Abstract
The design of N-phenylbenzo[d]oxazolamines as CYP26A1 inhibitors involved ligand docking experiments using molecular modeling (FlexX) and analysis of ligand interactions at the binding domain. The synthesis of the benzooxazol-2-yl-[phenyl-imidazol-1-yl-methyl)phenyl]amines was achieved by cyclisation of the corresponding isothiocyanates with subsequent introduction of the haem-binding heterocycle. Triazole and tetrazole derivatives were also prepared for comparison with the lead imidazole derivative. The benzooxazol-2-yl-[phenyl-imidazol-1-yl-methyl)phenyl]amines with small substituents in the phenyl ring were moderately potent CYP26A1 inhibitors (IC50 8 and 12 μM) and comparable with liarozole (IC50 7 μM).
Introduction
Retinoic acid (RA) is a naturally occurring retinoid that exists in three isomeric forms: all-trans-RA (atRA), 9-cis-RA (9cRA) and 13-cis-RA (13cRA) (). RA is responsible for growth and differentiation of mammalian epithelial tissues [Citation1], and exerts activity by binding to transcription-regulatory factors in the cell nucleus known as RAR (retinoic acid receptor) and RXR (retinoid X receptor), each having subtypes α, β and γ [Citation2]. The antiproliferative activity of RA has resulted in its therapeutic use for the treatment of acne, psoriasis [Citation3,Citation4], acute promyelocytic leukaemia (APL) [Citation5,Citation6] and Kaposi's sarcoma (KS) [Citation7] (). However, RA is rapidly metabolised by P450 enzymes [Citation8], a key player being CYP26A1, which is capable of both 4-hydroxylation and 18-hydroxylation resulting in the generation of several inactive hydroxylated forms of atRA [Citation9]. An inhibitor of the metabolism of endogenous RA would be expected to have a beneficial effect on epithelial differentiation and proliferation as a RA-mimetic, with potential use as an agent for hormone-independent cancers and various skin conditions.
Table I. Structure and therapeutic application of retinoic acid isomers used clinically.
The absence of crystallographic data on the 3-dimensional structure of CYP26A1 led us to perform homology modelling using human P450 templates to derive a theoretical CYP26A1 model [Citation10]. Using this CYP26A1 homology model, molecular docking was performed to design a compound with optimal binding interaction at the CYP26A1 ligand-binding domain (LBD). Several potent CYP26A1 inhibitors have been described by us Citation11, Citation12, Citation13, Citation14 and others [Citation15] allowing evaluation through SAR of important pharmacophores. We have recently described a series of novel azolyl-(phenylmethyl)]aryl/heteroarylamines, from this study the benzoxazole group (, R = H, X = CH) was shown to be important for activity (IC50 0.9 μM) with the phenyl and methyl groups resulting in reduced inhibitory activity (5 and 35 μM respectively) [Citation14]. To explore the optimal substitution of the phenyl ring, docking experiments were performed, resulting in the design of substituted imidazole and triazole N-phenylbenzo[d]oxazolamines ().
Figure 1. Structure and key binding interactions of designed N -phenylbenzo[d]oxazolamines.
![Figure 1. Structure and key binding interactions of designed N -phenylbenzo[d]oxazolamines.](/cms/asset/b805bbb7-c3af-4a1e-bde0-a699265307b7/ienz_a_322000_f0001_b.gif)
The benzooxazol-2-yl-[phenylimidazol-1-yl and triazol-1-ylmethyl)phenyl]amines could be envisaged from the corresponding benzophenone (). This benzophenone (C) could be transformed by a reduction of the ketone to give the corresponding alcohol (B) followed by the addition of the imidazole or triazole ring (A ). The reaction of the isothiocyanate (D) and 2-hydroxyaniline with nickel peroxide as a catalyst could form the benzooxazole ring (C ). Then, the isothiocyanate (D) could be prepared by the action of thiophosgene on the aminobenzophenone (E).
Figure 2. Disconnective strategy toward substituted N-phenylbenzo[d]oxazolamines.
![Figure 2. Disconnective strategy toward substituted N-phenylbenzo[d]oxazolamines.](/cms/asset/a466685b-0539-49f1-900f-5535fa026a41/ienz_a_322000_f0002_b.gif)
Materials and Methods
Chemistry
Instrumentation
1H and 13C NMR spectra were recorded with a Brucker Avance DPX500 spectrometer operating at 500 and 125 MHz, with Me4Si as internal standard. Mass spectra were determined by the EPSRC mass spectrometry centre (Swansea, UK). Microanalyses were determined by Medac Ltd (Surrey, UK). Flash column chromatography was performed with silica gel 60 (230–400 mesh) (Merck) and TLC was carried out on precoated silica plates (kiesel gel 60 F254, BDH). Compounds were visualised by illumination under UV light (254 nm) or by the use of vanillin stain followed by charring on a hotplate. Melting points were determined on an electrothermal instrument and are uncorrected. All solvents were dried prior to use as described by the handbook Purification of Laboratory Chemicals [Citation16] and stored over 4 Å molecular sieves, under nitrogen. The numbering of compounds for 1H- and 13C-NMR characterisation is shown below.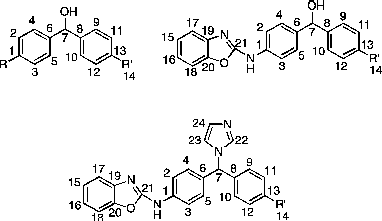
(4-Aminophenyl)-(4-fluorophenyl)methanol (2a)
To a cooled (0°C) solution of 4-fluoro-4-aminobenzophenone [Citation17] (1.5 g, 7 mmol) in MeOH (21 mL) was added NaBH4 (0.26 g, 7 mmol), then the mixture was stirred at room temperature for 1 h. The solvent was concentrated under reduced pressure and aqueous HCl (1 M, 50 mL) added to the residue. The oil that formed was extracted with EtOAc (100 mL) and washed with H2O (50 mL). The aqueous layer was basified (pH 9/10) with 1 M aq. KOH and extracted with EtOAc (2 × 75 mL). The organic layer was dried with MgSO4 and concentrated under reduced pressure to give a pale yellow syrup (0.91 g, 60%). Rf 0.47 (petroleum ether-EtOAc 1:1 v/v); 1H-NMR (CDCl3): δ7.35 (dd, J = 5.5, 14.1 Hz, 2H Ar), 7.12 (d, J = 8.5 Hz, 2H Ar), 7.03 (t, J = 8.9 Hz, 2H Ar), 6.65 (d, J = 8.5 Hz, 2H Ar), 5.73 (s, CH), 3.68 (s, 2H, NH2), 2.40 (s, OH). 13C-NMR (CDCl3): δ162.98 (C, C-13), 161.03 (C-F), 146.01 (C, C-6), 139.99 (C, C-1), 134.04 (C, C-8), 128.07 (CH, C-9 and C-10), 128.01 (CH, C-4 and C-5), 115.13 (CH, C-2 and C-3), 115.00 (CH, C-11 and C-12), 75.30 (CH, C-7). HRMS (EI) calcd for (M)+217.0897, found, 217.0896.
Methyl 4-[(4-aminophenyl)(hydroxy)methyl]benzoate (2b)
Prepared as previously described [Citation18].
(4-Aminophenyl)-(4-methylphenyl)methanol (2c)
To a solution of 1c [Citation19] (25 mmol) in ethanol (150 mL) was added Pd/C catalyst (1 g) and the reaction stirred under a H2 atmosphere (H2 balloon) for 1 h. The reaction was filtered through celite and the filtrate concentrated under reduced pressure. The resulting oil was extracted with CH2Cl2 (100 mL), washed with H2O (2 × 100 mL), dried (MgSO4) and concentrated in vacuo to give a yellow solid. Purification by column chromatography (petroleum ether-EtOAC 70:30 v/v) gave the product as a white crystalline solid (48%). Rf 0.21 (petroleum ether-EtOAc 3:2 v/v), m.p. 97–98 °C; 1H-NMR (CDCl3): δ 7.13 (d, 2H, J = 8.8 Hz, Ar), 7.03 (m, 4H, Ar), 6.50 (d, 2H, J = 8.8 Hz, Ar), 5.59 (s, 1H, CH), 3.11 (bs, 3H, NH2 + OH), 2.21 (s, 3H, CH3). 13C-NMR (CDCl3): δ 144.22 (C, C-1), 140.74 (C, C-8), 132.43 (C, C-13), 131.82 (C, C-6), 124.57 (CH, C-11, C-12), 122.49 (CH, C-4, C-5), 117.17 (CH, C-9, C-10), 113.57 (CH, C-2, C-3). HRMS (EI) calcd for (M-H2O)+196.1125, found, 196.1126.
Synthesis of isocyanates (3a-c)
To a solution of 2 (34.5 mmol) in CH2Cl2 (100 mL) was added a mixture of ice (100 g) and H2O (50 mL). Thiophosgene (41.40 mmol) was then added dropwise with vigorous stirring and the mixture stirred for 2 h at 0 °C and kept overnight in a refrigerator. The organic layer was separated and extracted successively with H2O (2 × 50 mL), 10 % aqueous NaHCO3 (50 mL) and H2O again (50 mL), dried (MgSO4) and concentrated under reduced pressure to obtain the pure product.
(4-Fluorophenyl)-(4-isothiocyanatophenyl)methanol (3a)
Yield: 60%, yellow amorphous solid. Rf 0.68 (petroleum ether-EtOAc 2:1 v/v); 1H-NMR (CDCl3): δ7.4 (d, J = 8.4 Hz, ArH), 7.3 (dd, J = 5.4, 8.5 Hz, ArH), 7.2 (d, J = 8.5 Hz, ArH), 7.05 (t, J = 8.65 Hz, ArH). 13C-NMR (CDCl3): δ 163.36 (C, C-13), 161.39 (C-F), 142.83 (C, C-6), 139.07 (C = N), 135.65 (C, C-8), 130.56 (C, C-1), 128.33 (CH, C-9 and C-10), 128.26 and 127.59 (2 x CH, C-4 and C-5), 125.84 (CH, C-2 and C-3), 115.67 and 115.50 (2 x CH, C-11 and C-12), 74.97 (CH, C-7). HRMS (EI) calcd for (M-H)+258.0383, found, 258.0379.
(4-Isothiocyanatophenyl)-4-(methylphenyl)methanol (3b)
Yield: 88%, brown syrup. Rf 0.59 (petroleum ether-EtOAc 2:1 v/v). 1H-NMR (CDCl3): δ7.33 (m, 2H, Ar), 7.22 (m, 4H, Ar), 7.16 (m, 2H, Ar), 5.57 (s, 1H, CH), 3.56 (bs, 1H, OH), 2.44 (s, 3H, CH3). 13C-NMR (CDCl3): δ143.43 (C, C-6), 140.48 (C, C-8), 137.70 (C, C-13), 135.42 (C, C-15), 130.07 (C, C-1), 129.48 (CH, C-11, C-12), 127.68 (C, C-4, C-5), 126.71 (C, C-9, C-10), 125.77 (C, C-2, C-3), 75.36 (CH, C-7), 21.37 (CH3, C-14).HRMS (EI) calcd for (M)+255.0718, found, 255.0718.
Methyl 4-[hydroxy(4-isothiocyanatophenyl)methyl]benzoate (3c)
Yield: 89%, yellow solid; m.p. 114–115 °C. Rf 0.59 (petroleum ether-EtOAc 1:1 v/v). 1H-NMR (CDCl3): δ7.90 (d, 2H, J = 7.9 Hz, Ar), 7.31 (d, 2H, J = 7.9 Hz, Ar), 7.25 (d, 2H, J = 8.1 Hz, Ar), 7.09 (d, 2H, J = 8.1 Hz, Ar), 5.75 (s, 1H, H-7), 3.80 (s, 3H, CH3), 2.51 (bs, 1H, OH).). 13C-NMR (CDCl3): δ 166.83 (C, C-14), 148.17 (C, C-8), 142.48 (C, C-6), 135.72 (C, C-16), 130.71 (C, C-13), 129.96 (CH, C-11, C-12), 129.90 (C, C-1), 128.62 (CH, C-4, C-5), 126.57 (CH, C-9, C-10), 125.92 (CH, C-2, C-3), 75.18 (CH, C-7), 52.19 (CH3, C-15). HRMS (EI) calcd for (M)+299.0610, found, 299.0610.
Synthesis of benzoxazoles (4a-c and 7)
2-Aminophenol (0.27 g, 2.5 mmol) was added to a solution of 3 (2.5 mmol) in ethanol (32 mL). After stirring overnight at room temperature, HgO (1.08 g, 5 mmol) and sulphur (0.02 g, 0.5 mmol) were added and the reaction was refluxed for 2 h. The cooled mixture was filtered through a celite pad and the solvent evaporated. The product was purified by column chromatography (petroleum ether-EtOAc 80:20 v/v to 60:40 v/v) to give the pure product.
[4-(1,3-Benozoxazol-2-ylaminol)phenyl]-(4-fluorophenyl)methanol (4a)
Yield: 51%, pale orange solid; m.p. 54–56° C. Rf 0.71 (petroleum ether-EtOAc 1:1 v/v). 1H-7 NMR (CDCl3): δ 10.59 (S, 1H, NH ex), 7.69 (d, J = 7.1 Hz, 2H, Ar), 7.46 (dd, J = 7.7, 16.3 Hz, 2H, Ar), 7.38 (m, 4H, Ar), 7.21 (t, J = 7.4 Hz, 1H, Ar), 7.13 (m, 3H, Ar), 5.93 (s, 1H, OH ex), 5.70 (s, 1H, H-1). 13C-NMR (CDCl3): δ161.90 (C, C-13), 158.00 (C-C-21), 147.00 (C, C-19), 142.40 (C, C-20), 139.4 (C, C-1), 137.40 (C, C-8), 128.10 (CH, C-4 and C-5), 128.00 (CH, C-9 and C-10), 126.80 (C, C-6), 123.90 (CH, C-16), 121.60 (CH, C-15), 117.40 (CH, C-2 and C-3), 114.80 (CH, C-18), 114.60 (CH, C-11 and C-12), 108.90 (CH, C-17), 73.20 (CH, C-7).
[4-(1,3-Benzoxazol-2-ylamino)phenyl]-(4-methylphenyl)methanol (4b)
Yield: 35%, orange solid; m.p. 157–158°C. Rf 0.76 (petroleum ether-EtOAc 1:1 v/v). 1H-NMR (CDCl3): δ 8.88 (bs, 1H, NH), 7.86 (m, 4H, Ar), 7.47 (m, 2H, Ar), 7.28 (m, 2H, Ar), 7.12 (m, 1H, Ar), 6.98 (m, 5H, Ar), 5.59 (s, 1H, H-7), 3.34 (bs, 1H, OH), 2.22 (s, 3H, CH3). 13C-NMR (CDCl3): δ 147.89 (C, C-21), 141.84 (C, C-1), 141.00 (C, C-18), 139.17 (C, C-19), 137.25 (C, C-8), 137.16 (C, C-6), 135.29 (CH, C-13), 129.21 (CH, C-11, C-12), 127.59 (CH, C-4, C-5), 126.55 (CH, C-9, C-10), 124.39 (CH, C-15), 121.78 (CH, C-16), 118.77 (CH, C-2, C-3), 116.81 (CH, C-17), 109.23 (CH, C-16), 75.63 (CH, C-7), 21.17 (CH3, C-14). HRMS (EI) calcd for (M)+331.1447, found,: 331.1443.
Methyl-4-[(4-(benzo[d]oxazol-2-ylamino)phenyl)(hydroxy)methyl]benzoate (4c)
Yield: 61%, yellow syrup. Rf 0.45 (petroleum ether-EtOAc 1:1 v/v). 1H-NMR (CDCl3): δ7.92 (bs, 1H, NH), 7.91 (d, 2H, J = 8.1 Hz, Ar), 7.48 (d, 2H, J = 8.1 Hz, Ar), 7.38 (m, 3H, Ar), 7.25 (m, 3H, Ar), 7.12 (m, 1H, Ar), 7.03 (m, 1H, Ar), 5.77 (s, 1H, H-7), 3.81 (s, 3H, CH3), 2.60 (bs, 1H, OH). 13C-NMR (CDCl3): δ166.94 (C, C-14), 158.13 (C, C-22), 148.70 (C, C-20), 142.10 (C, C-8), 138.32 (C, C-1), 137.53 (C, C-21), 129.81 (CH, C-11, C-12), 129.27 (C, C-6), 127.81 (CH, C-4, C-5), 126.31 (CH, C-9, C-10), 124.35 (CH, C-17), 122.01 (CH, C-16), 118.65 (CH, C-2, C-3), 117.18 (CH, C-19), 109.14 (CH, C-18), 75.45 (CH, C-7), 52.10 (CH3, C-15). HRMS (EI) calcd for (M)+375.1339, found, 375.1337.
[4-(Benzo[d]oxazol-2-ylamino)phenyl](4-methoxyphenyl)methanone (7)
Yield: 46%, orange solid; m.p. 143–144°C. Rf 0.41 (petroleum ether-EtOAc 2:1 v/v). 1H-NMR (CDCl3): δ 8.62 (bs, 1H, NH), 7.80 (m, 4H, Ar), 7.67 (d, 2H, J = 8.5 Hz, Ar), 7.46 (d, 1H, J = 7.5 Hz, Ar), 7.32 (d, 1H, J = 8.0 Hz, Ar), 7.20 (d, 1H, J = 8.5 Hz, Ar), 7.11 (m, 1H, Ar), 6.92 (m, 2H, Ar), 6.88 (d, 2H, J = 9.0 Hz, Ar), 3.83 (s, 3H, CH3). 13C-NMR (CDCl3): δ188.04 (C, C-7), 163.09 (C, C-13), 152.63 (C, C-21), 149.35 (C, C-20), 141.36 (C, C-1), 139.65 (C, C-19), 132.43 (CH, C-9, C-10), 131.83 (CH, C-4, C-5), 130.46 (C, C-8), 129.52 (C, C-6), 125.44 (CH, C-15), 124.79 (CH, C-16), 120.96 (CH, C-17), 117.17 (CH, C-2, C-3), 113.57 (CH, C-11, C-12), 110.07 (CH, C-18), 55.53 (CH3, C-14). HRMS (EI) calcd for (M)+345.1239, found, 345.1236.
Synthesis of imidazoles (5a-c, e-f)
To a solution of 4 (6 mmol) in acetonitrile (40 mL) was added imidazole (18 mmol) and 1,1′-carbonyldiimidazole (9 mmol). The mixture was then heated under reflux for 2–12 h. Then the reaction mixture was allowed to cool and the solvent removed under reduced pressure. The oil formed was then extracted with CH2Cl2 (50 mL) and washed with water (2 × 50 mL). The recombined organic layers were dried (MgSO4), filtered and reduced in vacuo. Purification by column chromatography (CH2Cl2-MeOH 100:0 v/v increasing to 96:4 v/v) gave the product.
Benzoxazol-2-yl-(4-[(fluorophenyl)imidazol-1-yl-methyl]phenyl)amine (5a)
Yield: 50%, white solid; m.p. 226–228 °C. Rf 0.49 (CH2Cl2-MeOH 9:1 v/v). 1H-NMR (DMSO-d6): δ 10.73 (s, 1H, NH), 7.8(d, J = 8.4 Hz, 2H, Ar), 7.7(s, 1H, imid), 7.5(dd, J = 7.8 Hz, 20.5 Hz, 2H, Ar), 7.19(m, 9H, Ar), 7.12(s, 1H, CH-imid), 7.0(s, 1H, CH imid), 6.9(s,CH, H1). 13C-NMR (DMSO-d6): δ162.53 (C, C-13), 160.58 (C, C-21), 157.85 (C, C-19), 146.99 (C, C-20), 142.26 (C, C-1), 138.49 (CH, C-22), 136.46 (CH, C-9 and C-10), 133.24 (C, C-8), 129.73 (CH, C-5 and C-5), 128.72 (C, C-6), 128.62 (CH, C-24), 124.02 (CH, C-16), 121.76 (CH, C-2 and C-3), 119.06 (CH, C-11 and C-9 12), 117.79 (CH, C-15), 116.64 (CH, C-23), 115.57 (CH, C-18), 108.97 (CH, C-17), 62.27 (C, C-7). Anal. Calcd. for C23H17N4OF.0.3H2O (389.81646): C, 70.87; H, 4.61; N,14.57%. Found: C, 70.80; H, 4.29; N, 14.31%.
Benzoxazol-2-yl-(4-[(methylphenyl)imidazol-1-yl-methyl]phenyl)amine (5b)
Yield: 66%, yellow syrup. Rf 0.08 (EtOAc). 1H-NMR (CDCl3): δ10.27 (bs, 1H, NH), 7.58 (m, 2H, Ar), 7.41 (s, 1H, Ar), 7.36 (m, 1H, Ar), 7.18 (m, 1H, Ar), 7.08 (m, 1H, Ar), 7.02 (m, 3H, Ar), 6.95 (m, 3H, Ar), 6.88 (m, 2H, Ar), 6.76 (s, 1H, Ar), 6.37 (s, 1H, H-7), 2.25 (s, 3H, CH3). 13C-NMR (CDCl3): δ 157.00 (C, C-21), 148.82 (C, C-20), 141.23 (C, C-1), 137.29 (C, C-19), 137.25 (C, C-8), 135.09 (C, C-13), 132.63 (C, C-6), 129.87 (CH, C-22), 128.55 (CH, C-11, C-12), 127.96 (CH, C-4, C-5), 127.79 (CH, C-24), 126.91 (CH, C-9, C-10), 123.24 (CH, C-15), 122.06 (CH, C-23), 120.98 (CH, C-16), 117.43 (CH, C-2, C-3), 116.24 (CH, C-17), 108.08 (CH, C-18), 63.49 (CH, C-7), 21.96 (CH3, C-14). HRMS (EI) calcd for (M)+381.1715, found, 381.1712.
4-{[4-(Benzoxazol-2-ylamino)phenyl]imidazol-1-yl-methyl}benzoic acid methyl ester (5c)
Yield: 90%, yellow syrup. Rf 0.21 (EtOAc). 1H-NMR (CDCl3): δ 8.52 (bs, 1H, NH), 7.96 (d, 2H, J = 8.2 Hz, Ar), 7.59 (d, 2H, J = 8.5 Hz, Ar), 7.38 (d, 2H, J = 7.5 Hz, Ar), 7.22 (d, 1H, J = 7.9 Hz, Ar), 7.18 (m, 1H, Ar), 7.08 (m, 6H, Ar), 6.79 (m, 1H, Ar), 6.45 (s, 1H, H-7), 3.86 (s, 3H, CH3). 13C-NMR (CDCl3): δ 166.47 (C, C-14), 157.82 (C, C-22), 147.78 (C, C-20), 144.19 (C, C-8), 142.21 (C, C-1), 139.82 (CH, C-23), 138.67 (C, C-21), 132.46 (C, C-6), 130.28 (C, C-13), 130.18 (CH, C-11, C-12), 129.57 (CH, C-4, C-5), 127.78 (CH, C-9, C-10), 125.94 (CH, C-25), 124.32 (CH, C-17), 122.63 (CH, C-24), 122.13 (CH, C-16), 119.33 (CH, C-2, C-3), 117.36 (CH, C-19), 109.12 (CH, C-18), 64.37 (CH, C-7), 52.29 (CH3, C-15). HRMS (EI) calcd for (M)+425.1608, found, 425.1610.
Benzooxazol-2-yl{4-[imidazole-1-yl-(4-methoxyphenyl)methyl]phenyl}amine (5e)
Yield: 38%, yellow syrup. Rf 0.09 (EtOAc). 1H-NMR (CDCl3): δ 9.62 (bs, 1H, NH), 7.60 (d, 2H, J = 8.5 Hz Ar), 7.45 (s, 1H, Ar), 7.39 (d, 1H, J = 7.5 Hz, Ar), 7.23 (d, 1H, J = 8.0 Hz, Ar), 7.16 (m, 1H, Ar), 7.08 (m, 1H, Ar), 7.01 (m, 6H, Ar), 6.78 (d, 2H, J = 9.0 Hz, Ar), 6.39 (s, 1H, H-7), 3.75 (s, 3H, CH3). 13C-NMR (CDCl3): δ 158.59 (C, C-13), 157.28 (C, C-21), 146.77 (C, C-20), 141.29 (C, C-1), 139.84 (CH, C-22), 137.56 (C, C-19), 132.51 (C, C-8), 130.00 (C, C-6), 128.34 (CH, C-9, C-10), 127.72 (CH, C-4, C-5), 125.92 (CH, C-24), 123.18 (CH, C-15), 122.61 (CH, C-23), 120.86 (CH, C-16), 117.47 (CH, C-2, C-3), 116.11 (CH, C-17), 113.24 (CH, C-11, C-12), 108.05 (CH, C-18), 63.31 (CH, C-7), 54.33 (CH3, C-14). HRMS (EI) calcd for (M)+397.1665, found, 397.1667.
Benzooxazol-2-yl{4-[imidazole-1-yl-(4-hydroxyphenyl)methyl]phenyl}amine (5f)
Yield: 74%, colourless syrup. Rf 0.10 (EtOAc). 1H-NMR (CDCl3): δ7.56 (d, 2H, J = 8.6 Hz, Ar), 7.45 (s, 1H, Ar), 7.26 (d, 1H, J = 7.4 Hz, Ar), 7.20 (d, 1H, J = 7.4 Hz, Ar), 7.07 (m, 1H, Ar), 6.98 (m, 5H, Ar), 6.80 (d, 2H, J = 8.2 Hz, Ar), 6.63 (d, 2H, J = 8.6 Hz, Ar), 6.46 (s, 1H, H-7). 13C-NMR (CDCl3): δ 159.89 (C, C-13), 158.76 (C, C-20), 148.98 (C, C-19), 143.50 (C, C-1), 139.90 (CH, C-18), 135.41 (C, C-8), 131.37 (C, C-6), 130.58 (CH, C-9, C-10), 130.25 (CH, C-21), 130.13 (CH, C-23), 129.74 (CH, C-4, C-5), 125.28 (CH, C-14), 123.28 (CH, C-15), 122.68 (CH, C-23), 119.68 (CH, C-2, C-3), 117.82 (CH, C-16), 116.68 (CH, C-11, C-12), 110.01 (CH, C-17), 65.48 (CH, C-7). HRMS (EI) calcd for (M-imidazole)+315.1134, found, 315.1130.
Synthesis of 4-{[4-(Benzoxazol-2-ylamino)phenyl]imidazol-1-yl-methyl}benzoic acid (5d)
To a suspension of 5c (0.7 mmol) in MeOH (6 mL) was added a 2M solution of aqueous NaOH (3 mL) and the mixture was refluxed for 30 min. The mixture was then allowed to cool and was diluted with H2O (5 mL), cHCl was added until pH 1. After removing the solvent under reduced pressure, MeOH (5 mL) was added to the mixture to precipitate the NaCl formed during the reaction. The solid was removed by filtration and the filtrate reduced in vacuo to give the product. Yellow syrup (62%). Rf 0.16 (CH2Cl2-MeOH 98:2 v/v). 1H-NMR (DMSO-d6): δ 13.11 (bs, 1H, COOH), 11.02 (bs, 1H, NH), 9.21 (s, 1H, Ar), 8.01 (m, 2H, Ar), 7.87 (m, 2H, Ar), 7.79 (m, 2H, Ar), 7.50 (m, 1H, Ar), 7.43 (m, 1H, Ar), 7.32 (m, 5H, Ar), 7.22 (m, 1H, Ar), 6.95 (s, 1H, H-7). 13C-NMR (DMSO-d6): δ 169.14 (C, C-14), 159.69 (C, C-21), 148.96 (C, C-19), 143.75 (C, C-8), 143.45 (C, C-1), 141.13 (C, C-20), 132.74 (C, C-6), 131.72 (C, C-13), 131.60 (CH, C-22), 130.65 (CH, C-11, C-12), 129.03 (CH, C-4, C-5), 125.33 (CH, C-9, C-10), 123.34 (CH, C-24), 123.22 (CH, C-16), 122.89 (CH, C-15), 120.23 (CH, C-23), 120.23 (CH, C-2, C-3), 117.89 (CH, C-18), 110.11 (CH, C-17), 67.19 (CH, C-7). HRMS (EI) calcd for (M)+411.1449, found, 411.1452.
Synthesis of [4-(1,3-Benzoxazol-2-ylamino)phenyl]-(4-hydroxyphenyl)methanone (8)
A mixture of 7 (1.33 mmol) in 20 mL of a 48% solution of HBr in acetic acid and 10 mL of acetic acid was heated at reflux for 6 h and then left to stir overnight at room temperature. The mixture was then evaporated to one-half the original volume and the residue diluted with H2O (30 mL) and extracted with EtOAc (50 mL). The organic extract was washed with aq. NaHCO3 (2 × 30 mL) and evaporated to give a yellow oil. Purification by column chromatography (petroleum ether – EtOAc 60:40 v/v) gave the pure product as a yellow syrup (77%). Rf 0.37 (petroleum ether–EtOAc 1:1 v/v). 1H-NMR (CD3OD): δ 7.76 (d, 2H, J = 9.0 Hz, Ar), 7.71 (d, 2H, J = 9.0 Hz, Ar), 7.62 (d, 2H, J = 8.6 Hz, Ar), 7.39 (d, 1H, J = 7.8 Hz, Ar), 7.33 (d, 1H, J = 8.2 Hz, Ar), 7.18 (m, 1H, Ar), 7.10 (m, 1H, Ar), 6.81 (d, 2H, J = 8.6 Hz, Ar). 13C-NMR (CD3OD): δ 196.81 (C, C-7), 163.43 (C, C-13), 152.79 (C, C-20), 144.04 (C, C-19), 143.91 (C, C-1), 139.06 (C, C-18), 133.94 (CH, C-9, C-10), 133.26 (C, C-8), 132.57 (CH, C-4, C-5), 130.36 (C, C-6), 125.37 (CH, C-14), 125.12 (CH, C-15), 123.53 (CH, C-16), 118.18 (CH, C-2, C-3), 116.14 (CH, C-11, C-12), 110.11 (CH, C-18). HRMS (EI) calcd for (M)+330.1004, found, 330.1004.
Synthesis of carbinols (9a-b)
To a cooled (0 0C) solution of 7 or 8 (20 mmol) in anhydrous methanol (100 mL) was added sodium borohydride (20 mmol), then the mixture was allowed to stir at room temperature under nitrogen for 1–12 h. The solvent was concentrated under reduced pressure and aqueous HCl (1M, 10 mL) added to the residue. The oil formed was extracted with diethyl ether (2 × 50 mL) and washed with H2O (2 × 50 mL), the organic layers were combined and dried with MgSO4 and the solvent concentrated under reduced pressure to give the product.
[4-(1,3-Benzoxazol-2-ylamino)phenyl]-(4-methoxyphenyl)methanol (9a)
Yield: 91%, orange solid; m.p. 123–124° C. Rf 0.38 (petroleum ether–EtOAc 2:1 v/v). 1H-NMR (CDCl3): δ 8.74 (bs, 1H, NH), 7.38 (d, 2H, J = 8.0 Hz, Ar), 7.33 (d, 1H, J = 8.0 Hz, Ar), 7.20 (m, 3H, Ar), 7.15 (m, 2H, Ar), 7.10 (m, 1H, Ar), 7.01 (m, 1H, Ar), 6.76 (d, 2H, J = 8.5 Hz, Ar), 5.68 (s, 1H, H-7), 3.69 (s, 3H, CH3). 13C-NMR (CDCl3): δ 157.67 (C, C-13), 156.74 (C, C-21), 146.76 (C, C-20), 142.56 (C, C-1), 140.73 (C, C-19), 138.18 (C, C-8), 135.11 (C, C-6), 129.00 (CH, C-15), 127.21 (CH, C-9, C-10), 126.87 (CH, C-4, C-5), 123.31 (CH, C-16), 120.84 (CH, C-17), 117.87 (CH, C-2, C-3), 112.99 (CH, C-11, C-12), 108.10 (CH, C-18), 54.20 (CH3, C-14). HRMS (EI) calcd for (M)+347.1396, found, 347.1395.
[4-(1,3-Benzoxazol-2-ylamino)phenyl]-(4-hydroxyphenyl)methanol (9b)
Yield: 42%, yellow syrup. Rf 0.51 (petroleum ether–EtOAc 1:1 v/v). 1H-NMR (CD3OD): δ 9.96 (bs, 1H, OH), 9.07 (bs, 1H, NH), 7.48 (d, 2H, J = 8.6 Hz, Ar), 7.22 (m, 4H, Ar), 7.11 (m, 3H, Ar), 6.95 (m, 1H, Ar), 6.65 (d, 2H, J = 8.6 Hz, Ar), 5.60 (s, 1H, H-7), 4.58 (bs, 1H, OH). 13C-NMR (CD3OD): δ 160.34 (C, C-13), 157.68 (C, C-20), 148.98(C, C-19), 143.50 (C, C-1), 140.98 (C, C-18), 138.72 (C, C-8), 136.96 (C, C-6), 129.18 (CH, C-9, C-10), 128.46 (CH, C-4, C-5), 125.25 (CH, C-14), 123.02 (CH, C-15), 119.48 (CH, C-16), 117.54 (CH, C-2, C-3), 116.07 (CH, C-11, C-12), 109.94 (CH, C-17), 76.34 (CH, C-7). HRMS (EI) calcd for (M)+333.1239, found, 333.1238.
Synthesis of N-(4-(phenyl(1H-1,2,4-triazol-1-yl)methyl)phenyl)benzo[d]oxazol-2-amine (11) and N-(4-(phenyl(4H-1,2,4-triazol-4-yl)methyl)phenyl)benzo[d]oxazol-2-13 amine (12)
To a cooled (0 0C) solution of 1,2,4-triazole (3.20 mmol) in CH3CN (10 mL) was added thionyl chloride (1.60 mmol) in CH3CN (6 mL) and the reaction stirred at 10° C under N2 for 1 h. Potassium carbonate (1.60 mmol) was then added followed by a solution of [4-(1,3-benzoxazol-2-ylamino)phenyl]-(phenyl)methanol [Citation14] (10) (0.80 mmol) in CH3CN (10 mL) and the reaction allowed to stir at room temperature for 3 days. The suspension was filtered and the solution concentrated under reduced pressure to produce a mixture, which was extracted with EtOAc (2 × 25 mL) and washed with H2O (2 × 25 mL). The organic layer was dried (MgSO4) and the solvent evaporated to give a yellow solid. Purification by flash-chromatography (EtOAc) gave the 1H-1,2,4-triazole compound 11. Purification with flash-chromatography (CH2Cl2-MeOH 9:1 v/v) gave the 4H-1,2,4-triazole compound 12.
N-(4-(phenyl(1H-1,2,4-triazol-1-yl)methyl)phenyl)benzo[d]oxazol-2-amine (11)
Yield: 35%, white solid; m.p. 150–151 °C. Rf 0.32 (petroleum ether–EtOAc 2:1 v/v). 1H-NMR (CDCl3): δ 8.18 (bs, 1H, NH), 8.10 (s, 1H, Ar), 7.99 (s, 1H, Ar), 7.67 (m, 2H, Ar), 7.48 (m, 1H, Ar), 7.39 (m, 4H, Ar), 7.28 (m, 1H, Ar), 7.17 (m, 5H, Ar), 6.80 (s, 1H, H-7). 13C-NMR (CDCl3): δ 157.80 (C, C-20), 152.31 (CH, C-21), 147.81 (C, C-18), 143.59 (CH, C-22), 142.10 (C, C-1), 138.30 (C, C-8), 138.03 (C, C-19), 132.46 (C, C-6), 129.35 (CH, C-4, C-5), 128.99 (CH, C-11, C-12), 128.62 (CH, C-13), 127.98 (CH, C-9, C-10), 124.39 (CH, C-15), 122.16 (CH, C-14), 118.50 (CH, C-2, C-3), 117.34 (CH, C-17), 109.17 (CH, C-16), 67.45 (CH, C-7). HRMS (EI) calcd for (M)+368.1510, found, 368.1511.
N-(4-(phenyl(4H-1,2,4-triazol-4-yl)methyl)phenyl)benzo[d]oxazol-2-amine (12)
Yield: 12%, brown-orange solid; m.p. 118–119°C. Rf 0.08 (EtOAc). 1H-NMR (CDCl3): δ 9.73 (bs, 1H, NH), 8.04 (s, 2H, Ar), 7.68 (m, 2H, Ar), 7.47 (m, 1H, Ar), 7.26 (m, 3H, Ar), 7.18 (m, 1H, Ar), 7.10 (m, 1H, Ar), 7.00 (m, 5H, Ar), 6.48 (s, 1H, H-7). 13C-NMR (CDCl3): δ 158.23 (C, C-20), 147.72 (C, C-18), 142.22 (C, C-1), 139.42 (C, C-8), 137.76 (C, C-19), 131.26 (C, C-6), 129.27 (CH, C-21, C-22), 128.97 (CH, C-4, C-5), 128.86 (CH, C-11, C-12), 128.67 (CH, C-13), 128.44 (CH, C-9, C-10), 124.34 (CH, C-15), 121.96 (CH, C-14), 118.78 (CH, C-2, C-3), 117.12 (CH, C-17), 109.10 (CH, C-16), 63.57 (CH, C-7). HRMS (EI) calcd for (2M + H+)+735.2940, found, 735.2944.
Synthesis of N-(4-(phenyl(1H-tetrazol-1-yl)methyl)phenyl)benzo[d]oxazol-2-amine (13)
Thionyl chloride (4.89 mmol) in CH3CN (5 mL) was added dropwise to a stirred solution of 1H-tetrazole (0.45 M in CH3CN, 22 mL) at a temperature of 10° C. The white suspension formed was allowed to stand under N2 for 1 h at 10° C. A solution of [4-(1,3-benzoxazol-2-ylamino)phenyl]-(phenyl)methanol [Citation14] (10) (2.44 mmol) in CH3CN (5 mL) was added to the mixture followed by activated potassium carbonate (0.68 g, 4.89 mmol). The suspension was stirred under nitrogen at room temperature for 3 days. The resulting suspension was filtered and the filtrate was evaporated in vacuo to yield a brownish oil. The oil was extracted with CH2Cl2 (50 mL) and H2O (3 × 50 mL). The organic layer was dried with (MgSO4), filtered and reduced in vacuo to give an orange oil. Purification by flash column chromatography (petroleum ether–EtOAc 70:30 v/v) gave the product. Yield: 32%, brown solid; m.p. 116–118° C. Rf 0.68 (petroleum ether–EtOAc 2:1 v/v). 1H-NMR (CDCl3): δ8.49 (s, 1H, tetrazole), 7.52 (m, 2H, Ar), 7.34 (m, 1H, Ar), 7.20 (m, 4H, Ar), 7.14 (m, 3H, Ar), 7.07 (m, 3H, Ar), 7.01 (s, 1H, H-7), 6.85 (bs, 1H, NH). 13C-NMR (CDCl3): δ 157.28 (C, C-20), 152.01 (CH, C-21), 147.02 (C, C-18), 140.46 (C, C-1), 137.52 (C, C-8), 135.94 (C, C-19), 130.35 (C, C-6), 128.44 (CH, C-4, C-5), 127.81 (CH, C-11, C-12), 127.74 (CH, C-13), 126.94 (CH, C-9, C-10), 123.46 (CH, C-15), 121.02 (CH, C-14), 117.51 (CH, C-2, C-3), 115.83 (CH, C-17), 108.27 (CH, C-16), 69.66 (CH, C-7). HRMS (EI) calcd for (M)+369.1468, found, 369.1464.
Enzyme Assay
[11,12-3H] All trans-retinoic acid (37 MBq/mL) was purchased from Amersham (UK). Acetic acid, ammonium acetate and Optisafe 3 scintillation fluid were obtained from Fisher Scientific (UK). All solvents used for chromatography were HPLC grade from Fisher Scientific (UK).
MCF-7 (CYP26A1) assay for inhibition of metabolism of atRA
Performed as previously described [12], MCF-7 cells were seeded in 12-well cell culture plates (Cornings Inc., New York, USA) at 2.5 × 105 cells per well in a total volume of 1.5 mL. Cells were allowed to adhere to the well for 24 h. After 24 h, the medium from each well was removed, washed once with Phosphate Buffer Saline (PBS) and replaced by fresh medium plus 10 μL inhibitor/solvent (acetonitrile) and 10 μL of atRA (to give final concentration of 1 × 10− 7 M atRA and 0.1 μCi [11,12-3H] all-trans retinoic acid). The plates were foil wrapped and incubated at 37 °C for 24 h. Each treatment was performed in duplicate. The incubation was stopped by addition of 1% acetic acid (100 μL/well), the medium was removed into separate glass tubes. 200 μL distilled water was added to each well and the cells scrapped off and the contents added to the appropriate glass tube. This procedure was repeated with a further 400 μL water but without scraping. Ethyl acetate containing 0.05% (w/v) butylated hydroxyanisole (2 × 2 mL) was added to each tube. After vortexing for 15 s, the tubes were spun down at 3000 rpm for 15 min. The organic layer was then evaporated using a Christ centrifuge connected to a vacuum pump and a multitrap at – 80 °C.
High Performance Liquid Chromatography (HPLC)
The HPLC system was equipped with a high pressure pump (Milton-Roy pump), injector with a 50 μL loop connected to a beta-RAM radioactivity detector, connected to a Compaq™ computer running Laura® data acquisition and analysis software. This enabled on-line detection and quantification of radioactive peaks. The HPLC column (10 μM C18 μBondapak™ 3.9 × 300 mm HPLC column from Waters, UK) operating at ambient temperature was used to separate the metabolites, which were eluted with acetonitrile/1% ammonium acetate in water/acetic acid (75:25:0.1 v/v/v) at a flow rate of 1.9 mL/min. The Ecoscint™ was used as the flow scintillation fluid.
Molecular Docking
Ligands were docked within the active site of the CYP26A1 homology model [Citation10] using the FlexX docking programme of SYBYL [Citation20], performed with the default values. The active site was defined by all the amino acid residues within a 6.5 Å distance from TRP112, VAL116, THR304, VAL370 and GLY373, including the heme in a heteroatom file. Subsequent manipulation and interaction evaluation was performed with MOE (Molecular Operating Environment) software [Citation21].
Results and Discussion
Molecular Docking
Substrates were docked within the active site of the homology model (built using CYP3A4 template) using the FlexX docking programme of SYBYL [Citation20]. Subsequent manipulation and interaction evaluation was performed with MOE (Molecular Operating Environment) software [Citation21]. The position of the imidazole ring from the haem was first evaluated. In the well docked compounds, the distance between the imidazole N and the iron of the haem should be 3 Å or less thereby allowing the coordination bond to be formed. Secondly, the effect of the phenyl substituents R () were assessed for their ability to form hydrogen and/or hydrophobic bonds with different residues at the active site, both with respect to direct interaction of the phenyl substituent and indirectly as a result on the effect of the substituent on the orientation of the ligand within the active site. Ligands with OH, OMe, OAc, CN, CH2OH, COOH, COOMe and CONH2 as substituents showed interesting results.
In the example shown in with COOH as a substituent, hydrogen bonds on both sides of the active site are observed with both enantiomers. The key interactions noted were as follows: The R-enantiomer forms a hydrogen bond between the oxygen of the benzoxazole ring and SER115 on one side and between the carboxylic acid group and THR479, PRO369, SER307 and GLU303 on the other side; the S-enantiomer also displayed interesting results with hydrogen bonds observed on one side between the benzoxazole nitrogen and SER115 and on the other side between the carboxylic acid and MET217, HIS302 and PHE299. Both enantiomers interact closely with the haem with a distance of 2.88 Å and 2.29 Å for the R- and S-enantiomer respectively.
Figure 3. Diagram showing both enantiomers R and S of 4-{[4-(benzoxazol-2-ylamino)phenyl]imidazol-1-yl-methyl}benzoic acid docked at the active site region of the CYP26A1 model. Hydrogen bonding interactions are shown as green lines and co-ordination with the transition metal is indicated with a purple line. Colour coding of the atoms: grey = C, red = O, blue = N, brown = Fe and yellow = S. Amino-acid residues identified are involved in hydrophobic interactions.
![Figure 3. Diagram showing both enantiomers R and S of 4-{[4-(benzoxazol-2-ylamino)phenyl]imidazol-1-yl-methyl}benzoic acid docked at the active site region of the CYP26A1 model. Hydrogen bonding interactions are shown as green lines and co-ordination with the transition metal is indicated with a purple line. Colour coding of the atoms: grey = C, red = O, blue = N, brown = Fe and yellow = S. Amino-acid residues identified are involved in hydrophobic interactions.](/cms/asset/4662d6ef-d963-49bd-9b1e-c109aadc28a4/ienz_a_322000_f0003_b.gif)
Chemistry
The synthesis of the fluoro, methyl, methyl ester and carboxylic acid substituted phenyl derivatives (5a-d) is described in Scheme . Preparation of the aminoalcohol 2 was achieved either by NaBH4 reduction of the corresponding ketone (1a) or by a mild reduction of the corresponding nitro (1b/c) using Pd/C with a H2 balloon (∼ 20–30 psi).
Scheme 1. Reagents and Conditions: (i) NaBH4, MeOH, 1 h, rt, 60% (ii) H2, Pd/C, EtOH, 1 h, 48% (iii) CSCl2, CH2Cl2, H2O, 18 h, 0°C, 60–89% (iv) (a) 2-aminophenol, EtOH, o/n, rt, then (b) HgO, S, reflux, 2 h, 35–61% (v) 1,1'-carbonyldiimidazole, imidazole, CH3CN, reflux 2-12 h, 50–90% (vi) 2 M aq. NaOH, MeOH, reflux, 30 min, 62%.
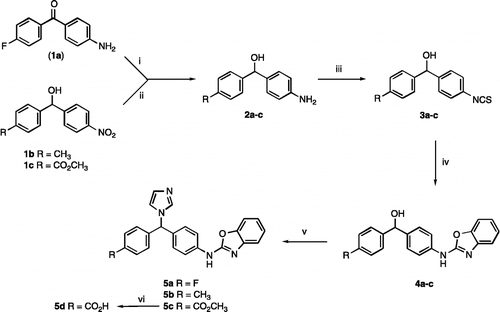
The isothiocyanate 3 was obtained by reaction of 2 with thiophosgene in a mixture of dichloromethane, ice and water, the reaction was stirred overnight at 0 °C then the thiophosgene residue and the hydrochloric acid formed during the reaction removed by washing the organic layer thoroughly with water and sodium bicarbonate. The product was obtained in high yield and was pure enough for use in the next step. Cyclisation of the isocyanate 3 to form the benzoxazole 4 was achieved by reaction with 2-aminophenol in the presence of mercury oxide and catalytic sulphur [Citation22]. Introduction of the imidazole to give the required product 5 involved reaction with carbonyldiimidazole (CDI) and imidazole [Citation23] with subsequent purification by column chromatography. The carboxylic acid 5d was obtained by saponification of the ester 5c using a 2M aq. NaOH solution in MeOH at reflux for 30 min (Scheme ).
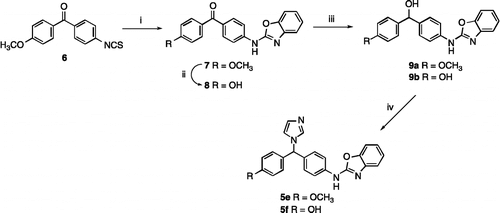
The synthesis of the methoxy and hydroxy substituted phenyl derivatives (5e-f) is described in Scheme . Commencing from the isothiocyanate (6), the benzoxazole ring was formed as described in Scheme to give the methoxy benzoxazole (7) in moderate yield. The hydroxy benzoxazole (8) was prepared from 7 by reaction with HBr in acetic acid in 77% yield. Subsequent reduction with sodium borohydride gave the carbinols (9), which were converted in the usual manner to the required imidazole derivatives (5e and 5f) (Scheme ).
Scheme 2. Reagents and Conditions (i) (a) 2-aminophenol, EtOH, o/n, rt, then (b) HgO, S, reflux, 2 h, 46% (ii) HBr, AcOH, reflux 6 h then o/n rt, 77% (iii) NaBH4, MeOH, 1–12 h, rt, 42–91% (iv) 1,1′-carbonyldiimidazole, imidazole, CH3CN, reflux 2–12 h, 38–74%.
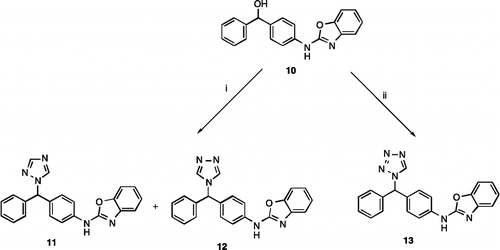
To determine the effect of the nitrogen heterocycle on inhibitory activity, the triazole and tetrazole derivatives of the potent benzoxazole (, R = H, IC50 0.9 μM) were prepared. Reaction of (4-(benzo[d]oxazol-2-ylamino)phenyl)(phenyl)methanol [Citation14] (10) with either 1,1'-sulfinylbis(1H-1,2,4-triazole) or 1,1'-sulfinylbis(1H-tetrazole), generated in situ from thionyl chloride and traizole or tetrazole, as previously described [Citation24], gave the corresponding 1H-1,2,4-triazole (11), 1H-1,2,4-triazole (12) and 1H-tetrazole (13) (Scheme ),
Scheme 3. Reagents and Conditions (i) 1H-1,2,4-triazole, SOCl2, K2CO3, CH3CN, 72 h, rt,11 35%, 12 12% (ii) 1H-tetrazole, SOCl2, K2CO3, CH3CN, 72 h, rt, 32%.
Enzyme Inhibition
The imidazole derivatives (5) were evaluated for their retinoic acid metabolism inhibitory activity using a MCF-7 cell assay [Citation12], using radiolabelled [11,12-3H] all-trans retinoic acid as the substrate and liarozole [Citation25,Citation26] (IC50 = 7 μM) and R115866 [Citation27] (IC50 = 5 nM) as standards for comparison.
Substitution in the phenyl ring was not well tolerated with a loss of inhibitory activity (R = H, IC50 = 0.9 μM), however small substituents e.g. R = CH3 (IC50 = 8.0 μM) and R = OH (IC50 = 12.0 μM) displayed comparable activity with liarozole (IC50 = 7 μM) (). Further substitution and altering the position of substitution with CH3 has been explored by our group, however no improvement in activity was observed (unpublished data), suggesting that any substitutions are unfavourable suggesting steric limitations. Docking studies indicated a favourable interaction of the carboxylic acid derivative (5d) with the enzyme active site, it is possible that the poor inhibitory activity observed (IC50 = 50 μM) was owing to reduced uptake across the cell membrane, a more accurate value of CYP26A1 inhibition would require a microsomal assay.
Table II. IC50 data for the novel imidazole N-phenylbenzo[d]oxazolamines (5) .
.
The imidazole was optimal for inhibition ((IC50 = 0.9 μM), with reduced activity observed for the triazole ((IC50 = 18 μM and >20 μM) and the tetrazole (IC50 = > 20 μM) derivatives (). This result is consistent with previous studies correlating inhibitory activity with the coordination potential of the nitrogen heterocycle with the haem [Citation28].
Table III. IC50 data for the novel triazole and tetrazole derivatives compared with imidazoleA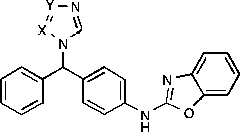
Conclusions
Inhibitors with activity comparable with liarozole have been prepared, however substitution in the phenyl ring is sterically limited with very little scope for modification. Introduction of a more flexible alkyl substituted chain in place of the phenyl ring may allow greater interaction with the amino acids within this region of the enzyme active site and improve binding and inhibitory activity.
Acknowledgements
For ASA we acknowledge the Embassy of the Arab Republic of Egypt for the award of a PhD scholarship. We thank the technical staff of the Tenovus Cancer Centre, Welsh School of Pharmacy, Cardiff University, UK for the preparation of cells and media, and the EPSRC Mass Spectrometry Centre, Swansea, UK for mass spectroscopy data.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- MB Sporn, AB Roberts, and DS Goodman, editors. The Retinoids, Biology, Chemistry and Medicine. New York: Academic Press; (1984) . p 424.
- E Sonneveld, CE Van den Brink, BJ Van der Leede, RK Schulkes, M Petkovich, B Van der Burg, and PT Van der Saag. (1998). Human retinoic acid (RA) 4-hydroxylase (CYP26) is highly specific for all-trans-RA and can be induced through RA receptors in human breast and carcinoma cells. Cell Growth Differ 9:629–637.
- N Ahmad, and H Mukhtar. (2004). Cytochrome P450: A target for drug development for skin diseases. J Investig Dermatol 123:417–425.
- AR Brecher, and SJ Orlow. (2003). Oral retinoid therapy for dermatologic conditions in children and adolescents. J Am Acad Dermatol 49:171–182.
- EH Estey, F Giles, H Kantarjian, S O'Brien, J Cortes, E Freireich, G Lopez-Berestein, and M Keating. (1999). Molecular remissions induced by liposomal-encapsulated all-trans retinoic acid in newly diagnosed acute promyelocytic leukaemia. Blood 94:2230–2235.
- P Fenaux, C Chastang, S Chevret, M Sanz, H Dombret, E Archimbaud, M Fey, C Rayon, F Huguet, JJ Sotto, C Gardin, PC Makhoul, P Travade, E Solary, N Fegueux, D Bordessoule, J San Miguel, H Link, B Desablens, A Stamatoullas, E Deconinck, F Maloisel, S Castaigne, C Preudhomme, and L Degos. (1999). A randomized comparison of all trans retinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukaemia. Blood 94:1192–1200.
- WX Guo, PS Gill, and T Antakly. (1995). Inhibition of AIDS-Kaposi's sarcoma cell proliferation following retinoic acid receptor activation. Cancer Res 55:823–829.
- J Marill, T Cresteil, M Lanotte, and GG Chabot. (2000). Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol Pharmacol 58:1341–1348.
- WJ Ray, G Bain, M Yao, and DI Gottlieb. (1997). CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J Biol Chem 272:18702–18708.
- MS Gomaa, SW Yee, CE Milbourne, M-C Barbara, A Brancale, and C Simons. (2006). Homology model of human retinoic acid metabolising enzyme cytochrome P450 26A1 (CYP26A1): Active site architecture and ligand binding. J Enz Inhib Med Chem 21:361–369.
- AJ Kirby, R LeLain, P Mason, F Maharlouie, PJ Nicholls, HJ Smith, and C Simons. (2003). Inhibition of retinoic acid metabolising enzymes by 2-(4-aminophenylmethyl)-6-hydroxy-3,4-dihydronaphthalen-1(2H)-one and related compounds. J Enz Inhib Med Chem 18:27–33.
- SW Yee, L Jarno, MS Gomaa, C Elford, L-L Ooi, MP Coogan, R McClelland, RI Nicholson, BAJ Evans, A Brancale, and C Simons. (2005). Novel tetralone-derived retinoic acid metabolism blocking agents: Synthesis and in vitro evaluation with liver microsomal and MCF-7 CYP26A1 cell assays. J Med Chem 48:7123–7131.
- S Pautus, SW Yee, M Jayne, MP Coogan, and C Simons. (2006). Synthesis and CYP26A1 inhibitory activity of 1-[benzofuran-2-yl-(4-alkyl/aryl)-methyl]-1H-triazoles. Bioorg Med Chem 14:3643–3653.
- MS Gomaa, B Bobillon, JL Armstrong, GJ Veal, A Brancale, CPF Redfern, and C Simons. (2008). Synthesis and in vitro evaluation of novel, 4-[(imidazol-1-yl and triazol-1-yl)(phenyl)methyl]arylamines: Potent CYP26 inhibitors and inducers of retinoic acid in Neuroblastoma cells. Bioorg Med Chem in press
- VCO Njar, L Gediya, P Purushottamachar, P Chopra, TS Vasaitis, A Khandelwal, J Mehta, C Huynh, A Belosay, and J Patel. (2006). Retinoic acid metabolism blocking agents (RAMBAs) for treatment of cancer and dermatological diseases. Bioorg Med Chem 14:4323–4340.
- DD Perrin, and WLF Armarengo. Purification of Laboratory Chemicals .3rd ed. New York: Pergamon Press; (1988).
- B Staskun. (1964). Nuclear acylation of arylamides. J Org Chem 29:2856–2860.
- GJ Bridger, Y Zhou, and R Skerlj. (2005). Preparation of piperidines as chemokine receptor, particularly CCR5, modulators for the treatment of inflammatory and autoimmune disease. PCT Int Appl 384.
- S Massa, G Stefancich, F Corelli, R Silvestri, A Mai, M Artico, S Panico, and N Simonetti. (1989). Researches on antibacterial and antifungal agents. X. Synthesis and antifungal activities of 1-[p-methyl-α-[4-(1H-pyrrol-1-yl)phenyl]benzyl]azoles and some related products. Archiv Pharm 322:369–373.
- Tripos SYBYL 7.0; Tripos Inc., 1699 South Hanley Rd, St. Louis, Missouri 63144, USA. http://www. tripos.com.
- Molecular Operating Environment (MOE). Chemical Computing Group, Inc. Montreal, Quebec, Canada: http://www.chemcomp.com.
- F Janssens, J Torremans, M Janssen, RA Stokbroekx, M Luyckx, and PAJ Janssen. (1985). New antihistaminic N-heterocyclic 4-piperidinamines. Synthesis and antihistaminic activity of N-(4-piperidinyl)-1H-benzimidazole-2-amines. J Med Chem 28:1925–1933.
- W Aelterman, Y Lang, B Willemsens, I Vervest, S Leurs, and F De Knaep. (2001). Conversion of the laboratory synthetic route of the N-aryl-2-benzothiazolamine R116010 to a manufacturing method. Org Proc Res Dev 5:467–471.
- TK Vinh, M Ahmadi, PO Lopez Delgado, S Fernandez Perez, HM Walters, HJ Smith, PJ Nicholls, and C Simons. (1999). 1-[(Benzofuran-2-yl)-triazoles and -tetrazoles -potent competitive inhibitors of aromatase. Bioorg Med Chem Lett 9:2105–2108.
- GPH Lucker, AMC Heremans, PJ Boegheim, PCM Van de Kerkhof, and PM Steijlen. (1997). Oral treatment of ichthyosis by the cytochrome P450 inhibitor liarozole. Brit J Dermatol 136:71–75.
- L Denis, F Debruyne, P De Porre, and J Bruynseels. (1998). Early clinical experience with Liarozole (Liazal (TM)) in patients with progressive prostate cancer. Eur J Cancer 34:469–475.
- P Stoppie, M Borgers, P Borghgraef, L Dillen, J Goossens, G Sanz, H Szel, C Van Hove, G Van Nyen, G Nobels, H Vanden Bossche, M Venet, G Willemsens, and J Van Wauwe. (2000). R115866 inhibits all-trans-retinoic acid metabolism and exerts retinoidal effects in rodent. J Pharmacol Exp Ther 293:304–312.
- MR Saberi, K Shah, and C Simons. (2005). Benzofuran- and furan-2-yl-(phenyl)-3-pyridylmethanols: Synthesis and inhibition of human placental aromatase (CYP19). J Enz Inhib Med Chem 20:135–141.