Abstract
The involvement of tau phosphorylation in apoptosis resembling Alzheimer's disease (AD) was investigated using a cell model of P19 cells stably expressing human tau441 (tau/P19 cells). Apoptotic cell death was observed specifically in tau/P19 cells during neural differentiation with retinoic acid (RA) treatment. A CaM kinase II inhibitor, KN-93, protected tau/P19 cells from apoptosis, although it stimulated the cell death of wild-type P19 cells (wt/P19 cells). W-7 and calmidazolium, calmodulin antagonists, also specifically inhibited the apoptosis of tau/P19 cells. LiCl, an inhibitor of glycogen synthase 3, a tau kinase, was effective in protecting tau/P19 cells from apoptosis, but the protective effect was less than that of CaM kinase II inhibitor and calmodulin antagonists. Tau in the nuclei of tau/P19 cells was phosphorylated at the sites for CaM kinase II detected by an antibody recognizing a phosphorylated form of tau. These results indicated that CaM kinase II was involved in the apoptosis of tau/P19 cells induced by RA treatment.
Introduction
Alzheimer's disease (AD), a progressive neurodegenerative disorder, is characterized by two hallmark lesions: amyloid plaques and neurofibrillary tangles (NFTs). The core peptide of amyloid plaques is Amyloid-β (Aβ), which is processed from the amyloid precursor protein (APP), and NFTs are composed of tau in a hyperphosphorylated state (NFT-tau) relative to normal tau in the adult brain. Hyperphosphorylation of tau is a crucial step in the development of NFTs and one of the earliest signs of neuronal degeneration Citation1, Citation2, Citation3. Tau is a family of proteins that modulates the dynamics of microtubules, and is an important component of the neuronal cytoskeleton. The relationship between the phosphorylation of tau and neuronal cell death in AD can be addressed using cell models where tau is overexpressed. A good correlation between the amount of hyperphosphorylated tau and the clinical severity of AD patients has been demonstrated [Citation4]. It has been reported that a MEK inhibitor prevents fibrillar β-amyloid-induced tau phosphorylation and degeneration in cultured hippocampal neurons [Citation5]. Pseudohyperphosphorylation of tau induces apoptotic cell death in cultured cortical neurons [Citation6], and in organotypic hippocampal slices [Citation7]. Site-specific phosphorylation of tau is regulated by concerted and sequential actions of many protein kinases and protein phosphatases [Citation3,Citation8].
In studies of AD pathogenesis, it is important to understand the kinase(s) involved in enhanced tau phosphorylation and neuronal cell death. Tau protein can be phosphorylated in vitro at many specific sites of NFT-tau by tau kinases such as glycogen synthase kinase 3 (GSK3) and cyclin-dependent protein kinase 5 (CDK5) [Citation9,Citation10] as well as second messenger-dependent protein kinases such as cyclic AMP-dependent protein kinase (PKA) [Citation11], Ca2 + /calmodulin-dependent protein kinase II (CaM kinase II) [Citation12,Citation13] and protein kinase C (PKC) [Citation14]. We have established P19 and NG108-15 cell lines that allow the stable expression of human tau Citation15, Citation16, Citation17, and demonstrated that P19 cells stably expressing human tau441 (tau/P19 cells) specifically undergo extensive apoptosis during neural differentiation with retinoic acid (RA) treatment [Citation16]. In the present study, we demonstrated, using CaM kinase II inhibitor, a calmodulin antagonist, and an antibody against phosphorylated tau at the site of CaM kinase II, that CaM kinase II is involved in the apoptosis of tau/P19 cells induced by RA treatment.
Materials and methods
Materials
Mouse embryonal carcinoma cell line P19 cells overexpressing human tau441 (tau/P19 cells) was established previously [Citation16,Citation17]. Human tau441 (hTau441) is the longest central nervous system isoform of tau among splice forms. hTau441 was purified as described previously [Citation13]. Monoclonal anti-tau antibody 12E8 recognizing phosphorylated Ser262 and Ser356 in hTau441 [Citation18] was provided by Dr. Peter Seubert, Elan Pharmaceutical Inc. (South San Francisco, U.S.A). Polyclonal anti-tau antibody clone H-150 recognizing total tau was purchased from Santa Cruz Biotech. Inc. (Santa Cruz, U.S.A.). Indirubin-3′-monoxime was purchased from Merck Ltd. Japan (Tokyo, Japan). KN-93, W-7, calmidazolium, and LiCl were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Enhanced chemiluminescence reagents for immunoblotting were from PerkinElmer Life Sci. (Boston, USA).
Cell culture and analysis
P19 cells (wt/P19 and tau/P19 cells) were cultured and induced to differentiate with RA treatment as described previously [Citation16,Citation17]. When the effect of various inhibitors on cell viability was examined, these reagents were added to culture medium simultaneously with RA. Cells were cultured in the presence of RA alone or both inhibitor and RA for the indicated period. RA was dissolved in ethanol, and W-7, calmidazolium and indirubin-3′-monoxime were dissolved in dimethyl sulfoxide (DMSO). The final concentration of ethanol and DMSO was less than 0.5% in the culture medium. DMSO and ethanol were added at the same concentrations in control experiments. KN-93 and LiCl were dissolved in water and added to the culture medium. Cell viability was assayed by trypan blue exclusion, and expressed as a percent of living cells at indicated times. Cytosolic and nuclear fractions of cells were isolated as described previously [Citation16]. Immunoblot analysis was carried out as described previously [Citation16]. Briefly, proteins were separated by SDS-PAGE and electrophoretically transferred to a nitrocellulose membrane. The membrane was blocked with 5% non-fat dry skim milk, and then incubated with anti-tau antibody H-150 (diluted 1:500) or anti-phosphorylated tau antibody12E8 (1:500) overnight at 4°C. After washing, the membrane was incubated with horseradish peroxidase-coupled anti-mouse or anti-rabbit IgG antibody (diluted 1: 2000). Immunoreactive bands were detected using an enhanced chemiluminescence technique.
Results
KN-93 specifically protects apoptosis of tau/P19 cells
We previously demonstrated that tau/P19 cells specifically underwent extensive apoptosis during neural differentiation with RA treatment [Citation16]. Apoptosis was demonstrated by caspase-3 activation, DNA fragmentation, and chromatin condensation. In the present study, we investigated the involvement of CaM kinase II in the apoptosis of tau/P19 cells induced by RA. The viabilities of tau/P19 and wt/P19 cells were compared during neural differentiation with RA treatment ((A)). The viability of tau/P19 cells was greatly reduced with RA treatment, whereas that of wt/P19 cells was gradually reduced ((A), left panel). The tau/P19 cells abundantly underwent cell death in the early period of RA treatment; the cell viability was about 40% and 30% at 24 and 48 h, respectively, during neural differentiation induced by RA treatment. KN-93, a selective inhibitor of CaM kinase II, increased the viability of tau/P19 cells about 1.7- and 2.1-fold at 24 and 48 h, respectively, as compared with that in its absence. The viability of wt/P19 cells was about 80% and 55% at 24 and 48 h, respectively, during neural differentiation with RA treatment. KN-93 reduced the viability of wt/P19 cells to about 50% and 30% at 24 and 48 h, respectively, during the neural differentiation with RA treatment ((A), right panel).
Figure 1. Effect of KN-93 on the apoptosis induced during neural differentiation with RA treatment. (A), Time course of apoptosis in the presence or absence of KN-93. Tau/P19 or wt/P19 cells were differentiated with treatment with 0.3 μM RA in the absence (left panel) or presence (right panel) of 5 μM KN-93. RA and KN-93 were added to the culture medium at 0 time, and cell viability was determined at the indicated times. (B), Effect of the concentration of differentiated cells with treatment with 0.3 μM RA in the presence or absence of the indicated amounts of KN-93 at 24 h (left panel) and 48 h (right panel) after induction of differentiation. Cell viability was determined at the indicated concentrations of KN-93.
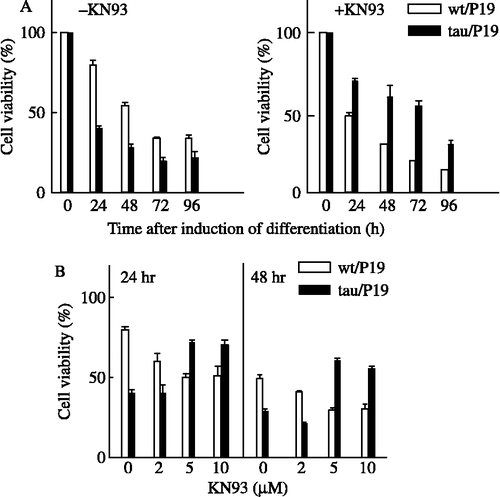
The effect of KN-93 concentrations on cell viability was examined ((B)). Optimum concentration of KN-93 to inhibit the apoptosis of tau/P19 cells was 5-10 μM. At 2 μM, KN-93 did not show any protective effect against the apoptosis of tau/P19 cells, although it stimulated the apoptosis of wt/P19 cells. These results showed that KN-93 inhibited normal differentiation and stimulated apoptosis of wt/P19 cells, but protected tau/P19 cells from apoptosis, suggesting that CaM kinase II was required for the survival of differentiating wt/P19 cells, but participated in apoptosis of tau/P19 cells via excessive tau phosphorylation.
Calmodulin antagonists specifically protect apoptosis of tau/P19 cells
To confirm that the apoptosis of tau/P19 cells was mediated by calmodulin and the CaM kinase II pathway, the effect of calmodulin antagonists was examined, as shown ((A)). W-7 and calmidazolium inhibited the apoptosis of tau/P19 cells and increased cell viability about 1.9- and 1.8-fold, respectively, as compared with that in the absence of the inhibitors. W-7 and calmidazolium did not inhibit, but slightly stimulated the apoptosis of wt/P19 cells. These results indicated that the CaM kinase II pathway was specifically involved in the apoptosis of tau/P19 cells induced by RA treatment.
Figure 2. Effect of calmodulin antagonists or tau kinase inhibitors on apoptosis, and phosphorylation of nuclear tau at CaM kinase II site(s). (A), Effect of calmodulin antagonists or tau kinase inhibitors on the apoptosis during neural differentiation induced by RA treatment. Tau/P19 or wt/P19 cells were differentiated with treatment with 0.3 μM RA in the presence or absence of 10 μM W-7, 10 μM calmidazolium, 5 mM LiCl, and 50 nM indirubin-3′-monoxime. Cell viability was measured at 24 h after induction of differentiation. left panel, tau/P19 cells; right panel, wt/P19 cells. Abbreviations: C, control in the absence of inhibitors; Calmi, calmidazolium; Indi, Indirubin-3′-monoxime. (B), Immunoblot analysis of phosphorylation of nuclear tau at CaM kinase II site(s). Cytosolic and nuclear fractions of tau/P19 cells without RA treatment were subjected to immunoblot analysis. Each lane was applied with 50 μg of protein. Lane Tau441 was applied with purified hTau441 (100 ng). Tau was detected with H-150 antibody recognizing total tau and with 12E8 antibody recognizing phosphorylated Ser262 and Ser365. Upper panel, tau/P19 cells; lower panel, wt/P19 cells; left panel, H-150 antibody (H-150 Ab); right panel, 12E8 antibody (12E8 Ab). Abbreviations: Cyt, cytosolic fraction; Nuc, nuclear fraction.
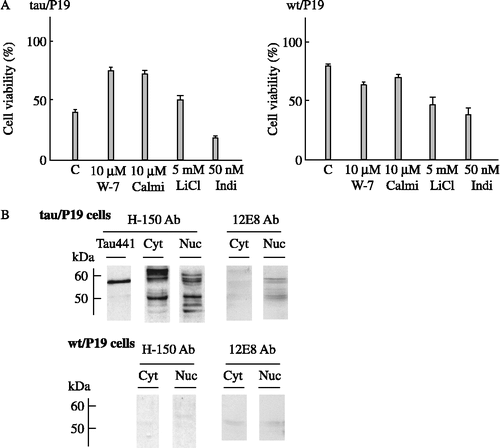
The effect of inhibitors of tau kinases, such as GSK3 and CDK5, was also examined ((A)). LiCl, an inhibitor of GSK3, increased the viability of tau/P19 cells 1.25-fold, but stimulated the apoptosis of wt/P19 cells. The protective effect of LiCl on apoptosis of tau/P19 cells was weaker than that of CaM kinase II inhibitor and calmodulin antagonists. Indirubin-3′-monoxime, an inhibitor of CDK5, greatly stimulated the apoptosis of both wt/P19 and tau/P19 cells, indicating that CDK5 played an important role in the survival of cells under neural differentiation with RA treatment.
Nuclear tau is phosphorylated at CaM kinase II site(s)
KN-93 and calmodulin antagonists are known to inhibit calmodulin-dependent protein kinases and calmodulin pathway including CaM kinase I, II and IV. To confirm the involvement of CaM kinase II in the apoptosis of tau/P19 cells, we next investigated the phosphorylation of tau at the site of CaM kinase II using anti-tau antibody 12E8 ((B)). Tau in the cytosol and nuclei of tau/P19 cells detected by H-150 antibody showed several bands around 60 kDa depending on the phosphorylation states, and some degradation products were also detected around 50 kDa or smaller. Tau in the nuclei of tau/P19 cells, as detected by 12E8 antibody, was the phosphorylated form with a molecular mass of about 60 kDa and its degradation product of about 50 kDa, indicating that nuclear tau was phosphorylated at Ser262 and/or Ser356. Cytosolic tau did not react with 12E8 antibody, indicating that it was not phosphorylated at these sites. Significant bands were not detected in wt/P19 cells with H-150 and 12E8 antibodies. These results indicted that tau in the nuclei of tau/P19 cells was phosphorylated at CaM kinase II site(s) before RA treatment.
Discussion
In studies of AD pathogenesis, the underlying molecular mechanisms responsible for cell death are unsolved. The concept of apoptosis in AD was initially demonstrated by Cotman's group that synthetic Aβ peptides trigger the degeneration of cultured neurons through activation of an apoptotic pathway [Citation19]. Postmortem analysis of the human brain has also shown apoptotic DNA fragmentation [Citation20]. In an animal model of AD, the toxicity of tau is required in its mutation in some inherited diseases, such as familial frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). The P301L mutation of human tau is the most common mutation linked to FTDP-17 and is pathologically characterized by the presence of NFTs and neuronal loss in the forebrain [Citation21]. The overexpression of P301L mutation of human tau in transgenic mice leads to increased tau phosphorylation [Citation22]. The main cytotoxic effects of tau are not exerted by NFTs but by lower molecular mass aggregates of tau in P301L mutant mice [Citation23]. Cytoplasmic c-jun N-terminal kinase (JNK) activation may also play an important role in the abnormal tau hyperphosphorylation associated with R406W tau mutation and in AD [Citation24]. This mutation also causes AD-like dementia and tauopathy in humans. On the other hand, an animal model of AD expressing human mutated tau protein provides evidence for nonapoptotic neurodegeneration [Citation25]. Some AD characterized by tau hyperphosphorylation and neurodegeneration are directly attributable to mutations in the tau gene, whereas in others, tau hyperphosphorylation occurs in the absence of tau mutations in this study [Citation26]; therefore, we used tau/P19 cells as a cell model to investigate apoptosis related to tau expression. P19 is a widely used embryonic stem cell line and a well-documented system for the analysis of neural differentiation: RA induces P19 cells to differentiate into neurons [Citation27].
Previously, we demonstrated the involvement of CaM kinase II in neuronal differentiation of P19 cells induced by RA treatment [Citation28]. In the present study, we demonstrated, using CaM kinase II inhibitor, a calmodulin antagonist, and an antibody against phosphorylated tau at the site of CaM kinase II, that the calmodulin pathway through CaM kinase II is involved in the apoptosis of tau/P19 cells induced during neural differentiation with RA treatment ( and ). Under experimental conditions, KN-93 and calmodulin antagonists protected the apoptosis of tau/P19 cells, but stimulated that of wt/P19 cells ().
Neuronal CaM kinase II is one of the most abundant protein kinases in the brain and phosphorylates a broad range of brain proteins to regulate important neuronal functions [Citation29]. One of its major substrates is cytoskeletal protein, including tubulin, microtubule-associated protein 2 (MAP2) and tau. Among abnormal phosphorylation sites of NFTs, 4 sites, such as Thr212, Ser214, Ser262, and Ser356, were phosphorylated by CaM kinase II ((A)) [Citation13]. Some protein kinases other than CaM kinase II phosphorylate these sites, such as Thr212, which is phosphorylated by GSK3 and MAPK, Ser214 by PKA and PKC, Ser262 by GSK3, p110K, PKA, and PKC, and Ser356 by GSK3, PKA and p110K Citation9, Citation10, Citation11, Citation14, Citation30–32; however, only CaM kinase II can phosphorylate all four sites. If CaM kinase II had been abnormally activated, due to a breakdown of the normal regulatory mechanisms, it would be responsible for the phosphorylation of tau at NFT sites. Ser262 and Ser356 residues are present at tubulin-binding sites, and the phosphorylation of tau at these sites reduces the ability of tau to bind MTs and to promote their assembly [Citation33,Citation34].
Figure 3. Schematic representation of specific phosphorylation sites of htau441 found in PHF-tau, and involvement of CaM kinase II in apoptosis of tau/P19 cells. (A), Specific phosphorylation sites of htau441 found in only PHF-tau of the AD brain. Non-fetal-type phosphorylation sites found in PHF-tau, namely PHF-tau specific phosphorylation sites, are shown, although PHF-tau is also phosphorylated at the same sites of fetal-type phosphorylation. *, tubulin binding site; P, PHF-tau-specific phosphorylation site; P in circle, phosphorylation site with CaM kinase II. (B), Involvement of CaM kinase II in apoptosis of tau/P19 cells. In tau/P19 cells, tau was distributed throughout the cytoplasm and co-localized with microtubules. Some tau dissociated from MTs was phosphorylated by CaM kinase II and/or GSK3, and then translocated into the nucleus. Nuclear tau may disrupt RA signaling during neural differentiation resulting in apoptosis. Abbreviations; MTs, microtubules; Mt, mitochondria, Nuc, nuclei; RAR, retinoic acid receptor.
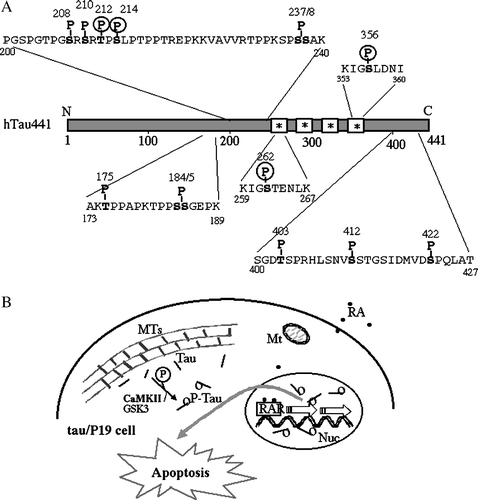
In tau/P19 cells, tau is distributed throughout the cytoplasm and is co-localized with microtubules, but some tau is localized inside nuclei separated from MTs [Citation16]. Some tau dissociated from MTs is phosphorylated by protein kinases, including CaM kinase II and GSK3, and then translocated into the nucleus (). Nuclear tau may disrupt RA signaling during neural differentiation, resulting in apoptosis. It has been reported that RA signaling stimulates the expression of TrkB, and regulates neurogenesis in neural stem cells, suggesting that it may promote cell survival [Citation35]. RA signaling may be disrupted by increased nuclear tau, resulting in the reduction of TrkB followed by cell death. CaM kinase II may be involved in the apoptosis of tau/P19 cells during neural differentiation with RA treatment.
In the AD brain, the concentration of tau is elevated, and hyperphosphorylated tau polymerizes into NFTs, [Citation36]; however, several lines of evidence indicate that aberrant tau phosphorylation in the absence of tau aggregates can cause a neurodegenerative phenotype similar to AD [Citation37]. It is also observed that elevated tau moves into the nucleus, and serves as an apoptotic substrate of caspase-3 [Citation38]. These observations suggest that tau phosphorylated at tubulin binding sites by CaM kinase II dissociates from MTs and translocates into the nucleus, and that nuclear tau promotes the apoptosis of tau/P19 cells resembling those in the AD brain, as shown in (B). Our observations provide new insight into the possible cause of AD and other neurodegenerative diseases.
Acknowledgements
We thank Dr. K. Iqubal of New York State Institute for Basic Research in Developmental Disabilities, Staten Island, New York, U.S.A., for providing the plasmid containing the cDNA clone of human tau441. We thank Dr. P. Seubert of Elan Pharmaceutical Inc., South San Francisco, California, U.S.A., for providing monoclonal anti-tau antibody 12E8. This work was supported in part by a grant-in-aid for the National Project on Protein Structure and Functional Analysis from the Ministry of Education, Culture, Sport, Science and Technology of Japan.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- L Buee, T Bussiere, V Buee-Scherrer, A Delacourte, and PR Hof. (2000). Tau protein isoforms, phosphorylation, and role in neurodegerative disorders. Brain Res Rev 33:95–130.
- VMY Lee, M Goedert, and JQ Trojanowski. (2001). Neurogenerative tauopathies. Annu Rev Neurosci 24:1121–1159.
- K Iqbal, AC Alonso, S Chen, MO Chohan, E El-Akkad, CX Gong, S Khatoon, B Li, F Liu, A Rahman, H Tanimukai, and I Grundke-Iqbal. (2005). Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739:198–210.
- M Holzer, HP Holzapfel, D Zedlick, MK Bruckner, and T Arendt. (1994). Abnormally phosphorylated tau protein in Alzheimer's disease: Heterogeneity of individual regional distribution and relationship to clinical severity. Neuroscience 63:499–516.
- M Rapoport, and A Ferreira. (2000). PD98059 prevents neurite degeneration induced by fibrillar β-amyliod in mature hippocampal neurons. J Neurochem 74:125–133.
- N Shahani, S Subramaniam, T Wolf, C Tackenber, and R Brand. (2006). Tau aggregation and progressive neuronal degeneration in the absence of changes in spine density and morphology after targeted expression of Alzheimer's disease-relevant tau constructs in organotypic hippocampal slices. J Neurosci 26:6103–6114.
- J Leschik, A Welzel, C Weissmann, A Eckert, and R Brandt. (2007). Inverse and distinct modulation of tau-dependent neurodegeneration by presenilin 1 and amyloid-β in cultures cortical neurons: Evidence that tau phosphorylation is the limiting factor in amyloid-β-induced cell death. J Neurochem 101:1303–1315.
- WH Stoothoff, and GV Johsnon. (2005). Tau phosphorylation: Physiological and pathological consequences. Biochim Biophys Acta 1739:280–297.
- K Ishiguro, A Shiratsuchi, S Sato, A Omori, M Arioka, S Kobayashi, T Uchida, and K Imahori. (1993). Glycogen synthase kinase 3β is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett 325:167–172.
- ET Lund, R McKenna, DB Evans, SK Sharma, and WR Mathews. (2001). Characterization of the in vitro phosphorylation of human tau by tau protein kinase II (cdk5/p20) using mass spectrometry. J Neurochem 76:1221–1232.
- CW Scott, RC Spreen, JL Herman, FP Chow, MD Davison, J Young, and CB Caputo. (1993). Phosphorylation of recombinant tau by cAMP-dependent protein kinase. Identification of phosphorylation sites and effect on microtubule assembly. J Biol Chem 268:1166–1173.
- B Steiner, EM Mandelkow, J Biernat, N Gustke, HE Meyer, B Schmidt, G Mieskes, HD Söling, D Drechsel, MW Kirschner, M Goedert, and E Mandelelkow. (1990). Phosphorylation of microtubule-associated protein tau: Identification of the site for Ca2+-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J 9:3539–3544.
- Y Yoshimura, T Ichinose, and T Yamauchi. (2003). Phosphorylation of tau protein to sites found in Alzheimer's disease brain is catalyzed by Ca2+/calmodukin-dependent protein kinase II as demonstrated tandem mass spectrometry. Neurosci Lett 353:185–188.
- I Correas, J Diaz-Nido, and J Avila. (1992). Microtubule-associated protein tau is phosphorylated by protein kinase C on its tubulin binding domain. J Biol Chem 267:15721–15728.
- C Yoshizaki, M Tsukane, and T Yamauchi. (2004). Overexpression of tau leads to the stimulation of neurite outgrowth, the activation of caspase 3 activity, and accumulation and phosphorylation of tau in neuroblastoma cells on cAMP treatment. Neurosci Res 49:363–371.
- M Tsukane, and T Yamauchi. (2006). Increase in apoptosis with neural differentiation and shortening of the lifespan of P19 cells overexpressing tau. Neurochem Int 48:243–254.
- M Tsukane, C Yoshizaki, and T Yamauchi. (2007). Development and specific induction of apoptosis of cultured cell models overexpressing human tau during neural differentiation: Implication in Alzheimer's disease. Anal Biochem 360:114–122.
- P Seubert, M Mawal-Dewan, R Barbort, R Jakes, M Goedert, GVW Johnson, JM Litersky, D Schenk, I Lieberburg, JQ Trojanowski, and VMY Lee. (1995). Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem 270:18917–18922.
- DT Loo, A Copani, CJ Pike, ER Whittemore, AJ Walencewicz, and CW Cotman. (1993). Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA 90:7951–7955.
- G Smale, NR Nichols, DR Brady, CE Finch, and WE HortonJr. (1995). Evidence for apoptotic cell death in Alzheimer's disease. Exp Neurol 133:225–230.
- M Hutton, CL Lendon, P Rizzu, M Baker1, S Froelich, H Houlden, S Pickering-Brown, S Chakraverty, A Isaacs, A Grover, J Hackett, J Adamson, S Lincoln, D Dickson, P Davies, RC Petersen, M Stevens, E de Graaff, E Wauters, J van Baren, M Hillebrand, M Joosse, JM Kwon, P Nowotny, LK Che, J Norton, JC Morris, LA Reed, J Trojanowski, H Basun, L Lannfelt, M Neystat, S Fahn, F Dark, T Tannenberg, PR Dodd, N Hayward, JBJ Kwok, PR Schofield, A Andreadis, J Snowden, D Craufurd, D Neary, F Owen, BA Oostra, J Hardy, A Goate, J van Swieten, D Mann, T Lynch, and P Heutink. (1998). Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705.
- J Lewis, E McGowan, J Rockwood, H Melrose, P Nacharaju, M van Slegtenhorst, K Gwinn-Hardy, MP Murphy, M Baker, X Yu, K Duff, J Hardy, A Corral, WL Lin, SH Yen, DW Dickson, P Davies, and M Hutton. (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25:402–405.
- S Le Corre, HW Klafki, N Plesnila, G Hubinger, A Obermeier, H Sahagun, B Monse, P Seneci, J Lewis, J Eriksen, C Zehr, M Yue, E McGowan, DW Dickson, M Hutton, and HM Roder. (2006). An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci USA 103:9673–9678.
- Y Takabayashi, E Planel, DH Chui, S Sato, T Miyasaka, N Sahara, M Maruyama, N Kikuchi, K Yoshioka, R Rivka, and A Takashima. (2006). c-jun N-terminal kinase hyperphosphorylates R406W tau at the PHF-1 site during mitosis. FASEB J 20:762–764.
- B Allen, E Ingram, M Takao, MJ Smith, R Jakes, K Virdee, H Yoshida, H Yoshida, M Holzer, M Craxton, PC Emson, C Atzori, A Migheli, RA Crowther, B Ghetti, MG Spilantini, and M Goedert. (2002). Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22:9340–9351.
- J Hardy, K Duff, KG Hardy, J Perez-Tur, and M Hutton. (1998). Genetic dissection of Alzheimer's disease and related dementias: Amyloid and its relationship to tau. Nat Neurosci 1:355–358.
- EM Jones-Villeneuv, MA Rudnucki, JF Harris, and MW McBurney. (1983). Retinoic acid-induced neural differentiation of embryonal carcinoma cells. Mol Cell Biol 3:2271–2279.
- H Donai, T Murakami, T Amano, Y Sogawa, and T Yamauchi. (2000). Induction and alternative splicing of δ isoform of Ca2+/calmodulin-dependent protein kinase II during neural differentiation of P19 embryonal carcinoma cells and during brain development. Mol Brain Res 85:189–199.
- T Yamauchi. (2005). Neuronal Ca2+/calmodulin-dependnet protein kinase II—Discovery, progress in a quarter of a century, and perspective: Implication for learning and memory. Biol Pharm Bull 28:1342–1354.
- DP Hanger, JC Betts, TLF Loviny, WP Blackstock, and BH Anderton. (1998). New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J Neurochem 71:2465–2476.
- M Morishima-Kawashima, M Hasegawa, K Takio, M Suzuki, H Yoshida, K Titani, and Y Ihara. (1995). Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem 270:823–829.
- A Watanabe, M Hasegawa, M Suzuki, K Takio, M Morishima-Kawashima, K Titani, T Arai, KS Kosik, and Y Ihara. (1993). In vivo phosphorylation sites in fetal and adult rat tau. J Biol Chem 268:25712–25717.
- G Drewes, B Trinczek, S Illenberger, J Biernat, G Schmitt-Ulms, HE Meyer, EM Mandelkow, and E Mandelkow. (1995). Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at Alzheimer-specific serine 262. J Biol Chem 270:7679–7688.
- TJ Singh, JZ Wang, M Novak, E Kontzekova, I Grundke-Iqubal, and K Iqubal. (1996). Calcium/calmodulin-dependent protein kinase II phosphorylates tau at Ser-262 but only partially inhibits its binding to microtubules. FEBS Lett 387:145–148.
- J Takahashi, TD Palmer, and FH Gage. (1999). Retinoic acid and neurotrophins collaborate to regulate neurogenesis in adult-derived neural stem cell cultures. J Neurobiol 38:65–81.
- S Khatoon, I Grundke-Iqbal, and K Iqbal. (1992). Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: A radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem 59:750–753.
- R Brandt, M Hundelt, and N Shahani. (2005). Tau alteration and neuronal degeneration in tauopathies: Mechanisms and models. Biochim Biophys Acta 1739:331–354.
- TC Gamblin, F Chen, A Zambrano, A Abraha, S Lagalwar, AL Guillozet, M Lu, Y Fu, F Garcia-Sierra, N LaPointe, R Miller, RW Berry, LI Binder, and VL Cryns. (2003). Caspase cleavage of tau: Linking amyloid and neurofibirillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA 100:10032–10037.