptide as an inhibitor of cysteine proteases. Biochemistry 1996; 35: 8149–8157
- Olah GA, Narang SC, Fung AP, Gupta BG. Synthetic methods and reactions; part 82: Cyanuric chloride, a mild dehydrating agent in the preparation of nitriles from amides. Synthesis 1980; 657–658