Abstract
A new series of 12 N4-substituted isatin-3-thiosemicarbazones 2a-l has been synthesized, characterized and screened for in vitro cytotoxic, phytotoxic and urease inhibitory effects. All the compounds proved to be active in the brine shrimp bioassay; 2a, 2b, 2d, 2f and 2h-l exhibited a high degree of cytotoxic activity (LD50 = 1.10 × 10− 5 M–3.10 × 10− 5 M). In urease-inhibition assay, compounds 2a, 2b, 2e, 2f, 2h-j and 2l proved to be potent inhibitors displaying relatively much greater inhibition of the enzyme with IC50 values ranging from 20.6 μM to 50.6 μM. Amongst these, 2a and 2f were found to be the most potent ones exhibiting pronounced inhibition with IC50 value 20.6 μM. All the synthetic compounds showed weak to moderate (10–40%) phytotoxicity at the highest tested concentration (500 μg/mL) indicating their usefulness as inhibitors of soil ureases.
Introduction
The biological activities of isatin and its derivatives have been known for a long time. Isatin itself exhibited a range of actions such as CNS-MAO inhibition, sedative, anticonvulsant and anxiogenic activities [Citation1]. Similarly, isatin derivatives are known to possess a wide spectrum of pharmacological properties including anthelmintic, antibacterial, anticonvulsant, antifungal, antineoplastic, antiviral, cysticidal, herbicidal, hypotensive and enzymatic inhibition Citation1,Citation2,Citation3,Citation4. Among these, isatins-derived thiosemicarbazones have raised considerable interest Citation5,Citation6,Citation7,Citation8,Citation9,Citation10,Citation11,Citation12. Stimulated by this and in continuation of our drug discovery programme Citation13,Citation14,Citation15,Citation16,Citation17,Citation18, we have introduced very recently a number of N4-substituted isatin-3-thiosemicarbazones as urease inhibitors [Citation19,Citation20]. Urease, a nickel-dependent metalloenzyme, catalyzes the hydrolysis of urea in plants, algae, fungi and several microorganisms in the final step of organic nitrogen mineralization to produce ammonia and carbamate [Citation21]. The carbamate then spontaneously hydrolyses, at physiological pH, to form bicarbonate and a second molecule of ammonia Citation22,Citation23,Citation24,Citation25. The hydrolysis of the reaction products results in an abrupt net increase in pH, the major cause for the negative effects of the action of urease both for human and animal health, and for agriculture.
Urease serves as a virulence factor in certain human and animal infections of the urinary and gastrointestinal tracts, being involved in the development of kidney stones, urinary catheter encrustation, pyelonephritis, peptic ulceration, ammonia encephalopathy, hepatic coma and urinary tracts infections [Citation22,Citation25,Citation26]. Several classes of molecules including hydroxamic acids may be an answer for the treatment of these conditions [Citation27]. However, earlier studies [Citation22,Citation25,Citation28,Citation29] show many side effects, including depressed bone marrow biosynthesis and inhibition of DNA synthesis, and high doses have shown to be teratogenic. Therefore, need for the development of more potent and non- or less toxic urease inhibitors is evident and continues to be of a considerable interest.
In an agricultural setting, high soil bacterial ureases activity causes significant environmental and economic problems by releasing abnormally large amounts of ammonia into the atmosphere during urea fertilization. This further induces damage to germinating seeds, seedlings and young plants primarily by depriving them of their nutrition by the essential nutrient and secondly by ammonia toxicity, increasing the pH of the soil Citation30,Citation31,Citation32,Citation33. Urease inhibitors have been proposed to control urea hydrolysis in soil Citation22, Citation34,Citation35,Citation36,Citation37,Citation38,Citation39. Particularly, phophoramides have received considerable attention as urease inhibitors [Citation37,Citation39]. Studies have shown that urease inhibitors can be combined with fertilizers in small amounts to increase the overall efficiency of nitrogen utilization [Citation38]. However, it has been found that the inhibitors can induce some phytotoxicity at higher concentrations [Citation40]. These findings have emphasized the need to search for some efficient urease inhibitors, which could be combined with fertilizers in small amounts, so as to reduce environmental pollution and increase activity of urea nitrogen uptake by plants.
The exact knowledge with regard to the regions of urease involved in the binding of the substrates or inhibitors is a starting point in designing effective inhibitors capable of complementing all the structural requirements for a close interaction. In all the enzyme-inhibitor complexes examined so far, the urease active site is found to have pseudooctahedral and paramagnetic dinuclear nickel ions. It has been unambiguously proved by spectroscopic and X-ray crystallographic studies that hydroxamic acids (HXA) and phosphorodiamidates (PPD) represent the classes of synthetic inhibitors, which are good metal chelators and their mechanism of interaction involves binding to the metal ions of the active site of enzyme. Nonetheless, regardless of the class of the compounds, it is reported [Citation41] that a few functionalities with electronegative atoms such as oxygen, nitrogen and sulphur act either as bidentate (mostly), tridentate (rarely), or as ligand chelator to form octahedral complexes with the two slightly distorted octahedral nickel ions of the enzyme. Prompted by these findings, we very recently synthesized [Citation19] a number of isatin-thiosemicarbazones (derivatives of thiourea, a substrate-like urease inhibitor) and tested them as urease inhibitors [Citation20], as they were expected to act as chelating agents coordinating with the two nickel ions of the enzyme through carbonylo oxygen, imino nitrogen and thiolato sulphur atoms, thus inhibiting its activity. Some of these thiosemicarbazones showed promising activity and are proposed to be potential candidates for orally effective therapeutic agents used for the treatment of certain clinical conditions caused by bacterial ureases. The present work is an extension to such studies and is mainly based on the optimization of new urease inhibitors with enhanced efficacy. It deals with the synthesis and evaluation of urease inhibitory properties of a new series of 12 N4-substituted isatin-3-thiosemicarbazones and describes the effects of the nature of phenyl group at N4 (modified by placement of one, two or three substituents about the ring) on the cytotoxic, phytotoxic and urease inhibitory properties of these compounds.
Materials and methods
General
All reagents and solvents were used as obtained from the suppliers or recrystallized/redistilled as necessary. Melting points were taken on a Fisher-Johns melting point apparatus and are uncorrected. Elemental analyses were performed on a Leco CHNS-9320 elemental analyzer. Infrared spectra (KBr discs) were run on Shimadzu Prestige-21 FT-IR spectrometer. The 1H-NMR spectra were recorded in CDCl3 and C2D6SO on Bruker (Rhenistetten-Forchheim, Germany) AM 300 and AM 400 spectrometers operating at 300 MHz and 400 MHz, respectively, using TMS as an internal standard. 1H chemical shifts are reported in δ (ppm) and coupling constants in Hz. The electron impact mass spectra (EIMS) were determined with a Finnigan MAT-312 and a JEOL MSRoute mass spectrometer. The progress of the reaction and the purity of the products were checked on TLC plates coated with Merck silica gel 60 GF254 and the spots were visualized under ultraviolet light at 254 and 366 nm and/or spraying with iodine vapour. In vitro biological testing of the synthesized compounds was made at the Department of Chemistry, The Islamia University of Bahawalpur, Pakistan and Dr.Panjwani Center for Molecular Medicine & Drug Research, H.E.J. Research Institute of Chemistry, University of Karachi, Pakistan.
Synthesis
General procedure for the preparation of isatin-thiosemicarbazones (2a-l)
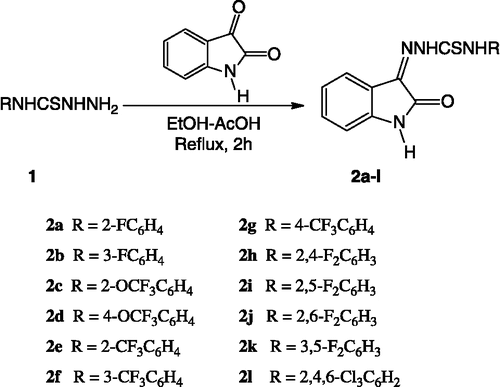
To a hot solution of isatin (0.005 mol) in ethanol (10 mL) containing a few drops of glacial acetic acid was added the appropriate thiosemicarbazide (0.005 mol) dissolved in ethanol (10 mL) under stirring. The reaction mixture was then refluxed for 2 h. The crystalline solid formed during refluxing was collected by suction filtration. Thorough washing with hot ethanol followed by ether afforded the desired compounds 2a-l in pure form.
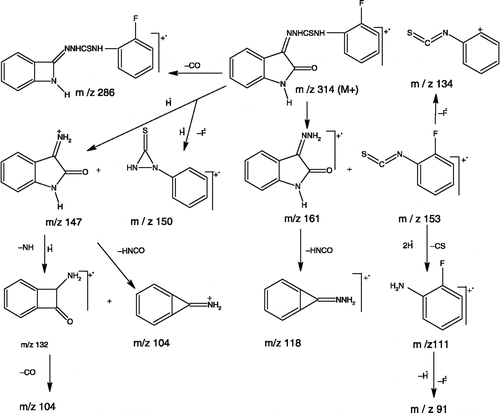
N-(2-Fluorophenyl)-2-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1-hydrazinecarbothioamide (2a). Yield 76% as yellow crystals; m.p. 232-234°C (d); IR (KBr, cm− 1): 3311,3186, 3147 (NH stretching),1691 (C = O), 1618(C = N), 1531 (NH bending), 1163 (C = S); 1H-NMR (CDCl3, δ, ppm): 6.91 (d, J = 7.9 Hz, 1H, indole C7-H), 7.01-7.20 (m, 4H, indole C5-H,C6-H and phenyl C4-H, C5-H), 7.30-7.37 (m, 2H, phenyl C3-H,C6-H), 7.62 (d, J = 7.5 Hz, 1H, indole C4-H), 8.57 (s, 1H, CS-NH), 9.69 (s, 1H, indole NH), 12.86 (s, 1H, N-NH); EIMS (70 eV) m/z (%): 316 ([M+]+2, 3), 315 ([M+]+1, 8), 314 ([M+], 42), 286 (100), 161 (19), 153(13), 150 (16), 147(3),134 (18), 132 (16), 118(13), 104 (15), 91 (6), 77 (6); (Found: C, 57.44; H, 3.51; N, 17.84. Calc. for C15H11FN4OS: C, 57.32; H, 3.50; N, 17.83%).
N-(3-Fluorophenyl)-2-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1-hydrazinecarbothioamide (2b). Yield 71% as light orange crystals; m.p. 240°C (d); IR (KBr, cm− 1): 3282, 3246 (NH stretching),1695 (C = O),1593 (C = N), 1535 (NH bending),1145 (C = S); 1H-NMR (DMSO, δ, ppm): 6.92 (d, J = 7.8 Hz, 1H, indole C7-H), 7.07-7.13 (m, 2H, indole C5-H, phenyl C5-H), 7.34-7.51 (m, 2H, indole C6-H, phenyl C6-H), 7.59-7.63 (m, 2H, phenyl C2-H, C4-H), 7.75 (d, J = 7.4 Hz, 1H, indole C4-H), 10.86 (s, 1H, CS-NH), 11.26 (s, 1H, indole NH), 12.85 (s, 1H, N-NH); EIMS (70 eV) m/z (%):314 ([M+], 37), 285 (100), 203 (5), 161 (25), 160 (4),153 (19), 150 (30),147 (3),145 (14), 132 (30), 118 (34), 104 (73), 91 (14), 77 (25); (Found: C, 57.42; H, 3.51; N, 17.82. Calc. for C15H11FN4OS: C, 57.32; H, 3.50; N, 17.83%).
2-(2-Oxo-1,2-dihydro-3H-indol-3-ylidene)-N-[2-(trifluoromethoxy)phenyl]-1-hydrazinecarbothioamide (2c). Yield 80% as orange crystals; m.p. 244°C (d); IR (KBr, cm− 1): 3320, 3200, 3110 (NH stretching), 1705 (C = O), 1605 (C = N), 1541 (NH bending), 1193 (C = S); 1H-NMR (CDCl3, δ, ppm): 6.95 (d, J = 7.8 Hz, 1H, indole C7-H), 7.14-7.24 (m, 2H, indole C5-H, phenyl C4-H), 7.34-7.42 (m, 3H, indole C6-H, phenyl C5-H, C6-H), 7.60 (d, J = 7.5 Hz, 1H, indole C4-H), 7.89 (s, 1H, phenyl C3-H), 8.98 (s, 1H, CS-NH), 10.13 (s, 1H, indole NH), 12.85 (s, 1H, N-NH); EIMS (70 eV) m/z (%): 380 ([M+], 3), 352 (100), 295 (94), 219 (53), 203 (17), 161 (29), 144 (34), 134 (18), 104 (17), 91 (11), 77 (9), 69 (12); (Found: C, 50.70; H, 2.90; N, 14.73. Calc. for C16H11F3N4O2S: C, 50.53; H, 2.89; N, 14.74%).
2-(2-Oxo-1,2-dihydro-3H-indol-3-ylidene)-N-[4-(trifluoromethoxy)phenyl]-1-hydrazinecarbothioamide (2d). Yield 69% as yellow crystals; m.p. 228°C (d); IR (KBr, cm− 1): 3310, 3244 (NH stretching), 1695 (C = O), 1615 (C = N), 1544 (NH bending), 1157 (C = S); 1H-NMR (CDCl3, δ, ppm): 6.92 (d, J = 7.8 Hz, 1H, indole C7-H), 7.11 (t, J = 7.6 Hz, 1H, indole C5-H), 7.26 (d (overlaped with the solvent), 2H, phenyl C2-H, C6-H), 7.35 (t, J = 7.6 Hz, 1H, indole C6-H), 7.61 (d, J = 7.5 Hz, 1H, indole C4-H), 7.75 (d, J = 8.8 Hz, 2H, phenyl C3-H, C5-H), 7.98 (s,1H,CS-NH), 9.46 (s, 1H, indole NH), 12.87 (s, 1H, N-NH); EIMS (70 eV) m/z (%): 382 ([M+]+2, 7), 381 ([M+]+1, 18), 380 ([M+], 88), 352 (100), 295 (10), 220 (35), 177 (56), 161 (49), 150 (62), 147 (17), 132 (48), 118 (34), 104 (44), 91 (11), 77 (15), 65 (5); (Found: C, 50.65; H, 2.90; N, 14.74. Calc. for C16H11F3N4O2S: C, 50.53; H, 2.89; N, 14.74%).
2-(2-Oxo-1,2-dihydro-3H-indol-3-ylidene)-N-[2-(trifluoromethyl)phenyl]-1-hydrazinecarbothioamide (2e). Yield 83% as yellow crystals; m.p. 256°C (d); IR (KBr, cm− 1): 3350, 3250, 3150 (NH stretching), 1683 (C = O), 1589 (C = N), 1521 (NH bending), 1159 (C = S); 1H-NMR (CDCl3, δ, ppm): 6.94 (d, J = 8.1 Hz, 1H, indole C7-H), 7.16 (t, J = 7.5 Hz, 1H, indole C5-H), 7.34-7.42 (m, 2H, indole C6-H, phenyl C4-H), 7.61-7.69 (m, 3H, phenyl C3-H, C5-H, C6-H), 7.72 (d, J = 7.5 Hz, 1H, indole C4-H), 8.49 (s, 1H, CS-NH), 9.96 (s, 1H, indole NH), 12.87 (s, 1H, N-NH); EIMS (70 eV) m/z (%): 366 ([M+]+2, 5), 365 ([M+]+1, 15), 364 ([M+], 71), 336 (100), 295 (5), 203 (26), 184 (69), 161 (50), 160 (15), 150 (24), 147 (7), 132 (43), 118 (23), 104 (34), 91 (10), 77 (12), 65 (3); (Found: C, 52.90; H, 3.03; N, 15.39. Calc. for C16H11F3N4OS: C, 52.75; H, 3.02; N, 15.38%).
2-(2-Oxo-1,2-dihydro-3H-indol-3-ylidene)-N-[3-(trifluoromethyl)phenyl]-1-hydrazinecarbothioamide (2f). Yield 55% as deep orange crystals; m.p. 228°C (d); IR (KBr, cm− 1): 3261, 3244 (NH stretching), 1695 (C = O), 1598 (C = N), 1544 (NH bending), 1165 (C = S); 1H-NMR (CDCl3, δ, ppm): 6.97 (d, J = 7.8 Hz, 1H, indole C7-H), 7.17 (ddd, J = 7.5, 7.5, 0.6 Hz, 1H, indole C5-H), 7.71 (ddd, J = 7.8, 7.8, 1.2 Hz, 1H, phenyl C5-H), 7.52-7.60 (m, 2H, indole C6-H, phenyl C6-H), 7.67 (d, J = 7.8 Hz, 1H, indole C4-H), 7.99 (br.s, 2H, phenyl C2-H, C4-H), 8.10 (s, 1H, CS-NH), 9.59 (s, 1H, indole NH), 12.93 (s, 1H, N-NH); EIMS (70 eV) m/z (%): 364([M+], 27), 336 (100), 219 (4), 203 (18), 161 (37), 160 (12), 150 (29), 147 (4), 145 (56), 134 (2), 132 (34), 118 (24), 104 (45), 91 (12), 77 (22), 65 (6), 51 (15); (Found: C, 52.88; H, 3.03; N, 15.38. Calc. for C16H11F3N4OS: C, 52.75; H, 3.02; N, 15.38%)
2-(2-Oxo-1,2-dihydro-3H-indol-3-ylidene)-N-[4-(trifluoromethyl)phenyl]-1-hydrazinecarbothioamide (2g). Yield 84% as yellow crystals; m.p. 270°C (d); IR (KBr, cm− 1): 3284, 3244, 3184, 3124 (NH stretching), 1691(C = O), 1600 (C = N), 1544 (NH bending), 1163 (C = S); 1H-NMR (CDCl3, δ, ppm): 6.91-7.16 (m, 3H, indole C7-H, phenyl C2-H, C6-H), 7.27-7.67 (m, 4H, indole C5-H, C6-H and phenyl C3-H, C5-H), 7.91 (d, J = 8.2 Hz, 1H, indole C4-H), 8.50 (s,1H,CS-NH), 9.59 (s, 1H, indole NH), 12.91 (s, 1H, N-NH); EIMS (70 eV) m/z (%):365([M+]+1, 5), 364([M+], 26), 336 (100), 219 (4), 203 (24), 161 (40), 160 (12), 150 (32), 147 (3), 145 (59), 133 (21), 132 (38), 118 (32), 104 (55), 91 (19), 77 (28), 65 (7), 51 (20); (Found: C, 52.91; H, 3.01; N, 15.37. Calc. for C16H11F3N4OS: C, 52.75; H, 3.02; N, 15.38%).
N-(2,4-Difluorophenyl)-2-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1-hydrazinecarbothioamide (2h). Yield 68% as deep yellow crystals; m.p. 252°C (d); IR (KBr, cm− 1): 3282, 3244 (NH stretching), 1695 (C = O), 1618 (C = N), 1544 (NH bending), 1159 (C = S); 1H-NMR (CDCl3, δ, ppm): 6.89-6.95 (m, 3H, indole C7-H, phenyl C5-H, C6-H), 7.12 (t, J = 7.6 Hz, 1H, indole C5-H), 7.36 (t, J = 7.7 Hz, 1H, indole C6-H), 7.58 (s, 1H, phenyl C3-H), 7.61 (d, J = 7.6 Hz, 1H, indole C4-H), 8.37 (s, 1H, CS-NH), 9.48 (s,1H, indole NH), 12.88 (s, 1H, N-NH); EIMS (70 eV) m/z (%): 334 ([M+]+2, 3), 333 ([M+]+1, 6), 332 ([M+], 22), 304 (100), 171 (44), 161 (25), 150 (10), 147 (8), 132 (13), 118 (25), 104 (22), 91 (9), 77 (10), 65 (5); (Found: C, 54.35; H, 3.02; N, 16.86. Calc. for C15H10F2N4OS: C, 54.22; H, 3.01; N, 16.87%).
N-(2,5-Difluorophenyl)-2-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1-hydrazinecarbothioamide (2i). Yield 69% as deep yellow crystals; m.p. 256°C (d); IR (KBr, cm− 1): 3307, 3246 (NH stretching), 1695 (C = O), 1620 (C = N), 1544 (NH bending), 1165 (C = S); 1H-NMR (DMSO, δ, ppm): 6.94 (d, J = 7.8 Hz, 1H, indole C7-H), 7.12 (t, J = 7.5 Hz, 1H, indole C5-H), 7.23-7.52 (m, 4H, indole C6-H, phenyl C3-H, C4-H, C6-H), 7.68 (d, J = 7.5 Hz, 1H, indole C4-H), 10.73 (s,1H, CS-NH), 11.28 (s, 1H, indole NH), 12.92 (s, 1H, N-NH); EIMS (70 eV) m/z (%):334 ([M+]+2, 3), 333 ([M+]+1, 8), 332 ([M+], 38), 304 (100), 203 (8), 171 (54), 161 (27), 154 (60), 144 (41), 129 (32), 118 (26), 104 (15), 91 (8), 76 (8), 63 (12); (Found: C, 54.37; H, 3.02; N, 16.86. Calc. for C15H10F2N4OS: C, 54.22; H, 3.01; N, 16.87%).
N-(2,6-Difluorophenyl)-2-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1-hydrazinecarbothioamide (2j). Yield 69% as deep yellow crystals; m.p. 250°C (d); IR (KBr, cm− 1): 3350, 3235 (NH stretching),1695 (C = O), 1620 (C = N), 1557 (NH bending),1182 (C = S); 1H-NMR (DMSO, δ, ppm): 6.94 (d, J = 7.8 Hz, 1H, indole C7-H), 7.12 (ddd, J = 7.5, 7.5, 0.6 Hz, 1H, indole C5-H), 7.25 (t, J = 8.1 Hz, 1H, phenyl C4-H), 7.39 (ddd, J = 7.8, 7.8, 1.2 Hz, 1H, indole C6-H), 7.46-7.52 (m, 2H, phenyl C3-H, C5-H), 7.67 (d, J = 7.5 Hz, 1H, indole C4-H), 10.57 (s, 1H, CS-NH), 11.28 (s,1H, indole NH), 12.96 (s, 1H, N-NH); EIMS (70 eV) m/z (%): 334 ([M+]+2, 6), 333 ([M+]+1, 18), 332 ([M+], 92), 304 (100), 171 (29), 161 (39), 150 (49), 147 (6), 132 (54), 118 (37), 104 (50), 91 (11), 77 (19), 65 (4); (Found: C, 54.38; H, 3.02; N, 16.85. Calc. for C15H10F2N4OS: C, 54.22; H, 3.01; N, 16.87%).
N-(3,5-Difluorophenyl)-2-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1-hydrazinecarbothioamide (2k). Yield 84% as orange crystals; m.p. 270°C (d); IR (KBr, cm− 1): 3304, 3203, 3188 (NH stretching), 1699 (C = O), 1604 (C = N), 1548 (NH bending), 1155 (C = S); 1H-NMR (DMSO, δ, ppm): 6.93 (d, J = 7.8 Hz, 1H, indole C7-H), 7.09-7.18 (m, 2H, indole C5-H, phenyl C4-H), 7.38 (ddd, J = 7.8, 7.8, 1.1 Hz, 1H, indole C6-H), 7.56, 7.58 (2d, J = 2.2 Hz, 2H, phenyl C2-H, C6-H), 7.75 (d, J = 7.4 Hz, 1H, indole C4-H), 10.89 (s, 1H, CS-NH), 11.28 (s, 1H, indole NH), 12.91 (s, 1H, N-NH); EIMS (70 eV) m/z (%):333 ([M+]+1, 7), 332 ([M+], 48), 304 (100), 187 (5),171 (19), 161 (19),160 (4), 150 (20),147 (2), 145 (22), 133 (13), 132 (30), 118 (26), 104 (57), 91 (14), 77 (37), 51 (28); (Found: C, 54.40; H, 3.02; N, 16.86. Calc. for C15H10F2N4OS: C, 54.22; H, 3.01; N, 16.87%).
2-(2-Oxo-1,2-dihydro-3H-indol-3-ylidene)-N-(2,4,6-trichlorophenyl)-1-hydrazinecarbothioamide (2l). Yield 81% as light orange crystals; m.p. 261°C (d); IR (KBr, cm− 1): 3300, 3143, 3132 (NH stretching), 1683 (C = O),1585 (C = N),1527 (NH bending), 1168 (C = S); 1H-NMR (DMSO, δ, ppm): 6.93 (d, J = 7.8 Hz, 1H, indole C7-H), 7.10 (t, J = 7.5 Hz, 1H, indole C5-H), 7.38 (t, J = 7.6 Hz, 1H, indole C6-H), 7.67 (d, J = 7.1 Hz, 1H, indole C4-H), 7.82 (s, 2H, phenyl C3-H, C5-H), 10.80 (s, 1H, CS-NH), 11.24 (s, 1H, indole NH), 12.95 (s, 1H, N-NH); EIMS (70 eV) m/z (%): 400 ([M+]+2, 3), 399 ([M+]+1, 2), 398 ([M+], 7), 371 (6), 297 (1), 244 (10), 196 (56), 138 (95), 111 (100), 102 (43), 89 (50), 75 (83), 63 (23), 50 (41); (Found: C, 45.20; H, 2.25; N, 14.03. Calc. for C15H9Cl3N4OS: C, 45.06; H, 2.25; N, 14.02%).
Biological testing
Cytotoxicity (in vitro)
Brine shrimp (Artemia salina leach) eggs were hatched in a shallow rectangular plastic dish (22 × 32 cm) filled with artificial sea water, which was prepared with a commercial salt mixture (Instant Ocean, Aquarium System, Inc., Mentor, Ohio, USA) and double-distilled water. An unequal partition was made in the plastic dish with the help of a perforated device. Approximately 50 mg of eggs were sprinkled into the large compartment, which was darkened, while the smaller compartment was opened to ordinary light. After two days, nauplii were collected by a pipette from the lighted side. A sample of the test compound was prepared by dissolving 2 mg of each compound in 2 mL of methanol. From this stock solution, 500, 50 and 5 μL were transferred to 9 vials, three for each dilution, and one vial was kept as control having 2 mL of methanol. The solvent was allowed to evaporate overnight. After two days, when shrimp larvae were ready, 1 mL of sea water and 10 shrimps were added to each vial (30 shrimps/dilution) and the volume was adjusted with sea water to 5 mL per vial. After 24 h, the number of survivors was counted. Data were analyzed by a Finney computer program to determine the LD50 values [Citation42].
Phytotoxicity (in vitro)
This bioassay was performed according to a modified protocol [Citation43]. The test compounds were incorporated with sterilized E-medium at different concentrations i.e. 5, 50 and 500 μg/mL in MeOH. Sterilized conical flasks were inoculated with compounds of desired concentrations prepared from the stock solution and allowed to evaporate overnight. Each flask was inoculated with 20 mL of sterilized E-medium and ten Lemna aequinocitalis Welv, each containing a rosette of three fronds. Other flasks were supplemented with MeOH serving as negative control and a reference inhibitor i.e. paraquat serving as positive control.Treatments were replicated three times and flasks incubated at 30°C in Fisons Fi-Totron 600 H growth cabinet for 7d, 9000 lux light intensity, 56 ± 10 rh (relative humidity), and 12 h day length. Growth of Lemna aequinocitalis in compound-containing flask was determined by counting the number of fronds per dose and growth inhibition was calculated with reference to negative control.
Urease inhibitory activity (in vitro)
Reaction mixtures comprising 25 μL of enzyme (Jack bean urease) solution and 55 μL of buffers containing 100 mM urea were incubated with 5 μL of test compounds (0.1 mM concentration) at 30°C for 15 min in 96-well plates. Urease activity was determined by measuring ammonia production using the indophenol method as described by Weatherburn [Citation44]. Briefly, 45 μL each of phenol reagent (1% w/v phenol and 0.005% w/v sodium nitroprusside) and 70 μL of alkali reagent (0.5% w/v NaOH and 0.1% active chloride NaOCl) were added to each well. The increasing absorbance at 630 nm was measured after 50 min, using a microplate reader (Molecular Device, USA). All reactions were performed in triplicate in a final volume of 200 μL. The results (change in absorbance per min) were processed by using SoftMax Pro software (Molecular Device, USA). All the assays were performed at pH 8.2 (0.01 M K2HPO4. 3H2O, 1 mM EDTA and 0.01 M LiCl). Percentage inhibitions were calculated from the formula 100-(ODtestwell/ODcontrol) × 100. Thiourea was used as the standard inhibitor of urease.
Results and discussion
The present work describes the synthesis and in vitro determination of the cytotoxic, phytotoxic and urease inhibitory effects of twelve new N4-substituted isatin-3-thiosemicarbazones 2a-l.
Chemistry
The synthesis of isatin-thiosemicarbazones was straightforward. A mixture of isatin, appropriate thiosemicarbazide and ethanol containing a few drops of glacial acetic acid was heated under reflux for 2 h (). The crystalline solid formed during heating in each case was filtered. Thorough washing with hot ethanol followed by ether provided the desired compounds 2a-l in moderate to good yields (55-84%).
Scheme 1.. Synthesis of title compounds.
The synthesized thiosemicarbazones were identified by elemental analysis, IR, 1H-NMR and EI mass spectra. Satisfactory elemental analysis ( ± 0.4% of calculated values) was obtained for all compounds. The IR spectra of 2 showed two separate bands resulting from the NH stretchings of indole and thioamide functions in the 3350-3200 and 3188-3124 cm− 1 regions. The lactam C = O, azomethine C = N and thioamide C = S stretchings were observed in the 1705-1683, 1620-1585 and 1193-1145 cm− 1 regions, respectively Citation45,Citation46,Citation47. The 1H-NMR spectra of 2 exhibited three singlets at δ 7.98-10.89, δ 9.42-11.95 and δ 12.75-12.96 for the thiosemicarbazone N4-H, indole NH and thiosemicarbazone N2-H, respectively [Citation45,Citation48,Citation49]. The indole C7-H appeared as doublet at δ 6.87-6.97, while the indole C5-H and C6-H appeared at δ 7.10-7.17 and 7.35-7.39, respectively, as a triplet or a doublet of double doublet. Indole C4-H, experienced a deshielding effect due to the inductive effect of the C = N function and resonated as a doublet at δ 7.60-7.91 Citation49,Citation50,Citation51. In certain cases, however, overlapping of the first three signals, particularly that of indole C5-H and C6-H were observed as multiplets. These signals appeared in combination with different aromatic protons of the N4-substituents. The EI mass spectra of 2 showed molecular ions of different intensity, which confirmed their molecular weights. The major fragmentation pathway involved the cleavage of the exocyclic N-N, NH-CS and endocyclic NH-CO bonds. The proposed fragmentation pattern of 2a is depicted in .
Figure 1. The proposed fragmentation pattern of the compound 2a.
Biological testing
Cytotoxicity (in vitro)
The synthetic thiosemicarbazones 2a-l having one, two or three substituents about the phenyl ring substituted at N4 were tested for their cytotoxicity by the brine shrimp bioassay. All the compounds of this series displayed promising cytotoxicity (LD50 = 1.10 × 10− 5 M–1.36 × 10− 4 M) against Artemia salina (). The remaining compound 2m with no substituent in the phenyl ring gave a value of LD50 >3.38 × 10− 4 M in our earlier assay [Citation20] and, therefore, can be considered to be almost inactive. It may be noted that compound 2l, the only chloro-substituted compound tested in the assay, showed maximum activity (LD50 = 1.10 × 10− 5 M) and, therefore, proved to be the most potent compound in the present series. Next potent was 2b (LD50 = 1.60 × 10− 5 M) having a fluoro substituent at meta position of the phenyl ring. Compound 2a with fluoro substituent at ortho position was found to be relatively less potent, giving a value of LD50 = 3.10 × 10− 5 M. To the contrary, all the difluoro-substituted compounds 2h-k, regardless of the positions of substitution in the phenyl ring, displayed about the same values (LD50 = 2.00 or 2.10 × 10− 5 M). In the case of trifluoromethyl-substituted compounds 2e-g, the meta-substituted compound 2f showed about six-fold larger cytotoxicity (LD50 = 2.20 × 10− 5 M) than the ortho-substituted 2e (LD50 = 1.36 × 10− 4 M) and five-fold larger cytotoxicity than the para-substituted 2g (LD50 = 1.17 × 10− 4 M). Similarly, amongst the two trifluoromethoxy-substituted compounds tested in the assay, compound 2d with a para substituent displayed about seven-fold larger cytotoxicity (LD50 = 1.80 × 10− 5 M) than the ortho-substituted 2c (LD50 = 1.21 × 10− 4 M). These structure-activity relationships may serve as a basis for chemical modifications aimed at the development of certain cytotoxic agents of clinical interest.
Table I.. Brine shrimp bioassay for compounds 2a-l.
Phytotoxicity (in vitro)
All the presently synthesized thiosemicarbazones 2a-l were further screened for their phytotoxic effects at three different concentrations i.e. 500, 50 and 5 μg/mL. Compound 2m, the synthesis of which has been reported elsewhere [Citation19], serves as a reference point to evaluate the effects of substituents about the phenyl ring on the inhibition potential of these synthetic compounds. At the highest concentration tested (500 μg/mL), compounds 2b-m exhibited weak or non-significant (10-30%) plant growth inhibition, whereas 2a displayed moderate (40%) inhibition (). On the other hand, at the lowest concentration (5 μg/mL), eight compounds i.e. 2a-c, 2e, 2f, 2h, 2i and 2m, out of the thirteen compounds tested, showed no plant growth inhibition, whereas the rest i.e. 2d, 2g and 2j-l exhibited non-significant (5-15%) inhibition. From the results obtained in this assay, it may thus be concluded that our compounds, in general, displayed either non-significant or no plant growth inhibition at much higher levels of concentration as compared to the standard paraquat, which showed 100% inhibition at a concentration of 0.015 μg/mL. Moreover, the percent inhibition values revealed that the type and position of the substituents did not affect the phytotoxic activity to a larger extent. In deed, very small difference in activity of certain compounds was observed, particularly at the lowest level of concentration (5 μg/mL).
Table II.. Percent growth inhibition of Lemna aequinocitalis by compounds 2a-l at different concentrations.*
Urease inhibiton (in vitro)
All the synthesized thiosemicarbazones 2a-l were also subjected to urease inhibitory studies. The results collected in revealed that as compared to compound 2m with no substituent in the phenyl ring, substitution of one, two or three inductively electron-withdrawing substituents at different positions of the phenyl ring either induced or enhanced the urease inhibitory activity of the compounds. This conclusion receives support from the results obtained by us in an earlier assay [Citation20]. For example, compounds 2c and 2d having trifluoromethoxy substituents at ortho and para positions, and 2f and 2g with trifluoromethyl substituents at meta and para positions are found to show enhanced activity (40.7%, 49.6%, 78.2% and 44.6%, respectively) when compared with the corresponding compounds having methoxy- and methyl- substituents exhibiting 12.85%, 28.67%, 13.98% and 7.58% inhibition, respectively. Much pronounced enhancement was observed in the case of 2f (13.98% → 78.2%). Similarly, compound 2e with trifluoromethyl group at the ortho position displayed 63.1% inhibition of the enzyme, whereas the corresponding compound having methyl substituent was found to exhibit no inhibitory activity at the tested concentration (100 μM). This indicated that the thiosemicarbazone moiety having electron-withdrawing substituents about the phenyl ring substituted at N4 might interfere with the enzyme activity more efficiently. Of the twelve compounds 2a-l tested in this assay, eight i.e. 2a, 2b, 2e, 2f, 2h-j and 2l proved to be potent inhibitors demonstrating relatively a higher degree of urease inhibitory activity with IC50 values ranging from 20.6 μM to 50.6 μM. Compounds 2a and 2f showed pronounced urease inhibition with IC50 value 20.6 μM, which was even better than the standard thiourea (IC50 value 21.0 μM) and may act as lead compounds. However, in comparison to compound 2a, compound 2b with a fluoro substituent at meta position of the phenyl ring exhibited much enhanced IC50 value (50.6 μM). This clearly indicated that 2b as compared to 2a interfered with the enzyme activity differently, resulting into decreased inhibitory potential. Compound 2l having three chloro substituents at ortho and para positions of the phenyl ring also exhibited much better inhibition of the enzyme (79.6%) with an IC50 value 28.6 μM. However, in our earlier assay [Citation20], compounds having only one chloro substituent at ortho or para position of the phenyl ring exhibited relatively much lower inhibition (43.66% and 25.58%, respectively). Chlorine is both electron-donating by resonance effect ( + M) and electron-withdrawing by inductive effect ( − I). Much higher percentage of inhibition exhibited by compound 2l in the present assay clearly indicates that the ultimate or overall electro-withdrawing effect of the three chloro substituents increases the inhibitory potential of the compound to a much larger extent. Compounds 2e and 2j having one trifluoromethyl and two fluoro substituents at ortho positions of the phenyl ring, respectively, were also proved to be potent inhibitors in the present series, as they displayed relatively much greater activity with IC50 value 31.1 μM. However, these compounds were found to be less potent than compounds 2f and 2a having trifluoromethyl and fluoro substituents at meta and ortho positions of the phenyl ring, respectively. These results indicate that steric hindrance, particularly in the case of 2e plays an important role in decreasing the inhibitory potential of the compound. Amongst the rest difluoro-substituted compounds 2h and 2i, compound 2h having substituents at the ortho and para positions exhibited slightly more inhibition (71.9%) with an IC50 value 47.6 μM. Compared to this, 2i having substituents at the ortho and meta positions demonstrated 66.6% inhibition with an IC50 value 47.9 μM.
Table III.. Inhibition of Jack bean urease by compounds 2a-l.
Ureases obtained from different sources contain, in addition to two Lewis acid nickel ions, one to three protein subunits present in varied stoichiometric ratios [Citation25]. A urease inhibitor can, therefore, interfere with the enzymatic activity by interacting either with the metal ions or the protein component. Sulphenamide, for example, has been reported to inhibit activity of the H. pylori urease by interacting with the sulphydryl (S-H) group present in its protein components [Citation52,Citation53]. Many mechanisms, including competitive and non-competitive are known to be involved in the interaction of an inhibitor with an enzyme. Though the exact mechanism of urease inhibition by our test compounds 2a-l is not known, it is intriguing to carry out investigations with regard to detailed kinetics of such interaction. Apparently, these compounds employ a mechanism of action by exploiting a common transition catalysis state and acting as chelating agents to form octahedral complexes with the two slightly distorted octahedral nickel ions of the enzyme. Further, since our test compounds showed non-significant phytotoxic activity at the highest tested concentration, therefore, they could be combined with fertilizers in small amounts for inhibiting the soil ureases, thus increasing the overall efficiency of nitrogen utilization.
In summary, we have demonstrated the potential of N4-substituted isatin-3-thiosemicarbazones to exhibit selected biological activities i.e. cytotoxicity, phytotoxicity and more importantly the urease inhibitory activity. All the synthesized thiosemicarbazones proved to be active in brine shrimp bioassay; most of them exhibited promising cytotoxic activity. In urease-inhibition bioassay, eight out of twelve tested compounds showed a high degree of activity; two of these i.e. 2a and 2f demonstrated pronounced urease inhibition and may act as lead compounds. Extensive mechanism-based studies are required to contribute to the better understanding of the mechanism of action of these compounds. In phytotoxicity assay, almost all the synthetic compounds demonstrated non-significant activity at the highest tested concentration, thus inviting attention to their usefulness as potent inhibitors of soil ureases. Structure-activity relationship studies revealed that the type and position of the substituents about the phenyl ring substituted at N4 of the thiosemicarbazone moiety did not affect the phytotoxic activity to a larger extent. Based on the data presented in, and in terms of further development and structure-activity relationship studies, combination of the substitution at position-1 and/or -5 of the isatin scaffold with attachment of different aryl groups at N4 of the thiosemicarbazone moiety is considered worth pursuing. Work in this regard along with extended structure-activity relationship studies will be reported in the near future.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- SN Pandeya, S Smitha, M Jyoti, and SK Sridhar. (2005). Biological activities of isatin and its derivatives. Acta Pharm 55:27–46. (and references therein)
- JFM da Silva, SJ Garden, and AC Pinto. (2001). The chemistry of isatins: A review from 1975 to 1999. J Braz Chem Soc 12:273–324. (and references therein)
- A Beauchard, Y Ferandin, S Frere, O Lozach, M Blairvacq, L Meijer, V Thiery, and T Besson. (2006). Synthesis of novel 5-substituted indirubins as protein kinases inhibitors. Bioorg Med Chem 14:6434–6443.
- JL Hyatt, T Moak, JM Hatfield, L Tsurkan, CC Edwards, M Wierdl, MK Danks, RM Wadkins, and PM Potter. (2007). Selective inhibition of carboxylesterases by isatins,indole-2,3-diones. J Med Chem 50:1876–1885.
- N Karali. (2002). Synthesis and primary cytotoxicity evaluation of new 5-nitroindole-2, 3-dione derivatives. Eur J Med Chem 37:909–918.
- I Chiyanzu, E Hansell, J Gut, PJ Rosenthal, JH McKerrow, and K Chibale. (2003). Synthesis and evaluation of isatins and thiosemicarbazone derivatives against cruzain falcipain-2 and rhodesain. Bioorg Med Chem Lett 13:3527–3530.
- TR Bal, B Anand, P Yogeeswari, and D Sriram. (2005). Synthesis and evaluation of anti-HIV activity of isatin β-thiosemicarbazone derivatives. Bioorg Med Chem Lett 15:4451–4455.
- I Chiyanzu, C Clarkson, PJ Smith, J Lehman, J Gut, PJ Rosenthal, and K Chibale. (2005). Design, synthesis and anti-plasmodial evaluation in vitro of new 4-aminoquinoline isatin derivatives. Bioorg Med Chem 13:3249–3261.
- A Rai, SK Sengupta, and OP Pandey. (2005). Lanthanum (III) and praseodymium (III) complexes with isatin thiosemicarbazones. Spectrochim Acta 61A:2761–2765.
- N Terzioglu, N Karali, A Gursoy, C Pannecouque, P Leysen, J Paeshuyse, J Neyts, and E de Clercq. (2006). Synthesis and primary antiviral activity evaluation of 3-hydrazono-5-nitro-2-indolinone derivatives. Arkivoc 109–118.
- DC Quenelle, KA Keith, and ER Kern. (2006). In vitro and vivo evaluation of isatin-β-thiosemicarbazone and marboran against vaccinia and cowpox virus infections. Antiviral Res 71:24–30.
- SS Karki, S Thota, SY Darj, J Balzarini, and E de Clercq. (2007). Synthesis, anticancer and cytotoxic activities of some mononuclear Ru(II) compounds. Bioorg Med Chem 15:6632–6641.
- ZH Chohan, H Pervez, A Rauf, A Scozzafava, and CT Supuran. (2002). Antibacterial Co(II), Cu (II), Ni (II) and Zi (II) complexes of thiadiazole-derived furanyl, thiophenyl and pyrrolyl Schiff bases. J Enz Inhib Med Chem 17:117–122.
- ZH Chohan, H Pervez, KM Khan, A Rauf, and CT Supuran. (2004). Binding of transition metal ions [cobalt, copper, nickel and zinc] with furanyl-, thiophenyl-, pyrrolyl-, salicylyl- and pyridyl-derived cephalexins as potent antibacterial agents. J Enz Inhib Med Chem 19:51–56.
- ZH Chohan, H Pervez, KM Khan, A Rauf, GM Maharvi, and CT Supuran. (2004). Antifungal cobalt (II), copper (II), nickel (II) and zinc (II) complexes of furanyl-, thiophenyl-, pyrrolyl-, salicylyl- and pyridyl-derived cephalexins. J Enz Inhib Med Chem 19:85–90.
- ZH Chohan, H Pervez, KM Khan, A Rauf, GM Maharvi, and CT Supuran. (2004). Antifungal mono- and di- substituted symmetrical and unsymmetrical tirazine-derived Schiff bases and their transition metal complexes. J Enz Inhib Med Chem 19:161–168.
- ZH Chohan, H Pervez, KM Khan, A Rauf, and CT Supuran. (2004). Isatin-derived antibacterial and antifungal compounds and their transition metal complexes. J Enz Inhib Med Chem 19:417–423.
- ZH Chohan, H Pervez, A Rauf, KM Khan, and CT Supuran. (2006). Antibacterial cobalt (II), copper (II), nickel (II) and zinc (II) complexes of mercaptothiadiazole- derived furanyl, thienyl, pyrrolyl, salicylyl and pyridinyl Schiff bases. J Enz Inhib Med Chem 21:193–201.
- H Pervez, MS Iqbal, MY Tahir, MI Choudhary, and KM Khan. (2007). Synthesis of some N4-substituted isatin-3-thiosemicarbazones. Nat Prod Res 21:1178–1186.
- H Pervez, MS Iqbal, MY Tahir, FH Nasim, MI Choudhary, and KM Khan. (2007). In vitro cytotoxic, antibacterial, antifungal and urease inhibitory activities of some N4-substituted isatin-3-thiosemicarbazones. J Enz Inhib Med Chem iFirst:1–7. 10.1080/14756360701746179
- RP Hausinger. (1987). Nickel utilization by microorganisms. Microbiol Rev 51:22–46.
- HLT Mobley, and RP Hausinger. (1989). Microbial urease: Significance, regulation and molecular characterization. Microbiol Rev 53:85–108.
- RP Hausinger. Biochemistry of nickel. New York: Plenum; (1993). p 23–57.
- RK Andrews, RL Blakeley, and B. Zerner. In: LR LancasterJr.. editor. The bioinorganic chemistry of nickelNew York: VCH; (1988). p 141–165.
- HLT Mobley, MD Island, and RP Hausinger. (1995). Molecular biology of microbial ureases. Microbiol Rev 59:451–480.
- CM Collins, and SEF D'Orazio. (1993). Helicobacter pylori nickel binding protein. Mol Microbiol 9:907–913.
- S Ciurli, S Benini, WR Rypniewski, KS Wilson, S Miletti, and S Mangani. (1999). Structural properties of the nickel ions in urease: Novel insights into the catalytic and inhibition mechanism. Coord Chem Rev 190–192:331–355. (and references therein)
- DP Griffith. (1978). Struvite stones. Kidney Int 13:372–382.
- E Sim, A Jones, and L Stanley. (1985). Acetohydroxamate, a urease inhibitor, inhibits the covalent binding reaction of complement proteins C3 and C4. Acta Pharmacol Toxicol 57:304–306.
- RJ Radel, J Gautney, and GE Peters. Urease inhibitor developments In: B Bock, and D Kissel. Ammonia volatilization from urea fertilizers. Muscle Shoals, AL: Tennessee Valley Authority; (1988). p 111–136.
- GE Peters, RJ Radel, and RJ Medina. (1988). Synthesis of phenoxyaminocyclo-triphosphazatrienes. J Agric Food Chem 36:384–390.
- JKR Gasser. (1964). Urea as fertilizer. Soils Fert 27:175–180.
- TE Tomlinson. (1970). Urea: Agronomic applications. Proc Fert Soc 113:1–76.
- KL Sahrawat. (1980). Control of urea hydrolysis and nitrification in soils by chemicals: Properties and problems. Plant Soil 57:335–352.
- RL Mulvaney, and JM Bremner. (1981). Control of urease transformation in soils. Soil Biochem 5:153–196.
- RD. Hauck. In: RD Hauck. Nitrogen in crop production. Madison, WI: American Society of Agronomy; (1984). p 551–560.
- DA Martens, and JM Bremner. (1984). Effectiveness of phosphoroamides for retardation of urea hydrolysis in soils. Soil Sci Soc Am J 48:302–305.
- JM Bremner, and MJ Krogmeier. (1988). Elimination of the adverse effects of urea fertilizer on seed germination,seedling growth, and early plant growth in soil. Proc Natl Acad Sci USA 85:4601–4604.
- GW McCarthy, JM Bremner, and JS Lee. (1990). Inhibition of plant and microbial ureases by phosphoroamides. Plant Soil 127:269–283.
- MJ Krogmeier, GW McCarthy, and JM Bremner. (1989). Potential phytotoxicity associated with the use of soil urease inhibitors. Proc Natl Acad Sci USA 86:1110–1112.
- Z Amtul, AU Rahman, RA Siddiqui, and MI Choudhry. (2002). Chemistry and mechanism of urease inhibition. Curr Med Chem 9:1323–1348. (and references therein)
- DJ Finney. Probit analysis 3rd ed. London: Cambridge University Press; (1971).
- JL McLaughlin, CJ Chang, and DL. Smith. Studies in natural products chemistry; Bench-top bioassays for the discovery of products: Structure and chemistry (Part B)'AU Rahman. Elsevier, Amsterdam, (1991) 383.
- MW Weatherburn. (1967). Phenol-hypochlorite reaction for determination of ammonia. Anal Chem 39:971–974.
- AME Omar, NH Eshba, and HM Salama. (1984). Synthesis of some substituted isatin-β-thiosemicarbazones and isatin- β-hydrazonothiazoline derivatives as potential antiviral and antimicrobial agents. Arch Pharm (Weinheim) 317:701–709.
- I Petrov, O Grupce, and T Stafilov. (1986). The nitrogen–hydrogen stretching region of some imides and thioimides. J Mol Struct 142:275–278.
- P Naumov, and F Anastasova. (2001). Experimental and theoretical vibrational study of isatin, its 5-(NO2, F, Cl, Br, I, CH3) analogues and the isatinato anion. Spectrochim Acta 57A:469–481.
- N Karali, and A Gursoy. (1994). Synthesis and anticonvulsant activity of some new thiosemicarbazone and 4-thiazolidone derivatives bearing an isatin moiety. Farmaco 49:819–822.
- H Laatsch, RH Thomson, and PJ Cox. (1984). Spectroscopic properties of violacein and related compounds: Crystal structure of tetramethyl violacein. J Chem Soc Perkin Trans 2 1331–1339.
- NH Eshba, HM Salama, IM Labouta, and AME Omar. (1987). Synthesis of some substituted 1,2,4-triazino [5,6-b] indole derivatives as potential antiviral anticancer agents. Pharmazie 42:664–666.
- ML Baron, LL Martin, ID Rae, PM Simmonds, and ML Woolcock. (1990). Relaxation processes in aromatic methyl groups. II. Methyl-methyl nuclear overhauser enhancements. Aust J Chem 43:741–747.
- K Nagata, H Satoh, T Iwahi, T Shimoyama, and T Tamura. (1993). Potent inhibitory action of the gastric proton pump inhibitor lansoprazole against urease activity of Helicobacter pylori: Unique action selective for H. pylori cells. Antimicrob Agents Chemother 37:769–774.
- TC Kuhler, J Fryklund, NA Bergman, J Weilitz, A Lee, and H Larsson. (1995). Structure-activity relationship of omeprazole and analogues as Helicobacter pylori urease inhibitor. J Med Chem 38:4906–4916.