Abstract
In the present study we report the simple synthesis and antitumour activity of novel stilbene derivatives 13–22. The key synthetic strategies involved Wadsworth-Horner-Emmons condensation and coupling reactions in high yields. All compounds showed significant growth inhibition on human tumour cell lines, with the most potent compound (19) exhibiting an IC50 of 5.7 μM – 11.4 μM in vitro.
Introduction
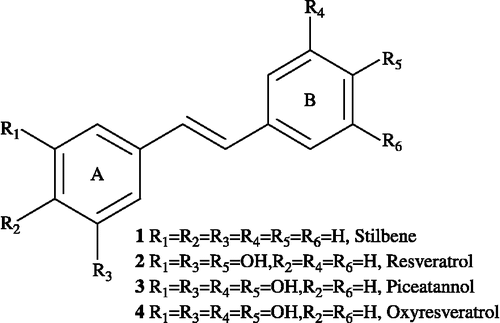
Naturally occurring resveratrol (trans-3,4,5′-trihydroxystilbene, see ) isolated from grapes is the most representative compound of stilbene analogues, and it exhibits a wide range of intriguing biological activities, such as antibacterial [Citation1], antitumour [Citation2], antiviral [Citation3], antioxidant [Citation4], and antihypertensive [Citation5] activities, and it inhibits the activity of platelet–activating factor (PAF) [Citation6], Ca2 + channels [Citation7] and cAMP phosphodiesterase [Citation8] Furthermore, stilbene moieties have the potential to be useful intermediates for many industrial products and may have numerous agrochemical applications, such as in liquid crystals [Citation9], color photography [Citation10], dyestuffs [Citation11], and herbicides [Citation12]. These stilbene derivatives have received a great deal of attention from synthetic chemists, biologists, and pharmacologists due to their various pharmacological activities and unique structural features, such as the bis–phenyl groups between E-olefin or Z-olefin. Since it was first isolated as a constituent of the roots of white hellebore in 1940, several novel synthetic approaches and medicinal developments involving stilbene derivatives that have two different aryl groups or identical aryl groups have been reported in the literature [Citation13]. Recent laboratory studies indicate that resveratrol and its analogues have promising therapeutic activities for various cancers, including breast, prostate, lung, and colon cancer [Citation14].
Figure 1. Chemical structures of trans-stilbene and its analogues.
The Nakamura group [Citation15] recently reported the synthesis and biological evaluation of boronic acid containing cis-stilbenes as apoptotic tubulin polymerization inhibitors. The Macchia and Ghidoni groups [Citation16] introduced resveratrol analogues with high ceramide-mediated proapoptotic activity on human breast cancer cells. Likhitwitayawuid and co‐workers [Citation17] chemically transformed oxyresveratrol (trans-2,4,3′,5′-tetrahydroxystilbene) into a potent tyrosinase inhibitor and a strong cytotoxic agent. The Andrus group [Citation18] recently developed a technique for the synthesis of polyhydroxylated ester analogues of the stilbene resveratrol using decarbonylative Heck couplings. The Simoni group [Citation19] reported the synthesis of resveratrol analogues, which are stilbene-based agents that show anticancer activity on HL60 leukemic cells with a non-specific phase mechanism.
In a preliminary article [Citation20], we reported the design, synthesis, and biological activity of resveratrol derivatives as protein tyrosine phosphatase 1B inhibitors. In a continuation of our medicinal chemistry program aimed at the efficient synthesis of new stilbene derivatives and the evaluation of their cytotoxicities, we describe the simple synthesis of ten novel stilbene derivatives based on resveratrol and determined the anticancer properties of each derivative. We used the SRB assay for the growth inhibition (IC50) of six tumour cell lines in vitro. In the present study, we report the convenient synthesis and evaluation of the anticancer activities of novel stilbene derivatives 13–22.
Experimental
General
All other commercial reagents and solvents were used as received without further purification. Reaction solvents were distilled from calcium hydride for dichloromethane and from sodium metal and benzophenone for tetrahydrofuran. The reactions were monitored and the Rf values determined using analytical thin layer chromatography (TLC) with Merck silica gel 60, F-254 precoated plates (0.25 mm thickness). Spots on the TLC plates were visualized using ultraviolet light (254 nm), a basic potassium permanganate solution or cerium sulfate/ammonium dimolybdate/sulfuric acid solution followed by heating on a hot plate. Flash column chromatography was performed with Merck silica gel 60 (230–400 mesh). 1H NMR spectra were recorded on Bruker DPX-250 or Varian Unity-Inova 500 Spectrometers. Proton chemical shifts are reported in ppm (δ) relative to internal tetramethylsilane (TMS, δ 0.00) or with the solvent reference relative to TMS employed as the internal standard (CDCl3, δ7.26 ppm; d4-CD3OD, δ3.31ppm). Data are reported as follows: Chemical shift {multiplicity [singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m)], coupling constants [Hz], integration}. 13C NMR spectra were recorded on Bruker DPX-250 (62.9 MHz) or Varian Unity-Inova 500 (125.8 MHz) spectrometers with complete proton decoupling. Carbon chemical shifts are reported in ppm (δ) relative to TMS with the respective solvent resonance as the internal standard (CDCl3, δ 77.0 ppm; d4-CD3OD, δ 49.0 ppm). Infrared (IR) spectra were recorded on a Nicolet Model Impact FT-IR 400 spectrometer. Data are reported in wave numbers (cm− 1). High resolution mass spectrometer (HRMS) was recorded on an Applied Biosystems 4700 proteomics analyzer spectrometer.
Synthesis
(E)-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]etheny}-benzoic acid methyl ester (8)
Method A
To a solution of 30 (1.0 g, 3.5 mmol) in dry CH2Cl2 (5 mL) was added dropwise a suspension of NaH (10 mg, 60% dispersion in parafilm liquid, washed successively with n-pentane) in dry CH2Cl2 (5 mL) at 0 °C and stirred for 1 h. A solution of protecting aldehyde 14 (1.0 g, 2.7 mmol) in CH2Cl2 (5 mL) was added dropwise and stirred at room temperature for 15 h. The reaction mixture was quenched by slow addition of water (10 mL) and extracted with ethyl acetate (30 mL × 2). The combined organic layer was washed with NaHCO3 and brine. The organic layer was dried (anhydrous MgSO4), filtered and concentrated under reduced pressure. The crude product was purified by flash silica chromatography (hexanes/ethyl acetate, 5/1, v/v) to yielded stilbene 8 (1.4 g, 80%) as a white solid. Rf = 0.41 (hexanes/ethyl acetate, 10:1, v/v), IR νmaxcm− 1 (CHCl3) 2941, 2854, 1729, 1672, 1601, 1515, 1464, 1429, 1281, 1179, 1113 1H-NMR (250 MHz, CDCl3) δ 8.02 (d, 2H, J = 8.2 Hz, aromatic H), 7.55 (d, 2H, J = 8.2 Hz, aromatic H), 7.13 (d, 1H, J = 16.0 Hz, vinyl CH), 7.00 (m, 3H, aromatic H), 6.94 (d, 1H, J = 16.0 Hz, vinyl CH), 6.84 (m, 1H, aromatic H), 3.91(s, 3H, OCH3), 1.01–0.99 (m, 18H, tert-butyl H), 0.23–0.22 (m, 12H, dimethyl H); 13C-NMR (63 MHz, CDCl3) δ 167.3, 147.5, 143.0, 132.3, 130.9, 130.5, 130.4, 130.0, 128.1, 126.4, 126.0, 122.8, 121.7, 121.6, 120.8, 52.4, 26.3 (3), 26.2 (3), 18.9, 18.7, − 3.7 (2), − 3.87 (2). HRMS (FAB) 499.2230 m/z: ([M + H]+, obsd), 499.2622 (calcd for C28H42O4Si2)
Method B
To a solution of 30 (1.0 g, 3.5 mmol) in dry THF (5 mL) was added dropwise a suspension of n-BuLi (2.2 mL, 1.6 M in THF) in dry THF (5 mL) at 20 °C and stirred for 1 h. A solution of protecting aldehyde 14 (1.0 g, 2.7 mmol) in CH2Cl2 (5 mL) was added dropwise and stirred at room temperature for 12 h. The reaction mixture was quenched by slow addition of water (10 mL) and extracted with ethyl acetate (30 mL × 2). The combined organic layer was washed with NaHCO3 and brine. The organic layer was dried (anhydrous MgSO4), filtered and concentrated under reduced pressure. The crude product was purified by flash silica chromatography (hexane/ethyl acetate, 15/1, v/v) to yielded stilbene 8 (1.5 g, 86%) as a white solid.
(E)-4-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]-etheny}-benzoic acid (10)
Method A
To a solution of oxalyl chloride (0.08 g, 0.62 mmol) in dichloromethane (20 mL) was added and stirred for 1 h. Alcohol 16 (0.29 g, 0.61 mmol) in dichloromethane (3 mL) was added dropwise at − 78 °C, and resulting mixture was stirred for 45 min. The reaction mixture was quenched by slow addition of triethylamine (0.31 g, 3.1 mmol) at − 78 °C, and the reaction mixture was allowed to warm to 0 °C. After 15 min, ether (30 mL) was added followed by saturated aqueous NH4Cl solution (30 mL). The organic layer was separated, and aqueous phase was extracted with ether (20 mL). The combined organic layer was washed with brine (30 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the aldehyde (0.25 g, 89%).
Method B
To a solution of alcohol 9 (0.29 g, 0.61 mmol) in dichloromethane (20 mL) were added molecular sieves (0.29 g, 4 Å, powdered) and 4-methylmorpholine N-oxide (0.11 g, 0.92 mmol), followed by addition of tetrapropylammonium perruthenate(0.011 g, 0.031 mmol) at 0 °C. The resulting mixture was stirred for 15 min. The reaction mixture was filtered through a short pad of silica (hexanes/ethyl acetate, 10/1, v/v) to give aldehyde (0.26 g, 90%). Then, to a stirred solution of aldehyde (89 mg, 0.19 mmol) in tert-butanol (3 mL) and 2-methyl-2-butene (5 mL) was added a solution of NaClO2 (52mg, 0.57 mmol) and NaH2PO4 (0.2 g, 1.70 mmol) in water (1 mL) at 0 °C and the mixture was stirred at 0 °C for 16 h. The reaction mixture was diluted with ethyl acetate (10 mL), washed with brine (10 mL). The organic phase was separated, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude mixture was purified by flash chromatography (silica gel, hexanes/ethyl acetate, 1/1, v/v) to yield pure acid 18 (88 mg, 96%). Rf = 0.26 (hexanes/ethyl acetate, 2:1, v/v), IRυmax (CHCl3) 2928, 2855, 1681, 1605, 1595, 1564, 1513, 1421, 1286, 1251, 1177, 1126, 916, 837 cm− 1;1H-NMR (250 MHz, DMSO) δ 7.98 (d, 2H, J = 8.0 Hz, aromatic H), 7.75 (d, 2H, J = 8.1 Hz, aromatic H), 7.41 (d, 1H, J = 16.2 Hz, vinyl CH), 7.23 (3H, m, aromatic H, vinyl CH), 6.94 (d, 1H, aromatic H), 1.02–1.01(m, 18H, tert-butyl H), 0.25–0.24 (m, 12H, dimethyl H); 13C-NMR(63 MHz, DMSO) δ167.6, 147.1, 146.8, 131.3, 131.1, 130.2 (2), 129.6, 126.7 (2), 126.0, 121.5, 120.9, 119.9, 26.2 (6), 18.7 (2), − 3.7 (4). HRMS (FAB) 484.1861 m/z: ([M]+, obsd), 484.2465 (calcd for C27H40O4Si2).
General procedure for the amide coupling of acid with various amines (12a–12j)
A stirred solution of acid 18 (0.1 g, 0.21 mmol), HATU (236 mg, 0.62 mmol) in CH2Cl2 (5 mL) was added appropriate amines (19a–19n) at room temperature, and the reaction mixture was stirred at same temperature for 16 h. The reaction mixture was quenched with slow addition of water (5 mL), extracted with ethyl acetate (10 mL) and washed with brine (10 mL) an anhydrous MgSO4, filtered, and concentrated under reduced pressure to give crude product, which was purified by flash silica chromatography to afford pure amide derivatives. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to give crude product, which was purified by flash silica chromatography to afford pure amide derivatives.
(E)-4-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]vinyl}-N-allylbenzamide (12a)
Yield: 66%. Rf = 0.89 (hexanes/ethyl acetate, 1:1, v/v); IR υmax (CHCl3) 3324, 2928, 2857, 1638, 1607, 1596, 1542, 1510, 1472, 1421, 1299, 1253, 1124, 992, 962, 907, 839, 781 cm− 1; 1H NMR (250 MHz, CDCl3) δ7.79 (d, 2H, J = 8.3 Hz, aromatic H), 7.54 (d, 2H, J = 8.3 Hz, aromatic H), 6.84 (d, 2H, J = 8.8 Hz, aromatic H), 6.01–5.86 (m, 1H, vinyl CH), 5.30–5.15 (m, 2H, vinyl CH), 4.10 (t, 2H, J = 5.7 Hz, N-CH2), 1.02–1.01 (m, 18H, tert-butyl H), 0.23–0.21 (m, 12H, dimethyl H); 13C NMR (63 MHz, CDCl3) δ167.1, 147.5, 147.2, 141.0, 134.4, 132.8, 130.6, 127.5 (2), 126.4 (2), 125.6, 121.3, 120.4, 119.4, 116.7, 42.5, 26.1 (3), 26.0 (3), 18.6 (2), − 4.0 (4). HRMS (FAB) 524.2736 m/z: ([M + H]+, obsd), 524.2938 (calcd for C30H45NO3Si2).
(E)-4-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]vinyl}-N-decylbenzamide (12b)
Yield: 73%. Rf = 0.23 (hexanes/ethyl acetate, 5:1, v/v); IR υmax(CHCl3) 3320, 2928, 2856, 1634, 1608, 1957, 1548, 1511, 1471, 1421, 1298, 1253, 1221, 1164, 1125, 982, 953, 907, 839, 782 cm− 1; 1H NMR (250 MHz, CDCl3) δ7.77 (d, 2H, J = 8.2 Hz, aromatic H), 7.52 (d, 2H, J = 8.2 Hz, aromatic H), 7.08–6.98 (m, 3H, aromatic H, vinyl CH), 6.93 (d, 1H, J = 16.3 Hz, vinyl CH), 6.83 (d, 1H, J = 8.8 Hz, aromatic H), 3.46–3.39 (m, 2H, N-CH2), 1.60–1.58 (m, 2H, alkyl chain), 1.60–1.58 (m, 12H, alkyl chain), 1.02–0.97 (m, 18H, tert-butyl H), 0.93–0.88 (m, 5H, alkyl chain), 0.23–0.22 (m, 12H, dimethyl H); 13C NMR (63 MHz, CDCl3) δ167.3, 147.5, 147.2, 140.8, 133.2, 130.7, 130.5, 127.4 (2), 126.1 (2), 125.7, 121.3, 120.4, 119.4, 40.6, 32.0, 29.8, 29.7 (2), 29.5, 29.4, 27.1, 26.1 (3), 26.0 (3), 22.8, 18.6 (2), 14.2, − 4.0 (4). HRMS (FAB) 624.4006 m/z: ([M + H]+, obsd), 624.4190 (calcd for C37H61NO3Si2)
(E)-4-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]vinyl}-N-[3-(2-oxopyrrolidin-1-yl)propyl]benzamide (12c)
Yield: 50%. Rf = 0.54 (CH2Cl2/Methanol, 10:1, v/v); IR υmax (CHCl3) 3329, 2954, 2929, 2857, 1667, 1607, 1510, 1471, 1295, 1253, 1229, 1124, 981, 906, 840, 782 cm− 1; 1H NMR (250 MHz, CDCl3) δ 7.92 (d, 2H, J = 8.0 Hz, aromatic H), 7.54 (d, 2H, J = 8.0 Hz, aromatic H) 7.08–6.99 (m, 3H, aromatic H, vinyl CH), 6.93 (d, 1H, J = 16.3 Hz, vinyl CH), 6.83 (d, 1H, J = 8.8 Hz, aromatic H), 3.41–3.39 (m, 6H, NH-CH2, CH2N-CH2), 2.44–2.41 (m, 2H, pyrrolidone H), 2.09–2.04 (m, 2H, pyrrolidone H), 1.78–1.69. (m, 2H, -CH2), 1.01–0.98 (m, 18H, tert-butyl H), 0.23–0.21 (m, 12H, dimethyl H); 13C NMR (63 MHz, CDCl3) δ176.4, 166.9, 147.4, 147.1, 140.7, 132.8, 130.8, 130.3, 127.6 (2), 126.3 (2), 125.9, 121.3, 120.4, 119.4, 39.7, 35.7, 31.0, 29.8, 26.3, 26.3 (6), 18.6 (2), 18.1, − 3.9 (4). HRMS (FAB) 609.3529 m/z: ([M + H]+, obsd), 609.3466 (calcd for C34H52N2O4Si2).
(E)-4-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]vinyl}phenyl-cyclohexylamide (12d)
Yield: 61%. Rf = 0.83 (hexanes/ethyl acetate, 2:1, v/v); IR υmax (CHCl3) 3313, 2928, 2856, 1630, 1541, 1510, 1463, 1298, 1254, 1124, 982, 907, 839, 782 cm− 1; 1H NMR (250 MHz, CDCl3) δ7.36 (d, 2H, J = 8.20 Hz, aromatic H), δ7.52 (d, 2H, J = 8.09 Hz, aromatic H), 7.08–6.80 (m, 5H, aromatic H, vinyl CH), 3.99–3.96 (m, 1H, NH-CH), 2.04–1.99 (m, 2H, cyclohexane), 1.78–1.63 (m, 4H, cyclohexane), 1.26–1.18 (m, 4H, cyclohexane), 1.02–0.99 (m, 18H, tert-butyl H), 0.23–0.22 (m, 12H, dimethyl H); 13C NMR (63 MHz, CDCl3) δ166.4, 147.5, 147.2, 140.7, 133.5, 130.7,130.5, 127.4 (2), 126.2 (2), 125.7, 121.3, 120.4, 119.4, 48.8, 33.4 (2), 25.7, 26.1 (6), 25.1 (2), 18.6 (2), − 3.9 (4). HRMS (FAB) 566.3291 m/z: ([M + H]+, obsd), 566.3407 (calcd for C33H51NO3Si2).
(E)-4-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]vinyl}-N-[(furan-2-yl)methyl]benzamide (12e)
Yield: 91%. Rf = 0.63 (hexanes/ethyl acetate, 2:1, v/v); IR υmax (CHCl3) 3318, 2929, 2857, 11641, 1606, 1596, 1510, 1471, 1421, 1299, 1253, 1220, 1125, 982, 906, 839, 781 cm− 1; 1H NMR (250 MHz, CDCl3) δ 7.79 (d, 2H, J = 8.3 Hz, aromatic H), 7.54 (d, 2H, J = 8.3 Hz, aromatic H), 7.39–7.37 (m, 1H, furfuran H), 7.10–6.81 (m, 5H, aromatic H, vinyl CH), 6.35–6.30 (m, 2H, furfuran H), 4.66 (d, 2H, J = 5.4 Hz, N-CH2), 1.01–1.00 (m, 18H, tert-butyl H), 0.23–0.19 (m, 12H, dimethyl H); 13C NMR (63 MHz, CDCl3) δ 167.0, 151.4, 147.6, 147.2, 142.4, 141.1, 132.5, 130.7, 130.6, 127.6 (2), 126.4 (2), 125.6, 121.3, 120.5, 119.4, 110.6, 107.8, 37.1, 26.1 (6), 18.6 (2), − 4.0 (4). HRMS (FAB)568.3083 m/z: ([M + H]+, obsd), 568.3200 (calcd for C32H49NO4Si2).
(E)-4-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]vinyl}-N-(2-fluoro)-benzamide (12f)
Yield: 50%. Rf = 0.85 (hexanes/ethyl acetate, 1:1, v/v); IR υmax (CHCl3) 3333, 2957, 2929, 2895, 2858, 1647, 1610, 1594, 1487, 1421, 1315, 1204,1277, 1237, 1124, 985, 967, 909, 839, 857, 781 cm− 1; 1H NMR (250 MHz, CDCl3) δ7.78 (d, 2H, J = 8.3 Hz, aromatic H), δ 7.51 (d, 2H, J = 8.3 Hz, aromatic H), 7.43–7.37 (m, 1H, aromatic H) 7.27–7.21 (m, 1H, aromatic H), 7.21–6.80 (m, 7H, aromatic H, vinyl CH), 4.69 (d, 2H, J = 5.8 Hz), 1.02–0.99 (m, 18H, tert-butyl H), 0.23–0.22 (m, 12H, dimethyl H); 13C NMR (63 MHz, CDCl3) δ 167.2, 147.5, 147.2, 141.1, 132.5, 130.6, 130.5, 130.4, 129.5, 129.3, 127.5 (2), 126.4 (2), 125.6, 121.3, 120.5, 119.4, 115.6, 115.3, 38.1, 26.1 (6), 18.6 (2), − 3.9 (4). HRMS (FAB) 592.2626 m/z: ([M +H]+, obsd), 592.3000 (calcd for C34H46NO3Si2).
(E)-ethyl-3-(4-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]vinyl})-benzamino benzoate (12g)
Yield: 73%. Rf = 0.37 (hexanes/ethyl acetate, 5:1, v/v), IR υmax (CHCl3) 3336, 2930, 2858, 1721, 1652, 1596, 1546, 1512, 1488, 1472, 1433, 1299, 1253, 1223, 1165, 1124, 982, 906, 840, 782, 757 cm− 1; 1H NMR (250 MHz, CDCl3) δ8.19–8.08 (m, 2H, aromatic H), 7.87–7.80 (m, 3H, aromatic H), 7.57 (d, 2H, J = 8.3 Hz, aromatic H), 7.46 (t, J = 7.9 Hz, aromatic H), 7.12–7.00 (m, 3H, aromatic H, vinyl CH), 6.94 (d, 1H, J = 16.3 Hz, vinyl CH), 6.85 (d, 1H, J = 8.8 Hz, aromatic H), 4.39 (2H, J = 7.13 Hz, O-CH2), 1.40 (m, J = 7.13 Hz, CH3), 1.02–1.00 (m, 18H, tert-butyl H), 0.24–0.23 (m, 12H, dimethyl H); 13C NMR (63 MHz, CDCl3) δ 166.4, 165.8, 147.6, 147.2, 141.5, 138.4, 132.77, 131.3, 131.0, 129.2, 127.7 (2), 126.5 (2), 125.5, 125.4, 124.9, 121.3 (2), 120.5, 119.5, 61.2, 26.1, 26.0, 18.6 (6), 14.4 (2), − 3.9 (4). HRMS (FAB) 632.3063 m/z: ([M + H]+, obsd), 632.3149 (calcd for C36H49NO5Si2).
(E)-4-{2-[3,4-Bis-(tert-butyldimethylsilanyloxy)-phenyl]vinyl}-N,N-(1-benzoyl)-piperazinamide (12h)
Yield: 74%. Rf = 0.56 (CH2Cl2/Methanol, 10:1, v/v); IR νmax (CHCl3) 2955, 2928, 2857, 1633, 1511, 1461, 1422, 1287, 1252, 1125, 1003, 906, 839, 782 cm− 1; 1H NMR (250 MHz, CDCl3) δ7.54 (d, 2H, J = 8.0 Hz, aromatic H), 7.41–7.38 (m, 7H, aromatic H), 7.07–8.80 (m, 5H, aromatic H, vinyl CH), 3.66 (s, 8H, piperazine H), 1.02–1.00 (m, 18H, tert-butyl H), 0.23–0.22 (m, 12H, dimethyl H); 13C NMR (63 MHz, CDCl3) δ 170.8, 170.7, 147.5, 147.1, 139.8, 135.2, 133.4, 130.6, 130.4, 130.2, 128.7 (2), 127.8 (2), 127.2 (2), 126.4 (2), 125.6, 121.3, 120.3, 119.4, 26.0 (6), 18.6 (2), − 4.0 (4). HRMS (FAB) 657.3398 m/z: ([M + H]+, obsd), 657.3466 (calcd for C33H52N2O4Si2).
(E)-4-{2-[3,4-bis-(tert-butyl-dimethyl-silanyloxy)phenyl]vinyl}-(4-methylpiperidin-1-yl)methanone (12i)
%Yield: 63%. Rf = 0.72 (hexanes/ethyl acetate, 1:1, v/v), IR υmax (CHCl3) 2954, 2927, 2857, 1635, 1568, 1508, 1471, 1458, 1425, 1303, 1275, 1251, 1124, 970, 906, 839, 782 cm− 1; 1H NMR (250 MHz, CDCl3) δ7.51 (d, 2H, J = 8.2 Hz, aromatic H), 7.39 (d, J = 8.2 Hz, aromatic H), 7.05–6.98 (m, 3H, aromatic H, vinyl CH), 6.92 (d, 1H, J = 16.3 Hz, vinyl CH), 6.84 (d, 1H, J = 8.8 Hz, aromatic H), 4.67 (s, 1H, piperidine H), 3.90 (s, 1H, piperidine H), 2.96–2.82 (m, 2H, piperidine H); 13C NMR (63 MHz, CDCl3) δ 170.31, 147.34, 147.14, 139.00, 135.04, 130.83, 129.83, 127.54 (2), 126.3 (2), 125.9, 121.3, 120.3, 119.4, 31.3 (2), 29.8, 26.1 (6), 21.9 (2), 18.6 (2), − 3.9 (4). HRMS (FAB) 566.3105 m/z: ([M + H]+, obsd), 566.3407 (calcd for C33H51NO3Si2)
(E)-(4-{2-[3,4-Bis-(tert-butyl-dimethyl-silanyloxy)phenyl]vinyl}-(4-benzylpiperidin-1-yl)methanone (12j)
Yield: 85%. Rf = 0.65 (hexanes/ethyl acetate, 2:1, v/v); IR νmax (CHCl3) 2951, 2929, 2884, 2857, 1721, 1632, 1512, 1471, 1429, 1293, 1253, 1164, 1126, 982, 840, 782 cm− 1; 1H NMR (250 MHz, CDCl3) δ7.53 (d, 2H, J = 8.1 Hz, aromatic H), 7.41 (d, J = 8.2 Hz, aromatic H), 7.32–7.27 (m, 2H, aromatic H), 7.23–7.14 (m, 3H, aromatic H), 7.08–7.00 (m, 3H, aromatic H), 6.93 (d, 1H, J = 16.4 Hz, vinyl CH), 6.84 (d, 1H, J = 8.8 Hz, aromatic H), 4.71 (s, 1H, piperidine H), 3.80 (s, 1H, piperidine), 2.92–2.74 (m, 2H, piperidine H), 2.60 (d, 2H, J = 6.7 Hz, CH2-Ph), 1.81–1.66 (m, 4H, piperidine H), 1.38–1.28 (m, 1H, piperidine H), 1.02–1.00 (m, 18H, tert-butyl H), 0.23–0.20 (m, 12H, dimethyli H); 13C NMR (63 MHz, CDCl3) δ 170.2, 147.3, 147.1, 140.0, 139.0, 134.8, 130.8, 129.8, 129.1 (2), 128.4 (2), 127.5 (2), 126.2 (2), 125.9, 121.3, 120.2, 119.35, 43.1, 38.4, 26.0 (6), 18.6, 18.5, − 4.0 (4). HRMS (FAB) 642.3265 m/z: ([M + H]+, obsd), 642.3720 (calcd for C39H55NO3Si2).
General procedure for the preparation of deprotected dihydroxy stilbenes (13–22)
Method A
To a stirred solution of 12a–12j (0.10 mmol) in dry THF (5 mL) was added dropwise TBAF (0.25 mmol, 0.25 ml, 1 M solution in THF) at 0 °C under nitrogen atmosphere and the mixture was stirred at room temperature for 30 min. The reaction mixture was quenched with water and acidified with 10% HCl. The mixture is diluted with ethyl acetate (10 mL) and washed with brine (7 mL). The organic phase was separated, and the aqueous phase was extracted with ethyl acetate (2 × 10 mL). The combined organic phases were washed with brine (15 mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield dihydroxy stilbenes, which were purified by flash column chromatography (silica gel, CH2Cl2/Methanol, 15/1–10/1, v/v) to give 13–22.
Method B
To a stirred solution of 12a–12j(0.10mmol) in dry CH2Cl2(5 mL) was added dropwise TFA (1mL) at room temperature under nitrogen atmosphere and the mixture was stirred for 1 h. The residue TFA was removed by reduced pressure. The resulting mixture was purified by flash column chromatography (silica gel, CH2Cl2/Methanol, 15/1-10/1, v/v) to give 13–22.
(E)-4-[2-(3,4-Dihydroxyphenyl)vinyl]-N-allylbenzamide (13)
Yield: 64%. Rf = 0.06 (CH2Cl2/Methanol, 20:1, v/v); 1H NMR (250 MHz, CD3OD) δ 7.82 (d, 2H, J = 8.3 Hz, aromatic H), 7.57 (d, 2H, J = 8.2 Hz, aromatic H), 7.20–6.89 (m, 4H, aromatic H, vinyl CH), 6.63 (d, 1H, J = 8.2 Hz, aromatic H), 6.01–5.86 (m, 1H, vinyl CH), 5.27–5.11 (m, 2H, vinyl CH), 4.00–3.98 (m, 2H, N-CH2); 13C NMR (63 MHz, CD3OD) δ 169.7, 147.1, 146.6, 142.8, 135.6, 133.4, 132.1, 130.6, 128.7 (2), 127.1 (2), 125.5, 120.7, 116.4, 116.2, 114.1, 43.2.; HRMS (FAB): m/z 296.1238 ([M + H]+, obsd), 296.1208 (calcd for C18H17NO3).
(E)-4-[2-(3,4-Dihydroxyphenyl)vinyl]-N-decylbenzamide (14)
Yield: 60%. Rf = 0.47 (CH2Cl2/Methanol, 10:1, v/v); IR νmax (CHCl3) 3317, 2951, 2923, 2851, 1734, 1630, 1602, 1541, 1524, 1466, 1439, 1269, 963, 859 cm− 1; 1H NMR (250 MHz, CD3OD) δ 7.79 (d, 2H, J = 8.3 Hz, aromatic H), 7.58 (d, 2H, J = 8.3 Hz, aromatic H), 7.17 (d, 1H, J = 16.3 Hz, vinyl CH), 7.05–6.89 (m, 3H, aromatic H, vinyl CH), 6.77 (d, 1H, J = 8.2 Hz, aromatic H), 3.39–3.36 (m, 2H, N-CH2), 1.62–1.59 (m, 2H, alkyl chain), 1.34–1.29 (m, 12H, alkyl chain), 1.00–0.87 (m, 5H, alkyl chain); 13C NMR (63 MHz, CD3OD) δ169.7, 147.10, 146.59, 142.7, 133.8, 132.1, 130.2, 128.7 (2), 127.0 (2), 125.5, 120.7, 116.4, 114.1, 41.1, 33.1, 30.2 (2), 30.5, 30.5 (2), 28.1, 23.7, 14.5: HRMS (FAB): m/z 396.2532 ([M + H]+, obsd), 396.2460 (calcd for C25H33NO3).
(E)-4-[2-(3,4-Dihydroxyphenyl)vinyl]-N-[3-(2-oxopyrrolidin-1-yl)propyl]benzamide (15)
Yield: 66%. Rf = 0.41 (CH2Cl2/Methanol, 10:1, v/v); IR νmax (CHCl3) 3444, 2924, 2958, 2853, 1732, 1646, 1455, 1383, 1242, 1123, 1086, 1025 cm− 1; 1H NMR (250 MHz, CD3OD) δ 7.82 (d, 2H, J = 8.4 Hz, aromatic H), 7.59 (d, 2H, J = 8.4 Hz, aromatic H), 7.17 (d, 1H, J = 16.3 Hz, vinyl CH), 7.05–6.89 (m, 3H, aromatic H, vinyl CH), 6.77 (d, 1H, J = 8.2 Hz, aromatic H), 3.51–3.46 (m, 2H, NH-CH2,), 3.40–3.29 (m, 4H, CH2N-CH2), 2.40–2.36 (m, 2H, pyrrolidone H), 2.11–2.02 (m, 2H, pyrrolidone H), 1.87–1.80 (m, 2H, CH2); 13C NMR (63 MHz, CD3OD) δ 178.1, 169.8, 147.1, 146.6, 142.8, 133.5, 132.2, 130.6, 128.7 (2), 127.1 (2), 125.5, 120.7, 116.5, 114.1, 41.2, 38.1, 32.0, 30.8, 27.9, 18.8: HRMS (FAB): m/z 380.1146 ([M]+, obsd), 380.1736 (calcd for C22H24N2O4).
(E)-4-[2-(3,4-Dihydroxyphenyl)-vinyl]phenyl-cyclohexylamide (16)
Yield: 63%. Rf = 0.28 (CH2Cl2/Methanol, 10:1, v/v); IR υmax (CHCl3) 3393, 2930, 2854, 1724, 1628, 1603, 1526, 1507, 1447, 1374, 1257 cm− 1; 1H NMR (250 MHz, CD3OD) δ7.78 (d, 2H, J = 8.3, aromatic H),7.55 (d, 2H, J = 8.3, aromatic H), 7.15 (d, 1H, J = 16.3 Hz, vinyl CH), 7.04–6.89 (m, 3H, aromatic H, vinyl CH), 6.78 (d, 1H, J = 8.2 Hz, aromatic H), 3.87–3.85 (m, 1H, NH-CH), 2.02–1.93 (m, 2H, cyclohexane), 1.79–1.66 (m, 4H, cyclohexane), 1.42–1.35 (m, 4H, cyclohexane); 13C NMR (63 MHz, CDCl3) δ160.2, 138.1, 137.6, 133.6, 125.0, 123.0, 121.6, 120.9, 119.7 (2), 118.0 (2), 116.5, 111.7, 107.5, 105.1, 41.5, 24.7 (2), 17.6, 17.4 (2): HRMS (FAB): m/z 338.1785 ([M + H]+, obsd), 338.1678 (calcd for C21H23NO5).
(E)-4-[2-(3,4-Dihydroxyphenyl)vinyl]-N-[(furan-2-yl)methyl]benzamide (17)
Yield: 73%. Rf = 0.1 (CH2Cl2/Methanol, 20:1, v/v); IR νmax (CHCl3) 3431, 2924, 2958, 2853, 1726, 1634, 1601, 1523, 1442, 1289, 1189, 1112, 1043, 1101 cm− 1; 1H NMR (250 MHz, CD3OD) δ 7.81 (d, 2H, J = 8.4 Hz, aromatic H), 7.55 (d, 2H, J = 8.4 Hz, aromatic H), 7.42 (m, 1H, furfuran H), 7.15 (d, 1H, J = 16.3 Hz, vinyl CH), 7.05–6.88 (m, 3H, aromatic H, vinyl CH), 6.78 (d, 1H, J = 8.2 Hz, aromatic H), 6.35–6.28 (m, 2H, furfuran H), 4.55 (m, 2H, NCH2); 13C NMR (63 MHz, CD3OD) δ169.7, 153.2, 147.1, 146.6, 143.2, 142.8, 133.2, 130.6, 128.8 (2), 127.0 (2), 125.4, 120.7, 116.4, 114.1, 111.4, 108.1, 37.6: HRMS (FAB): m/z 336.1097 ([M + H]+, obsd), 335.1158 (calcd for C20H17NO4).
(E)-4-[2-(3,4-Dihydroxyphenyl)vinyl]-N-(2-fluoro)-benzamide (18)
Yield: 64%. Rf = 0.28 (CH2Cl2/Methanol, 10:1, v/v); IR νmax (CHCl3) 3349, 2924, 2853, 1726, 1636, 1601, 1541, 1505, 1456, 1374, 1270, 1190, 1108, 962, 757 cm− 1; 1H NMR (250 MHz, CD3OD) δ 7.83 (d, 2H, J = 8.0, aromatic H), 7.57 (d, 2H, J = 8.6 Hz, aromatic H), 7.42–7.23 (m, 3H, aromatic H) 7.15–6.89 (m, 6H, aromatic H, vinyl CH), 6.78 (d, 1H, J = 8.1 Hz, aromatic H), 4.63 (s, 2H, CH2);13C NMR (63 MHz, CD3OD) δ170.0, 147.1, 146.6, 142.9, 133.3, 132.2, 130.6, 130.2, 130.0, 128.8 (2), 128.3, 127.1 (2), 125.5, 125.3, 120.7, 116.5, 116.3, 115.9, 114.1, 38.3: HRMS (FAB): m/z 364.1 ([M + H]+, obsd), 364.1271 (calcd for C22H18FNO3).
(E)-Ethyl-3-{4-[2-(3,4-dihydroxyphenyl)vinyl]}-benzamino benzoate (19)
Yield: 74%. Rf = 0.26 (CH2Cl2/Methanol, 10:1, v/v); IR νmax 3345, 2924, 2853, 1716, 1651, 1598, 1544, 1437, 1290, 1259, 1108, 1112, 1025, 1002, 957, 756 cm− 1; 1H NMR (250 MHz, CD3OD) δ 8.40–8.39 (m, 1H, aromatic H), 7.99–7.90 (m, 3H, aromatic H), 7.80 (d, 1H, J = 8.3 Hz, aromatic H), 7.63 (d, 2H, J = 8.3 Hz, aromatic H), 7.49 (t, 1H, J = 7.9 Hz, aromatic H), 7.20 (d, 1H, J = 16.3 Hz, vinyl CH), 7.06–6.90 (m, 3H, aromatic H, vinyl CH), 6.78 (d, 1H, J = 8.2 Hz, aromatic H), 4.41 (q, 2H, J = 7.1 Hz, O-CH2), 1.42 (t, 3H, J = 7.1 Hz, CH3); 13C NMR (63 MHz, CD3OD) δ 168.6, 167.8, 147.2, 146.6, 143.2, 140.4, 133.7, 130.5, 132.6, 132.2, 129.9 (2), 129.1 (2), 127.1, 126.5, 126.2, 125.4, 123.0, 120.8, 116.5, 114.1, 62.3, 14.6: HRMS (FAB): m/z 404.1436 ([M + H]+, obsd), 404.1420 (calcd for C24H21NO5).
(E)-4-[2-(3,4-Dihydroxyphenyl)vinyl]-N,N-(1-benzoyl)-piperazinamide (20)
Yield: 64%. Rf = 0.46 (CH2Cl2/Methanol, 10:1, v/v); IR νmax (CHCl3) 3403, 2924, 2853, 1734, 1560, 1508, 1460, 1431, 1372, 1256, 1002 cm− 1; 1H NMR (250 MHz, CD3OD) δ 7.60 (d, 2H, J = 8.1 Hz, aromatic H), 7.46–7.39 (m, 7H, aromatic H), 7.15 (d, 1H, J = 16.3 Hz, vinyl CH), 7.03–6.88 (m, 3H, aromatic H, vinyl CH), 6.77 (d, 1H, J = 8.1 Hz, aromatic H), 3.91–3.61 (m, 8H, piperazine H); 13C NMR (63 MHz, CDCl3) δ 172.8 (2), 147.1, 146.6, 141.6, 136.4, 134.2, 131.9, 131.4, 130.6, 129.8 (2), 128.8 (2), 128.2 (2), 127.2 (2), 125.4, 120.6, 116.4, 114.1, 30.8 (4): HRMS (FAB): m/z 429.1660 ([M + H]+, obsd), 429.1736 (calcd for C26H24N2O4).
(E)-4-[2-(3,4-Dihydroxy-phenyl)vinyl]-(4-methylpiperidin-1-yl)methanone (21)
Rf = 0.49 (CH2Cl2/Methanol, 10:1, v/v), IR υmax (CHCl3) 3408, 2957, 2924, 2854, 1736, 1601, 1508, 1446, 1373, 1273, 1251, 1197, 1114, 1045, 969 cm− 1; 1H NMR (250 MHz, CD3OD); δ7.58 (d, 2H, J = 8.0 Hz, aromatic H), 7.37 (d, 2H, J = 8.0 Hz, aromatic H), 7.17–6.88 (m, 4H, aromatic H, vinyl CH), 6.77 (d, 1H, J = 8.2 Hz, aromatic H), 4.61–4.65 (m, 1H, piperidine H), 3.59–3.50 (m, 1H, piperidine H), 2.83–2.78 (m, 2H, piperidine H), 1.72–1.67 (m, 4H, piperidine H), 1.26 (s, 3H, -CH3); 13C NMR (63 MHz, CD3OD) δ172.4, 147.0, 146.6, 141.1, 135.3, 130.7, 131.7, 128.4 (2), 127.1 (2), 125.6, 120.6, 116.5, 114.0. 32.2, 22.0: HRMS (FAB): m/z 348.1210 ([M+Na] +, obsd), 348.1314 (calcd for C19H19NO4).
(E)-4-[2-(3,4-Dihydroxy-phenyl)vinyl]-(4-benzylpiperidin-1-yl)methanone (22)
Yield: 68%. Rf = 0.46 (CH2Cl2/Methanol, 10:1, v/v); IR υmax (CHCl3) 3395, 2923, 2851, 1593, 1558, 1445, 1838, 1237, 1086, 1021, 963 cm− 1; 1H NMR (250 MHz, CD3OD) δ7.59 (d, 2H, J = 8.2 Hz, aromatic H), 7.37 (d, J = 8.2 Hz, aromatic H), 7.32–7.27 (m, 2H, aromatic H), 7.23–7.14 (m, 3H, aromatic H), 7.29–6.88 (m, 9H, aromatic H), 6.77 (d, 1H, J = 8.2 Hz, aromatic H), 4.37 (m, 1H, piperidine H), 3.57 (m, 1H, piperidine H), 2.71–2.60 (2H, piperidine H), 2.60 (d, 2H, J = 6.6 Hz, CH2-Ph), 1.74–1.55 (m, 4H, piperidine H), 1.21–1.18 (m, 1H, piperidine H); 13C NMR (63 MHz, CDCl3) δ 168.8, 145.9, 145.5, 140.1, 138.7, 134.5, 130.1, 129.0 (2), 128.4, 128.2 (2), 127.3 (2), 125.9 (2), 124.1, 118.9, 115.7, 113.5, 42.1, 37.6: HRMS (FAB): m/z 414.2081 ([M + H]+, obsd), 414.1991 (calcd for C27H27NO3).
Measurement of anticancer activity
Human cancer cell lines of the lung (A549), the ovarian (SK-OV-3), the melanoma (SK-MEL-2), the brain (XF498) and the colon (HCT15) were used for cytotoxicity test in vitro using SRB (sulforhodamin B) assay [Citation21]. They were maintained as stocks in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum (Gibco). Cell cultures were passaged once or twice weekly by using trypsin-EDTA to detach the cells from their culture flasks. The rapidly growing cells were harvested, counted, and incubated at the appropriate concentration (1–2 × 104 cells/well) in 96-well plates. After incubation for 24 h, the compounds dissolved in culture medium were applied to the culture wells in triplicate and incubated for 48 h at 37°C under 5% CO2 /95% air atmosphere in a humidified incubator. The culture cells were fixed with 10% cold TCA and stained with 0.4% SRB dissolved in 1% acetic acid. After solublizing the bound stain with 10 mM of unbuffered Trisma base solution (pH 10.5) using gyratory shaker, the absorbance at 520 nm was measured spectrophotometrically in a microplate reader. Cytotoxic activity was evaluated by measuring the concentration of a compound which was required to inhibit the protein synthesis by 50% (IC50) and compared with that of adriamycin.
Results and discussion
Chemistry
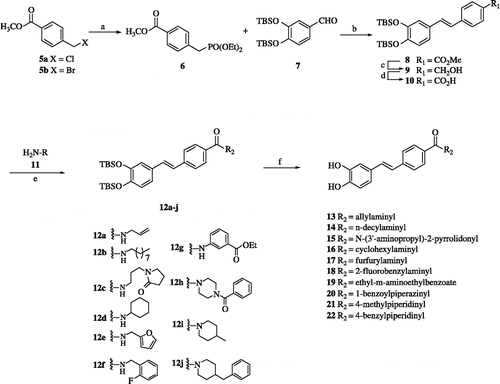
A series of stilbene derivatives (13–22) was prepared in 6 steps (Scheme ) using commercially available methyl 4-(chloro or bromomethyl)benzoate (5a or 5b) as a starting material. Compounds 5a and 5b were converted to phosphonate 6 by treatment with triethyl phosphite at 160 °C for 3 h in 85% yield [Citation22] Compound 6 was coupled with freshly prepared aldehyde 7 through a Wadsworth-Horner-Emmons reaction in order to obtain ester 8 in good yield. The n-butyllithium (n-BuLi) used as a base resulted in the production of ester 8, mostly the E isomer, while sodium hydride (NaH, 60% dispersion) afforded ester 8 in a 25:1 ratio of E:Z, respectively, which were cleanly separated by flash column chromatography. The reduction of ester 8 by diisobutylaluminum hydride (DIBAL-H) in dichloromethane afforded alcohol 9 in 77% yield. The oxidation of alcohol 9 through Swern oxidation [Citation23] or tetrapropylammonium perruthenate (TPAP)/N-methylmorpholine (NMO) [Citation24] gave aldehydes in 89% and 90% yields, respectively, which was subsequently treated with sodium chlorite (NaClO2) and sodium dihydrogen phosphate (NaH2PO4) in t-BuOH to give acid 10 in high yield. Acid 10 was coupled with several amines [allyl amine, N-decyl amine, N-(3′-aminopropyl)-2-pyrrolidone, N-cyclohexyl amine, furfuryl amine, 2-fluorobenzylamine, ethyl-m-aminoethylbenzoate, 1-benzoylpiperazine, 4-methylpiperidine, 4-benzylpiperidine] in the presence of 2-(7-aza-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorophosphate (HATU)/CH2Cl2 [Citation25] to afford amides 12a–j, which were subjected to the removal of TBS from dihydroxy groups to produce new trans-stilbene derivatives 13–22 in good yields (Scheme ).
Scheme 1. Reagents and conditions: (a) P(OEt)3, 160 °C, 3 h, 85%; (b) 7, n-BuLi (1.3 equiv), THF, − 20 °C, 1 h; then rt, 12 h, 86%, or 7, NaH (1.3 equiv), CH2Cl2, 0 °C, 16 h, 80% (E:Z = 25:1); (c) DIBAL-H (2.5equiv), CH2Cl2, − 78 °C, 1 h, 77%; (d) DMSO, (COCl)2, TEA, CH2Cl2, − 78 °C, 1 h, 89% or TPAP (5 mol %), NMO, THF, rt 30 min, 90%, then NaClO2 (3.0 equiv), NaH2PO4 (3.0 equiv), 2-methyl-2-butene, t-BuOH, 0 °C, 16 h, 96%; (e) HATU (1.5 equiv), CH2Cl2, rt, 16 h, 50%–91%; (f) 20% TFA, CH2Cl2, rt, 1 h, 50–80%, or TBAF (2.5 equiv), THF, 0 °C, 30 min, (64%–73%).
Biological activity
The in vitro cytotoxicities of stilbene derivatives 13–22 were evaluated against five human cancer cell lines, A549 (non-small cell lung carcinoma), SK-OV-3 (ovarian carcinoma), SK-MEL-2 (melanoma), XF498 (CNS carcinoma) and HCT-15 (colon carcinoma) using the SRB (sulforhodamine B) method (). Compounds 16 and 19 showed more cytotoxic activity than the other compounds, and the cytotoxic activity of compound 19 was especially superior to that of resveratrol against the human tumour cell lines tested. The IC50 value of compound 19 was 5.7 μM against the SK-OV-3, whereas that of adriamycin was 4.3 μM ().
Table I. In vitro anticancer activity of new stilbene derivatives 13–22.
Conclusion
A series of new stilbene derivatives 13–22 was prepared, and the anticancer activities of the derivatives were evaluated in vitro. The synthetic strategies involved the use of the well-known Wadsworth-Horner-Emmons condensation and coupling reactions. We found that compound 19 exhibited the most potent anticancer activity with an IC50 value of 5.7 μM-14.4 μM, and compound 16 and 19 showed efficacies comparable to the anticancer activity of adriamycin in vitro in the SK-OV-3 cell line.
Acknowledgements
This work has been supported by a KOSEF Brain Neurobiology Grant (2007), Ewha Global Challenge (BK21) grant, a research grant from the Ministry of Health & Welfare (Project No B040002), the Republic of Korea, E. L and Y. L the fellowship of the BK 21 program from the Ministry of Education and Human Resources Development and the Seoul Science Fellowship Program, and a grant (20070301-034-026-007-04-00) from BioGreen 21 Program, Rural Development Administration, Republic of Korea. Hyung-In Moon and Ill-Min Chung equally contributed to this work.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- GR Pettit, CR Anderson, DL Herald, MK Jung, DJ Lee, E Hamel, and RK Pettit. (2003). Antineoplastic agents. 487. Synthesis and biological evaluation of the antineoplastic agent 3,4-methylenedioxy-5,4′-dimethoxy-3′-amino-Z-stilbene and derived amino acid amides. J Med Chem 46:525–531.
- H Nakamura, H Kuroda, H Saito, R Suzuki, T Yamori, K Maruyama, and T Haga. (2006). Synthesis and biological evaluation of boronic acid containing cis-stilbenes as apoptotic tubulin polymerization inhibitors. Chem Med Chem 1:729–740.
- YQ Li, ZL Li, WJ Zhao, RX Wen, QW Meng, and Y Zeng. (2006). Synthesis of stilbene derivatives with inhibition of SARS coronavirus replication. Eur J Med Chem 41:1084–1089.
- JM Merillon, B Fauconneau, PW Teguo, L Barrier, J Vercauteren, and F Huguet. (1997). Antioxidant activity of the stilbene astringin, newly extracted from Vitis vinifera cell cultures. Clin Chem 43:1092–1093.
- Y Inamori, M Kubo, M Ogawa, H Tsujibo, Y Miki, and S Takemura. (1987). Biological activities of 3,3′-dihydroxy-alpha,beta-diethylstilbene and its isomer, diethylstilbestrol. Chem Pharm Bull 35:3502–3506.
- R Nieuwland, OL Wijburg, G van Willigen, and JW Akkerman. (1994). Alpha 2A-adrenergic receptors activate protein kinase C in human platelets via a pertussis toxin-sensitive G-protein. FEBS Lett 339:79–83.
- NN Kasri, G Bultynck, JB Parys, G Callewaert, L Missiaen, and H De Smedt. (2005). The N-terminal Ca2+-independent calmodulin-binding site on the inositol 1,4,5-trisphosphate receptor is responsible for calmodulin inhibition, even though this inhibition requires Ca2+. Mol Pharmacol 68:241–250.
- F Hakimuddin, G Paliyath, and K Meckling. (2004). Selective cytotoxicity of a red grape wine flavonoid fraction against MCF-7 cells. Breast Cancer Res Treat 85:65–79.
- A Carella, A Castaldo, R Centore, A Fort, A Sirigu, and A Tuzi. (2002). Synthesis and second order nonlinear optical properties of new chromophores containing 1,3,4-oxadiazole and thiophene rings. J Chem Soc Perkin Trans 2 11:1791–1795.
- S Gester, F Wuest, B Pawelke, R Bergmann, and J Pietzsch. (2005). Synthesis and biodistribution of an 18F-labelled resveratrol derivative for small animal positron emission tomography. Amino Acids 29:415–428.
- XM Wang, YF Zhou, WT Yu, C Wang, Q Fang, MH Jiang, H Lei, and HZ Wang. (2000). Two-photon pumped lasing stilbene-type chromophores containing various terminal donor groups: Relationship between lasing efficiency and intramolecular charge transfer. J Mater Chem 10:2698–2703.
- E Christian, M Bernd, N Manfred, R Verena, L Barbara, B Peter, and S Joachim. (2003). Covalent binding of chloroacetamide herbicides to the active site cysteine of plant type III polyketide synthases. Phytochemistry 64:1045–1054.
- M Alami, F Liron, M Gervais, JF Peyrat, and JD Brion. (2002). Ortho substituents direct regioselective addition of tributyltin hydride to unsymmetrical diaryl (or heteroaryl) alkynes: An efficient route to stannylated stilbene derivatives. Angew Chem Int Edit 41:1578–1580.
- V Cardile, R Chillemi, L Lombardo, S Sciuto, C Spatafora, and C Tringali. (2007). Antiproliferative activity of methylated analogues of E- and Z-resveratrol. Z Naturforsch [C] 62:189–195.
- H Nakamura, H Kuroda, H Saito, R Suzuki, T Yamori, K Maruyama, and T Haga. (2006). Synthesis and biological evaluation of boronic acid containing cis-stilbenes as apoptotic tubulin polymerization inhibitors. Chem Med Chem 1:729–740.
- F Minutolo, G Sala, A Bagnacani, S Bertini, I Carboni, G Placanica, G Prota, S Rapposelli, N Sacchi, M Macchia, and R Ghidoni. (2005). Synthesis of a resveratrol analogue with high ceramide-mediated proapoptotic activity on human breast cancer cells. J Med Chem 48:6783–6786.
- K Likhitwitayawuid, A Sornsute, B Sritularak, and P Ploypradith. (2006). Chemical transformations of oxyresveratrol (trans-2,4,3′,5′-tetrahydroxystilbene) into a potent tyrosinase inhibitor and a strong cytotoxic agent. Bioorg Med Chem Lett 16:5650–5653.
- MB Andrus, and J Liu. (2006). Synthesis of polyhydroxylated ester analogs of the stilbene resveratrol using decarbonylative Heck couplings. Tetrahedron Lett 47:5811–5814.
- D Simoni, M Roberti, FP Invidiata, E Aiello, S Aiello, P Marchetti, R Baruchello, M Eleopra, A Di Cristina, S Grimaudo, N Gebbia, L Crosta, F Dieli, and M Tolomeo. (2006). Stilbene-based anticancer agents: Resveratrol analogues active toward HL60 leukemic cells with a non-specific phase mechanism. Bioorg Med Chem Lett 16:3245–3248.
- M Jung, Y Lee, M Park, H Kim, H Kim, E Lim, J Tak, M Sim, D Lee, N Park, WK Oh, KY Hur, ES Kang, and HC Lee. (2007). Design, synthesis, and discovery of stilbene derivatives based on lithospermic acid B as potent protein tyrosine phosphatase 1B inhibitors. Bioorg Med Chem Lett 17:4481–4486.
- LV Rubinstein, RH Shoemaker, KD Paull, RM Simon, S Tosini, P Skehan, D Scudiero, A Monks, and MR Boyd. (1990). Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst 82:1113.
- AC Freydank, MG Humphrey, RW Friedrich, and B Luther-Davies. (2002). Synthesis and optical characterization of unsymmetrical oligophenylenevinylenes. Tetrahedron 58:1425–1432.
- AJ Mancuso, SL Huang, and D Swern. (1978). Oxidation of Long-chain and Related Alcohols to Carbonyls by Dimethyl Sulfoxide “Activated” by Oxalyl Chloride. J Org Chem 43:2480–2482.
- JC Jung, R Kache, KK Vines, YS Zheng, P Bijoy, M Valluri, and MA Avery. (2004). Total syntheses of epothilones B and d. J Org Chem 69:9269–9284.
- SK Shannon, MJ Peacock, SA Kates, and G Barany. (2003). Solid-phase synthesis of lidocaine and procainamide analogues using backbone amide linker (BAL) anchoring. J Comb Chem 5:860–868.