Abstract
Cancer cells in hypoxic areas of solid tumors are to a large extent protected against the action of radiation as well as many chemotherapeutic drugs. There are, however, two different aspects of the problem caused by tumor hypoxia when cancer therapy is concerned: One is due to the chemical reactions that molecular oxygen enters into therapeutically targeted cells. This results in a direct chemical protection against therapy by the hypoxic microenvironment, which has little to do with cellular biological regulatory processes. This part of the protective effect of hypoxia has been known for more than half a century and has been studied extensively. However, in recent years there has been more focus on the other aspect of hypoxia, namely the effect of this microenvironmental condition on selecting cells with certain genetic prerequisites that are negative with respect to patient prognosis. There are adaptive mechanisms, where hypoxia induces regulatory cascades in cells resulting in a changed metabolism or changes in extracellular signaling. These processes may lead to changes in cellular intrinsic sensitivity to treatment irrespective of oxygenation and, furthermore, may also have consequences for tissue organization. Thus, the adaptive mechanisms induced by hypoxia itself may have a selective effect on cells, with a fine-tuned protection against damage and stress of many kinds. It therefore could be that the adaptive mechanisms may take advantage of for new tumor labeling/imaging and treatment strategies. One of the Achilles’ heels of hypoxia research has always been the exact measurements of tissue oxygenation as well as the control of oxygenation in biological tumor models. Thus, development of technology that can ease this control is vital in order to study mechanisms and perform drug development under relevant conditions. An integrated EU Framework project 2004–2009, termed EUROXY, demonstrates several pathways involved in transcription and translation control of the hypoxic cell phenotype and evidence of cross-talk with responses to pH and redox changes. The carbonic anhydrase isoenzyme CA IX was selected for further studies due to its expression on the surface of many types of hypoxic tumors. The effort has led to marketable culture flasks with sensors and incubation equipment, and the synthesis of new drug candidates against new molecular targets. New labeling/imaging methods for cancer diagnosing and imaging of hypoxic cancer tissue are now being tested in xenograft models and are also in early clinical testing, while new potential anti-cancer drugs are undergoing tests using xenografted tumor cancers. The present article describes the above results in individual consortium partner presentations.
Table of Contents
1. Introduction 3
2. The EUROXY Program 4
3. Technical aspects of measuring oxygen tensions in vivo and in vitro 4
3.1. Introduction 4
3.2. Oxygen microsensors 5
3.3. Sensing cell culture flask 5
3.4. Oxygen sensor array strips 6
3.5. Conclusion and outlook 6
4. Responses to hypoxia in normal and malignant cells 6
4.1. Cellular oxygen sensors 6
4.2. Pre-mRNA splicing 8
4.3. Transcription and regulators at moderate hypoxia 9
4.4. Targeting tumor hypoxia through interference with mTOR and the UPR signaling 10
4.4.1. Introduction 10
4.4.2. The role of mTOR signaling during hypoxia 10
4.4.3. Hypoxia and the unfolded protein response 11
4.4.4. Changes in gene expression through regulation of translation 11
4.4.5. Conclusions 11
4.5. Importance of ROS signaling in tumors 12
4.6. pH Regulation at the cellular and tissue level 13
4.7. Cellular metabolism and respiration 14
4.8. Proliferation and regulation of cell cycle transit 14
4.9. Conserved energy saving responses in hypoxia tolerant animal and cancer cells 15
5. Clinical consequences of tumor hypoxia 16
5.1. Introduction 16
5.2. Tumor progression 17
5.3. Tumor resistance to conventional drug therapies 17
5.4. Tumor resistance to irradiation 17
6. Taking advantage of the hypoxic tumor phenotype 18
6.1. Gene signatures for prognosis and stratification 18
6.1.1. How to select patients for treatment 18
6.1.2. Oxygen needle electrode 18
6.1.3. 2-Nitroimidazole binding agents 18
6.1.4. Tumor immunohistochemistry 18
6.1.5. Imaging 19
6.1.6. Gene expression profiling 19
6.2. Renal carcinomas and HIF-1/HIF-2 22
6.3. Imaging acute and chronic tumor hypoxia 23
6.4. Bioreductive drugs 24
6.4.1. Introduction 24
6.4.2. Prototype agents in the development of bioreductive drugs 25
6.4.3. Clinical lead compounds 26
6.4.4. New kids on the block 26
6.4.5. Conclusions 26
6.5. Synthesis of anti-carbonic anhydrase IX (CA IX) drugs 27
6.6. Targeting CA IX in tumors 29
6.7. ROS and redox measurements and research potential in oncology 30
7. Concluding remarks 31
References 31
1. Introduction
Due to lower or more erratic overall blood flow in malignant compared to normal tissues, solid tumors generally contain smaller or larger areas where cells have far less than normal access to oxygenCitation1. This was first indicated as early as the 1930sCitation2, and was related to a possible relevance to radiotherapy, since earlier observationsCitation3 had indicated that oxygen increased radiosensitivity in developing eggs of the parasitic nematode Ascaris.
Within this field, the term “hypoxia” became a standard notion when referring to cells having a microenvironment with less oxygen than that of normal tissues (40–50 μM). The definition is, however, vague, since even the level of oxygenation in normal tissues may vary, and this variation by itself is of importance for normal tissue regulation.
Traditionally, the finding of hypoxic areas in solid human cancer tumors, as reported by Thomlinson and GrayCitation4, was viewed as being primarily a problem for radiation therapyCitation5. The hypothesis was that not all hypoxic cancer cells die due to low oxygen, since some are necessarily located near the rim of the hypoxic areas and it takes some time for cells to die even during extremely low oxygenation. The radioresistance of hypoxic cells was later found to be related to the high electron affinity of the moleculeCitation6, which enables even small amounts of oxygen to fixate radiation-induced macromolecular damage before the damage can be repaired by naturally occurring radical scavengers in the cells. Since tissue hypoxia is restricted primarily to malignant and not normal tissues, it was postulated that hypoxia represents a specific protection against radiation for malignant compared to normal cells.
Later it was seen that this specific protection was not limited to radiation, but even included some chemotherapeutic drugsCitation7,Citation8. This has partly been attributed to the fact that hypoxic cells are located far from the nearest blood vessel, and therefore may experience limited influx of the drug, but it has also turned out to be due to a specifically reduced sensitivity to some drugs under hypoxic conditionsCitation9.
In order to increase the radiosensitivity of hypoxic cells without affecting that of well-oxygenated (i.e. normal) cells, drugs denoted hypoxic cell sensitizers were developed. These should preferentially have some of the same electron-affinity as oxygen itselfCitation10. There were, however, two important differences compared to oxygen: (1) these drugs should not be metabolized by the cells: in that way they would be able to diffuse into the hypoxic areas; (2) the drugs should have some of the same electron-affinity as oxygen and therefore also the ability to fixate radiation-induced damage. They should be somewhat less effective than oxygen, though: in that way they would not increase the radiosensitivity of well-oxygenated (i.e. normal) cells.
Although the sensitizer compounds developed should in principle be non-toxic to cells, some of them turned out to be interesting from a chemotherapeutic (and diagnostic) point of view. The hypoxic cell sensitizers acted as targets for one-electron reductases such as cytochrome P450Citation11. The addition of an electron started a progression toward the development of a toxic compound. Thus, the hypoxic cell sensitizer acted as a non-toxic prodrugCitation12. The positive thing was that the toxicity only developed under hypoxic conditions since oxygen, if present, captured the electron and reversed the process back to the non-toxic prodrug. Thus, for the first time there were drugs developed that had a potential specificity against severely hypoxic and not aerobic cells. Another possibility that was opened by these hypoxia-specific compounds was that they could be used as markers to detect and even visualize hypoxic regions in tumor tissue.
A large part of the survival and treatment resistance of deoxygenated cells is conferred by signaling pathways that are specifically active under hypoxic pO2. During the last 15 years the cellular oxygen sensing mechanism of metazoan cells and the cascade of regulatory mechanisms that is triggered by the sensing of reduced oxygenation has been intensively studied, particularly in regard to the predominant hypoxia-sensing machinery: that of the hypoxia-inducible transcription factor HIFCitation13. Regulatory processes in the HIF casacde involve, as a first step, deactivation of prolyl hydroxylase domain (PHD) proteins by the reduction in oxygen concentration, and are triggered at oxygen levels within the range experienced even by normal cellsCitation14. Although the oxygen levels where PHD deactivation takes place are close to that of normal tissue, they are nevertheless denoted hypoxia, and the term is often used and understood as synonymous to the term hypoxia used to describe the cancer-specific low oxygenation causing resistance to therapy. It could, however, be that tumor hypoxia and the PHD-sensing hypoxia are so different that the two concepts should not be mixed. The reason why this is uncertain is that the scientific work within the field of hypoxia is still largely done without exact knowledge of the pericellular oxygenation. Measurement as well as maintenance of oxygenation in cellular microenvironments is so challenging and technically time-consuming that exact pericellular measurements are usually not performed. The problem of experimental reproducibility is instead taken care of by other means, for example culturing of cells in glove-boxes with a known and well-controlled atmospheric oxygen concentration. If cell culturing techniques are standardized with respect to timing and density of cells, one may then obtain reproducible conditions within one laboratory without any direct measurement of oxygen at the cell membrane. It is far from obvious, however, that microenvironmental oxygenation between different laboratories can be compared.
Several mechanisms in the regulatory cascades starting by deactivation of PHD and stabilization of HIFα may have potential as mechanisms for new cancer drug development. There is still some uncertainty with respect to the cancer-specificity of these regulations.
Later still it was, however, experienced that low oxygen generally has profound consequences with regard to cell behavior, both in the form of the role that molecular oxygen plays in the regulation/deregulation of key cellular enzymes (i.e. dihydro-orotate dehydrogenase or ribonucleotide reductase, which are vital for supply of precursors for DNA synthesis) and in terms of the role it plays in regulation of gene transcription (HIF) and translation.
Mostly due to the fact that normal tissue in most cancer patients is not hypoxic, hypoxia offers a broad range of cancer-related mechanisms with the potential for improvement of therapy and diagnosis. The theme of the EUROXY project is: “Taking advantage of tumor cell adaptations to hypoxia for developing new tumor markers and treatment strategies”. More than 20 research groups covering different areas of research within this vast field have collaborated over the last 5 years and have supported each other with ideas and possibilities. The research has covered:
development of new technology for continuous monitoring of pericellular oxygen concentration and respiration rate during in vitro cell culturing
studies of the role of the cancer-specific carbonic anhydrase (CA) IX in pH regulation with the aim to develop CA IX as a possible target for therapy as well as diagnosis
studies of design and synthesis of new drugs for CA IX inhibition as well as bioreductive drugs
fundamental studies of cell cycle control under hypoxia
studies of new types of radiation delivery to selectively increase the radiosensitivity of hypoxic cells
fundamental studies giving new knowledge concerning PHD oxygen sensing on concomitant regulatory cascades through HIF
fundamental studies concerning protein synthesis (translation) and splicing.
In the following these different themes are described under separate headings.
2. The EUROXY Program
A characteristic of EUROXY is the many years of building up the necessary scientific and collaborative strength to take on an integrated European Union (EU) project. Many of the EUROXY members were previously members of two hypoxia-focused projects supported by regional (Scandinavian) sources, then members of an EU-funded infrastructure project (Oxnorm), and now the integrated project EUROXY.
This continuity in scientific focus and scientific partners made each effort a stepping stone for the next joint project. Actually, already after the first 2 years of work on EUROXY, a decision to seek extension of the collaboration beyond the 5 years was taken. The proposal was to take the preclinical EUROXY results and seek funding for their translation into clinical testing. After the inclusion of strong clinical groups, EU funding of the successor program was ensured.
The EUROXY predecessor Oxnorm included a development of concepts and nomenclature. One attempt to explicitly define what hypoxia should stand for in cellular biology was not to relate it to measured pericellular oxygen tension in absolute terms but to consider hypoxia as a variable, i.e. the pericellular tension which, for particular tissues/cells, makes the cells switch on their adaptive metabolic mechanisms induced by the lowered oxygen tension. However, no consensus was reached on the definition, but there was agreement on the need for pericellular oxygen tension measurements in order to facilitate reproducibility and comparison of results.
The follow-up study EUROXY was largely shaped by the recent finding of a central role of the transcription regulator HIF and its effect on many downstream gene functionsCitation15, and the emerging evidence of a role of HIF in such varied areas as microbiology, immunology, cardiology, etc. Our general working hypothesis became that the cellular responses to hypoxia allowing tumor cells to survive the low oxygen tension in solid tumors, which at the same time shielded the tumor cells against ionizing irradiation, and many cytostatics might themselves represent novel targets for tumor imaging/labeling and anti-tumor therapy.
The means to take advantage of this concept was a concerted basic cellular and molecular study of hypoxic response pathways, and, when promising targets were identified, find the labeling/therapeutic agents in preparation for future clinical testing.
Our focus on HIF-related pathways was soon found to be insufficient, because the growing number of downstream functions governed by HIF indicated that a high number of side effects would result if one made HIF itself the therapeutic target. Our focus was widened, and more effort went into unraveling the upstream oxygen-sensing hydroxylases and later also into translation research on the unfolded protein response (UPR) and mammalian target of rapamycin (mTOR).
An important consideration in the last year of EUROXY has been to ensure optimal utilization of obtained results. A part of this effort is the present article. Another part is the participating companies’ representation in the market of antibodies, sensors, incubators, and compounds for imaging. Third, we have secured a continuation program, Metoxia, funded by the EU.
3. Technical aspects of measuring oxygen tensions in vivo and in vitro
3.1. Introduction
Oxygen sensors for in vivo and in vitro application mainly use two principles: optical sensor probes, consisting of fluorescence dyes, and electrochemical sensor electrodes. Both in vivo and in vitro, there is an undisputed need for more practicable and cost-effective means of monitoring pericellular oxygen tension, as compared to the currently commercially available brittle and expensive oxygen probes. This vision has guided us toward the development of sensor chips permanently mounted in a cell culture flask, as well as a polymeric oxygen sensor array strip with a stability of sensitivity of 2 weeks, in a cost-effective fabrication flow.
3.2. Oxygen microsensors
Optical sensors use a dye to measure oxygen tension by evaluating the quenching effect due to the presence of oxygen moleculesCitation16. As dyes usually tend to bleach during operation, a modulated excitation light is used, enabling performing lifetime analysis to obtain sensor readings independent of the bleachingCitation17. This principle allows stable readings over time, but has no defined zero-point, which hampers the calibration and application of such sensors for low oxygen situations as often found in hypoxic cell cultures. The main advantage of the optical approach is that the measurement does not consume any oxygen.
Electrochemical oxygen sensors are usually amperometric sensors, where the oxygen is reduced at an appropriate metal electrode. There are also some potentiometric dissolved oxygen sensorsCitation18, which have—like the optical sensors—no defined zero-point. Amperometric oxygen sensors can be distinguished as Clark-type and direct amperometric sensors. The main improvement that Clark made regarding oxygen sensors was to cover the electrode arrangement with a gas-permeable membraneCitation19. This was later often referred to as the Clark-type sensor, where the measurement region was separated from the electrolyte chamber by a gas-permeable membrane. An example of realization of such a sensor type in microtechnology was made by Jobst et al.Citation20. This sensor type has the advantage that degradation of the sensor’s response due to substances from the measurement medium is avoided, overcoming either the need for frequent recalibration or the implementation of advanced operational procedures.
On the other hand, the fabrication of Clark-type sensors in microtechnology is cumbersome, and so far such devices have failed to provide the desired operational lifetime. Therefore, nowadays cell culture sensors deal with direct amperometric setupsCitation21–23.
To overcome the effect of performance-degrading substances from the cell culture medium, chronoamperometric protocols are used to ensure signal stability over a range of weeks. These protocols for application with platinum electrodes comprise cleaning steps, with which a platinum oxide layer is formed on the electrode and removed immediately before the reduction of oxygen, forcing the rapid establishment of long-term stable surface conditions for oxygen reduction. Furthermore, by setting an appropriately short on-time, the inherent disadvantage of amperometric oxygen sensors—consumption of the analyte—can be minimized.
To obtain pericellular oxygen readings in vitro, two approaches have been realized. One is with a chip sensor, where the cells settle directly on the chip surface. The second approach is to use a sensor array strip, which can be inserted through the neck or an additional opening into the cell culture flask. These strips measure the oxygen concentration at different height levels simultaneously, which allows—assuming stable diffusion profiles—extrapolation of pericellular oxygen levels and the cells’ oxygen consumption at the same time. Alternatively, conventional single-electrode dip-in sensors can be used to measure the oxygen partial pressure at different height levels by means of an automated motorized stageCitation24.
For both approaches, the sensor materials must not show any cytotoxic effects with the used cell cultures. A sensor chip, where adherent cells have to settle directly on the chip surface, must comprise a top layer that is not only non-cytotoxic, but further allows cells to adhere well. The cells should behave during proliferation similar to the situation in an ordinary cell culture flask. We found that a plasma-deposited silicon oxide fulfills these requirements for the tested human breast cancer cell lines (MCF-7 and T-47D).
3.3. Sensing cell culture flask
During the EUROXY project, the concept and prototypes for a sensing cell culture flask (SCCF) were developed (). The SCCF is based on a conventional 50 mL cell culture flask, where a sensor chip is integrated in a milled opening in the bottom of the flask in such a way that the chip surface is on a plane with the surface of the cell culture area in the flask.
Figure 1. Sensing cell culture flask (SCCF). A sensor chip, comprising oxygen and optional pH and NO sensors, is integrated in a standard cell culture flask.

The SCCF chip comprises four platinum oxygen electrodes along with a common counter and a silver/silver chloride reference electrode. Further, the SCCF chip can be equipped with metal oxide electrodes for pH-respective acidification measurements and working electrodes for a NO sensor.
The SCCF chip is fabricated with thin-film platinum electrodes on a Pyrex wafer. The platinum is insulated with an inorganic plasma-deposited (plasma enhanced chemical vapor deposition, PECVD) multilayer. These layers are opened at the electrode positions by reactive ion etching (RIE). The layer setup is, despite the region of the electrodes and connection lines, optically transparent, and therefore allows inspection of the cell culture by inverted microscopy. The electrodes are covered with a dispensed poly(2-hydroxyethylmethacrylate) (pHEMA)-based hydrogel to ensure that cells which adhere on the chip do not settle directly on the electrode surface. At the four working electrodes for oxygen, this hydrogel also acts as a region with lower oxygen diffusion coefficient, to confine the major drop of the oxygen gradient toward the electrodes inside the hydrogel. This minimizes disturbance of the pericellular oxygen profile. The integrated reference electrode is made by electrodeposition of silver and subsequent partial electrochemical conversion of silver in a chloride solution to silver chloride.
3.4. Oxygen sensor array strips
The all-polymeric devices are made by patterned metallization of a polyimide foil, subsequent insulation, galvanic processing, and final laser cutting. Inspired by biocompatibility considerations, originally an insulation scheme via lamination of a laser-cut thermoset adhesive-coated polyimide foil as insulation was used. Though these devices performed very well on the timescale of single days, upon prolonged continuous use over several days sensitivity of the devices increased to an unacceptable extent. This effect originates from degradation of the insulation properties. While other all-polymeric devices for long-term use—such as connectors for cochlear or retina implants—are insulated by means of elaborate and expensive processes, applying these technologies for our application would sacrifice any vision of a cost-effective device. Our technological innovation now provides us with probes having insulation properties preserved for at least 2 weeks. Even when fabricated in moderate quantities, costs below 10 euros per device seem feasible.
As one can easily imagine from , the pericellular oxygen concentration is calculated from the linear fit to the readings of the four oxygen sensors extrapolated to the flask bottom. From this fit, additionally quantification of the oxygen flux or the oxygen consumption by the cells is obtained from its slope.
Figure 2. Micrograph of the sensing tip of a flexible oxygen sensor array indicating the mean distance of the individual 200 μm diameter oxygen sensors from the cells.

The intrinsic precision of the photo-lithographic method for patterning of the insulation layer and improved hydrogel membrane composition and deposition even make it possible to fabricate devices offering the prospect of overcoming the need for initial calibration—which would drastically improve the acceptance of such devices by potential customers.
While the strip format of the described oxygen sensor array is dedicated to in vitro work, translation of the fabrication technology into a needle format is easy, and already demonstrated with a 400 μm wide and 50 mm long biosensor probe for in vivo use.
Handling of the probe is another major factor influencing customer acceptance, as well as measurement reliability. Since we consider this a very critical factor, a variety of access schemes was realized and presented to the end users. The most widely accepted is a permanent mounting of the probe through the sidewall of a cell culture flask that allows handling of the flask almost like one without an oxygen sensor array.
3.5. Conclusion and outlook
The vision of a robust, easily handled, and cost-effective probe for monitoring of the pericellular oxygen concentration in a hypoxic environment was realized by two approaches. Currently, cost-effectiveness and ease of handling of the system are still compromised by the assembly and interfacing—which will be the focus of future work. This work, ideally done with the cooperation of a cell culture flask manufacturer, will also provide the platform for the integration of additional features aiming at oxygen tension and metabolite control at individual flask level.
4. Responses to hypoxia in normal and malignant cells
4.1. Cellular oxygen sensors
One of the most sensitive (and most studied) physiological responses to hypoxia is the massive up-regulation of the hematopoietic growth factor erythropoietin (Epo) by acute reductions in blood oxygen availability. In the early 1990s, work on transcriptional control of Epo unexpectedly revealed that the underlying system of oxygen sensing and transcriptional control operates widely in cells, and is conserved in essentially all animal species, even those without red cell or vascular systems.
Whereas Epo production may increase several hundred-fold over a matter of hours in response to acute anemia, many effects of hypoxia on metabolism and growth/differentiation occur over longer timescales, with lower amplitude, and (within the intact organism) at apparently different tissue oxygen tensions (see above). Despite these contrasts, genetic evidence indicates that a surprisingly large number of these responses are dependent on the integrity of the same system of oxygen sensing and signaling, involving the post-translational hydroxylation of the transcription factor HIF. This system is activated in cancer by a range of genetic and microenvironmental mechanisms, and hence has been one of the main focuses of enquiry by the EUROXY consortium.
The pathways regulated by the HIF transcriptional cascade have been reviewed in detail elsewhereCitation25–28. In outline, HIF is a heterodimer of α and β subunits. Oxygen sensitive signaling is mediated by the α subunits, of which there are three—HIF-1α, HIF-2α, and HIF-3α—each encoded at a distinct genetic locus. Oxygen-dependent post-translational hydroxylation of two conserved prolyl residues in the central degradation domain of HIF-1α and HIF-2α (and of one prolyl residue in HIF-3α) targets the HIFα polypeptide to the von Hippel–Lindau (pVHL) ubiquitin E3 ligase and hence destruction by the ubiquitin–proteasome pathway (for review see reference 13). In a second oxygen-regulated step, hydroxylation of an asparaginyl residue that is conserved in the C-terminal activation domains of HIF-1α and HIF-2α reduces transcriptional activity at least in part by blocking the physical association of co-activators with this domain. These hydroxylations are all catalyzed by non-heme Fe(II) enzymes belonging to the 2-oxoglutarate (2-OG) dependent dioxygenase superfamily. These enzymes couple oxidation of the HIFα polypeptide to the oxidative decarboxylation of 2-OG in an “oxygen splitting” reaction that directly consumes molecular oxygen (for review see references 29 and 30). Three closely similar enzymes termed PHD (prolyl hydroxylase domain) 1, 2, and 3 catalyse HIF prolyl hydroxylation. A fourth enzyme more closely related to the procollagen prolyl hydroxylases (PHD4) has been shown to have in vitro hydroxylase activity for HIFα substrates, though whether it directly targets HIFα in vivo is uncertainCitation31. To date, a single enzyme FIH (factor inhibiting HIF) has been identified that catalyzes HIF asparaginyl hydroxylation.
In most members of the Fe(II) and 2-OG dependent oxygenase superfamily, including the HIF hydroxylases, three of the six available coordination positions at the catalytic iron center are utilized for (relatively labile) binding to the apo-enzyme. This occurs via a 2-histidine-1-carboxylate “facial triad” presented by residues on the second and seventh strands of the eight-stranded β-barrel jelly-role conformation of the catalytic domain (for review see reference 32). This iron coordination arrangement contrasts, for instance, with heme enzymes where four of the coordination positions are generally occupied by the heme. Such differences may be relevant to the selection of members of this family of enzymes as physiological oxygen sensors. In catalysis, two of the remaining three coordination positions are used to bind 2-OG whilst the sixth is used to bind molecular oxygen. The reaction proceeds via a radical mechanism in which the catalytic center is activated (most likely involving the creation of a high reactive ferryl species (FeIV=O)), which then oxidizes the HIFα polypeptide substrate. Ascorbate is required for full catalytic activity, and is believed to operate as an alternative substrate reducing the catalytic iron center in the event of an uncoupled cycle, in which oxidation of HIFα fails to occur. It appears likely that these diverse co-factor and co-substrate requirements are important for the physiological function of these enzymes as cellular oxygen sensors, allowing a more flexible interface between the availability of molecular oxygen and the rate of catalysis, than might be the case with other types of enzyme.
In vitro studies of the kinetic behavior of recombinant HIF hydroxylases have indicated that both the PHDs and FIH can operate as high turnover enzymes, with a maximum rate of enzymatic metabolism (VMax) in the range of 50–200 mol/mol/minCitation33–35. Such studies have also defined apparent Michaelis constant (Km) values for Fe(II), ascorbate, 2-OG, and oxygen. In keeping with predictions from kinetic analyses of other members of the 2-OG oxygenase superfamily that it is an E.2-OG substrate species that interacts with molecular oxygen, the apparent KmO2 varies with the HIFα polypeptide substrate used in the assayCitation36,Citation37. Thus, for short HIFα polypeptides in the range 15–30 residues, KmO2 values of approximately 200 μM have been measured for the PHDs whilst measurements using HIFα polypeptides in the range of 150–250 residues have yielded values of approximately 80–100 μM. All these values are well above measured tissue pO2, and (given that oxygen is not produced by animal cells) they can be predicted to be above the concentration available to the enzymes within their subcellular compartment. Thus, the availability of oxygen is predicted to be limiting for activity over the entire physiological range—fulfilling a condition for their operation as oxygen sensors.
HIF hydroxylase co-factors also appear to be limiting for catalytic activity under physiological or pathophysiological conditions, and thus act to modulate the “oxygen sensing” function.
Lability of iron-binding accounts for the classical property of the HIF system of being activated by iron chelation as well as hypoxia. Tissue culture cells, especially rapidly growing cells with activated oncogenic pathways, accumulate HIFα in a non-hydroxylated form even under well-oxygenated conditionsCitation38,Citation39. Addition of iron (or ascorbate) to such cells promotes HIFα prolyl hydroxylation and down-regulates HIF, suggesting that iron, ascorbate, or both are limiting under these conditionsCitation39,Citation40. The extent of which iron/ascorbate limitation of HIF hydroxylation occurs physiologically in vivo is of substantial interest. For instance, human studies of pulmonary responses to hypoxia have recently demonstrated not only that infusions of iron chelators can mimic responses to hypoxia but also that iron infusions to apparently normal subjects can blunt responses to hypoxiaCitation41. Whether the observed effects are in fact mediated by effects on HIF hydroxylases is not proven, but taken together with the cell culture studies, the findings raise the possibility that physiological changes in cellular availability of iron, as well as oxygen, may modulate these pathways. Given the frequency of clinical iron deficiency, particularly in malignant disease, this is now an important avenue for further investigation.
The HIF hydroxylase co-substrate 2-OG plays a pivotal role in metabolism. It is a Krebs cycle intermediate that is a substrate for nicotinamide adenine dinucleotide (NAD)-linked oxidation by α-ketoglutarate (2-OG) dehydrogenase, a co-substrate/product in (reversible) reductive amination/oxidative deamination by glutamate dehydrogenase, and the major amino group acceptor for transaminases. In addition, the HIF hydroxylases have been reported to be inhibited by citrate, isocitrate, succinate, fumarate, malate, oxaloacetate, and pyruvateCitation42–46. For at least some of these metabolites, in vitro kinetic studies have indicated that they act by competitive inhibition of 2-OG binding. Given the difficulties of measurement of these metabolites in different intracellular compartments (e.g. cytosolic versus mitochondrial), the effects of altered intermediary metabolism in vivo are difficult to predict from these data. Nevertheless, the possibility that hydroxylase co-substrate requirements for both molecular oxygen and 2-OG link oxygen and “metabolic” sensing is appealing.
Both succinate dehydrogenase (SDH) and fumarate hydrate (FH) have been identified as tumor suppressors. In SDH- and FH-associated cancer, intracellular accumulation of succinate and fumarate results in impairment of HIF hydroxylationCitation44,Citation47. The resultant increase in HIF can be reduced by extracellular provision of esterified (cell penetrating) 2-OGCitation48. Taken together with in vitro kinetic data demonstrating competitive inhibition of PHD enzymes by fumarate and succinate, these findings indicate that at least under these conditions, fumarate and succinate act as endogenous modulators of HIF hydroxylase activityCitation43,Citation45. Whether this can occur in other situations in which the Krebs cycle is inhibited, and whether 2-OG is itself limiting under other conditions, are unclear.
Overall, these findings have brought into focus two important considerations in relation to the work of the EUROXY consortium. First, they raise the possibility that therapeutic interventions might be targeted at increasing, as well as reducing, HIF hydroxylase activity and suggest that the clinical importance of iron/ascorbate status and metabolic energy provision in cancer patients should be re-evaluatedCitation40,Citation49–51. Second, they provide potential insights into the mechanisms by which a wide range of redox active stimuli that do not appear to affect the cellular availability of oxygen directly may impact on the regulation of HIFCitation52,Citation53.
As is detailed elsewhere in the cited references, a range of genetic and pharmacological interventions that affect the intracellular production of reactive oxygen species (ROS) modulate the HIF systemCitation54–57. Accumulating data indicate that many of these interventions affect HIF hydroxylase activity. Possible modes of action include direct effects on the HIF hydroxylase catalytic center, effects on ascorbate/dehydroascorbate and Fe(II)/Fe(III) redox couples, effects on the Krebs cycle through ROS-mediated inactivation of susceptible enzymes such as aconitase, or effects on the many NAD-linked or nicotinamide adenine dinucleotide phosphate (NADP)-linked redox couples that could directly or indirectly affect 2-OG availability.
Though obstacles remain, particularly in the measurement of relevant metabolites within appropriate subcellular compartments, and in the accurate measurement of the extent of hydroxylation at specific sites in HIFα polypeptides, these advances provide a new framework for understanding biological responses to hypoxia and how they might be manipulated in cancer and other diseases.
4.2. Pre-mRNA splicing
The coding regions (exons) of most human genes are interrupted by non-coding intervening sequences (introns) that are removed from pre-mRNA molecules to produce mature mRNAs through the process of RNA splicing. RNA splicing takes place in the nucleus and occurs co-transcriptionallyCitation58,Citation59.
Exons are defined by rather short and degenerate classical splice-site sequences at the intron/exon borders (5’ splice site, 3’ splice site, and branch site) (). Components of the basal splicing machinery bind to the classical splice-site sequences and promote assembly of the multicomponent splicing complex known as the spliceosome. Intron removal must be performed with the precision of up to one nucleotide. The spliceosome performs the two primary functions of splicing: recognition of the intron/exon boundaries and catalysis of the cut-and-paste reactions that remove introns and join exonsCitation58,Citation59.
Figure 3. Elements involved in alternative splicing of pre-mRNA. Exons are indicated as boxes, introns as thin lines. The 5’ splice-site (CAGguaagu) and 3’ splice-site cagG, as well as the branch point (ynyurac) and polypyrimidine tract (yyyyy), are indicated (y: c or u, n: a, g, c, or u, r: a or g). Upper case letters refer to nucleotides that remain in the mature mRNA.
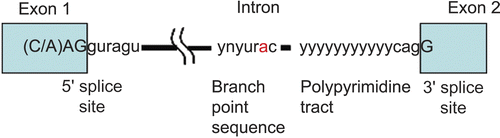
The spliceosome is made up of five small nuclear ribonucleoproteins (snRNPs) and > 100 proteins. Each snRNP is composed of a single uridine-rich small nuclear RNA (snRNA) and multiple proteins. The U1 snRNP binds the 5’ splice-site, and the U2 snRNP binds the branch site via RNA:RNA interactions between the snRNA and the pre-mRNA. Spliceosome assembly is highly dynamic. However, it is not fully understood how it is accomplishedCitation58,Citation59.
The typical human gene contains an average of eight exons. Internal exons average 145 nucleotides in length, and introns average more than 10 times this size and can be much larger. Alternative splicing pathways generate different mRNAs encoding distinct protein products, thus increasing the coding capacity of genes. Recent genome-wide analyses of alternative splicing indicate that up to 70% of human genes may have alternative splice forms, suggesting that alternative splicing together with various post-translational modifications plays a major role in the production of proteome complexityCitation60.
Pre-mRNA splicing is a sophisticated and ubiquitous nuclear process, which is a natural source of cancer-causing errors in gene expression. Changes in splice-site selection have been observed in various types of cancer, and may affect genes implicated in tumor progression and in susceptibility to cancerCitation61. In cancer, there are examples of every kind of alternative splicing, which include the use of alternative individual splice sites, alternative exons, and alternative introns. Splicing changes have been observed in genes implicated in both the susceptibility and the progression of cancer (oncogene KIT, neurofibromatosis type 1 (NF1) protein, rac1, oncogene crk, Kruppel-like Zn finger transcripion factor KLF6, androgen receptor, sex hormone-binding globulin, DNA methyltransferase DNMT3b, nucleocytosolic adapter protein Bin1). The mechanisms leading to splicing defects in cancer are poorly understoodCitation62–65.
Hypoxia has long been recognized as a common feature of solid tumors and a negative prognostic factor for response to treatment and survival of cancer patients. The discovery of HIF-1, a molecular determinant of the response of mammalian cells to hypoxia, has led to the identification of a “molecular target” of hypoxia suitable for the development of cancer therapeuticsCitation66,Citation67.
It has been demonstrated that in mice the protein IPAS (inhibitory PAS protein), a dominant negative regulator of hypoxia-inducible gene expression, is generated from HIF-3α pre-mRNA by an alternative splicing mechanism. At normal atmospheric oxygen tension, IPAS expression is restricted (in mice) to the avascular cornea epithelium. Inactivation of the IPAS transcript leads to neovascularization of the cornea, suggesting that IPAS is an important mechanism of anti-angiognenesis in this tissue. Strikingly, IPAS mRNA expression is induced in heart and lung tissue samples from hypoxic miceCitation68. IPAS expression in hepatoma cells selectively impairs the induction of hypoxia-inducible genes regulated by HIF-1 and results in retarded tumor growth and tumor vascular density in vivo. In mice, IPAS was selectively expressed in Purkinje cells of the cerebellum and in the corneal epithelium of the eye. Moreover, the expression of IPAS in the cornea correlates with low vascular endothelial growth factor (VEGF) gene expression under hypoxic conditionsCitation68.
Recently in mice, another hypoxia-inducable negative regulator NEPAS (neonatal and embryonic PAS protein) was foundCitation69. Its mRNA, as in the IPAS case, derives from the HIF-3α gene by alternative splicing, where the first exon of HIF-3α is replaced with the IPAS first exon. NEPAS can dimerize with Arnt, and shows only low levels of transcriptional activity. NEPAS is expressed in the late embryonic and early postnatal stages and its expression is predominant in the lung and heart. It is reported that NEPAS suppresses gene expression driven by HIF-1α and HIF-2αCitation69.
In addition, multiple HIF α-subunit isoforms, generated by alternative pre-mRNA splicing and expressed in different tissues, have been identified in humansCitation70–72. For instance, three isoforms have been identified for HIF-1α protein: the HIF-1α827 (at exon 1–2 junction containing an additional arginine residue), HIF-1α736 (lacking exon 14), and HIF-1α516 (lacking exons 11 and 12) isoforms. It has been shown that the HIF-1α736 isoform is three-fold less active compared to the full length HIF-1α protein, and the HIF-1α516 isoform suppresses hypoxic induction of HIF-1-regulated genes. All these isoforms are ubiquitously expressed in mammalian tissues. Also, five additional HIF-3α protein isoforms generated by alternative pre-mRNA splicing (HIF-3α2 (also referred to as human IPAS), HIF-3α3, HIF-3α4, HIF-3α5, HIF-3α6) have been identifiedCitation72. It has been suggested that some of these isoforms can act as negative HIF-1α function regulators.
In conclusion, alternative splicing of the mouse HIF-3α locus generates NEPAS and IPAS mRNA species which appear to be tissue-specific and strictly regulated by hypoxia, defining a novel mechanism of hypoxia-dependent regulation of gene expression69,73. In tumors this mechanism is an attractive means to target HIF signaling.
Also, recently, a truncated isoform of human cancer-associated CA IX, lacking part of the catalytic domain, was discovered. It has been shown that the shorter CA IX isoform is produced from the same pre-mRNA as the long one, by an alternative pre-mRNA splicing. The short CA IX isoform is detectable in normal tissues, while the long CA IX isoform is detectable only in hypoxic tissuesCitation74. The appearance of the long CA IX isoform only in hypoxic cells indicates that CA IX pre-mRNA splicing is strictly regulated by cellular oxygen tension.
Cancer is a multistep process that involves severe changes in gene expression. The more we learn about regulatory pathways that are disturbed during tumorigenesis, the more we realize about the complexity of tissue homeostasis. Thus, elucidation of the molecular mechanisms of regulation of RNA processing by oxygen tension is critical for our understanding of the biology of these important regulatory proteins, in particular our understanding of their role in angiogenetic and tumorigenic processes.
4.3. Transcription and regulators at moderate hypoxia
Activation of transcription in response to low oxygen tension is mediated by the hypoxia-inducible factors (HIFs) 1–3. These transcription factors are heterodimeric complexes of two proteins: Arnt, which is constitutively expressed, and HIFα proteins 1–3, the stability and transactivation function of which are regulated by oxygen levels. In the HIFα proteins 1–2 there are two functional domains mediating transcriptional activation by interaction with transcriptional co-regulatory proteins and the transcriptional machinery. One of these two domains, the N-terminal transactivation domain (N-TAD), is located within the oxygen-regulated degradation domain, whereas the other transactivation domain, termed C-terminal activation domain (C-TAD), is located in the very C-terminus of the HIFα proteins 1–2.
CREB binding protein (CBP)/p300 is the major transcriptional co-regulatory factor participating in HIF-1α-dependent activation of transcription. The C-terminal activation domains (C-TADs) of both HIF-1α and HIF-2α have been shown to interact with the cysteine/histidine-rich region 1 (CH1) of CBP/p300 in a hypoxia-dependent manner. Within the EUROXY program we have demonstrated that the N-terminal activation domain (N-TAD) of either HIF-1α or HIF-2α is also able to interact with endogenous CBP, and that this interaction facilitates the transactivation function of the corresponding HIF complexes.
The HIFα protein 3 distinguishes itself from the HIFα proteins 1–2 by only having the N-TAD and not the C-TAD. Thus, HIF-3α also shows both oxygen-regulated protein stability and transactivation function but is a much weaker transactivator than HIFα proteins 1–2. This property of HIF-3α results in it being a negative regulator of the hypoxia-dependent gene regulatory response when HIF-3α is in excess over HIF-1α or HIF-2α.
We have observed in neuroblastoma tumor cells that HIF-1α stabilization under hypoxic (1% oxygen) conditions is primarily an acute response to hypoxia, and that HIF-1α protein levels become reduced or disappear at prolonged hypoxia. HIF-2α levels, on the other hand, continue to increase with time at hypoxia, and we have observed that hypoxia-driven genes, primarily VEGF, are a HIF-1α target during an acute phase, while HIF-2α becomes more important as an activator of VEGF gene expression during later phases of hypoxia. Based on our observations in neuroblastoma, HIF-1α and HIF-2α seem to have the capacity to transcribe most of the hypoxia-driven genes containing HRE sequences, but they do it in different contexts, for instance as a response to acute or prolonged hypoxia. Thus, the question of what genes are driven by either of the HIFs might simply be answered by a lack of target gene preferences and that the HIF dependence instead is context-dependent.
There is a clear link between hypoxia and regulation of the cell differentiation status. We have previously observed that hypoxia induces dedifferentiation of neuroblastoma tumor cells. In this process there is a clear integration between the HIF and Notch signaling pathways to mediate the hypoxia-dependent dedifferentiation process. In addition to this mode of regulation, hypoxia promotes the undifferentiated cell state in various stem and precursor cell populations. We have also shown that the latter process requires Notch signaling. Hypoxia blocks neuronal and myogenic differentiation in a Notch-dependent manner. Hypoxia activates Notch-responsive promoters and increases expression of direct Notch downstream genes. The Notch intracellular domain interacts with HIF-1α, and HIF-1α is recruited to Notch-responsive promoters upon Notch activation under hypoxic conditions. Taken together, these data provide molecular insights into how reduced oxygen levels control the cellular differentiation status and demonstrate a role for both HIF-1α and Notch in this process. We are now exploring whether regulation of the tumor cell differentiation status by the integrated HIF/Notch signaling pathways is a relevant new target for tumor therapy.
4.4. Targeting tumor hypoxia through interference with mTOR and the UPR signaling
4.4.1. Introduction
Mammalian cells utilize multiple mechanisms to adapt to changes in nutrient supply, energy, and oxygenCitation75. Often these mechanisms are exploited by tumor cells in order to survive the harsh conditions that exist within the microenvironment of solid human tumorsCitation76. The ability to adapt to hypoxia seems particularly important, as high levels of tumor hypoxia are associated with poor patient prognosisCitation77. This adaptation influences the behavior of tumor cells by activating a number of oxygen-sensitive pathways that have been reviewed recentlyCitation78. Of these, the best understood pathway is mediated by the HIF family of transcription factors. More recently, two other oxygen-sensitive pathways have been described that mediate changes in gene expression and important phenotypic tumor traits.
4.4.2. The role of mTOR signaling during hypoxia
The first of these newly identified pathways is regulated through the mammalian target of rapamycin (mTOR) kinase and its downstream effectors that coordinate processes such as the initiation of protein synthesis, autophagy, and apoptosis. Activity of mTORC1 is inhibited under moderate hypoxia (~1% O2), resulting in decreased protein synthesis and proliferation in cells of benign originCitation79–81. Severe hypoxia (≤ 0.1% O2) on the other hand inhibits mRNA translation in nearly all cell typesCitation82. Since protein synthesis is extremely adenosine triphosphate (ATP)-costly, this inhibition in mRNA translation may be essential in order to maintain the energy balance within the cell. Nevertheless, it is currently unclear to what extent mTORC1 inhibition is accountable for the translational repression that is observed under severe hypoxia.
Part of this translational repression is mediated through dephosphorylation of one of the mTORC1 downstream effectors, 4E binding protein 1 (4E-BP1). Initiation of cap-dependent mRNA translation requires binding of the eIF4F complex to the 59 cap structure of the mRNA. This complex consists of the cap-binding protein eIF4E, the scaffolding protein eIF4G, and the RNA helicase eIF4A. During hypoxia, 4E-BP1 becomes hypophosphorylated and scavenges eIF4E away from eIF4G, thereby preventing initiation of cap-dependent mRNA translation. We have shown that knockdown of 4E-BP1 by overexpression of a shRNA against 4E-BP1 does not noticeably alter the overall inhibition of translation in cancer cells during severe hypoxiaCitation83. Nevertheless, 4E-BP1 does play an important role by modulating mRNA translation of specific genesCitation83. Interestingly, we observed an increased sensitivity to hypoxia for the 4E-BP1 depleted cancer cells in clonogenic assays (Magagnin et al., unpublished results). Xenograft tumors established from 4E-BP1 deficient cells did not demonstrate any difference in overall tumor growth or in their hypoxic fraction (determined by pimonidazole) as compared with their wild-type controls. However the 4E-BP1 deficient tumors showed increased cell death in hypoxic regions and had decreased ATP levels compared with controls. This might explain the notable differences in radiosensitivity that we observed after single dose irradiation of 10 Gy. Xenograft tumors derived from 4E-BP1 knock-down cells required a significantly longer time to reach the initial tumor starting volume after irradiation (Magagnin et al., unpublished results). These data suggest that control of translation through 4E-BP1 acts as an important metabolic brake to preserve cell survival in tumors, resulting in an increased number of viable radioresistant hypoxic cells.
The fact that hypoxia can influence mTOR signaling and cap-dependent translation has potential implications for the use of a number of new agents that are directed against these pathways, including rapamycin. Our data suggest that these drugs may influence hypoxia tolerance, or at least be differentially effective against hypoxic cells. In support of this idea, we recently showed that although rapamycin causes a substantial tumor growth delay on its own, it does not improve the ability to cure tumors with fractionated radiotherapyCitation84.
4.4.3. Hypoxia and the unfolded protein response
The second oxygen-sensitive signaling pathway involves activation of the unfolded protein response (UPR). This is a program of transcriptional and translational changes that occur as a consequence of endoplasmic reticulum (ER) stress. Three distinct ER-stress sensors initiate the UPR: PERK, IRE1, and ATF6. PERK is activated upon hypoxia, resulting in phosphorylation of its main substrate eIF2α on serine residue 51. This is a rapid but reversible response that occurs within minutes when cells encounter severe hypoxic conditions and takes somewhat longer under more moderate oxygenation conditions (1%)Citation85. Phosphorylation of eIF2α upon hypoxia has been shown in a diverse panel of cell lines from either normal or neoplastic tissueCitation82,Citation85. PERK activation and subsequent downstream signaling is important for survival during hypoxic conditionsCitation82,Citation85. PERK knock-out mouse embryonic fibroblasts (MEFs) revealed increased hypoxia sensitivity and profoundly decreased tumor growth compared to their wild-type controls in xenograft studiesCitation86. Reduced hypoxia tolerance was also observed after disruption of PERK signaling by other means, such as expression of unphosphorylatable eIF2α (S51A), dominant negative PERK, or overexpression of eIF2α phosphatase GADD34Citation82,Citation85.
4.4.4. Changes in gene expression through regulation of translation
Hypoxic signaling through mTOR and the UPR both contribute to changes in gene-specific mRNA translation during hypoxiaCitation78,Citation87–89. Together with the transcriptional program mediated by HIF this regulation mediates important changes in protein expression that influence cell behavior in hypoxic tumors. Importantly, mTOR and UPR affect different subsets of genes compared to the well known transcriptional targets of the HIF pathway. This opens up new opportunities for the discovery of novel diagnostic and therapeutic targets in hypoxic cells.
To obtain better insight into the contribution of translational control on hypoxia-induced gene expression, we performed a microarray study to assess both transcriptional and translational changes when cells are exposed to hypoxia (van den Beucken et al., unpublished data). Both total RNA and efficiently translated mRNA were extracted from DU145 human prostate carcinoma cells after exposure to hypoxia for different lengths of time (0, 1, 2, 4, 8, 12, 16, and 24 hours). Expression profiles were determined for genes regulated either at the transcriptional (total RNA) or the translational (polysomal RNA > 5 ribosomes) level using Affymetrix technology. This study demonstrated that translational control significantly affects both hypoxia-induced and -repressed genes at all time points examined. The contribution is most pronounced during acute hypoxia (2–4 h), as the influence of transcription becomes more important after prolonged hypoxia. Our data indicate that translation influences gene expression during hypoxia on a scale comparable to that of transcription, and reveals many potentially interesting targets for diagnosis and/or therapy.
Translational regulation of one of the identified genes, Cited2, was characterized in more detailCitation89. Our data provide further evidence that Cited2 can antagonize the interaction between HIF-1 and its co-activator CBP/p300, and thus is able to prevent HIF-1 transcriptional activation. These data also suggest that substantial cross-talk exists between transcriptional and translational cellular response programsCitation89.
During our gene expression profiling studies we also discovered that induction of the HIF-regulated gene CA IX was severely compromised in cells derived from an eIF2α S51A knock-in mouseCitation89. This finding was reproduced in two isogenic human tumor cell lines that were defective for eIF2α activation. The underlying mechanism for this defect involved direct binding of the eIF2α-dependent translationally regulated gene ATF4 to the CA IX promoter. Furthermore, we were able to show that transcriptional activation of the CA IX promoter was associated with loss of the transcriptional repressive histone 3 lysine 27 (H3K27) tri-methylation mark. In xenograft studies, eIF2α activation impaired U373 cells and showed CA IX protein levels comparable to those in HIF knock-down cells. This study reveals that CA IX expression is mediated through independent activation of both the HIF and eIF2α phosphorylation. These data may have important implications for the use of CA IX as a diagnostic tumor marker or molecular drug target (unpublished results)Citation89.
Oxygen levels in human tumors are not static. Changes in blood flow result in periodic changes in oxygenation within a large fraction of the tumor. Exposure to cycles of hypoxia and reoxygenation causes different kinds of cellular stress, and therefore likely requires different stress response mechanisms. Using a proteomic approach we investigated changes in protein expression in tumor cells after reoxygenationCitation90. We successfully identified several proteins, including ribosomal protein P0, valosin-containing protein (VCP), and FUSE binding protein 2, which are involved in different cellular processes such as protein synthesis and degradation. Interestingly, VCP, which is involved in ER-associated degradation (ERAD), translocates from the nucleus to the cytoplasm upon reoxygenationCitation90. This contributes to the removal of potentially toxic misfolded proteins that accumulate during transient periods of hypoxia. Together, this study suggests that these newly identified proteins might contribute to the recovery of ER stress and protein synthesis during reoxygenationCitation90. Subsequently these proteins may thus be important determinants of survival after transient exposures to hypoxia.
4.4.5. Conclusions
In summary, the HIF, mTOR, and UPR pathways play important roles in determining the hypoxic cell phenotype, hypoxia tolerance, and tumor growth. Integral components or downstream targets of these oxygen-sensitive pathways have potential interest as either direct targets for therapy or as novel diagnostic markers for tumor hypoxia. In the future it will be important to further evaluate which of these pathways contributes most to the adverse phenotype and poor prognosis associated with hypoxia in human tumors.
4.5. Importance of ROS signaling in tumors
Oxygen free radicals such as superoxide anion and other reactive oxygen species (ROS) are derived from electron transfer on molecular oxygen. ROS have the potential, due to their oxidizing properties, to damage proteins, nucleic acids, and lipids, and thus have been considered for a long time as harmful agents. Indeed, ROS derived from exogenous sources such as toxins, radiation, UV light, and metals have been implicated in the development of cancer, in particular due to their damaging effects on DNACitation91.
However, ROS production is a genuine function of all aerobic cells. In fact, ROS are continuously produced in aerobic organisms as by-products of normal energy metabolism. During aerobic metabolism, electrons can escape from the mitochondrial electron transport chain, especially complexes I and III, and react with molecular oxygen to form the superoxide radical. This superoxide radical is then converted into hydrogen peroxide by superoxide dismutase 2 (SOD2). Whereas complete deficiency of SOD2 is embryonically lethal, the incidence of certain tumors has been shown to increase 100% in old heterozygous SOD2 mice compared with wild-type miceCitation92. In a liver-specific SOD2 knock-out model, early signs of liver cell transformation were observedCitation93.
A key feature of many tumor cells is their increased need for a high amount of energy to support their increased rate of cellular activity. Thus, the neoplastic phenotype of many tumors has been associated with an increased production of ROS. As superoxides are constantly produced during respiration, and are converted into other ROS, mitochondria are considered an important source of cellular ROS in cancerCitation94.
It has also been demonstrated in some cancers that the increased levels of ROS produced during the increased energy demands of a tumor can directly mutate the mitochondrial genome. Mitochondrial DNA appears to be highly susceptible to mutagenic ROS not only due to its close proximity to their source, but also due to the fact that the mitochondrial genome does not contain non-coding sequences. Thus, mitochondrial proteins may be synthesized that are not as efficient at containing the electrons during ATP production in the respiratory chain, contributing to increased ROS production in the mitochondrionCitation95.
In addition to mitochondria, a variety of enzymes are able to use molecular oxygen to generate ROS. In fact, low levels of ROS generated in response to distinct signals including growth factors, cytokines, hormones, coagulation factors, and other growth promoting agents have been implicated to act as cellular signals promoting proliferation of cellsCitation96–98. Thus, ROS signaling may be of major importance not only for tumor initiation and carcinogenesis, but also for tumor growth and progression.
Important targets of ROS are the iron protein tyrosine phosphatases, which can be oxidized by the iron ROS at specific cysteines, thus modulating their activityCitation99. In fact, ROS have been shown to increase the activity of a variety of kinases including mitogen-activated protein (MAP) kinases and Akt, which have been considered to stimulate tumor growth, prevent apoptosis, and enhance survival. In addition, important transcription factors including nuclear factor κB (NFκB), and activator protein 1 (AP1), which are known to be associated with cancer development, are regulated by ROSCitation100. Recently, HIFs, which promote tumor growth under hypoxic conditions, have been recognized to be induced and activated by ROS also under non-hypoxic conditionsCitation101,Citation102. Moreover, HIF-1α has been shown to be a transcriptional target of NFκBCitation103,Citation104. These findings open new avenues in our understanding of tumor cell growth and therapeutic sensitivity by linking hypoxia signaling with ROS signaling.
The notion that ROS can act as signaling molecules indicates that ROS should be generated in a controlled manner. Several enzymes have been associated with receptor-controlled ROS productions In this regard, NADPH oxidases have gained increasing attention. These enzymes catalyze the NADPH-dependent reduction of molecular oxygen to the superoxide radical. NADPH oxidases comprise a family of multiprotein enzymes consisting of transmembrane proteins called Nox proteins and the p22phox subunit and several regulatory cytoplasmic proteins including p47phox and p67phox and their homologs NoxO1 and NoxA1, as well as p40phox and the GTPase Rac-1. The Nox family of proteins consists of seven members (Nox1, Nox2, Nox3, Nox4, Nox5, DUOX1, and DUOX2). They all share a similar structure, with a six-transmembrane domain, a NADPH binding domain, and a cytoplasmic domain. Each member is differentially expressed in different tissues, and is regulated in different ways. As a consequence they have been suggested to serve different biological functions, although the exact role of each Nox is far from being elucidatedCitation105. Interestingly, Nox proteins have been found in several cancer forms and transformed cells, as f.e. in gastrointestinal tumors. However, this expression did not correlate strictly with the normal tissues, suggesting that Nox expression could be deregulated during carcinogenesisCitation106. NADPH oxidases have been shown to be important sources of ROS in various signaling cascades. They have been associated with the activation of different kinases in various cell types, among them the PI3 kinase/Akt pathway. Moreover, NADPH oxidases have been shown to regulate transcription factors important for tumor progression, including NFκB and AP1. Strikingly, NADPH oxidases can also regulate HIFα levels and HIF activity, thus linking them intimately to hypoxia signaling and sensitivity to therapies. Thus, increased Nox signaling may be of importance for permitting survival signals during tumorigenesis, and could be involved in signaling processes relevant to tumor formation and progressionCitation102.
In addition, evidence has been accumulated that ROS and NADPH oxidases are also involved in the formation of new vessels (angiogenesis), a process which is highly relevant for tumor growth and therapeutic sensitivityCitation97,Citation107.
However, tumor vessels are clearly distinct from functional normal vessels, and perfusion can be irregular or even absent. Thus, the tumor microenvironment is characterized by episodes of cycling hypoxia and fluctuating ischemia and reperfusion, events which have been intimately linked to enhanced ROS levels and NADPH oxidase activity in various ischemic disordersCitation108,Citation109. Although the exact importance of cyclic hypoxia and ischemia–reoxygenation for tumor progression and therapeutic sensitivity is not clear at the moment, initial studies suggest an important role of these events and thus ROS in tumor survival and therapeutic sensitivityCitation110.
Finally, the tumor microenvironment is characterized by the invasion of inflammatory cells, which are intrinsically able to generate ROS via the leukocyte NADPH oxidaseCitation111,Citation112. Thus, cells within a tumor may also be exposed to exogenous ROS. Several studies in vitro implicated that exogenous application of ROS such as H2O2 can activate kinase pathways, HIF and other transcription factors, proliferation, and angiogenesisCitation52,Citation113,Citation114. Thus, leukocyte-derived ROS may represent an additional way to activate cellular signaling cascades by ROS in tumor cells and to modify tumor growth.
Taken together, activated oxygen species generated in tumor cells and different tumor-associated cells under different microenvironmental conditions play an important role in tumor initiation, tumor progression, and therapeutic sensitivity. Thus, therapeutic strategies targeting tumor cells in their ever changing microenvironment will need to take ROS as drug targets into account.
4.6. pH Regulation at the cellular and tissue level
Inadequate oxygen delivery to hypoxic tumor regions restricts oxidative phosphorylation and limits energy production. Therefore, hypoxic tumor cells shift their metabolism toward glycolysis, which is less efficient in energy yield but does not depend on oxygen. However, glycolysis often sustains after reoxygenation, because its metabolic intermediates can be utilized for biosynthesis of certain amino acids, nucleotides, and lipids, providing selective advantage to proliferating tumor cells. This explains classical Warburg’s observation of high glucose consumption and high lactate production in tumor tissuesCitation115.
Lactate is the principal end product of glycolysis, but the oncogenic metabolism also generates an excess of protons and carbon dioxideCitation116,Citation117. Tumor cells eliminate these acidic catabolites in order to preserve neutral intracellular pH (pHi) that is optimal for cell proliferation and survivalCitation118. Lactate and protons are extruded by ion transporters and pumps including the H+/monocarboxylate transporter (MCT), the Na+/H+ exchanger (NHE), and the vacuolar H+/ATP pump. Acid export leads to a reduction of extracellular pH (pHe) that is typical for the tumor microenvironment. In addition, bicarbonate transporters, such as anion exchangers (AEs) and Na+/bicarbonate co-transporters (NBCs), import bicarbonate ions to cytoplasm where they react with protons and increase cellular production of CO2. This reaction consumes intracellular protons contributing to pHi neutralization, and CO2 diffuses to pericellular space, further reducing pHe, as illustrated in . The resulting microenvironmental acidosis has important biological consequences, including up-regulation of angiogenic factors and proteases, increased invasion, and impaired immune functions, and thereby contributes to tumor progression. Moreover, acidosis can modulate the uptake of anti-cancer drugs and modify the outcome of conventional therapyCitation119.
Figure 4. pH regulation in hypoxic tumor cells. Hypoxia triggers a metabolic shift to glycolysis via HIF-induced up-regulation of glucose transporters (mainly GLUT1) and glycolytic enzymes. Glycolysis produces an excess of lactate and protons that have to be exported out of the cell to prevent intracellular acidification that is incompatible with cell growth and survival. This extrusion is executed by the monocarboxylate transporter (MCT1 and 4) and the Na+/H+ exchanger (NHE1), both transcriptionally regulated by HIF. Acidic catabolites are accumulated in the extracellular microenvironment and cause extracellular acidosis that supports invasion. However, oncogenic metabolism also produces a high amount of CO2 that diffuses through the plasma membrane and contributes to extracellular acidosis. Hypoxia-induced transmembrane carbonic anhydrase CA IX (and CA XII) catalyze a CO2 conversion to bicarbonate ions and protons. Bicarbonate ions are taken by bicarbonate transporters (BTs) and imported to intracellular space where they contribute to neutralization of intracellular pH. Protons remain outside of the cell and further acidify the microenvironment.
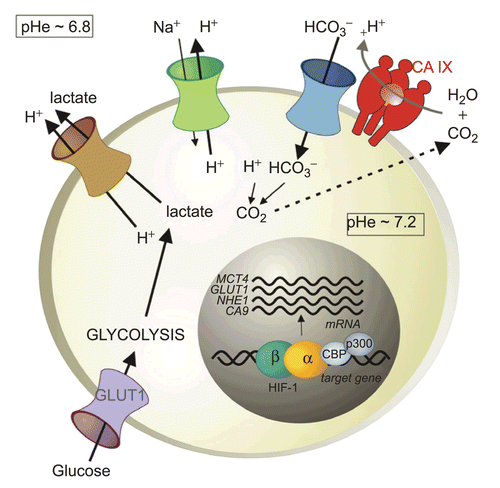
One of the mechanisms that enable hypoxic tumor cells to adapt to an acidic microenvironment involves a tumor-associated hypoxia-induced carbonic anhydrase IX, which is a direct transcriptional target of HIF and belongs to strongly hypoxia-dependent proteinsCitation120. CA IX is the member of a carbonic anhydrase (CA) family of enzymes that catalyze the reversible hydration of carbon dioxide to carbonic acid and participate in acid–base balanceCitation121. The 15 human CA isoforms are expressed at variable levels and differ by activity and kinetic properties, and subcellular and tissue distribution. They are mostly associated with differentiated cells of normal tissues, except CA XII that is elevated in some tumor types, and CA IX, which is predominantly expressed in diverse solid tumors including carcinomas of kidney, head and neck, lung, breast, uterine cervix, etc.Citation122.
CA IX is a transmembrane protein whose catalytic site faces the extracellular space and accelerates the pericellular metabolism of CO2 in a spatial and functional cooperation with bicarbonate transportersCitation123,Citation124. CA IX produces bicarbonate ions, which are actively transported to cytoplasm by bicarbonate transporters and contribute to neutralization of intracellular pH. In the same reaction, it also generates protons that remain in extracellular space and contribute to pericellular acidosis (). Functional involvement of CA IX in pH regulation in hypoxic cells has recently been demonstrated using cell models with ectopic expression of CA IXCitation125–127.
It has also been shown that hypoxia activates the catalytic performance of CA IX and that inhibitors of CA activity bind to hypoxic, but not to normoxic cellsCitation125,Citation128. On this basis it has been proposed that CA inhibitors can be potentially utilized for imaging of actually hypoxic tumors, in contrast to CA IX-specific monoclonal antibodies that visualize CA IX protein independently of hypoxia. Because CA IX is a stable protein (with a half-life of about 38 h), the antibodies detect both present and past hypoxiaCitation129. In that sense, inhibitors and antibodies might provide different prognostic information.
4.7. Cellular metabolism and respiration
A common feature of most malignant cells is their unique adaptation to the hypoxic tumor microenvironment, most obviously by angiogenesis and apoptotic resistanceCitation75. Another important strategy adopted from physiological hypoxia acclimatization is the alignment of oxygen availability with cellular metabolism, in particular by switching to anaerobic glycolysis, lowering energy-consuming processes such as protein synthesis, and adjusting mitochondrial respiration. In contrast to physiology, the glycolytic switch is often permanent, known as “aerobic glycolysis” or the Warburg effect, and kept up even when tumor cells are cultivated for a long time under oxygen excess conditions, suggesting (epi)genetic selection for hypoxia-tolerant cancer cells in a growing tumor.
Anaerobic glycolysis allows cells to maintain cellular ATP levels under oxygen-deprived conditions at the expense of acidosis and lactate accumulation. Virtually all steps of glycolysis, from glucose uptake (glucose transporter-1) to lactate production (lactate dehydrogenase), are controlled by HIFCitation130. Even the limitation of intracellular proton accumulation by CA IX-mediated acidification of the extracellular milieu is a HIF-dependent process that is especially pronounced in tumorsCitation131. As evident by the Warburg effect, these metabolic alterations are specific and stable features of tumor cells and thus represent prime targets for a general cancer therapy (see below).
Protein synthesis is a major energy-consuming process and hence a prime metabolic target to save energy under hypoxic conditions (see above). Indeed, general translation is down-regulated in hypoxic cells with the exception of the HIF signaling pathway, which remains operable even when translation of most other transcripts is down-regulatedCitation132.
Mitochondrial respiration is controlled in a complex manner. HIF-1 reciprocally controls the exchange of cytochrome c oxidase (COX) subunits by inducing the high oxygen-affinity subunit COX4-2 and simultaneously degrading the low oxygen-affinity COX4-1 subunit via induction of the mitochondrial LON proteaseCitation133. However, apart from this improved oxygen affinity, mitochondrial function can also be inhibited, probably when hypoxia becomes more severe, by limiting mitochondrial fueling or even by reducing mitochondrial mass. Impaired mitochondrial function is likely to re-direct non-consumed oxygen to cytosolic enzymes such as the PHD oxygen sensorsCitation134. Since the PHDs still operate under hypoxic conditionsCitation135, albeit at a lower rate, oxygen re-direction might limit the induction of HIF, establishing a negative feedback loop that limits respiratory and metabolic changes in a tumor cell.
Paradoxically, also mitochondrial inhibition is HIF-dependent. HIF induces the gene encoding pyruvate dehydrogenase kinase 1, thereby inhibiting pyruvate dehydrogenase and converting less pyruvate to acetyl-coenzyme A (acetyl-CoA), the fuel of the mitochondrial tricarboxylic acid (TCA) cycle. This TCA block was sufficient to actively suppress respiration, redirect both oxygen and glucose utilization toward cytosolic sinks, and rescue cells from hypoxia-induced apoptosisCitation136,Citation137. HIF was further reported to inhibit mitochondrial biogenesis and to eliminate mitochondria by autophagy, thereby definitively stopping oxygen-based production of ATPCitation138,Citation139. Interestingly, autophagy can also be induced by ATF-4, another hypoxia-inducible transcription factor up-regulated by preferential translation as well as by the PHD oxygen-sensing pathwayCitation140,Citation141. Future work will help to elucidate the role of this novel branch of the hypoxic signaling pathway in energy conservation under hypoxic conditions.
4.8. Proliferation and regulation of cell cycle transit
Proliferation of cancer cells under hypoxic conditions depends on the cell phenotype, i.e. which cell-cycle-regulatory genes are functional, as well as on the degree of hypoxia.
Generally, cells in S-phase immediately halt DNA synthesis when rendered extremely hypoxicCitation142,Citation143, while cells in G1 are either arrested at the retinoblastoma protein (pRb) checkpoint or proceed to late G1 before they become arrested at the so-called oxygen-dependent checkpoint in late G1Citation143,Citation144. Thus, there are, in reality, two different oxygen-dependent restriction points in G1, i.e. the pRB checkpoint in early or mid-G1, and another checkpoint in late G1. The molecular mechanisms regulating these two checkpoints are not clear. The pRb checkpoint can easily be stated to be a result of continued pRb activation under extremely hypoxic conditions, but it is not clear why low oxygen tensions have this effect on pRb. The molecular mechanism controlling the O2-dependent checkpoint in late G1 involves the cyclin-dependent kinase (cdk) inhibitor p27Kip1 Citation145–148. It has been shown that p27 regulates cell-cycle re-entry after hypoxiaCitation149. This checkpoint is particularly interesting from a cancer therapeutic viewpoint since it has been shown to be independent of both p53 and pRb and to operate in both normal and malignant cellsCitation143,Citation150. Thus, the oxygen-dependent checkpoint in late G1 is functional in all cell types so far studied. This is a finding which may have cancer-therapeutic relevance since mammalian cells seem to be particularly prone to become lethally damaged by hypoxia while in S-phaseCitation151,Citation152. In comparison, cells in G1 and G2 are far more resistantCitation148,Citation153. In fact, most cells rendered hypoxic while in G2 divide in spite of the lack of oxygen and become arrested in the subsequent G1.
Hypoxia-induced cell-cycle arrest in S phase has been shown to be a consequence of at least two levels of control. First, both DNA replication and DNA transcription are inhibited due to specific inhibition of oxygen-dependent enzymes and inhibition of replicon initiationCitation154–156. Second, the cell cycle checkpoint machinery is activated in all cell-cycle phases involving dephosphorylation of pRb and degradation of cyclinsCitation143,Citation144,Citation146,Citation157,Citation158. Exposure to moderate hypoxia (1300 ppm O2) resulted in arrest in S-phase for some cells containing active pRb, while cells not containing pRb were able to undergo some S-phase progressionCitation159.
A protein having a direct cell-cycle-regulatory effect which is found to be affected by low oxygen levels (i.e. below about 1000 ppm) is ribonucleotide reductase. Ribonucleotide reductase (RNR) is the enzyme responsible for conversion of the four standard ribonucleotides to their deoxyribonucleotide counterparts, and thereby provides the precursors needed for both synthesis and repair of DNACitation160,Citation161. The reduction of ribonucleotides is the rate-limiting step of DNA synthesis, which makes RNR an important target for cell growth control, and several RNR inhibitors are being used, or have been proposed, as drugs for chemotherapeutic treatment of cancer and a range of infections, such as malaria and acquired immunodeficiency syndrome (AIDS)Citation162–164.
4.9. Conserved energy saving responses in hypoxia tolerant animal and cancer cells
One major outcome of existing selection pressures under low oxygen that is common between lower species and human tumors is the gain of hypoxia tolerance. Normally, tumor angiogenesis, glycolysis, and apoptotic evasion are regarded as the main adaptations that aid the tumor in acquiring toleranceCitation75. Compared to these responses, metabolic depression as a physiological means to establish hypoxia tolerance, and HIF’s role in controlling it, is poorly understood with respect to neoplastic cells.
When focusing on invertebrates and some ectothermic vertebrates, the metabolic reprogramming that allows maintaining cellular energetics in homeostasis, albeit at a much depressed level, is well characterized as the ultimate hypoxia, ischemia, or hypothermia survival responseCitation165. However, a physiologically and metabolically based hypoxia tolerance also occurs in similar form in cells from the deeply hypoxic cores of solid human tumors. Regardless of cell transformation and phylogeny, this form of hypoxia tolerance always rests on energy conservation as a long-term survival strategy. Many lower animals, for example, acquire their remarkable stress resistance through the controlled, yet fully reversible, metabolic rate suppression down to a newly balanced ATP supply = ATP demand steady-state termed hypometabolismCitation166. Sustained throughout the entire stress, hypometabolism prevents lethal falls in cellular ATP levels and is the single most protective and unifying feature of hypoxia tolerant tissuesCitation167–169. During this physiological dormancy, O2 consumption rates equally drop to a small increment of normoxic uptake and become dependent on ambient pO2 (oxy-conformance). To match the synchronously declining ATP production in O2-depleted cells, hypometabolism requires the immediate and coordinated down-regulation of every major ATP-utilizing function in the cellCitation170, including: (1) macromolecular, notably protein and DNA synthesisCitation171; (2) protein degradationCitation172; (3) ion-motive ATPases, notably Na+/K+-ATPaseCitation173; and (4) gluconeogenesisCitation170. The further these activities can be suppressed, the better and longer the cells and organism can withstand deprivations of oxygenCitation165. In shifting remaining energy resources toward truly vital functions, a largely inhibited protein synthesis (translational arrest) and degradation allows, as tolerance hallmark, electrochemical gradients across membranes at reduced permeabilities to be maintained (channel arrest)Citation173,Citation174. As a further advantage of hypometabolic states, glycolytic fluxes only need to be elevated and provide for ATP as much as the residual energy expenditures require (i.e. weak to absent Pasteur effectCitation175,Citation176), explaining why this defense spares fermentable fuel, reduces metabolic waste, and extends survival time. Along with hypometabolism, cells also enter a non-replicative/non-proliferative “silent S-state” of quiescenceCitation177,Citation178. Together, hypometabolism, defined as pO2-conforming oxygen uptake rates, disengaged protein and DNA synthesis, and a non-cycling, quiescent phenotype, underlies the enormous resistance in lower organisms in response to a large variety of stresses, including hypoxia, ischemia, dehydration, and for mammalian hibernators, hypothermia. In all these cases and in hypoxic cancer cellsCitation75,Citation77, protective mechanisms act through critical checkpoints (e.g. translational arrest: via transcriptional or post-translational inhibition of initiation factors eIF2 and eIF4E, and elongation factor eEF2).
For the tumor, the degree of hypoxia tolerance may be an important determinant of the level of hypoxia that can be sustained within the mass. Thus, hypoxia tolerance can be viewed as a pro-tumor property that provides a molecular resistance toward effective therapy. Similar to animal models, hypometabolic defenses of severely hypoxic (0–0.5% O2) cancer cells include the (1) partly HIF-mediated suppression of respiration and switch into an oxy-conforming uptake modeCitation179,Citation180 that is known, in and of itself, to mediate enhanced hypoxia survival to multiple cancer cell lines; (2) general silencing of all ATP-costly macromolecular syntheses, including the rapid inhibition of replicon initiation in early S-phase of the cell cycle (replicon arrestCitation153,Citation181,Citation182) and the marked reduction in the overall rate of protein synthesis (translational arrestCitation183–185); and (3) the growth-stalling block of cell cycle proliferation, often occurring independent of p53 functionCitation150 at the G1/S phase transitionCitation77,Citation186. This ultimate defense, cellular quiescence, was first reported from the interior regions of multiple spheroid models, where “silent-S” cells with greatly depressed O2 consumption rates increasingly emerged as a function of spheroid growthCitation180,Citation187. It was later noted that the development of cellular quiescence, and beyond that, of cell death, is not simply a consequence of a lack of energy. In fact, many of the spheroid and tumor models employed saw the emergence of quiescent or necrotic cells despite maintaining a normal steady-state level of high energy phosphatesCitation188–190. This disconnect between energetics and activity or survival strongly suggests that hypoxic/ischemic cells in the peri-necrotic regions of a spheroid or tumor mimic lower organisms by coordinating declining ATP production with suppressed energy demands, which maintains elevated steady states of high energy phosphates. To date, various tumor xenografts have been reported to comprise up to 25% silent-S cellsCitation191. Since their suppressed metabolism is sustained on minimal O2 deliveries, quiescent cells in the tumor center are refractory to anti-angiogenic treatments. Their occurrence also correlates with extreme resistance to radiotherapyCitation192,Citation193.
We believe, therefore, that the players and pathways (including HIF) behind this physiological hypoxia tolerance carry a vast potential for improved therapeutic interventions. By targeting these hypoxia tolerance mechanisms, more selective diagnostic markers and anti-cancer therapies can be anticipated, since almost all healthy tissues in the human body are homeostatic in their oxygenation and, arguably, do not, or less so, recruit hypometabolic defenses when deoxygenated. Together with EUROXY partners Brad Wouters (Wp 10B) and Philippe Lambin (Wp 10A), we recently provided proof-of-principle for the validity of a fly-to-cancer translational approach to understand and interfere with hypometabolic functions. Our own genome-wide survey of the hypoxia transcriptome in hypoxia tolerant cells from Drosophila embryos, a state able to survive several days in a N2 atmosphere, recorded Thor as one of the most strongly hypoxia-induced fly genes (~15 times elevated mRNA levels at 0.2% O2). Thor is Drosophila’s single copy homolog of the mammalian translation repressors 4E-binding proteins 1–3 (4E-BP1–3). When active, 4E-BP’s act by sequestering the rate-limiting, cap-binding protein eIF4E from the key translation initiation complex, eIF4F. Thus, induction of Thor in hypoxic fly cells might contribute to inhibiting this branch of the ATP-costly protein synthesis. Indeed, fly genetics (collaboration: Prof. Pablo Wappner; Leloir Inst., Buenos Aires) was used to show: (1) the pronounced induction of Thor mRNA in hypoxic flies; (2) the strict requirement of HIF for in vivo Thor expression, and (3) the requirement of Thor function for Drosophila to adapt to, and recover from, hypoxia. Observation “2” implicates HIF for the first time in the control of a candidate hypoxia tolerance factor, and thus, in the down-regulation of ATP-costly activities in hypoxia. Next, we assessed the RNAi-based loss-of-function of 4E-BP1 for the hypoxic survival and treatment resistance of human cancer cells and tumor models (Wouters/Lambin/Gorr collaboration). Stable expression of a short hairpin interfering RNA (shRNA) specific for 4E-BP1 in two human cancer cells (HeLa, U87) significantly reduced the viability of both lines to prolonged (48 h) and severe (< 0.02% O2) hypoxia exposure. Moreover, a single dose irradiation (10 Gy) of U87 tumor xenografts, made from 4E-BP1 knock-down (kd) cells versus empty-vector (ev) transfected control cells, revealed a differential radiosensitivity. The post-radiation specific growth delay, a measure thought to closely reflect the relative number of cells killed by treatment, was always significantly higher in the kd tumors compared to ev control tumors, regardless of tumor end volumeCitation194. These data strongly suggested a reduction in the viable fraction of radioresistant hypoxic cells in the kd tumors. A reduced tolerance to a limiting supply of oxygen in the 4E-BP1 kd tumors was indeed evident by increased levels of cleaved Caspase-3 despite similar extent or severity of intratumoral hypoxia in both tumor types (pimonidazole IHC staining). Thus, 4E-BP1 loss-of-function increases radiosensitivity by decreasing the hypoxia tolerance of tumor xenografts, without affecting the hypoxic fractions of the xenografts. Diminished hypoxia viability in cells of 4E-BP1 kd tumors was, however, associated with a three-fold drop in steady-state ATP concentrationsCitation194. Thus, from fly to cancer cells, regulating the rates of protein synthesis via the control of cap-dependent, eIF4F-driven translation is required to facilitate energy conservation and to gain hypoxia tolerance. For tumors, this tolerance acquisition is associated with radioresistance. The results of this EUROXY effort show that targeting translational controls represents an effective new way to sensitize cells to hypoxia and solid cancers to radiotherapy.
5. Clinical consequences of tumor hypoxia
5.1. Introduction
Hypoxia is a reduction in the normal level of tissue oxygen tension and occurs in many disease processes including cancer. It results in the death of both cancer cells and normal cells if it is severe or prolonged, but cancer cells can adapt to this hostile environment by undergoing genetic changes that allow them to survive and even proliferate. It is in part this ability to adapt to a hostile environment that predicts the malignant potential and aggressive phenotype of a tumorCitation195.
Hypoxia can be broadly categorized into two types: acute and chronic. “Acute” or “transient” hypoxia occurs due to aberrant blood vessels shutting down and then reopening, thus reperfusing hypoxic tissue with oxygenated blood leading to an increase in free radical concentrations and tissue damage. “Chronic” or “diffusion-limited” hypoxia results from tumor angiogenesis lagging behind tumor growth. The perinecrotic regions of a tumor are located at a median distance of 130 μm from blood vessels, reflecting the limit in oxygen diffusionCitation196.
HIF signaling is a key mediator of response to hypoxia and regulates the transcription of several genes involved in biological processes such as angiogenesis, cell proliferation and survival, glucose metabolism, pH regulation, and apoptosisCitation195,Citation197. Erythropoietin was the first gene discovered to be under HIF regulationCitation198. Besides erythropoietin, another well-characterized HIF-regulated gene is VEGF, which is involved in regulating endothelial cell proliferation and blood vessel formation in both normal cells and cancer cells. Hypoxia also promotes the undifferentiated cell state in stem cells through interaction with the Notch signaling pathway. The Notch intracellular domain interacts with HIF-1α, leading to recruitment of HIF-1α to Notch-responsive promoters and activation of Notch downstream genesCitation199. HIF-1α and VEGF, which is a direct transcriptional target of HIF-1, have also been shown to up-regulate Delta 4, a ligand for the Notch receptor, important for physiological and pathological angiogenesisCitation200. Many of the known oncogenic signaling pathways overlap with hypoxia-induced pathways, and a recent review placed HIF at the center of the major oncogene and tumor-suppressor gene pathwaysCitation201.
5.2. Tumor progression
Genetic instability is induced in hypoxia by several mechanisms (reviewed in reference 202). Hypoxic cells defective in p53 and apoptosis have a greater propensity to acquire genomic instability during tumor progressionCitation203. Conversely, p53 mutated cells with diminished apoptotic potential are selected by tumor hypoxiaCitation204. Additionally, several genes involved in metastasis are induced such as lysyl oxidaseCitation205, metalloproteases, and urokinaseCitation206.
Many other pathways play a role, such as activation of mTOR, autophagy, and many different angiogenic factors (reviewed in reference 207).
These processes culminate in the observation that when genes regulated by hypoxia are up-regulated in cancers they are associated with a poor prognosis, independent of treatment modality, but also associated with resistance to each modality. Examples include urological cancers, head and neck cancer, lung cancer, colorectal cancerCitation208–212, and breast cancer, but all common cancers have been investigated and this found to be the case in most studies.
5.3. Tumor resistance to conventional drug therapies
The majority of anti-cancer chemotherapy drugs interact with DNA directly or indirectly, e.g. cross-linking with cisplatin and induction of double-strand breaks (DSBs) with topoisomerase 2 inhibitors such as epirubicin and etoposide. There is a very extensive literature on the ability of hypoxia to induce drug resistance, particularly to anthracyclines such as epirubicin, but also to other drugs including platinum. The mechanisms have not been well defined apart from anthracyclines where an acidic extracellular pH blocks uptake into the cells. This is certainly one of the major explanations, but Unruh et al.Citation213 showed in transformed mouse embryonic fibroblasts deficient in HIF-1α that there was a greater sensitivity to carboplatin, etoposide, and radiation. This was independent of p53, and in vivo experiments confirmed the in vitro results. Agents that did not cause DNA DSBs, such as DNA synthesis inhibitors, had equal effects on HIF-1α positive and HIF-1α negative cells. There was decreased repair of a fragmented reporter gene in normoxic HIF-1α deficient cells, suggesting that basal HIF-1α expression is required for DSB repair gene expression, although the detailed mechanism was not understood in these mouse embryonic fibroblasts.
More recently, silencing HIF-1α expression by RNAi in human non-small cell lung cancer cell lines was reported to decrease resistance to cisplatin and doxorubicin by modest degrees of hypoxia of 0.5% O2Citation214. This confirms the previous study, although, again, the mechanism was not described. It is important to also evaluate the P-glycoprotein in these studies of doxorubicin, because P-glycoprotein is known to be a HIF target and affects transport of drugs such as epirubicin and doxorubicin. Hypoxia also induced resistance to etoposide, which was multifactorialCitation215. This area of research needs more intensive investigation with detailed biochemical pathways linked to drug resistance and sensitivity studies.
5.4. Tumor resistance to irradiation
Hypoxia is one of the major mechanisms causing radioresistance, and it is mainly due to lack of O2 radicals generated by radiation to create DNA damage. However, there are many physiological processes regulated by HIF that could also contribute to radiation resistance. Moeller et al.Citation216,Citation217 have partly elucidated the complex role of radiation interaction with HIF. They showed that with conventional doses of radiation used therapeutically, HIF-1 was induced within 24 h in tumors of animal models, along with VEGF induction. One explanation is that oxygenated cells were killed by radiation therapy, and this allowed vessels to expand and reoxygenate the hypoxic areas. Although this would have been expected to switch off HIF, in fact free radicals produced by this pathway induced HIF. The source of the free radicals is not yet clear but may well be inflammatory cells or macrophages induced by the inflammatory response to radiation. The effects of HIF induction were complex in that HIF induction induced angiogenic factors that helped endothelial cells survive radiation damage, and therefore HIF appeared to cause radiation resistance from the point of view of allowing survival of the vasculature.
On the other hand, HIF-1α produces cell-cycle blockade by modifying p21 or p27 expression. In addition, it may interact with p53, so in the few tumor types that have wild-type p53, apoptosis may be enhanced, and this was shown also in appropriate p53 plus or minus control cell lines. Thus, from the tumor cell point of view, HIF-1α expression may enhance cell death through p53-dependent pathways and cell cycle regulation but enhance survival of vasculature, and the final effect will be complex depending on individual genetic factors in the tumor and in the interaction with the endothelial cells. Those studies also showed that HIF-1α on balance radiosensitizes tumors by keeping them proliferating when glucose levels are low because of the HIF effect on promoting glycolysis and maintaining ATP levels. Harada et al. also found HIF-mediated radioresistance in vivoCitation218.
However, using another model cell line of the mouse hepatoma HEPA1 with a HIF-deficient mutation (in ARNT), it was found that in vivo HIF-1 defective tumors were more readily responsive than the parental ones. The mutant cells induced less angiogenesis, but they did not have any difference in oxygenation status, and in this case radiosensitizers did not further sensitize these radioresponsive xenograftsCitation219. In contrast, HIF-1α proficient xenografts were sensitized by misonidazole. In this model, the lack of HIF was associated with radiosensitivity, but this did not appear to be due to O2 or vascular effects.
Despite being compatible with the previous reports in terms of the in vivo effect, this was a cell autonomous effect as shown by mixing experiments whereby the overall sensitivity depends on the proportion of HIF-deficient cells, and this implies that the mechanism differs and is not due to a paracrine vascular effect. Further work needs to be done to characterize the vascular response and free radical response in this model for comparative purposes.
6. Taking advantage of the hypoxic tumor phenotype
6.1. Gene signatures for prognosis and stratification
6.1.1. How to select patients for treatment
The evidence showing that hypoxia is important in tumor progression and prognosis has spurred research into developing therapies that target hypoxic cells. Therapeutic strategies include modification of the hypoxic environment or targeting components of the HIF-1 signaling pathwayCitation67,Citation76,Citation220. There is therefore a need for reliable methods to identify hypoxic tumors and those patients most likely to benefit from hypoxia-targeted therapy. Currently the level of tumor oxygenation is assessed by direct or indirect methods. The main direct approach is to measure intratumoral pO2 with needle electrodes. Indirect techniques include the immunohistochemical analysis of biopsies after injection of bioreductive drug markers of hypoxia, immunohistochemical analysis of expression of hypoxia-regulated proteins, imaging techniques, and more recently gene microarray analysis.
6.1.2. Oxygen needle electrode
The Eppendorf polarographic needle electrode is the main technique of direct pO2 measurement. Tissues are defined as hypoxic or normoxic based on the median pO2 level or the “hypoxic fraction” (percentage of pO2 values below a set level, typically, 2.5, 5, or 10 mmHg, designated HP2.5, HP5, and HP10 respectively)Citation221. The “hypoxic subvolume” (HSV) is another parameter of tumor oxygenation, defined as the hypoxic fraction multiplied by the total tumor volume, designed to more closely correlate with the absolute number of hypoxic cells in the tumorCitation222. However, use of the technique is limited by the tumor sites that are accessible to the probe (e.g. head and neck squamous cell carcinoma (HNSCC) and cervical cancer), dependency on a technically skilled user, and failure to differentiate between acute and chronic hypoxia.
6.1.3. 2-Nitroimidazole binding agents
Pimonidazole hydrochloride is a 2-nitroimidazole that has been used in human clinical trialsCitation223,Citation224. When injected intravenously, it undergoes enzymatic reduction and binding to macromolecules at low cellular oxygen tensions. Antibodies raised against the bound products are used to visualize hypoxia. Cells binding to pimonidazole are interpreted as existing at pO2 values <10 mmHg225. Pimonidazole binding assays provide hypoxia measurements with a high degree of spatial resolution. However, in a study of cervical cancer, there was no correlation between pimonidazole binding and oxygen electrode measurements of hypoxiaCitation226. Pimonidazole binding has been shown to relate to increased rates of early locoregional recurrence after radiation treatmentCitation224.
6.1.4. Tumor immunohistochemistry
Immunohistochemical detection of HIF-1α demonstrates patterns of expression similar to that of CA IX, with typical peri-necrotic staining, consistent with the distribution of diffusion-limited hypoxiaCitation227–229. The relationship between tumor HIF-1α expression and patient outcome has been discussed earlier. The difficulty in using tumor HIF-1α expression as an intrinsic marker of hypoxia is that its activation can occur in the presence of oxygen, for example in renal cell carcinoma (RCC) due to the loss of von Hippel–Lindau protein (pVHL)Citation230. Since other proteins, such as CA IX, can also be regulated by oxygen-independent pathways, it is imperative that these markers are assessed in the light of comparative studies with specific hypoxia markers.
CA IX is a zinc metalloenzyme that catalyzes the reversible hydration of carbon dioxide to bicarbonate and hydrogen ions and is involved in acid–base balance. The intratumoral expression of CA IX has been characterized in a variety of tumors, and high CA IX expression was associated with decreased survival in HNSCC, lung, cervix, and breast cancerCitation231–235. Koukourakis et al. showed that HNSCCs with high CA IX expression had a poorer complete response rate to chemoradiotherapy, whilst Loncaster et al. demonstrated that CA IX expression was an independent prognostic factor for disease-specific and metastasis-free survival but not related to local control after radiotherapy in cervical cancerCitation231–234.
Several studies have examined the correlation between the different techniques. In cervical cancer, there was a significant positive correlation between the level of tumor hypoxia (HP5) and the extent of CA IX expressionCitation234. In another study of cervical cancer, there was a weak negative correlation between HIF-1α expression and median pO2 but no relationship with HP5Citation236. However, Mayer et al. showed no correlation between tumor oxygenation and CA IX or HIF-1α expression in tissue samples originating from oxygen electrode tracks of locally advanced cervical cancersCitation237,Citation238. In cervical cancer, CA IX showed a substantial but incomplete overlap with pimonidazole in two studiesCitation120,Citation239. There was weak but significant correlation between pimonidazole and CA IX in HNSCC, with CA IX expression observed at shorter distances from blood vessels, implying that CA IX up-regulation occurs at pO2 levels higher than those required for pimonidazole bindingCitation224. In a study of HNSCC, no correlation was shown between HIF-1α and pimonidazoleCitation240, whilst in cervical cancer, there was a weak correlation between HIF-1α and pimonidazoleCitation236.
It is also possible that secreted markers of hypoxia could be monitored by a blood sample. Osteopontin (SPP1) is a marker that is associated with tumor hypoxia, and its plasma concentration is easily obtainableCitation241. A recent study showed that high plasma concentrations of osteopontin in patients with HNSCC are associated with a poor prognosis after radiotherapy, and that the prognosis in this group of patients can be improved by use of the hypoxia radiosensitizer, nimorazole, with radiotherapyCitation242.
6.1.5. Imaging
Positron emission tomography (PET) allows the in vivo measurement and quantification of physiological processes using short-lived positron emitting radiopharmaceuticals. Most radiopharmaceuticals under development for hypoxia detection use 2-nitroimidazole as the targeting moiety and a radioactive element, such as 18F, Citation67Cu, or Citation64Cu, amenable to nuclear medicine imaging. PET and PET/CT (computed tomography) offer the possibility of in vivo mapping of regional tumor hypoxia as well as monitoring of therapy through follow-up mapping of hypoxia. Additionally, PET/CT has the potential to be a single imaging modality for whole body staging. It also allows a more accurate estimation of the hypoxic tumor volume, which is important in planning radiation treatment. Numerous PET studies evaluating hypoxia in different tumor types have been conducted and were recently reviewed by Krause et al.Citation243.
6.1.6. Gene expression profiling
DNA microarrays are platforms on which several hundred or thousand oligonucleotides or cDNA of known genes are printed. Microarray technology allows simultaneous visualization of the expression of potentially all the genes within a cell population or tissue sample, revealing the “transcriptome.” The analysis of microarray data is commonly called “gene expression profiling” (GEP). “Prognostic signatures” can be obtained from GEP data, representing a relatively small number of genes that can be valuable in directing appropriate therapy and predicting outcomeCitation244. The ability to measure the expression of tens of thousands of genes in a single experiment is an enormous increase over the previously available techniques for measuring gene expression, which included northern blot and reverse transcription-polymerase chain reaction (PCR), where only a few genes could be studied at a time; however, these techniques are still required to confirm expression of genes after microarray analysis. The interpretation of microarray data is further complicated by the complexity of the biological systems, such as the relationship between mRNA levels and protein abundance.
The largest drawback of microarray analysis is the lack of well-defined standards for their use, interpretation, and validationCitation245. In an attempt to standardize the presentation and annotations associated with microarray experiments, the Minimum Information about Microarray Experiments (MIAME) criteria have been established in order that sufficient information is recorded about each experiment to interpret the results, enable comparisons, and permit replicationCitation246.
GEP can distinguish between normal, premalignant, and malignant epithelium and may be able to characterize more accurately the malignant or premalignant status of surgical resection marginsCitation247. Evidence for the extent of its use as a prognostic biomarker for response to therapy and survival is limited at present.
Hypoxia regulates a complex gene profile, and how the pattern of expression and extent of up-regulation of pathways relates to response to therapy and outcome is poorly understood. Defining gene profiles regulated by hypoxia and relating them to the above outcomes may provide a functional classification of tumors relevant to selection for specific therapies, e.g. bioreductive drugs, anti-angiogenic drugs, carbonic anhydrase and HIF inhibitors, and radiation. It is limited, however, by their variability of expression within a tumor, the lack of hypoxia specificity of individual proteins, and the complex inter-relationships between molecular pathways in different types of tumors. Establishing tumor type-specific or -independent hypoxia gene signatures would be a major advance in this area of research.
Therefore, we assessed the mRNA profile of HNSCC samples and defined an in vivo hypoxia metagene by clustering around the RNA expression of a set of known hypoxia-regulated genes. The validity of the method was confirmed by showing that the metagene contained many previously described in vitro derived hypoxia response genes and was prognostic for treatment outcome in independent datasets of many tumor types.
Affymetrix U133plus2 GeneChips were used to profile 59 head and neck squamous cell cancers. A hypoxia metagene was obtained by analysis of genes whose in vivo expression clustered with the expression of 10 well-known hypoxia-regulated genes (e.g. CA IX, GLUT1, VEGF). To minimize random aggregation, strongly correlated up-regulated genes appearing in more than 50% of clusters defined a signature comprising 99 genes, of which 27% were previously known to be hypoxia associated (). The median RNA expression of the 99 genes in the signature was an independent prognostic factor for recurrence-free survival in a publicly available head and neck cancer dataset, outperforming the original intrinsic classifier. In a published breast cancer series, the hypoxia signature was a significant prognostic factor for overall survival independent of clinicopathologic risk factors and a trained profile.
Figure 5. Gene array heatmap for hypoxia-regulated genes in head and neck ranked by degree of CA IX expression from left to right. The further to the right a tumor is, the greater is the degree of coordinated up-regulation of the profile. Note that every patient is different and no one marker is expressed in all hypoxic cases.

The up-regulated gene list HS-up () contained a number of genes previously described as induced by hypoxia, including those involved in glucose metabolism (e.g. aldolase A fructose-bisphosphonate, glyceraldehyde-3-phosphate dehydrogenase), angiogenesis (ANGPTL4, placental growth factor), cell migration (trophoblast glycoprotein), and the regulation of apoptosis (BNIP3). The known genes represent a broad spectrum of molecular pathways involved in cellular response to hypoxia. Some of the genes have also been described in RNA profiles of HNSCC, e.g. interleukin-8Citation245 and plasminogen activator urokinase. Genes not previously reported to be up-regulated under hypoxia include MTX1, BCAR1, HES2, PSMA7, and SLCO1B3.
Table 1. Gene list for hypoxia-clustered genes.
Hypoxia metagene as a prognostic factor in an independent HNSCC dataset As no treatment outcome data were available for our HNSCC dataset due to a short follow-up, it was not possible to train our hypoxia metagene on outcome or test its significance on the present dataset. Therefore, to explore whether our signature could provide prognostic information, it was applied to a publicly available datasetCitation248. The characteristics of our 59 metagene-generating and the 60 literature HNSCCs were compared using χCitation2. Patients in the Chung dataset were stratified according to HS-up quartiles or two groups (upper quartile vs. rest). High HS-up expression was an independent adverse prognostic factor for recurrence-free survival (); the hazard ratio (HR) for the upper quartile vs. the rest was 3.64 (95% confidence interval (CI) 1.43–9.31).
Figure 6. Cox multivariate analysis-derived hazard ratios (HRs) with 95% confidence intervals (CIs). Variables used in the original publication of an independent HNSCC patient dataset were entered in the model along with HS-up. HS-up was entered as a continuous variable (fractional rank varying from 0 to 1), and the HR reported represents the risk for an increasing HS-up quartile (the HR for the two ends of the spectrum was 14.83; 95% CI 1.80−122.35).
Hypoxia metagene as a prognostic factor in another cancer type The ability of HS-up to predict outcome was investigated in a breast cancer series. Increasing HS-up was a significant adverse prognostic factor for metastasis-free ( and ) and overall survival when patients were stratified by either quartiles or two groups. The HRs for metastasis-free survival were 1.62 (95% CI 1.33−1.98) for quartiles and 2.83 (95% CI 0.78−3.38) for two groups. In a multivariate analysis including HS-up with the clinical variables included in the original study, HS-up retained significance for metastasis-free (p = 0.003; ) and overall (p = 0.002) survival. In a multivariate analysis including the 70-gene trained intrinsic signature, HS-up retained significance for overall survival (p = 0.003). When a cell line-derived wound-response signature was included, HS-up retained independent prognostic significance for metastasis-free (HR = 2.80; 95% CI 0.09−7.22; p = 0.033) and overall (HR = 3.01; 95% CI 1.06−8.58; p = 0.038) survival.
Figure 7. Kaplan–Meier plots of metastasis-free survival in 295 patients with breast cancer. The data were taken from reference 249. (A) Stratified according to HS-up quartiles. The numbers of events and patients for increasing quartiles were 12/73, 18/73, 20/74, and 38/73. (B) Stratified by highest HS-up quartile (0.75−1) vs. the remaining three quartiles (0−0.75). The numbers of events and patients for the two arms were 50/220 and 38/73, respectively.
The work highlights the validity and potential of using data from analysis of in vitro stress pathways for deriving a biological metagene/gene signature in vivo. This also delivers a hypoxia gene profile for further clinical use and development.
Future plans Several key issues are as follows:
development of a reduced signature
comparison of metagenes between different cancer types
development of a common hypoxia metagene for all cancer types
These are currently being performed, and early results indicate that a common metagene of 34 genes can be derived. We have compared our results with our published metagenes for hypoxiaCitation250–252 and found superior performance in classifying outcome.
integration of other hypoxia markers, hypoxia-induced miRNA.
We recently showed that a hypoxia-induced miRNA, miR-210Citation253, provided strong independent prediction of poor outcome in breast cancer, and was correlated with the hypoxia metagene. However, since its targets are not regulated at the transcriptional level, this is likely to provide additional information to the metagene. We are currently analyzing this and also assessing other hypoxia-inducible miRNAs.
Validation of a reduced signature in randomized trials In a collaboration with Prof. Catharine West of Manchester University, a reduced common metagene plus miR-210 will be analyzed in RNA extracted from paraffin blocks from the following randomized trials:
Nimorazole trial in head and neck (H&N) cancer (n = 200). The Danish DAHANCA 5 trial was a randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma.
BCON trial in bladder cancer (n = 150). The BCON trial was designed to test whether hypoxia-modifying carbogen and nicotinamide with radiotherapy could improve local control compared with radiotherapy alone—based on results from a successful phase II trial. The phase III trial completed accrual in April 2006 with a final accrual of 333 patients.
ARCON trial in H&N cancer (n = 100). The ARCON phase II trial enrolled 215 patients with H&N cancer. Patients received accelerated radiotherapy with carbogen breathing and nicotinamide. The trial showed that ARCON yielded high locoregional control rates in advanced H&N cancer.
This will allow us to show whether the hypoxia metagene defines a subgroup that derives the most benefit from hypoxic modulation, and will provide the basis to be able to use this in prospective randomized trials and patient management.
6.2. Renal carcionmas and HIF-1/HIF-2
Kidney cancer is relatively common, tends to present late, and remains an important cause of morbidity and mortality. Illustrating this, the American Cancer Society estimated that there were more than 35,700 new cases of kidney cancer and 12,480 deaths from kidney cancer in the United States in 2004. Kidney cancer is categorized on histological grounds, and the commonest form is clear cell renal cell carcinoma (CCRCC). The majority of CCRCC (~70%) shows biallelic inactivation of the von Hippel–Lindau (VHL) tumor suppressor gene, resulting in constitutive activation of HIF. VHL defective CCRCC cell lines grown under standard conditions show high-level expression of the HIF-driven gene program, including GLUT1, VEGF, and CA IX. Complementation with VHL restores regulation of the HIF pathway by oxygen, and also suppresses tumor growth in xenograft assays. About 1 in 30,000 individuals carries a germline mutation in the VHL gene, resulting in the inherited tumor syndrome von Hippel–Lindau disease. This includes a ~70% lifetime risk of CCRCC, and tumors involve a somatic second hit leading to biallelic inactivation of VHL.
Understanding how VHL acts as a tumor suppressor gene in renal epithelium should provide an important insight both into kidney cancer, and more generally into the role of the HIF pathway in tumor biology. VHL has a number of other actions besides its role in the ubiquitin E3 ligase complex that captures HIF α-subunits. Nevertheless, several lines of evidence support the concept that suppression of HIF, and particularly HIF-2α, is central to VHL’s tumor suppressor action in the renal epithelium. A powerful system for examining the sequence of events leading from VHL inactivation to renal cancer is provided by the ability to detect microscopic foci of HIF activation in the kidneys of patients with a germline VHL defect using immunohistochemistry for CA IX. This shows that “second hits” are very common, and that they are not generally associated with increased proliferation, implying that further events are required for progression to malignancyCitation254. The earliest lesions detected by this method (seen as single cells in paraffin-embedded sections) express HIF-1α, with more complex lesions and tumors expressing both HIF-1α and HIF-2α. Very early lesions show loss of several epithelial characteristics (e.g. specialized junctions, see below), increased vascularization, clear cell morphological changes, and expression of the intermediate filament vimentin.
Experiments in rodents show that decreasing oxygen delivery to the kidney leads to tubular epithelial cells activating HIF-1α expression, but not HIF-2α expressionCitation255. In renal carcinoma cells, isoform-specific manipulation of HIF-1α and HIF-2α has shown that some changes in gene expression associated with CCRCC are driven by a specific HIF isoform: BNIP3 and CA IX were increased by HIF-1α and CCND1 by HIF-2αCitation256. Importantly, HIF-2α expression down-regulated HIF-1α, which may be important in effecting a switch between different types of HIF response. In xenograft experiments, expression of HIF-1α in 786-O cells, which express only HIF-2α, slowed tumor growth.
In an analysis of 42 clinical CCRCC specimens we found that 26 showed activation of HIF in all cells, consistent with loss of VHL function. In eight of these cases there were areas of tumor within which HIF-1α labeling was completely absent while expression of HIF-2α was preserved (Tran and Maxwell et al., unpublished results). Six of eight of these cases presented with metastatic disease or developed metastases within 6 months. In contrast, a single case of the 18 in which HIF-1α was present (6%) developed metastatic disease (χ2, p ≤ 0.001). With the caveat that this is a small series, it suggests that tumors with predominant expression of HIF-2α and reduced or absent HIF-1α are more likely to be associated with metastases.
In some cases a sharp boundary could be identified between a portion of the tumor expressing HIF-2α alone and adjacent tumor expressing both HIF α-subunits This allows direct comparison of a subpopulation in the tumor which has lost HIF-1α. Interestingly these areas had much lower CA IX expression, consistent with this being a HIF-1 target in CCRCC. Ki67 labeling showed a striking increase in proliferation index (p < 0.0001) in areas with predominant HIF-2α expression. Finally, we examined the key cell regulatory protein CCND1, which was expressed at a substantially higher level in the area that expressed only HIF-2α.
Taken together, these findings suggested that evolution to malignancy in VHL-defective cells involves evolution from an exclusive HIF-1α program in normal epithelium (which predisposes to apoptosis) to an exclusive HIF-2α program in advanced tumors, and that selective inhibition of HIF-2α might be an attractive treatment strategy.
Importantly, several findings show that HIF-2α activation in renal epithelium is not sufficient for malignant transformation. First, immunohistochemistry for the adherens junction protein E-cadherin reveals multiple foci of HIF-2α activation in VHL patients (Tran and Maxwell et al., unpublished results). These are distinct from those expressing CA IX; like the HIF-1α lesions identified on CA IX immunohistochemistry they show little evidence of increased proliferation. Second, analysis of mice with cell-specific biallelic inactivation of VHL in the renal epithelium shows that although the majority of cells activate HIF-1α, a subset activate HIF-2α. Third, expression of a transgene encoding constitutively active for HIF-2α does not result in striking proliferation (Wiesener et al., unpublished results).
The downstream consequences of VHL loss-of-function that contribute to the tumor phenotype are extensive and incompletely understood. Of particular interest, VHL loss-of-function results in a marked decrease in formation of adherens junctions and of tight junctions in premalignant foci in VHL patientsCitation257,Citation258. VHL re-expression rescues these characteristics in VHL-defective renal cancer cells. The effects on tight and adherens junctions are independent of each other (demonstrated by forced expression of E-cadherin, occludin, and claudin 1). HIF activation was shown to mediate the effects, at least in some cell backgrounds. In addition, VHL re-expression in renal carcinoma cells leads to formation of a primary cilium, which is a characteristic of normal renal epithelium, and this involves HIF suppressionCitation257. This aligns VHL disease with the “ciliary hypothesis” which suggests that defects in the primary cilium in a variety of non-malignant renal cystic conditions lead to cyst development. These findings link HIF activation to important aspects of cancer biology; decreased cell adhesion, and epithelial-to-mesenchymal transition. Interestingly, in the CCRCC cell lines studied, the effect on cilia and tight junctions appears to be more dependent on HIF-1α while adherens junctions are more influenced by HIF-2α.
Therefore, HIF activation plays a critical role in the majority of CCRCC and is responsible for many of the phenotypic characteristics of the tumor compared to normal epithelium. HIF activation has pleiotropic downstream consequences that are in part governed by which HIF α-isoform is expressed; which of these are most important in the malignant phenotype is not fully understood. The mechanisms governing the expression of HIF-1α vs. HIF-2α expression in renal epithelium remain to be determined; determining the factors that control this in normal cells and the genetic and epigenetic alterations that influence it in evolution of renal cancer will be important.
6.3. Imaging acute and chronic tumor hypoxia
The most important goal of cancer treatment is to offer patients an improved quality of life by achieving a cure for the disease, increasing their lifespan, or diminishing symptomatic side effects. Up to now the standard treatment options have included surgery, chemotherapy, and radiotherapy (RT). However, the effects of these treatments have been hampered by the presence of low oxygen regions heterogeneously spread within the tumor. These regions, also called hypoxia, are due to the unusual, chaotic, and insufficient organization of blood vessels within the tumors resulting in an inability to deliver oxygen and nutrientsCitation259,Citation260 and in a three-fold lower amount of lethal DNA lesions occurring upon irradiationCitation261. The importance of hypoxia has been demonstrated clinically, where it has been shown that patients with a hypoxic tumor have a worse outcomeCitation262–265. This has led to clinical efforts being made toward overcoming hypoxia, by focusing on eliminating hypoxia either by breathing high-oxygen gas mixtures or replacing oxygen with radiation-sensitizing drugsCitation220,Citation266.
This unique tumor microenvironment therefore makes hypoxia an attractive target for newly developed drugs to increase the radiation dose within these areasCitation267. To exploit this tumor characteristic, identification of hypoxia in tumors is essential. To date, various methods have been available to measure tumor oxygenation, including oxygen electrodesCitation268,Citation269, antibody-based detection of exogenousCitation270,Citation271 or endogenousCitation120,Citation272 hypoxia markers, and gene signaturesCitation273. However, these methods have been limited by practical challenges such as invasiveness and tumor accessibility. Furthermore, there is spatial as well as temporal heterogeneity, both in distribution of hypoxia and the hypoxia response, which results in a necessity for non-invasive three-dimensional imaging, which can be performed in a repeated fashion. The real power of molecular imaging goes beyond diagnosis by identifying different biologic processes within a tumor using tracers that characterize both genotypic and phenotypic signatures. PET with several hypoxia-specific tracers has the ability to quantify hypoxia, provides a basis for rational patient selection, and guides treatmentCitation274,Citation275.
[18F]Fluoromisonidazole ([18F]FMISO) was the first radiolabeled 2-nitroimidazole derivative proposed for hypoxia imaging with PETCitation276. This tracer has been evaluated extensively for the detection of tumor hypoxia both preclinically, using different animal modelsCitation277,Citation278, and clinically, for different cancer typesCitation279,Citation280. Our group was the first to validate the potential of this PET technique using a nitroimidazole related histopathology assayCitation281, which was confirmed by othersCitation282. In terms of impact of hypoxia on outcome, pre-therapy hypoxia values are important, because most biological changes occur early, persist throughout therapy, and are used to predict survivalCitation283. Several studies also investigated the post-therapy [18F]FMISO levels and found a decreased hypoxia distribution, suggesting that reoxygenation occurred as a consequence of radiationCitation284,Citation285. Tumor control probability models based on repeated [18F]FMISO-PET acquisitions during RT, combining the perfusion efficiency and the degree of hypoxia to estimate the reoxygenation time, are the key for future hypoxia image-guided dose escalation in RTCitation286 and in planning boost RT to persistent hypoxic subvolumesCitation267. Although [18F]FMISO provides a validated and reliable measurement of tissue hypoxia for individualized cancer treatment, it does not cope with different biological variables such as increased glycolysis. Therefore, it has been suggested to also include [18F]fluorodeoxyglucose (FDG)-PET in order to assess additional functional information during pre- and post-treatmentCitation287.
Although [18F]FMISO is extensively validated and used in clinical settings as a hypoxia marker, there are some concerns about the stability of the fluorin-18 linkage, the formation of metabolites in blood and urine, and the limited diffusion in tumor tissuesCitation288,Citation289. To overcome these problems, second generation hypoxia markers (e.g. FETNIM, FETA, EF3, EF5, FAZA, ATSM, etc.) have been developed that are more water soluble and have demonstrated a lower degradation, which results in higher tumor to background contrast, as reviewed in references 285, 287, and 290. These new markers have been extensively tested in order to replace [18F]FMISO, but results have not always been convincing. For example, our group has demonstrated that [18F]EF3 uptake in an experimental rat model was slightly faster cleared and showed a lower background compared with [18F]FMISO. However, the [18F]EF tumor uptake was significantly lower, resulting in not being superior over [18F]FMISOCitation288.
On the other hand, [18F]FMISO is probably not the most ideal tracer in imaging fluctuating tumor hypoxia, since it demonstrates slow clearance kinetics upon (radiation induced) reoxygenation once [18F]FMISO is trapped in the cellsCitation290. Therefore, instead of using bioreductive exogenous drugs, our laboratory is investigating the possibility of imaging the endogenous marker carbonic anhydrase (CA) IX. CA IX expression is dramatically increased in a variety of human tumors, whilst its expression in normal tissues is lowCitation291. This tumor-associated up-regulation of CA IX is the result of a strong transcriptional activation of the CA IX gene by HIF-1αCitation120. Expression of CA IX in tumors is associated with poor prognosis, tumor progression, and aggressiveness, suggesting that CA IX may be a good therapeutic targetCitation128. Furthermore, CA IX also seems to be implicated in tumorigenesis via its capacity to modulate cell adhesion and acidification of the tumor microenvironmentCitation125Citation.
Various sulfonamide inhibitors of CA IX (CAI) have been developed over recent yearsCitation293. Several of these inhibitors were found to be able to reduce the extracellular acidosis caused by hypoxia up-regulated CA IXCitation294 and could possibly be used as components of therapeutic strategies to increase extracellular pH, resulting in decreased tumor aggressiveness. Recently, a fluorescent derivative () with high affinity for CA IX has been developedCitation295, which has been shown to inhibit the hypoxia-mediated tumor acidificationCitation125,Citation128. The Pastoreková group demonstrated that this derivative was able to bind CA IX expressed under hypoxic conditions, but not under aerobic conditions, offering an attractive possibility of imaging CA IX non-invasively using sulfonamide-based compoundsCitation125.
Figure 8. Chemical structure of CA IX-selective sulfonamides: fluorescein-thioureido-homosulfanilamide. (Reprinted with permission of Elsevier.)
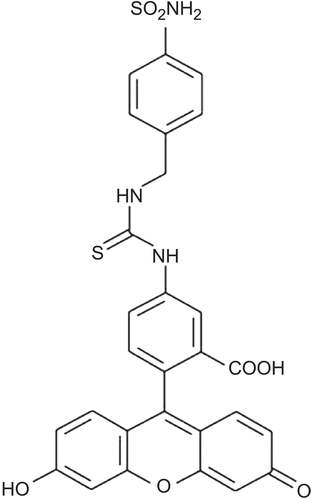
Based on these reports, our group further investigated the binding properties of those sulfonamides (CAI) in distinguishing cells that have been hypoxic and thus show an increase in hypoxia-regulated genes, but then reoxygenated. Immunofluorescence analysis demonstrated a significantly higher binding of CAI at cells exposed to hypoxia, compared with their normoxic counterparts, corresponding with an up-regulation of CA IX expression ( and ). Although CA IX expression levels remained high upon reoxygenation, CAI binding was dramatically reduced and no longer correlated with CA IX expressionCitation128. Furthermore, CAI that was bound under hypoxic conditions remained associated with cells during reoxygenation. This CAI bound fraction was gradually lost with time during reoxygenation, falling to near background levels after more than 1 hCitation128, which is much faster than the normal turnover of CA IXCitation129. Inhibitor binding is furthermore HIF-1α and cell-type independent, allowing for the distinction between actual and previously hypoxic cells. Recent studies indicate the possibility of using iodine-labeled CA IX antibody to monitor CA IX expression non-invasivelyCitation296,Citation297. Our data also demonstrate that antibodies against CA IX cannot discriminate between hypoxic and aerobic cells, since antibody binding is also observed upon reoxygenation, due to the normal turnover of CA IXCitation129. Co-localization between CA IX inhibitor and monoclonal antibody was only observed under hypoxic conditions, as seen by the yellowish color ()Citation128.
Figure 9. (Adapted from reference 125.) (A) Fluorescence analysis of HeLa cells treated with CAI during normoxia, hypoxia exposure, or upon reoxygenation. (B) Quantitative fluorescence activated cell sorting (FACS) analysis of CAI binding to HeLa cells treated with CAI under the respective conditions. (C) Immunofluorescence analysis of HeLa cells treated with CAI during hypoxia or upon reoxygenation (green). The presence of CA IX was assessed using the monoclonal CA IX (M75) antibody (red). Co-localization of CAI sulfonamide and CA IX monoclonal antibody was assessed by merging the images (yellow). Significant differences are indicated by asterisks (*p < 0.001; **p < 0.01). Scale bars are 25 μm. (Reprinted with permission of Elsevier.)
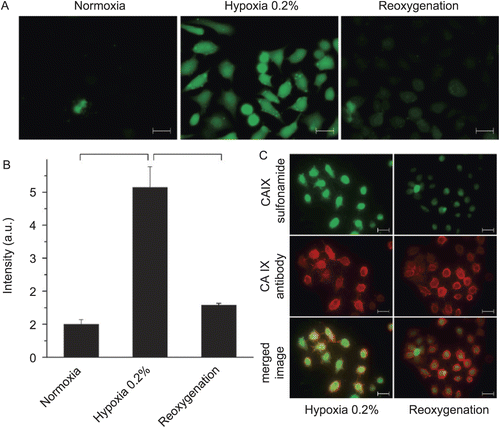
In summary, consequences of heterogeneously spread hypoxic areas within solid tumors are well known and specific treatments are under investigation. Therefore, adequate measurements of tumor oxygenation are the key for hypoxia image-guided dose escalation in radiotherapy. However, a lack of simple and efficient methods slows down the understanding of hypoxia. Recently, molecular markers of hypoxia such as CA IX have been under investigation, but no discrimination can be made between hypoxic and aerobic/reoxygenated cells based on antibody imaging. Therefore, CAI imaging probes are promising alternatives due to their ability to discriminate between such areas. Further in vivo investigations are ongoing within our institute. Our data demonstrate the possibility not only for imaging fluctuating hypoxia in vivo, but also for increasing radiotherapy efficiency, by specific targeting of CA IX, which will result in individualized patient treatment.
6.4. Bioreductive drugs
6.4.1. Introduction
The recognition of the importance of hypoxia in tumor response to therapy has precipitated a number of therapeutic approaches to overcome this resistant population. The potential use of bioreductive drugs was first proposed in the early 1970s. The concept was forwarded by Alan SartorelliCitation298 who suggested that hypoxia could be exploited in cancer therapy by using bioreductive prodrugs that would be activated specifically in hypoxic cells to yield cytotoxic product(s). As hypoxia is rare in normal tissues, the idea was that such agents would have limited normal tissue toxicity whilst killing the tumor cell population with a recognized refractory phenotype. The classical development pathway for bioreductive drugs was guided toward their use in combination with agents that show limited effectiveness against hypoxic cells. Radiation remains an obvious choice, where the need for oxygen to reveal cytotoxicity is well recognizedCitation4,Citation5. In addition, many other chemotherapy agents are preferentially more effective in the presence of oxygenCitation7. However, with our greater understanding of the cellular response to hypoxia, it is becoming increasingly apparent that targeting the hypoxic tumor population will not only overcome potential treatment resistance to conventional therapy, but will also selectively remove an aggressive subpopulation associated with the development of metastatic disease.
6.4.2. Prototype agents in the development of bioreductive drugs
Originally, two classes of bioreductive agents were developed simultaneously: the quinones and the nitroaromatics. The prototype quinone agent was mitomycin C (MMC). In clinical use from the 1950s, MMC was only recognized as having bioreductive properties three decades laterCitation299–301. The development of the nitroaromatic class began with the hypoxic cell radiosensitizer misonidazole, which was found to have equivalent or greater hypoxia-selectivity than MMC. For both agents (and all bioreductives developed subsequently), bio-activation depends not only on hypoxia but also on reductase enzymes present within the cell. Indeed the precise enzyme complement can markedly influence potency and toxicity. Both MMC and misonidazole can undergo one electron reduction to yield cytotoxic products. This reaction is readily reversed in the presence of oxygen. For MMC the one-electron product is the semi-quinone radical anion that causes DNA cross-links as the cytotoxic lesion. NADPH/cytochrome p450 reductase (P450R) has been implicated as a key bioactivating enzyme yielding this product under hypoxic conditions. However, MMC can also be reduced in an oxygen independent manner by the two-electron-reducing enzyme DT-diaphorase (DTD; NAD(P)H:quinone oxidoreductase, NQO1) that can compromise hypoxic selectivityCitation302–304. Nitroimidazole activation occurs via the step-wise addition of up to six electrons, with the reactions after the addition of the first electron being irreversible.
MMC and misonidazole showed early promise as bioreductive agents. However, the hypoxia-selectivity of both agents was deemed to be suboptimal (3–5-fold increased potency against hypoxic cells), with the clinical assessment of MMC in a “bioreductive context” being further hampered by its significant effects against well-oxygenated tumor cells. A methylated analog of MMC, porfiromycin, was developed that was a poorer substrate for DTD than the parental compound and showed greater hypoxic selectivityCitation305,Citation306. Although preclinical studies were encouragingCitation307, clinical trials showed little benefit over MMC when combined with radiotherapyCitation308. The indolequinone E09 was then developed. Like MMC, E09 is a substrate for both DTD and P450RCitation309–311. As for porfiromycin, preclinical data supported the clinical development of E09, but the clinical utility of the agent appeared compromised by very poor biodistribution and pharmacokineticsCitation312. However, E09 has had a recent clinical revival in superficial bladder cancer where intravesical administration circumvents the delivery and clearance issuesCitation313,Citation314.
Development of the nitroimidazoles progressed with the 2-nitroimidazole RSU1069: a modified misonidazole derivative that acts as a bi-functional alkylating agent by virtue of an aziridine ringCitation315. The hypoxia-selectivity of RSU1069 was markedly improved compared with misonidazole, and the agent showed excellent radioenhancing abilities in preclinical modelsCitation316,Citation317. As with many other bioreductive agents, one-electron reduction via P450R appears important in the bioactivation of RSU1069Citation318,Citation319. Despite the promising preclinical data, toxicity blocked the clinical development of this compoundCitation320. A prodrug variant of RSU1069 was produced (RB6145) to try and circumvent the toxicity issuesCitation321, but this gave rise to unexpected side effects in preclinical modelsCitation322, blocking further development.
6.4.3. Clinical lead compounds
Despite the initial interest in quinone and nitroimidazole compounds, the most clinically advanced bioreductive agents are not from these agent classes. Tirapazamine is a heteroaromatic N-oxide that progressed to phase III development. Tirapazamine shows marked hypoxia selectivity in vitro and significantly enhanced both radiation and chemotherapy response in animal models (reviewed by BrownCitation323). Further, tirapazamine in the neoadjuvant setting appears able to control metastatic dissemination from murine tumorsCitation324. Like MMC and the nitroimidazoles, one-electron reduction is important in the bioactivation of tirapazamine, with a role for P450R implied from preclinical studiesCitation318,Citation325 Clinically beneficial schedules were reported in combination with standard chemotherapy (particularly platinum-based) and both radio- and chemoradiotherapy in phase II trials (summarized in reference 326). However, not all of the results of phase III trials have concurred with the early phase successes, and the future development of tirapazamine is currently uncertain.
AQ4N (banoxantrone) belongs to the class of aliphatic N-oxides and is currently in phase I/II development. Upon reduction, AQ4N yields the cytotoxic product AQ4, via the AQ4M intermediate. Cytochrome p450s and inducible nitric oxide synthase reportedly catalyze this conversion, with little if any role implied for P450RCitation327–329. Unlike tirapazamine where the cytotoxic species is a short-lived free radical, AQ4 is a stable cytotoxin that can potentially diffuse from the cell in which it is bioactivated and elicit a bystander effect in adjacent cells. Preclinical studies demonstrated clear benefits of AQ4N in combination with chemo-, radio-, and chemoradiotherapyCitation330–334 (Williams et al., unpublished results), leading to clinical development. Importantly, significant investment has been made into the identification of hypoxic biomarkers that co-register with AQ4 accumulationCitation335. This may facilitate the identification of patients likely to show the greatest benefit from combined treatment with AQ4N. The ability to stratify treatment response based on the extent of tumor hypoxia is of great importance in the assessment of the clinical benefit of bioreductive drugs, and has been used successfully with tirapazamineCitation336 and trials using approaches to enhance oxygenationCitation224 (ARCON).
6.4.4. New kids on the block
NLCQ-1 (NSC 709257) is the lead agent from a rational approach to develop nitroaromatics with weak DNA-intercalating properties that may improve drug biodistribution (reviewed by Papadopoulou and BloomerCitation337). In vitro studies have established good hypoxia-selectivity ratios with NLCQ-1 and have highlighted the importance of one-electron reductases in its bioactivationCitation338 (Williams et al., unpublished observations). In vivo studies have shown that NLCQ-1 combines well with radiotherapy and a range of chemotherapy approaches and compares favorably with tirapazamineCitation339–343. Importantly, beneficial combinations with chemotherapy are not at the expense of any increase in normal tissue toxicityCitation337. As yet unpublished observations have also shown that NLCQ-1 is an excellent adjuvant to radiotherapy in the control of metastatic disease (Williams and Papadopoulou, manuscript in preparation).
PR-104 (Proacta) is the lead compound of a series of dinitrobenzamide (DNBM) mustards that are activated under severe hypoxiaCitation344. A bystander effect is elicited as the cytotoxic metabolites are relatively stable. DNBMs were initially developed as analogs of the weak monofunctional alkylating agent CB1954. PR-104 is effectively a “pre-prodrug” in that it has been developed as a water-soluble phosphate ester that, once administered, releases the corresponding alcohol via the action of systemic phosphatases. Preclinical evaluations described excellent hypoxic selectivity in vitro. Further, PR-104 exhibited single agent activity against a range of tumor xenografts, and enhanced both radio- and chemotherapeutic outcomeCitation345. PR-104 is currently in phase I trial in New Zealand and the USA in combination with docetaxel or gemcitabine in patients with solid tumors (NCT00459836, http://www.clinicaltrials.gov/).
RH1 is a novel aziridinylbenzoquinone alkylating agent that is also in phase I clinical trial in the USA (NCT00558727, http://www.clinicaltrials.gov/) and the UKCitation346. Like E09, RH1 is bioactivated by the oxygen-independent two-electron reductase DTD and the related NRH:quinone oxidoreductase 2 (NQO2)Citation347. Substantial activity against DTD-expressing tumors has been demonstrated in vivoCitation348. Importantly, side-by-side comparisons of E09 and RH1 suggest that the two drugs exhibit substantially different pharmacological properties that may favor a more general clinical utility of the latter compoundCitation348.
6.4.5. Conclusions
The potential of bioreductive agents to enhance the efficacy of standard therapies by targeting the most resistant tumor cells has long been recognized. Although numerous agents have proved excellent in preclinical models, translation to the clinic has been hampered by unforeseen side effects, pharmacological barriers, and/or apparently poor efficacy. One significant contributing factor in clinical analysis is patient stratification. Although most solid tumors exhibit regions of hypoxia, the extent clearly differs, and this impacts on response. There is a real need to ensure that clinical studies in some way analyze tumor hypoxia to enable responses to be correlated to the extent of the target population. Without this there is the possibility that extremely effective agents will fail, as their benefits in specific cohorts of patients will be masked by effectiveness against the population as a whole. There is certainly a shift toward biomarker analysis as a key input to early trial design, and this has particular relevance in the bioreductive arena for these agents to truly achieve their potential.
6.5. Synthesis of anti-carbonic anhydrase IX (CA IX) drugs
Unlike other CAs, which usually possess one polypeptide chain comprising just the catalytic domainCitation293,Citation349, CA IX is a multidomain, transmembrane protein possessing a more complex organization (), and consists of: (1) a small intracytosolic (IC) tail whose function is unknown; (2) a short transmembrane segment (TM); (3) the extracellular catalytic (CA) domain, which shows high sequence homolgy with that of other α-CAsCitation350; (4) a proteoglycan-like (PG) domain unique to this CA isozyme, which is critical to cell adhesion processes in which this protein is involvedCitation293,Citation350; and (5) a short signal peptide (SP). Many experiments have been performed with the PG-deleted (ΔPG) or catalytic domain-deleted (ΔCA) constructs of this protein in order to understand the role(s) of the various domains in its function and role in tumorigenesisCitation125,Citation350. The X-ray crystal structure of CA IX is unknown, but recent experiments led to the conclusion that it forms dimers linked by disulfide bondsCitation351.
Figure 10. Domain organization of the CA IX proteinCitation350. SP, signal peptide; PG, proteoglycan-like domain (residues 42–130); CA, catalytic, carbonic anhydrase domain (residues 167–405); TM, transmembrane segment; IC, intracellular tail.
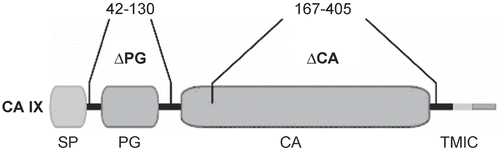
Many CAs present in the human body possess very high catalytic activity for the physiological reaction, i.e. hydration of carbon dioxide to bicarbonate and a proton, and CA IX is among such high-activity isoformsCitation293,Citation350, as seen from the data of . As with all other α-CAs investigated up to nowCitation293, the catalytic domain of CA IX contains a critically important Zn(II) ion coordinated by three histidine residues (i.e. His94, His96, and His119; CA I numberingCitation293) and a water molecule, which by deprotonation assisted by the active site residue His64 (CA I numbering for historical reasonsCitation293) leads to the zinc-hydroxide form that acts as the nucleophilic species in the catalytic cycleCitation293. CA IX together with CA II are highly active catalysts for the CO2 hydration reaction, being among the most effective enzymes known in nature, as shown in Citation351. CA IX is also susceptible to inhibition by anions and sulfonamides/sulfamates/sulfamides, as with all the other α-CAsCitation293, the inhibitors coordinating directly with the metal ion within the active site cavity and participating in various other favorable interactions with amino acid residues situated in both the hydrophobic and hydrophilic halves of the active siteCitation293. Many low-nanomolar CA IX inhibitors have been detected in the last several years ()Citation131,Citation293,Citation352,Citation353.
Figure 11. Structures of anti-carbonic anhydrase IX (CA IX) derivatives.
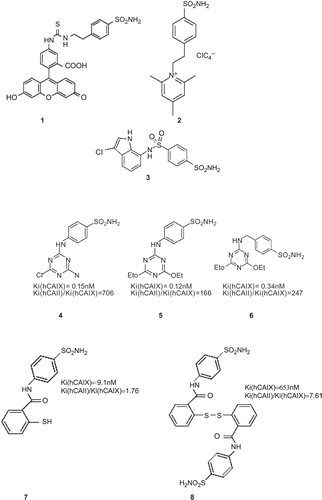
Table 2. Kinetic parameters for CO2 hydration reaction catalyzed by the 13 vertebrate catalytically active α-CA isozymes, at 20°C and pH 7.5, and their subcellular localizationCitation293.
The involvement of CAs and of their sulfonamide inhibitors in cancer has been investigated recently, as mentioned aboveCitation125,Citation293,Citation295. Many potent CA inhibitors derived from acetazolamide, ethoxzolamide, and other aromatic/heterocyclic sulfonamide scaffolds were shown to inhibit the growth of several tumor cell lines in vitro and in vivoCitation293.
Some of the most interesting CA IX inhibitors available at this time among the many such derivatives reportedCitation293 are the compounds investigated by Svastova et al.Citation125 (possessing structures 1 and 2) for their in vivo role in tumor acidification. These compounds present a special interest, because derivative 1 is a fluorescent sulfonamide with high affinity for CA IX (inhibition constant (Ki) of 24 nM)Citation293, which was shown to be useful as a fluorescent probe for hypoxic tumorsCitation293,Citation354. This inhibitor binds only to CA IX under hypoxia in vivoCitation125,Citation295. Although the biochemical rationale for this phenomenon is not understood at this point, these properties may be exploited for designing diagnostic tools for the imaging of hypoxic tumorsCitation125,Citation295. Compound 2 on the other hand, which is also a very strong CA IX inhibitor (Ki of 14 nM)Citation125,Citation295, belongs to a class of positively charged, membrane-impermeant compounds previously reported by one of our groupsCitation355, which are highly attractive for targeting CA IX with its extracellular active site, since such compounds do not inhibit intracellular CAs, and may thus lead to drugs with fewer side effects as compared to the currently available compounds (acetazolamide is the prototypical oneCitation293), which indiscriminately inhibit all CAsCitation293. The X-ray crystal structure of compound 2 in adduct with CA II (whose active site is very similar to that of CA IX, as shown recently by homology modeling) has been reported recentlyCitation356. It has been observed that the positively charged pyridinium derivative 2 favorably binds within the enzyme active site, coordinating with the deprotonated sulfonamide moiety to the catalytically critical Zn(II) ion. It also participates in many other favorable interactions with amino acid residues present in the active site cavity, among which is a stacking between the trimethylpyridinium ring of inhibitor 2 and the phenyl ring of Phe131, an amino acid important for the binding of inhibitors to CAsCitation356. A similar binding was subsequently reported for the fluorescein derivative 1Citation357. Thus, such structures can be used for the rational drug design of more isozyme IX-selective and potent CA inhibitorsCitation293. It should be mentioned that since the X-ray structure of CA IX is not yet available, most studies used the CA II structure for modeling and designing CA IX inhibitors. We stress again that positively charged compounds, of which 2 is a representative, may have the advantage of selectively inhibiting only CA IX in vivo, due to their membrane impermeabilityCitation356.
Indisulam 3 is a sulfonamide derivative (originally called E7070) discovered through extensive screening/synthetic studies, which showed powerful anticancer activity in vitro and in vivoCitation349. Indisulam has been also shown to act as a CA IX nanomolar inhibitorCitation358. However, its mechanism of action is not clearly elucidated, as the drug was shown to be involved in the perturbation of the cell cycle in the G1 and/or G2 phases, the down-regulation of cyclins, the reduction of CDK2 activity, the inhibition of pRb phosphorylation, and differential expression of molecules known to participate in cell adhesion, signaling, and immune response, in addition to its CA IX inhibitory properties. Indisulam showed in vivo efficacy against human tumor xenografts in nude mice and exhibited significant anti-tumor effect, and has progressed to phase I and phase II clinical trials for the treatment of solid tumorsCitation293.
Among the many compounds developed ultimately for targeting CA IX, one must also mention the 1,3,5-trazine derivatives incorporating benzenesulfonamide tails, of types 4–6, which showed excellent selectivities for inhibition of the tumor-associated isoform IX over the cytosolic, ubiquitous form CA II, with selectivity ratios in the range of 166–706Citation359.
The bioreductive, disulfide-type derivative 8, which is converted to the thiol 7 by reduction under hypoxic conditions, is also of great interest due to its excellent selectivity ratio for inhibition of the tumor-associated enzyme (CA IX) over the cytosolic ones, and for its properties of prodrugCitation360.
In conclusion, with its overexpression in many cancer tissues and not in their normal counterparts, CA IX constitutes an interesting target for novel approaches in the design of anticancer therapies. CA IX is crucial for tumor pH regulation, contributing both to the acquisition of metastasic phenotypes and to chemoresistance with some anticancer drugs. Consequently, further research needs to be done in the field of the tumor-associated CA IX in order to better understand its exact role in cancer. CA IX-selective inhibitors are now available, and they constitute interesting tools for studying the physiological and/or pathological effects of this enzyme. Indisulam, a sulfonamide anti-cancer drug, is actually in clinical trials, phase II. In addition to its hypothetical action in perturbing the cell cycle, it provides a potent inhibition of CA. However, given that multiple pathways contribute to tumor growth, anti-tumor activity may be increased by agents targeting multiple pathways, including CA IX, or by the combination of several agents to allow inhibition of multiple pathways. Recently, the design of CA IX-selective inhibitors containing a variety of scaffolds and with interesting physiocochemical properties has also been achieved. New sulfonamides, sulfamates, and sulfamides have been synthesized, with some of these derivatives strongly inhibiting CA IX, with inhibition constants in the low-nanomolar range, and also with excellent selectivity ratios for inhibition of the tumor-associated over the cytosolic CAs. Thus, many biochemical, physiological, and pharmacological novel data point to the possible use of inhibition of the tumor-associated isozyme CA IX in the management of hypoxic tumors which do not respond to the classical chemo- and radiotherapy. There are possibilities of developing both diagnostic tools for the non-invasive imaging of these tumors, as well as therapeutic agents that probably perturb the extratumoral acidification in which CA IX is involved. Much pharmacologic work is however warranted in order to understand whether a successful new class of anti-tumor drugs may be developed, starting from these preliminary but encouraging observations.
6.6. Targeting CA IX in tumors
CA IX possesses several properties determined by its distribution, localization, and function, which make it a suitable molecular target for cancer therapyCitation291. First, it is almost exclusively associated with solid tumors, predominantly those resistant to conventional chemotherapy and radiotherapy, as demonstrated by numerous studies of different tumor types. Second, it is exposed on the surface of cancer cells and is thus accessible to antibodies or inhibitors. Third, CA IX is a highly active enzyme that plays a role in hypoxic tumor phenotype and can therefore serve as a functional therapy target. This has been experimentally demonstrated using RNA interference or deletion of the catalytic domain, respectively, which resulted in decreased survival of CA IX-expressing tumor cells and reduced growth of tumor xenograftsCitation361 (Barathova et al., manuscript in preparation).
Tight association of CA IX with tumor phenotype represents a considerable advantage for immunotherapeutic targeting of CA IX-positive tumor cells. This approach has been extensively and thoroughly studied in preclinical renal cell carcinoma (RCC) models, as well as in RCC patients, using the CA IX-specific monoclonal antibody G250 and its chimeric and bispecific variantsCitation362,Citation363. Chimeric G250 (containing variable regions of the mouse G250 and constant regions of the human immunoglobulin G (IgG)), known under the commercial name Rencarex®, is currently in a phase III clinical trial as an adjuvant therapy of patients with non-metastatic RCC with high risk of recurrence.
Monoclonal antibodies are not the only immunotherapeutic tools developed against CA IX-expressing tumors. Other approaches include different types of vaccines (anti-idiotype, dendritic cell-based, oligopeptide, and chimeric protein vaccines) and genetically engineered cytotoxic cells (reviewed in reference 291). These approaches showed promising results when tested in preclinical settings.
Rationale for the use of CA inhibitors as anti-cancer drugs is based on the assumption that perturbation of pH control in tumor tissue via diminished bicarbonate production/import could lead to decreased survival of tumor cells or their increased capacity to accumulate therapeutic drugCitation293. Various CA inhibitors were studied using two-dimensional (2D) and 3D in vitro models and showed a capacity to block pH regulation in hypoxiaCitation125,Citation127. Although these promising drugs now attract considerable attention, their potential anti-cancer effects need further investigation.
6.7. ROS and redox measurements and research potential in oncology
The adequate supply of oxygen is mandatory for the function of diverse processes within all aerobic organisms. Thereby O2 can often be transformed into highly reactive derivates, called reactive oxygen species (ROS). In the eukaryotic cell, ROS can be generated through multiple sources including the electron transport chain in mitochondria, ionizing radiations, and through enzymes producing superoxide anion radicals. Superoxide anion radical formation is often the initial step in ROS generation. Given that superoxide anion radicals and ROS are cytotoxic, cells have developed antioxidant mechanisms which include enzymes that dismutate O2– into H2O2 (superoxide dismutases), or degrade H2O2 (catalase, glutathione peroxidases, and peroxiredoxins). When cellular production of ROS overwhelms its antioxidant capacity, a state of oxidative stress is reached, leading to serious cellular injuries contributing to the pathogenesis of several diseases. Compelling evidence exists that high levels of ROS can damage DNA, RNA, lipids, and proteins, thus helping to initiate tumors. Interestingly, ROS production is a mechanism shared by many non-surgical therapeutic approaches for cancers, including chemotherapy, radiotherapy, and photodynamic therapy, due to their implication in triggering cell death; therefore, ROS are also used to kill cancer cells. On the other hand, if generated in lower concentrations, ROS can act as second messengers in signal transduction and gene regulation. Indeed, ROS have been shown to act as important messengers in promoting the growth of tumor cells by activating important signaling pathways. ROS have also been shown to increase the growth of new vessels, angiogenesis, a process highly important to tumor growth. Finally, ROS can increase the activity of HIF, a major signaling element in the response to hypoxia. Since the formation of ROS requires molecular oxygen, it has been suggested that ROS may be involved in the response to hypoxia. Indeed, there has been a long-lasting debate about the role of ROS under hypoxic conditions stimulated by the ideas that: (1) a heme protein functioning as an oxidase generates ROS as signaling molecules; (2) ROS derived from the mitochondrial respiratory chain could contribute to oxygen signaling; or (3) ROS production may be linked to the activity of signaling components upstream of HIF, such as hydroxylases or kinases. A variety of sources contribute to cellular ROS generation, including the mitochondrial respiratory chain and NADPH oxidases, and both sources have been associated with tumor growthCitation364,Citation365.
Recent evidence suggests that in addition to chronic hypoxia, also periods of cycling or intermittent hypoxia occur in tumors, mostly due to the not fully functional tumor vasculature, which lead to heterogeneous oxygenation within the tumor. Moreover, reoxygenation events after hypoxia/ischemia have been associated with elevated ROS production in many disorders, suggesting that periods and regions of elevated ROS may also occur within a tumor. Both chronic as well as intermittent hypoxia have been associated with tumor progression and resistance to therapyCitation366,Citation367. In fact, intermittent hypoxia has been suggested to more effectively up-regulate the HIF system than chronic hypoxiaCitation368,Citation369 and to be of major importance for tumor progressionCitation370.
However, the importance of ROS either as therapeutic, cytotoxic molecule or as signaling molecule under the different oxygen conditions in a tumor and its specific relevance for the outcome to therapy are not clear. In fact, it has been convincingly demonstrated that ROS levels either increase or decrease under hypoxic conditionsCitation364,Citation365,Citation371–373.
These contradictory effects have been associated with differences in the methodological approaches, in the cell types investigated, in the metabolic state of the cells, in the disease context, and so on. However, no clear explanation has been provided for the divergent findings to date. Again, given the potential importance of ROS production for tumor growth and therapeutic sensitivity, it will be of major relevance to be able to assess ROS levels within the heterogeneous tumor microenvironment, to identify the sources and conditions of ROS production, and to investigate the specific role of ROS and the redox state for any therapeutic outcome. One may speculate that selectivity between tumor and non-tumor cells may depend on differences of their redox environments, since tumor cells have been associated with elevated levels of ROSCitation374.
A set of different cellular parameters including ROS levels, oxygenation, metabolic redox status, antioxidant enzyme expression, cell signaling, and transcription factor activation profiles, namely a “redox signaling signature,” is awaiting development. This could help to design ROS-elevating or ROS-depleting therapies specific to certain types of cancer cells in consideration of the detailed microenvironmental conditions. In a clinical setting, individualized choice of an optimal ROS-manipulation therapy would require in addition accurate and convenient measurements for ROS as well as the “redox signaling signature” for prediction of efficacy and systemic toxicity.
To date there are several methods in use, mainly detecting ROS in cultivated cells. Extracellular ROS levels can be detected using cytochrome c reduction or chemiluminescence. Intracellular ROS levels are mainly determined by fluorescent dyes, including dichlorofluorescein (DCF), dihydroethidine (DHE), or dihydrorhodamine (DHR), or by use of a redox-sensitive FRET probe. However, none of these latter methods is specific for a certain type of ROS. In addition, ROS can also be determined in frozen tissues by staining with the fluorescent dyes such as DCF or DHECitation364,Citation372,Citation373. However, to date, the most specific approach to detect superoxide anion radicals and other free radicals intra- or extracellularly is based on electron spin resonance (ESR). During the course of EUROXY, we developed an ESR method to specifically assess the ROS levels at any given oxygen concentration within the measurement tube by attaching a special gas mixture unit to the ESR device. This approach is rather specific for superoxide radicals dependent on the spin traps and compounds used. It can be used for cultivated cells, blood, and tissues.
However, although an important step forward, there is still no good approach to determine ROS levels in situ in a tumor or other tissue within the whole organ or organism. Important developments have been made regarding 3D in vivo imaging of oxygen distribution using electron paramagnetic resonance (EPR) within tumors and different organs. However, there is still no sufficient approach to reliably measure ROS in situ in a living organ with an adequate resolutionCitation375. This would be helpful in assessing the role of ROS in different microenvironments, under defined oxygen concentrations, within a tumor, in controlling tumor growth and therapeutic sensitivity, and a step forward toward an individualized optimal ROS-manipulation therapy.
7. Concluding remarks
The combined effort of the participating academic research groups and small companies has enabled us to indicate a few promising targets for tumor imaging and therapy among the major pathways active in tumor cell adaptation to hypoxia and to develop supporting equipment.
Moderate hypoxia seems to be where HIF is most active, while at deeper hypoxia (1300 ppm O2) cell arrest in S-phase is a central event, and at deep hypoxia (under 1000 ppm O2) the inhibition of ribonucleotide reductase activity and thus DNA synthesis arrest is a key event.
The working of the transcription factor HIF that is activated by both genetic and microenvironmental factors is reasonably well known. A particularly interesting fact is that the oxygen-sensing PHD enzymes themselves are under regulatory control. The same degree of detailed knowledge is not yet at hand for the two other major oxygen-sensitive pathways, UPR and mTOR, both of which, through translational control, may be as important for shaping cell phenotype in response to hypoxia. Cross-talk takes place between pathways, and other factors such as pH and redox processes also participate. Furthermore, many oncogenic signaling pathways overlap with hypoxia-induced pathways. The consortium’s gene expression profiling of hypoxic tumor tissues demonstrated the prognostic value of these patterns.
Much effort has successfully focused on improving imaging using positively charged sulfonamide CA IX inhibitor, binding to the extracellular CA IX domain of the CA IX. This splicing variant of CA IX is nearly exclusively expressed in the hypoxic and acidic tumor environment. This domain, therefore, appears to be a promising target for imaging the hypoxic areas of solid tumors. Our in vivo experiments support this notion.
CA IX inhibitors may also be found useful as cancer therapeutic agents in combination with proton pump inhibitors, working by decreasing intracellular pH. Another promising agent is the bioreductive compound NLCQ-1, which inhibits metastasis in animal tests.
Simultaneously with the work on molecular and cellular biology, participating small companies have developed new equipment: oxygen-sensing flasks, combined mini clean room and incubator, combined incubator and flow-through culture system, a redox sensor based on spectroscopy, and a new redox sensor based on electron spin resonance.
A general translation of our results into clinical studies will take place in the new EU-funded project Metoxia, running from 2009 to 2012. In this project, most of the EUROXY partners will continue and be complemented by a number of cancer clinics that have ongoing imaging and drug testing programs.
Acknowledgments
Declaration of interest: The authors report no conflicts of interest.
References
- Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res 1989;49:6449–65.
- Mottram JC. Factor of importance in radiosensitivity of tumors. Br J Radiol 1936;9:606–14.
- Holthusen H. Beitrage zu Biologie der Strahlenwirkung. Pfluger’s Arch. Ges. Physiol. 1921;187:1–24.
- Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer 1955;9:539–49.
- Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol 1953;26:638–48.
- Adams GE, Dewey DL. Hydrated electrons and radiobiological sensitization. Biochem Biophys Res Commun 1963;12:473–7.
- Teicher BA. Hypoxia and drug resistance. Cancer Metastasis Rev 1994;13:139–68.
- Cosse JP, Michiels C. Tumor hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med Chem 2008;8(7):790–7.
- Hill RP, Stanley JA. The response of hypoxic B16 melanoma cells to in vivo treatment with chemotherapeutic agents. Cancer Res 1975; 35:1147–53.
- Adams GE. Hypoxic cell sensitizers for radiotherapy. Int J Radiat Oncol Biol Phys 1978;4:135–41.
- Wouters BG, Brown JM. Cells at intermediate oxygen levels can be more important than the “hypoxic fraction” in determining tumor response to fractionated radiotherapy. Radiat Res 1997;147:541–50.
- Adams GE. Failla Memorial Lecture.Redox, radiation, and reductive bioactivation. Radiat Res 1992;132:129–39.
- Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008;30:393–402.
- Holmquist-Mengelbier L, Fredlund E, Löfstedt T, Noguera R, Navarro S, Nilsson H, et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 2006;10:413–23.
- Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008 May 23;30(4):393–402.
- Wolfbeis OS, Leiner MJP, Posch HE. A new sensing material for optical oxygen measurement, with the indicator embedded in an aqueous phase. Microchim Acta 1986;90(5):359–66.
- Lippitsch ME, Pusterhofer J, Leiner MJP, Wolfbeis OS. Fibre-optic oxygen sensor with the fluorescence decay time as the information carrier. Anal Chim Acta 1988;205:1–6.
- Martinez-Manez R, Soto J, Lizondo-Sabater J, Garcia-Breijo E, Gil L, Ibanez J, et al. New potentiometric dissolved oxygen sensors in thick film technology. Sens Actuators B Chem 2004;101:295–301.
- Clark LC Jr, Wolf R, Granger D, Taylor Z. Continuous recording of blood oxygen tensions by polarography. J Appl Physiol 1953;6(3):189–93.
- Jobst G, Urban G, Jachimowicz A, Kohl F, Tilado O. Thin-film Clark-type oxygen sensor-based on novel polymer membrane systems for in-vivo and biosensor applications. Biosens Bioelectron 1993;8(3–4):123–8.
- Kieninger J, Dannenberg A, Aravindalochanan A, Jobst G, Pettersen EO, Urban GA. Amperometric oxygen sensor array with novel chronoamperometric protocols for hypoxic tumor cell cultures. Presented at the International Conference on Solid-State Sensors, Actuators and Microsystems Conference, 2007: Transducers 2007, Lyon, France, pp 1907–10.
- Wittkampf M, Chemnitius G-C, Cammann K, Rospert M, Mokwa W. Silicon thin film sensor for measurement of dissolved oxygen. Sens Actuators B Chem 1997;43:40–4.
- Brischwein M, Motrescu ER, Cabala E, Otto AM, Grothe H, Wolf B. Functional cellular assays with multiparametric silicon sensor chips. Lab Chip 2003;3(4):234–40.
- Pettersen EO, Larsen LH, Ramsing NB, Ebbesen P. Pericellular oxygen depletion during ordinary tissue culturing, measured with oxygen microsensors. Cell Prolif 2005;38:257–67.
- Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumor regression. Nature 2006;441:437–43.
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003;9:677–84.
- Semenza GL. Life with oxygen. Science 2007;318:62–4.
- Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE 2005;(306):re12.
- Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nature Chem Biol 2008;4:152–6.
- Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 2004;5:343–54.
- Koivunen P, Tiainen P, Hyvarinen J, Williams KE, Sormunen R, Klaus SJ, et al. An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J Biol Chem 2007;282:30544–52.
- Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J Inorganic Biochem 2006;100:644–69.
- Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor HIF. J Biol Chem 2003;278:30772–80.
- Koivunen P, Hirsilä M, Günzler V, Kivirikko KI, Myllyharju J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004 Mar 12;279(11):9899–904.
- Tuckerman JR, Zhao Y, Hewitson KS, Tian YM, Pugh CW, Ratcliffe PJ, et al. Determination and comparison of specific activity of the HIF-prolyl hydroxylases. FEBS Lett 2004;576:145–50.
- Ehrismann D, Flashman E, Genn DN, Mathioudakis N, Hewitson KS, Ratcliffe PJ, et al. Studies on the activity of the hypoxia-inducible factor hydroxylases using an oxygen consumption assay. Biochem J 2007;401:227–34.
- Koivunen P, Hirsila M, Kivirikko KI, Myllyharju J. The length of peptide substrates has a marked effect on hydroxylation by the hypoxia-inducible factor prolyl 4 hydroxylases. J Biol Chem 2006;281:28712–20.
- Chan DA, Sutphin PD, Denko NC, Giaccia AJ. Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1a. J Biol Chem 2002;277:40112–17.
- Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 2004;118:781–94.
- Knowles HJ, Raval RR, Harris AL, Ratcliffe PJ. Effect of ascorbate on the activity of hypoxia inducible factor (HIF) in cancer cells. Cancer Res 2003; 63:1764–8.
- Smith TG, Balanos GM, Croft QPP, Talbot NP, Dorrington KL, Ratcliffe PJ, et al. The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J Physiol 2008;586:5999–6005.
- Dalgard CL, Lu H, Mohyeldin A, Verma A. Endogenous 2-oxoacids differentially regulate expression of oxygen sensors. Biochem J 2004; 380:419–24.
- Hewitson KS, Lienard BM, McDonough MA, Clifton IJ, Butler D, Soares AS, et al. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem 2007;282:3293–301.
- Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Merino M, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 2005;8:143–53.
- Koivunen P, Hirsila M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem 2007;282:4524–32.
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005;7:77–85.
- Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumors which result from germline FH and SDH mutations. Hum Mol Genet 2005;14:2231–9.
- Mackenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, et al. Cell permeating alpha-ketoglutarate derivatives alleviate pseudo-hypoxia in SDH deficient cells. Mol Cell Biol 2007;27:3282–9.
- Knowles HJ, Mole DR, Ratcliffe PJ, Harris AL. Normoxic stabilization of hypoxia-inducible factor-1alpha by modulation of the labile iron pool in differentiating U937 macrophages: effect of natural resistance-associated macrophage protein 1. Cancer Res 2006;66:2600–7.
- Pagé EL, Chan DA, Giaccia AJ, Levine M, Richard DE. Hypoxia-inducible factor-1alpha stabilization in nonhypoxic conditions: role of oxidation and intracellular ascorbate depletion. Mol Biol Cell. 2008 Jan;19(1):86–94.
- Vissers MC, Gunningham SP, Morrison MJ, Dachs GU, Currie MJ. Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Radic Biol Med 2007;42:765–72.
- Kietzmann T, Görlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol 2005;16:474–86.
- Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol 2007;27:912–25.
- Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signalling via reactive oxygen species production. J Cell Biol 2007;177:1029–36.
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005 Jun;1(6):401–8.
- Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol 2008;28:718–31.
- Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 2005;1:393–9.
- Matlin AJ, Clark F, Smith CWJ. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol 2005;6:386–98.
- Kramer A. The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem 1996;65:367–409.
- Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev 2003;17:419–37.
- Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci 2006;119:2635–41.
- Kalnina Z, Zyakin P, Silina K, Line A. Alterations of pre-mRNA splicing in cancer. Genes Chromosomes Cancer 2005;42:342–57.
- Veneables JP. Abberant and alternative splicing in cancer. Cancer Res 2004;64:7647–54.
- Stoss O, Stoilov P, Daoud R, Hartmann AM, Olbrich M, Stamm S. Misregulation of pre-mRNA splicing that causes human diseases.Concepts and therapeutic strategies. Gene Ther Mol Biol 2000; 5:9–30.
- Ge K, DuHadaway J, Du W, Herlyn M, Rodeck U, Prendergast GC. Mechanism for elimination of a tumor suppresor: aberant splicing of a brain-specific exon causes loss of function of Bin1 in melanoma. Proc Natl Acad Sci USA 1999;96:9689–94.
- Semenza G. HIF-1 and human disease: one highly involved factor. Genes Dev 2000;14:1983–91.
- Semenza G. Targeting HIF-1 for cancer therapy. Nat Rev 2003;3:721–32.
- Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001;414:550–4.
- Yamashita T, Ohneda O, Nagano M, Iemitsu M, Makino Y, Tanaka H, et al. Abnormal heart development and lung remodeling in mice lacking the hypoxia-inducible factor-related basic helix-loop-helix PAS protein NEPAS. Mol Cell Biol 2008;28:1285–97.
- Gothie E, Richard DE, Berra E, Pages G, Pouyssegur J. Identification of alternatively spliced variants of human hypoxia-inducible factor-1α. J Biol Chem 2000;275:6922–7.
- Chun Y-S, Choi E, Kim T-Y, Kim M-S, Park J-W. A dominant-negative isoform lacking exons 11 and 12 of the human hypoxia-inducible factor-1α gene. Biochem J 2002;362:71–9.
- Maynard MA, Qi H, Chung J, Lee EHL, Kondo Y, Hara S, et al. Multiple splice variants of the human HIF-3α locus are targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J Biol Chem 2003;278:11032–40.
- Makino Y, Kanopka A, Wilson WJ, Tanaka H, Poellinger L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3alpha locus. J Biol Chem 2002;277:32405–08.
- Barathova M, Takacova M, Holotnakova T, Gibadulinova A, Ohradanova A, Zatovicova M, et al. Alternative splicing variant of the hypoxia marker carbonic anhydrase IX expressed independently of hypoxia and tumor phenotype. Br J Cancer 2008;98:129–36.
- Wouters BG, van den Beucken T, Magagnin MG, Lambin P, Koumenis C. Targeting hypoxia tolerance in cancer. Drug Resist Updat 2004; 7(1):25–40.
- Brown JM, Wilson WR. Exploiting tumor hypoxia in cancer treatment. Nat Rev Cancer 2004;4(6):437–47.
- Hockel M, Vaupel P. Biological consequences of tumor hypoxia. Semin Oncol 2001;28(2 Suppl 8):36–41.
- Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008;8(11):851–64.
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004;18(23):2893–904.
- Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol Cell Biol 2006;26(10):3955–65.
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell 2006;21(4):521–31.
- Koritzinsky M, Rouschop KM, van den Beucken T, Magagnin MG, Savelkouls K, Lambin P, et al. Phosphorylation of eIF2alpha is required for mRNA translation inhibition and survival during moderate hypoxia. Radiother Oncol 2007;83(3):353–61.
- Magagnin MG, van den Beucken T, Sergeant K, Lambin P, Koritzinsky M, Devreese B, et al. The mTOR target 4E-BP1 contributes to differential protein expression during normoxia and hypoxia through changes in mRNA translation efficiency. Proteomics 2008;8(5):1019–28.
- Weppler SA, Krause M, Zyromska A, Lambin P, Baumann M, Wouters BG. Response of U87 glioma xenografts treated with concurrent rapamycin and fractionated radiotherapy: possible role for thrombosis. Radiother Oncol 2007;82(1):96–104.
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol 2002;22(21):7405–16.
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 2005;24(19):3470–81.
- Koritzinsky M, Magagnin MG, van den Beucken T, Seigneuric R, Savelkouls K, Dostie J, et al. Gene expression during acute and prolonged hypoxia is regulated by distinct mechanisms of translational control. EMBO J 2006;25(5):1114–25.
- Koritzinsky M, Seigneuric R, Magagnin MG, van den Beucken T, Lambin P, Wouters BG. The hypoxic proteome is influenced by gene-specific changes in mRNA translation. Radiother Oncol 2005;76(2):177–86.
- van den Beucken T, Magagnin MG, Savelkouls K, Lambin P, Koritzinsky M, Wouters BG. Regulation of Cited2 expression provides a functional link between translational and transcriptional responses during hypoxia. Radiother Oncol 2007;83(3):346–52.
- Magagnin MG, Sergeant K, van den Beucken T, Rouschop KM, Jutten B, Seigneuric R, et al. Proteomic analysis of gene expression following hypoxia and reoxygenation reveals proteins involved in the recovery from endoplasmic reticulum and oxidative stress. Radiother Oncol 2007;83(3):340–5.
- Goetz ME, Luch A. Reactive species: a cell damaging rout assisting to chemical carcinogens. Cancer Lett 2008;266:73–83.
- Lu W, Ogasawara MA, Huang P. Models of reactive oxygen species in cancer. Drug Discov Today Dis Models 2007;4:67–73.
- Lenart J, Dombrowski F, Görlach A, Kietzmann T. Deficiency of manganese superoxide dismutase in hepatocytes disrupts zonated gene expression in mouse liver. Arch Biol Biophys 2007;462:238–44.
- Hervouet E, Simonnet H, Godinot C. Mitochondria and reactive oxygen species in renal cancer. Biochimie 2007;89:1080–8.
- Penta JS, Johnson FM, Wachsman JT, Copeland WC. Mitochondrial DNA in human malignancy. Mutat Res, 2001;488:119–33.
- Sarti P, Avigliano L, Görlach A, Brüne B. Superoxide and nitric oxide—participation in cell communication. Cell Death Differ 2002;9:1160–2.
- Görlach A, Kietzmann T, Hess J. Redox signaling through NADPH oxidases – involvement in vascular proliferation and coagulation. Ann NY Acad Sci 2002;973:505–7.
- Forman HJ, Fukuto JM, Miller T, Zhang H, Rinna A, Levy S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch Biochem Biophys 2008;477:183–95.
- Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 2006;7:833–46.
- Clerkin JS, Naughton R, Quiney C, Cotter TG. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett 2008;266:30–6.
- Görlach A, Diebold I, Schini-Kerth VB, Berchner-Pfannschmidt U, Roth U, Brandes RP, et al. Thrombin activates the HIF-1 signaling pathway in vascular smooth muscle cells. Role of the p22phox-containing NADPH oxidase. Circ Res 2001;89:47–54.
- Görlach A, Kietzmann T. Superoxide and derived reactive oxygen species in the regulation of hypoxia-inducible factors. Methods Enzymol 2007;435:421–46.
- Bonello S, Zähringer C, BelAiba RS, Djordjevic T, Hess J, Michiels C, et al. Reactive oxygen species activate the HIF-1α promoter via a functional NFκB site. Arterioscler Thromb Vasc Biol 2007;27:755–61.
- BelAiba RS, Bonello S, Schmidt S, Diemer K, Kietzmann T, Hess J, et al. Hypoxia-induced HIF-1α mRNA level is mediated by NFκB via PI3K/AKT pathway activation in pulmonary artery smooth muscle cells. Mol Biol Cell 2007;18:4691–7.
- Lambeth JD. NOX enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 2007;43:332–47.
- Rokutan K, Kawahara T, Kuwano Y, Tominaga K, Sekiyama A, Teshima-Kondo S. NADPH oxidases in the gastrointestinal tract: a potential role of Nox1 in innate immune response and carcinogenesis. Antioxid Redox Signal 2006;8(9–10):1573–82.
- Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res 2006;71:226–35.
- Otani H. Reactive oxygen species as mediators of signal transduction in ischemic preconditioning. Antioxid Redox Signal 2004;6:449–69.
- Lambeth JD, Krause KH, Clark RA. NOX enzymes as novel targets for drug development. Semin Immunopathol 2008;30:339–63.
- Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 2008; 8:425–37.
- Robinson JM. Reactive oxygen species in phagocytic leukocytes. Histochem Cell Biol 2008;130:281–97.
- Ono M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci 2008;99:1501–6.
- BelAiba RS, Djordjevic T, Bonello S, Flügel D, Hess J, Kietzmann T, et al. Redox-sensitive regulation of the HIF pathway under non-hypoxic conditions in pulmonary artery smooth muscle cells. Biol Chem 2004;385:249–57.
- BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, et al. NOX5 variants are functionally active in endothelial cells. Free Rad Biol Med 2007;42:446–59.
- Brahimi-Horn MC, Pouysségur J. Hypoxia in cancer cell metabolism and pH regulation. Essays Biochem 2007;43:165–78.
- Walenta S, Mueller-Klieser WF. Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol 2004;14:267–74.
- Helmlinger G, Sckell A, Dellian M, Forbes NS, Jain RK. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin Cancer Res 2002;8:1284–91.
- Stubbs M, McSheehy PMJ, Griffiths JR, Bashford CL. Causes and consequences of tumor acidity and implications for treatment. Mol Med Today 2000;6:15–19.
- Raghunand N, Gatenby RA, Gillies RJ. Microenvironmental and cellular consequences of altered blood flow in tumors. Br J Radiol 2003;76:S11–22.
- Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res 2000;60:7075–83.
- Pastorekova S, Casini A, Scozzafava A, Vullo D, Pastorek J, Supuran CT. Carbonic anhydrase inhibitors: the first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Lett 2004;14:869–73.
- Potter CP, Harris AL. Diagnostic, prognostic and therapeutic implications of carbonic anhydrases in cancer. Br J Cancer 2003;89:2–7.
- Pastorek J, Pastorekova S, Callebaut I, Mornon JP, Zelnik V, Opavsky R, et al. Cloning and characterisation of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and putative helix-loop-helix DNA binding segment. Oncogene 1994;9:2877–88.
- Pastorekova S, Ratcliffe PJ, Pastorek J. Molecular mechanisms of carbonic anhydrase IX-mediated pH regulation under hypoxia. BJU Int 2008;101(Suppl 4):8–15.
- Svastova E, Hulikova A, Rafajova M, Zatovicova M, Gibadulinova A, Casini A, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004;577:439–45.
- Swietach P, Vaughan-Jones RD, Harris AL. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev 2007; 26:299–310.
- Swietach P, Wigfield S, Cobden P, Supuran CT, Harris AL, Vaughan-Jones RD. Tumor-associated carbonic anhydrase 9 spatially coordinates intracellular pH in three-dimensional multicellular growths. J Biol Chem 2008;283:20473–83.
- Dubois L, Douma K, Supuran CT, Chiu RK, van Zandvoort MA, Pastoreková S, et al. Imaging the hypoxia surrogate marker CA IX requires expression and catalytic activity for binding fluorescent sulfonamide inhibitors. Radiother Oncol 2007;83:367–73.
- Rafajova M, Zatovicova M, Kettmann R, Pastorek J, Pastorekova S. Induction by hypoxia combined with low glucose or low bicarbonate and high posttranslational stability upon reoxygenation contribute to carbonic anhydrase IX expression in cancer cells. Int J Oncol 2004;24:995–1004.
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev 1998;12:149–62.
- Pastorekova S, Zatovicova M, Pastorek J. Cancer-associated carbonic anhydrases and their inhibition. Curr Pharm Des 2008;14: 685–98.
- Görlach A, Camenisch G, Kvietikova I, Vogt L, Wenger RH, Gassmann M. Efficient translation of mouse hypoxia-inducible factor-1α under normoxic and hypoxic conditions. Biochim Biophys Acta 2000;1493:125–34.
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007;129:111–22.
- Wenger RH. Mitochondria: oxygen sinks rather than sensors? Med Hypoth 2006;66:380–3.
- Stiehl DP, Wirthner R, Köditz J, Spielmann P, Camenisch G, Wenger RH. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels.Evidence for an autoregulatory oxygen-sensing system. J Biol Chem 2006;281:23482–91.
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3:177–85.
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006;3:187–97.
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007;11:407–20.
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008;283:10892–903.
- Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 2004;24:7469–82.
- Köditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, et al. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 2007;110:3610–17.
- Pettersen EO, Lindmo T. Inhibition of cell-cycle progression by acute treatment with various degrees of hypoxia: modifications induced by low concentrations of misonidazole present during hypoxia. Br J Cancer 1983;48:809–17.
- åmellem O, Stokke T, Sandvik JA, Pettersen EO. The retinoblastoma gene product is reversibly dephosphorylated and bound in the nucleus in S and G2 phase during hypoxic stress. Exp Cell Res 1996;227:106–15.
- åmellem O, Stokke T, Sandvik JA, Karlsen F, Pettersen EO. The retinoblastoma protein-associated cell cycle arrest in S-phase under moderate hypoxia is disrupted in cells expressing HPV18 E7 oncoprotein. Br J Cancer 1998;77:862–72.
- Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV. Hypoxia inhibits G1/S transition through regulation of p27 expression. J Biol Chem. 2001 Mar 16;276(11):7919–26.
- Krtolica A, Krucher NA, Ludlow JW. Hypoxia-induced pRB hypophosphorylation results from downregulation of CDK and upregulation of PP1 activities. Oncogene 1998;17:2295–304.
- Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol 2003;23:359–69.
- Graff P, Seim J, Amellem Ø, Arakawa H, Nakamura Y, Andersson KK, Stokke T, Pettersen EO. Counteraction of pRb-dependent protection after extreme hypoxia by elevated ribonucleotide reductase. Cell Prolif. 2004 Oct;37(5):367–83.
- Green SL, Freiberg RA, Giaccia AJ. p21(Cip1) and p27(Kip1) regulate cell cycle reentry after hypoxic stress but are not necessary for hypoxia-induced arrest. Mol Cell Biol 2001;21:1196–206.
- Graeber TG, Peterson JF, Tsai M, Monica K, Fornace AJ Jr, Giaccia AJ. Hypoxia induces accumulation of p53 protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol Cell Biol 1994;14:6264–77.
- Pettersen EO, Lindmo T. Inhibition of cell-cycle progression by acute treatment with various degrees of hypoxia: modifications induced by low concentrations of misonidazole present during hypoxia. Br J Cancer. 1983 Dec;48(6):809–17.
- åmellem O, Pettersen EO. The role of protein accumulation on the kinetics of entry into S phase following extreme hypoxia. Anticancer Res 1991;11:1083–7.
- åmellem O, Pettersen EO. Cell inactivation and cell cycle inhibition as induced by extreme hypoxia: the possible role of cell cycle arrest as a protection against hypoxia-induced lethal damage. Cell Prolif 1991;24:127–41.
- åmellem O, Löffler M, Pettersen EO. Regulation of cell proliferation under extreme and moderate hypoxia: the role of pyrimidine (deoxy)nucleotides. Br J Cancer 1994;70:857–66.
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci USA 1995;92:12146–50.
- Probst G, Riedinger HJ, Martin P, Engelcke M, Probst H. Fast control of DNA replication in response to hypoxia and to inhibited protein synthesis in CCRF-CEM and HeLa cells. Biol Chem 1999;380:1371–82.
- Ludlow JW, Howell RL, Smith HC. Hypoxic stress induces reversible hypophosphorylation of pRb and reduction in cyclin A abundance independent of cell-cycle progression. Oncogene 1993;8:331–9.
- Krtolica A, Ludlow JW. Hypoxia arrests ovarian carcinoma cell cycle progression, but invasion is unaffected. Cancer Res 1996;56:1168–73.
- Graff P, åmellem ø, Andersson KK, Pettersen EO. Role of ribonucleotide reductase in regulation of cell cycle progression during and after exposure to moderate hypoxia. Anticancer Res 2002;22:59–68.
- Thelander L, Reichard P. Reduction of ribonucleotides. Annu Rev Biochem 1979;48:133–58.
- Eklund H, Uhlin U, Färnegårdh M, Logan DT, Nordlund P. Structure and function of the radical enzyme ribonucleotide reductase. Prog Biophys Mol Biol 2001;77:177–268.
- Bianchi V, Borella S, Calderazzo F, Ferraro P, Chieco BL, Reichard P. Inhibition of ribonucleotide reductase by 2’-substituted deoxycytidine analogs: possible application in AIDS treatment. Proc Natl Acad Sci USA 1994;91:8403–7.
- Nocentini G. Ribonucleotide reductase inhibitors: new strategies for cancer chemotherapy. Crit Rev Oncol Hematol 1996;22:89–126.
- Holland KP, Elford HL, Bracchi V, Annis CG, Schuster SM, Chakrabarti D. Antimalarial activities of polyhydroxyphenyl and hydroxamic acid derivatives. Antimicrob Agents Chemother 1998;42:2456–8.
- Guppy M, Withers P. Metabolic depression in animals: physiological perspectives and biochemical generalizations. Biol Rev Camb Philos Soc 1999;74:1–40.
- Gorr TA, Gassmann M, Wappner P. Sensing and responding to hypoxia via HIF in model invertebrates. J Insect Physiol 2006;52:349–64.
- Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA 1996;93:9493–8.
- Boutilier RG. Mechanisms of cell survival in hypoxia and hypothermia. J Exp Biol 2001;204:3171–81.
- Boutilier RG, Donohoe PH, Tattersall GJ, West TG. Hypometabolic homeostasis in overwintering aquatic amphibians. J Exp Biol. 1997 Jan;200(Pt 2):387–400.
- Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 1997;77:731–58.
- Land SC, Buck LT, Hochachka PW. Response of protein synthesis to anoxia and recovery in anoxia-tolerant hepatocytes. Am J Physiol 1993;265:R41–8.
- Land SC, Hochachka PW. Protein turnover during metabolic arrest in turtle hepatocytes: role and energy dependence of proteolysis. Am J Physiol 1994;266:C1028–36.
- Buck LT, Hochachka PW. Anoxic suppression of Na(+)-K(+)-ATPase and constant membrane potential in hepatocytes: support for channel arrest. Am J Physiol 1993;265:R1020–5.
- Krumschnabel G, Schwarzbaum PJ, Lisch J, Biasi C, Wieser W. Oxygen-dependent energetics of anoxia-tolerant and anoxia-intolerant hepatocytes. J Exp Biol 2000;203:951–9.
- Storey K. A re-evaluation of the Pasteur effect: new mechanisms in anaerobic metabolism. Mol Physiol 1985;8:439–61.
- Schmidt H, Kamp G. The Pasteur effect in facultative anaerobic metazoa. Experientia 1996;52:440–8.
- Padilla PA, Roth MB. Oxygen deprivation causes suspended animation in the zebrafish embryo. Proc Natl Acad Sci USA 2001;98:7331–5.
- Padilla PA, Nystul TG, Zager RA, Johnson AC, Roth MB. Dephosphorylation of cell cycle-regulated proteins correlates with anoxia-induced suspended animation in Caenorhabditis elegans. Mol Biol Cell 2002;13:1473–83.
- Froese G. The respiration of ascites tumor cells at low oxygen concentrations. Biochim Biophys Acta 1962;57:509–19.
- Freyer JP. Rates of oxygen consumption for proliferating and quiescent cells isolated from multicellular tumor spheroids. Adv Exp Med Biol 1994;345:335–42.
- Pettersen EO, Lindmo T. Inhibition of cell-cycle progression by acute treatment with various degrees of hypoxia: modifications induced by low concentrations of misonidazole present during hypoxia. Br J Cancer. 1983;48:809–817.
- Probst H, Schiffer H, Gekeler V, et al. Oxygen dependent regulation of DNA synthesis and growth of Ehrlich ascites tumor cells in vitro and in vivo. Cancer Res 1988;48:2053–60.
- Pettersen EO, Juul NO, Ronning OW. Regulation of protein metabolism of human cells during and after acute hypoxia. Cancer Res 1986;46:4346–51.
- Kraggerud SM, Sandvik JA, Pettersen EO. Regulation of protein synthesis in human cells exposed to extreme hypoxia. Anticancer Res 1995;15:683–6.
- Guppy M, Brunner S, Buchanan M. Metabolic depression: a response of cancer cells to hypoxia? Comp Biochem Physiol B Biochem Mol Biol 2005;140:233–9.
- Giaccia AJ. Hypoxic stress proteins: survival of the fittest. Semin Radiat Oncol 1996;6:46–58.
- Freyer JP, Sutherland RM. A reduction in the in situ rates of oxygen and glucose consumption of cells in EMT6/Ro spheroids during growth. J Cell Physiol 1985;124:516–24.
- Eskey CJ, Koretsky AP, Domach MM, Jain RK. Role of oxygen vs.glucose in energy metabolism in a mammary carcinoma perfused ex vivo: direct measurement by 31P NMR. Proc Natl Acad Sci USA 1993;90:2646–50.
- Walenta S, Doetsch J, Mueller-Klieser W, Kunz-Schughart LA. Metabolic imaging in multicellular spheroids of oncogene-transfected fibroblasts. J Histochem Cytochem 2000;48:509–22.
- Kunz-Schughart LA, Freyer JP. Phosphorous metabolites and steady-state energetics of transformed fibroblasts during three-dimensional growth. Am J Physiol Cell Physiol 2002;283:C1287–97.
- Zölzer F, Stüben G, Knühmann K, Streffer C, Sack H. Quiescent S-phase cells as indicators of extreme physiological conditions in human tumor xenografts. Int J Radiat Oncol Biol Phys 1999;45:1019–24.
- Masunaga S, Ono K, Takahashi A, Ohnishi T, Kinashi Y, Takagaki M. Radiobiological characteristics of solid tumors depending on the p53 status of the tumor cells, with emphasis on the response of intratumor quiescent cells. Eur J Cancer 2002;38:718–27.
- Masunaga S, Uto Y, Nagasawa H, Hori H, Nagata K, Suzuki M, et al. Evaluation of hypoxic cell radio-sensitizers in terms of radio-sensitizing and repair-inhibiting potential. Dependency on p53 status of tumor cells and the effects on intratumor quiescent cells. Anticancer Res 2006;26:1261–70.
- Dubois L, Magagnin MG, Cleven AH, Weppler SA, Grenacher B, Landuyt W, Lieuwes N, Lambin P, Gorr TA, Koritzinsky M, Wouters BG. Inhibition of 4E-BP1 Sensitizes U87 Glioblastoma Xenograft Tumors to Irradiation by Decreasing Hypoxia Tolerance. Int J Radiat Oncol Biol Phys. 2009 Mar 15;73(4):1219–27.
- Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer 2002;2(1):38–47.
- Beasley NJ, Wykoff CC, Watson PH, et al. Carbonic anhydrase IX, an endogenous hypoxia marker, expression in head and neck squamous cell carcinoma and its relationship to hypoxia, necrosis, and microvessel density. Cancer Res 2001;61(13):5262–7.
- Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med 2001;7(8):345–50.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995;92(12):5510–4.
- Gustafsson MV, Zheng X, Pereira T, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 2005;9(5):617–28.
- Patel NS, Li JL, Generali D, Poulsom R, Cranston DW, Harris AL. Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res 2005;65(19):8690–7.
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004;10(8):789–99.
- Huang LE, Bindra RS, Glazer PM, Harris AL. Hypoxia-induced genetic instability—a calculated mechanism underlying tumor progression. J Mol Med 2007;85(2):139–48.
- Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K, White E. Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes & development 2004;18(17):2095–107.
- Graeber TG, Osmanian C, Jacks T, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996;379(6560):88–91.
- Chan DA, Giaccia AJ. Hypoxia, gene expression, and metastasis. Cancer metastasis reviews 2007;26(2):333–9.
- Gort EH, Groot AJ, van der Wall E, van Diest PJ, Vooijs MA. Hypoxic regulation of metastasis via hypoxia-inducible factors. Current molecular medicine 2008;8(1):60–7.
- Cosse JP, Michiels C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med Chem 2008;8(7):790–7.
- Charlesworth PJ, Harris AL. Mechanisms of disease: angiogenesis in urologic malignancies. Nature clinical practice 2006;3(3):157–69.
- Koukourakis MI, Bentzen SM, Giatromanolaki A, et al. Endogenous markers of two separate hypoxia response pathways (hypoxia inducible factor 2 alpha and carbonic anhydrase 9) are associated with radiotherapy failure in head and neck cancer patients recruited in the CHART randomized trial. J Clin Oncol 2006;24(5):727–35.
- Koukourakis MI, Giatromanolaki A, Sivridis E, et al. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. British journal of cancer 2003;89(5):877–85.
- Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL. Lactate dehydrogenase 5 expression in operable colorectal cancer: strong association with survival and activated vascular endothelial growth factor pathway—a report of the Tumour Angiogenesis Research Group. J Clin Oncol 2006;24(26):4301–8.
- Marignol L, Coffey M, Lawler M, Hollywood D. Hypoxia in prostate cancer: a powerful shield against tumour destruction? Cancer Treat Rev 2008;34(4):313–27.
- Unruh A, Ressel A, Mohamed HG, Johnson RS, Nadrowitz R, Richter E, et al. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene 2003;22(21):3213–20.
- Song X, Liu X, Chi W, et al. Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1alpha gene. Cancer chemotherapy and pharmacology 2006;58(6):776–84.
- Sermeus A, Cosse JP, Crespin M, et al. Hypoxia induces protection against etoposide-induced apoptosis: molecular profiling of changes in gene expression and transcription factor activity. Mol Cancer 2008;7:27.
- Moeller BJ, Dewhirst MW. HIF-1 and tumor radiosensitivity. Br J Cancer 2006;95(1):1–5.
- Moeller BJ, Dreher MR, Rabbani ZN, Schroeder T, Cao Y, Li CY, et al. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell 2005;8(2):99–110.
- Harada H, Kizaka-Kondoh S, Li G, Itasaka S, Shibuya K, Inoue M, et al. Significance of HIF-1-active cells in angiogenesis and radioresistance. Oncogene 2007;26(54):7508–16.
- Williams KJ, Telfer BA, Xenaki D, et al. Enhanced response to radiotherapy in tumours deficient in the function of hypoxia-inducible factor-1. Radiother Oncol 2005;75(1):89–98.
- Wouters BG, Weppler SA, Koritzinsky M, Landuyt W, Nuyts S, Theys J, et al. Hypoxia as a target for combined modality treatments. Eur J Cancer 2002;38:240–57.
- Adam MF, Gabalski EC, Bloch DA, Oehlert JW, Brown JM, Elsaid AA, et al. Tissue oxygen distribution in head and neck cancer patients. Head Neck 1999;21(2):146–53.
- Stadler P, Becker A, Feldmann HJ, Hansgen G, Dunst J, Wurschmidt F, et al. Influence of the hypoxic subvolume on the survival of patients with head and neck cancer. Int J Radiat Oncol Biol Phys 1999;44(4):749–54.
- Raleigh JA, Chou SC, Calkins-Adams DP, Ballenger CA, Novotny DB, Varia MA. A clinical study of hypoxia and metallothionein protein expression in squamous cell carcinomas. Clin Cancer Res 2000;6(3):855–62.
- Kaanders JH, Wijffels KI, Marres HA, Ljungkvist AS, Pop LA, van den Hoogen JA, et al. Pimonidazole binding and tumor vascularity predict for treatment outcome in head and neck cancer. Cancer Res 2002;62:7066–74.
- Gross MW, Karbach U, Groebe K, Franko AJ, Mueller-Klieser W. Calibration of misonidazole labeling by simultaneous measurement of oxygen tension and labeling density in multicellular spheroids. Int J Cancer 1995;61(4):567–73.
- Nordsmark M, Loncaster J, Aquino-Parsons C, Chou SC, Ladekarl M, Havsteen H, et al. Measurements of hypoxia using pimonidazole and polarographic oxygen-sensitive electrodes in human cervix carcinomas. Radiother Oncol 2003;67(1):35–44.
- Beasley NJ, Wykoff CC, Watson PH, Leek R, Turley H, Gatter K, et al. Carbonic anhydrase IX, an endogenous hypoxia marker, expression in head and neck squamous cell carcinoma and its relationship to hypoxia, necrosis, and microvessel density. Cancer Res 2001;61(13):5262–7.
- Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, et al. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol 2000;157(2):411–21.
- Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 1999;59(22):5830–5.
- Lidgren A, Hedberg Y, Grankvist K, Rasmuson T, Vasko J, Ljungberg B. The expression of hypoxia-inducible factor 1alpha is a favorable independent prognostic factor in renal cell carcinoma. Clin Cancer Res 2005;11(3):1129–35.
- Koukourakis MI, Giatromanolaki A, Sivridis E, Simopoulos K, Pastorek J, Wykoff CC, et al. Hypoxia-regulated carbonic anhydrase-9 (CA IX) relates to poor vascularization and resistance of squamous cell head and neck cancer to chemoradiotherapy. Clin Cancer Res 2001;7(11):3399–403.
- Giatromanolaki A, Koukourakis MI, Sivridis E, Pastorek J, Wykoff CC, Gatter KC, et al. Expression of hypoxia-inducible carbonic anhydrase-9 relates to angiogenic pathways and independently to poor outcome in non-small cell lung cancer. Cancer Res 2001;61(21):7992–8.
- Swinson DE, Jones JL, Richardson D, Wykoff C, Turley H, Pastorek J, et al. Carbonic anhydrase IX expression, a novel surrogate marker of tumor hypoxia, is associated with a poor prognosis in non-small-cell lung cancer. J Clin Oncol 2003;21(3):473–82.
- Loncaster JA, Harris AL, Davidson SE, Loque JP, Hunter RD, Wykoff CC, et al. Carbonic anhydrase (CA IX) expression, a potential new intrinsic marker of hypoxia: correlations with tumor oxygen measurements and prognosis in locally advanced carcinoma of the cervix. Cancer Res 2001;61(17):6394–9.
- Chia SK, Wykoff CC, Watson PH, Han C, Leek RD, Pastorek J, et al. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J Clin Oncol 2001;19(16):3660–8.
- Hutchison GJ, Valentine HR, Loncaster JA, Davidson SE, Hunter RD, Roberts SA, et al. Hypoxia-inducible factor 1alpha expression as an intrinsic marker of hypoxia: correlation with tumor oxygen, pimonidazole measurements, and outcome in locally advanced carcinoma of the cervix. Clin Cancer Res 2004;10(24):8405–12.
- Mayer A, Hockel M, Vaupel P. Carbonic anhydrase IX expression and tumor oxygenation status do not correlate at the microregional level in locally advanced cancers of the uterine cervix. Clin Cancer Res 2005;11(20):7220–5.
- Mayer A, Wree A, Hockel M, Leo C, Pilch H, Vaupel P. Lack of correlation between expression of HIF-1alpha protein and oxygenation status in identical tissue areas of squamous cell carcinomas of the uterine cervix. Cancer Res 2004;64(16):5876–81.
- Olive PL, Aquino-Parsons C, MacPhail SH, Liao SY, Raleigh JA, Lerman MI, et al. Carbonic anhydrase 9 as an endogenous marker for hypoxic cells in cervical cancer. Cancer Res 2001;61(24):8924–9.
- Janssen HL, Haustermans KM, Sprong D, Blommestijn G, Hofland I, Hoebers FJ, et al. HIF-1A, pimonidazole, and iododeoxyuridine to estimate hypoxia and perfusion in human head-and-neck tumors. Int J Radiat Oncol Biol Phys 2002;54(5):1537–49.
- Le QT, Sutphin PD, Raychaudhuri S, Yu SC, Terris DJ, Lin HS, et al. Identification of osteopontin as a prognostic plasma marker for head and neck squamous cell carcinomas. Clin Cancer Res 2003;9(1):59–67.
- Overgaard J, Eriksen JG, Nordsmark M, Alsner J, Horsman MR. Plasma osteopontin, hypoxia, and response to the hypoxia sensitiser nimorazole in radiotherapy of head and neck cancer: results from the DAHANCA 5 randomised double-blind placebo-controlled trial. Lancet Oncol 2005;6(10):757–64.
- Krause BJ, Beck R, Souvatzoglou M, Piert M. PET and PET/CT studies of tumor tissue oxygenation. Q J Nucl Med Mol Imaging 2006;50(1):28–43.
- Bucca G, Carruba G, Saetta A, Muti P, Castagnetta L, Smith CP. Gene expression profiling of human cancers. Ann NY Acad Sci 2004;1028:28–37.
- Choi P, Chen C. Genetic expression profiles and biologic pathway alterations in head and neck squamous cell carcinoma. Cancer 2005;104(6):1113–28.
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat Genet 2001;29(4):365–71.
- Akervall J. Gene profiling in squamous cell carcinoma of the head and neck. Cancer Metastasis Rev 2005;24(1):87–94.
- Chung CH, Parker JS, Karaca G, Wu J, Funkhouser WK, Moore D, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell 2004;5(5):489–500.
- Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, Sorlie T, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA 2005;102(10):3738–43.
- Chi JT, Wang Z, Nuyten DS, Rodriguez EH, Schaner ME, Salim A, et al. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med 2006;3(3):e47.
- Nuyten DS, Kreike B, Hart AA, Chi JT, Sneddon JB, Wessels LF, et al. Predicting a local recurrence after breast-conserving therapy by gene expression profiling. Breast Cancer Res 2006;8(5):R62.
- Francis P, Namlos HM, Muller C, Eden P, Fernebro J, Berner JM, et al. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics 2007;8:73.
- Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H, et al. hsa-miR-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res 2008;14(5):1340–8.
- Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, et al. HIF activation identifies early lesions in VHL kidneys.Evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 2002;1:459–68.
- Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, et al. Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol 2002;13:1721–32.
- Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 2005;25:5675–86.
- Esteban MA, Harten SK, Tran MG, Maxwell PH. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J Am Soc Nephrol 2006;17:1801–6.
- Harten SK, Shukla D, Barod R, Hergovich A, Balda MS, Matter K, et al. Repression of occludin and claudin 1 contributes to tight junction abnormalities in von Hippel-Lindau defective renal cancer cells. Mol Biol Cell 2009;20:1089–101.
- Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res 1998;58:1408–16.
- Sutherland RM, Ausserer WA, Murphy BJ, Laderoute KR. Tumor hypoxia and heterogeneity: challenges and opportunities for the future. Semin Radiat Oncol 1996;6:59–70.
- Koch CJ. Oxygen effects in radiobiology. Adv Exp Med Biol 1982;157:123–44.
- Brizel DM, Sibley GS, Prosnitz LR, Scher RL, Dewhirst MW. Tumor hypoxia adversely affects the prognosis of carcinoma of the head and neck. Int J Radiat Oncol Biol Phys 1997;38:285–9.
- Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res 1996;56:4509–15.
- Nordsmark M, Alsner J, Keller J, Nielsen OS, Jensen OM, Horsman MR, et al. Hypoxia in human soft tissue sarcomas: adverse impact on survival and no association with p53 mutations. Br J Cancer 2001;84:1070–5.
- Vaupel P, Thews O, Kelleher DK, Hoeckel M. Oxygenation of human tumors: the Mainz experience. Strahlenther Onkol 1998;174 (Suppl 4):6–12.
- Magagnin MG, Koritzinsky M, Wouters BG. Patterns of tumor oxygenation and their influence on the cellular hypoxic response and hypoxia-directed therapies. Drug Resist Updat. 2006 Aug–Oct;9(4–5):185–97.
- Tatum JL, Kelloff GJ, Gillies RJ, Arbeit JM, Brown JM, Chao KS, et al. Hypoxia: importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Int J Radiat Biol 2006;82:699–757.
- Bussink J, Kaanders JH, Strik AM, Vojnovic B, van der Kogel AJ. Optical sensor-based oxygen tension measurements correspond with hypoxia marker binding in three human tumor xenograft lines. Radiat Res 2000;154:547–55.
- Vaupel P, Schlenger K, Knoop C, Hockel M. Oxygenation of human tumors: evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res 1991;51:3316–22.
- Koch CJ. Measurement of absolute oxygen levels in cells and tissues using oxygen sensors and 2-nitroimidazole EF5. Methods Enzymol 2002;352:3–31.
- Raleigh JA, Chou SC, Bono EL, Thrall DE, Varia MA. Semiquantitative immunohistochemical analysis for hypoxia in human tumors. Int J Radiat Oncol Biol Phys 2001;49:569–74.
- van Baardwijk A, Dooms C, van Suylen RJ, Verbeken E, Hochstenbag M, Dehing-Oberije C, et al. The maximum uptake of (18)F-deoxyglucose on positron emission tomography scan correlates with survival, hypoxia inducible factor-1alpha and GLUT-1 in non-small cell lung cancer. Eur J Cancer 2007;43:1392–8.
- Seigneuric R, Starmans MH, Fung G, Krishnapuram B, Nuyten DS, van Erk A, et al. Impact of supervised gene signatures of early hypoxia on patient survival. Radiother Oncol 2007;83:374–82.
- Chapman JD, Bradley JD, Eary JF, Haubner R, Larson SM, Michalski JM, et al. Molecular (functional) imaging for radiotherapy applications: an RTOG symposium. Int J Radiat Oncol Biol Phys 2003;55:294–301.
- Rajendran JG, Mankoff DA. Beyond detection: novel applications for PET imaging to guide cancer therapy. J Nucl Med 2007;48:855–6.
- Rasey JS, Grunbaum Z, Magee S, Nelson NJ, Olive PL, Durand RE, et al. Characterization of radiolabeled fluoromisonidazole as a probe for hypoxic cells. Radiat Res 1987;111:292–304.
- Bentzen L, Keiding S, Horsman MR, Gronroos T, Hansen SB, Overgaard J. Assessment of hypoxia in experimental mice tumors by [18F]fluoromisonidazole PET and pO2 electrode measurements. Influence of tumor volume and carbogen breathing. Acta Oncol 2002;41:304–12.
- Dubois L, Landuyt W, Haustermans K, Dupont P, Bormans G, Vermaelen P, et al. Evaluation of hypoxia in an experimental rat tumour model by [(18)F]fluoromisonidazole PET and immunohistochemistry. Br J Cancer 2004;91:1947–54.
- Gagel B, Reinartz P, Demirel C, Kaiser HJ, Zimny M, Piroth M, et al. [18F] fluoromisonidazole and [18F] fluorodeoxyglucose positron emission tomography in response evaluation after chemo-/radiotherapy of non-small-cell lung cancer: a feasibility study. BMC Cancer 2006;6:51.
- Rajendran JG, Schwartz DL, O’Sullivan J, Peterson ML, Ng P, Scharnhorst J, et al. Tumor hypoxia imaging with [F-18] fluoromisonidazole positron emission tomography in head and neck cancer. Clin Cancer Res 2006;12:5435–41.
- Dubois L, Landuyt W, Haustermans K, et al. Evaluation of hypoxia in an experimental rat tumour model by [(18)F]fluoromisonidazole PET and immunohistochemistry. Br J Cancer 2004;91:1947–1954.
- Troost EG, Bussink J, Kaanders JH, van Eerd J, Peters JP, Rijken PF, et al. Comparison of different methods of CAIX quantification in relation to hypoxia in three human head and neck tumor lines. Radiother Oncol 2005;76:194–9.
- Rajendran JG, Mankoff DA, O’Sullivan F, Peterson ML, Schwartz DL, Conrad EU, et al. Hypoxia and glucose metabolism in malignant tumors: evaluation by [18F]fluoromisonidazole and [18F]fluorodeoxyglucose positron emission tomography imaging. Clin Cancer Res 2004; 10:2245–52.
- Eschmann SM, Paulsen F, Bedeshem C, Machulla HJ, Hehr T, Bamberg M, et al. Hypoxia-imaging with (18)F-misonidazole and PET: changes of kinetics during radiotherapy of head-and-neck cancer. Radiother Oncol 2007;83:406–10.
- Lucignani G. PET imaging with hypoxia tracers: a must in radiation therapy. Eur J Nucl Med Mol Imaging 2008;35:838–42.
- Thorwarth D, Eschmann SM, Paulsen F, Alber M. A model of reoxygenation dynamics of head-and-neck tumors based on serial 18F-fluoromisonidazole positron emission tomography investigations. Int J Radiat Oncol Biol Phys 2007;68:515–21.
- Serganova I, Humm J, Ling C, Blasberg R. Tumor hypoxia imaging. Clin Cancer Res 2006;12:5260–4.
- Dubois L, Landuyt W, Cloetens L, Bol A, Bormans G, Haustermans K, et al. [(18)F]EF3 is not superior to [(18)F]FMISO for PET-based hypoxia evaluation as measured in a rat rhabdomyosarcoma tumour model. Eur J Nucl Med Mol Imaging 2009;36:209–18.
- Graham MA, Senan S, Robin H Jr, Eckhardt M, Lendrem D, Hincks J, et al. Pharmacokinetics of the hypoxic cell cytotoxic agent tirapazamine and its major bioreductive metabolites in mice and humans: retrospective analysis of a pharmacokinetically guided dose-escalation strategy in a phase I trial. Cancer Chemother Pharmacol 1997;40:1–10.
- Davda S, Bezabeh T. Advances in methods for assessing tumor hypoxia in vivo: implications for treatment planning. Cancer Metastasis Rev 2006;25:469–80.
- Pastorekova S, Parkkila S, Zavada J. Tumor-associated carbonic anhydrases and their clinical significance. Adv Clin Chem 2006;42:167–216.
- Svastova E, Zilka N, Zat’ovicova M, Gibadulinova A, Ciampor F, Pastorek J, et al. Carbonic anhydrase IX reduces E-cadherin-mediated adhesion of MDCK cells via interaction with beta-catenin. Exp Cell Res 2003;290:332–45.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Supuran CT. Development of small molecule carbonic anhydrase IX inhibitors. BJU Int. 2008 Jun;101 Suppl 4:39–40.
- Cecchi A, Hulikova A, Pastorek J, Pastorekova S, Scozzafava A, Winum JY, et al. Carbonic anhydrase inhibitors. Design of fluorescent sulfonamides as probes of tumor-associated carbonic anhydrase IX that inhibit isozyme IX-mediated acidification of hypoxic tumors. J Med Chem 2005;48:4834–41.
- Chrastina A, Zavada J, Parkkila S, Kaluz S, Kaluzova M, Rajcani J, et al. Biodistribution and pharmacokinetics of 125I-labeled monoclonal antibody M75 specific for carbonic anhydrase IX, an intrinsic marker of hypoxia, in nude mice xenografted with human colorectal carcinoma. Int J Cancer 2003;105:873–81.
- Divgi CR, Pandit-Taskar N, Jungbluth AA, Reuter VE, Gonen M, Ruan S, et al. Preoperative characterisation of clear-cell renal carcinoma using iodine-124-labelled antibody chimeric G250 (124I-cG250) and PET in patients with renal masses: a phase I trial. Lancet Oncol 2007;8:304–10.
- Lin AJ, Cosby LA, Shansky CW, Sartorelli AC. Potential bioreductive alkylating agents. 1.Benzoquinone derivatives. J Med Chem 1973;15:1247–52.
- Kennedy KA, Teicher BA, Rockwell S, Sartorelli AC. The hypoxic tumor cell: a target for selective cancer chemotherapy. Biochem Pharmacol 1980;29:1–8.
- Rockwell S. Effect of some proliferative and environmental factors on the toxicity of mitomycin C to tumor cells in vitro. Int J Cancer 1986;38:229–35.
- Stratford IJ, Adams GE, Godden J, Howells N. Induction of tumor hypoxia post-irradiation: a method for increasing the sensitizing efficiency of misonidazole and RSU 1069 in vivo. Int J Radiat Biol 1989;55:411–22.
- Traver RD, Horikoshi T, Danenberg KD, Stadlbauer TH, Danenberg PV, Ross D, et al. NAD(P)H:quinine oxidoreductase gene expression in human colon carcinoma cells: characterization of a mutation which modulates DT-diaphorase activity and mitomycin sensitivity. Cancer Res 1992;52:797–802.
- Gan Y, Mo Y, Kalns JE, Lu J, Danenberg K, Danenberg P, et al. Expression of DT-diaphorase and cytochrome P450 reductase correlates with mitomycin C activity in human bladder tumors. Clin Cancer Res 2001;7:1313–19.
- Cowen RL, Patterson AV, Telfer BA, Airley RE, Hobbs S, Phillips RM, et al. Viral delivery of P450 reductase recapitulates the ability of constitutive overexpression of reductase enzymes to potentiate the activity of mitomycin C in human breast cancer xenografts. Mol Cancer Ther 2003;2:901–9.
- Fracasso PM, Sartorelli AC. Cytotoxicity and DNA lesions produced by mitomycin C and porfiromycin in hypoxic and aerobic EMT6 and Chinese hamster ovary cells. Cancer Res 1986;46:3939–44.
- Keyes SR, Rockwell S, Sartorelli AC. Correlation between drug uptake and selective toxicity of porfiromycin to hypoxic EMT6 cells. Cancer Res 1987;47:5654–7.
- Rockwell S, Keyes SR, Sartorelli AC. Preclinical studies of porfiromycin as an adjunct to radiotherapy. Radiat Res 1988;116:100–13.
- Haffty BG, Wilson LD, Son YH, Cho EI, Papac RJ, Fischer DB, et al. Concurrent chemoradiotherapy with mitomycin C compared with porfiromycin in squamous cell cancer of the head and neck: final results of a randomized clinical trial. Int J Radiat Oncol Biol Phys 2005;61:119–28.
- Walton MI, Smith PJ, Workman P. The role of NAD(P)H: quinone reductase (EC1.6.99.2, DT-diaphorase) in the reductive bioactivation of the novel indoloquinone antitumor agent EO9. Cancer Commun 1991;3:199–206.
- Robertson N, Haigh A, Adams GE, Stratford IJ. Factors affecting sensitivity to EO9 in rodent and human tumor cells in vitro: DT-diaphorase activity and hypoxia. Eur J Cancer 1994;30A:1013–19.
- Saunders MP, Jaffar M, Patterson AV, Nolan J, Naylor MA, Phillips RM, et al. The relative importance of NADPH: cytochrome c (P450) reductase for determining the sensitivity of human tumor cells to the indolequinone EO9 and related analogues lacking functionality at the C-2 and C-3 positions. Biochem Pharmacol 2000;59:993–6.
- Loadman PM, Bibby MC, Phillips RM. Pharmacological approach towards the development of indolequinone bioreductive drugs based on the clinically inactive agent EO9. Br J Pharmacol 2002;137:701–9.
- Puri R, Palit V, Loadman PM, Flannigan M, Shah T, Choudry GA, et al. Phase I/II pilot study of intravesical apaziquone (EO9) for superficial bladder cancer. J Urol 2006;176:1344–8.
- Hendricksen K, Gleason D, Young JM, Saltzstein D, Gershman A, Lerner S, et al. Safety and side effects of immediate instillation of apaziquone following transurethral resection in patients with nonmuscle invasive bladder cancer. J Urol 2008;180:116–20.
- Stratford IJ, O’Neill P, Sheldon PW, Silver AR, Walling JM, Adams GE. RSU 1069, a nitroimidazole containing an aziridine group.Bioreduction greatly increases cytotoxicity under hypoxic conditions. Biochem Pharmacol 1986;35:105–9.
- Stratford IJ, Stephens MA. The differential hypoxic cytotoxicity of bioreductive agents determined in vitro by the MTT assay. Int J Radiat Oncol Biol Phys 1989;16:973–6.
- Stratford IJ, Williams KJ, Cowen RL, Jaffar M. Combining bioreductive drugs and radiation for the treatment of solid tumors. Semin Radiat Oncol 2003;13:42–52.
- Patterson AV, Saunders MP, Chinje EC, Talbot DC, Harris AL, Strafford IJ. Overexpression of human NADPH:cytochrome c (P450) reductase confers enhanced sensitivity to both tirapazamine (SR 4233) and RSU 1069. Br J Cancer 1997;76:1338–47.
- Patterson AV, Williams KJ, Cowen RL, Jaffar M, Telfer BA, Saunders M, et al. Oxygen-sensitive enzyme-prodrug gene therapy for the eradication of radiationresistant solid tumors. Gene Ther 2002;9:946–54.
- Horwich A, Holliday SB, Deacon JM, Peckham MJ. A toxicity and pharmacokinetic study in man of the hypoxic-cell radiosensitiser RSU-1069. Br J Radiol 1986;59:1238–40.
- Bremner JC. Assessing the bioreductive effectiveness of the nitroimidazole RSU1069 and its prodrug RB6145: with particular reference to in vivo methods of evaluation. Cancer Metastasis Rev 1993;12:177–93.
- Breider MA, Pilcher GD, Graziano MJ, Gough AW. Retinal degeneration in rats induced by Cl-1010, a 2-nitroimidazole radiosensitizer. Toxicol Pathol 1998;26:234–9.
- Brown JM. Tumor microenvironment and the response to anticancer therapy. Cancer Biol Ther 2002;1:453–8.
- Lunt SJ, Telfer BA, Fitzmaurice RJ, Stratford IJ, Williams KJ. Selective killing of hypoxic cells in primary tumors using tirapazamine reduces metastatic dissemination. Clin Cancer Res 2005;11:4212–16.
- Cowen RL, Williams KJ, Chinje EC, Jaffar M, Sheppard FC, Telfer BA, et al. Hypoxia targeted gene therapy to increase the efficacy of tirapazamine as an adjuvant to radiotherapy: reversing tumor radioresistance and effecting cure. Cancer Res 2004;64:1396–402.
- McKeown SR, Cowen RL, Williams KJ. Bioreductive drugs: from concept to clinic. Clin Oncol 2007;19:427–42.
- Raleigh SM, Wanogho E, Burke MD, Patterson LH. Rat cytochromes P450 (CYP) specifically contribute to the reductive bioactivation of AQ4N, an alkylaminoanthraquinone-di-N-oxide anticancer prodrug. Xenobiotica. 1999 Nov;29(11):1115–22.
- Fitzpatrick B, Mehibel M, Cowen RL, Stratford IJ. iNOS as a therapeutic target for treatment of human tumors. Nitric Oxide 2008;19:217–24.
- Nishida CR, Ortiz de Montellano PR. Reductive heme-dependent activation of the n-oxide prodrug AQ4N by nitric oxide synthase. J Med Chem 2008;5:5118–20.
- McKeown SR, Hejmadi MV, McIntyre IA, McAleer JJ, Patterson LH. AQ4N: an alkylaminoanthraquinone N-oxide showing bioreductive potential and positive interaction with radiation in vivo. Br J Cancer 1995;72:76–81.
- McKeown SR, Friery OP, McIntyre IA, Hejmadi MV, Patterson LH, Hirst DG. Evidence for a therapeutic gain when AQ4N or tirapazamine is combined with radiation. Br J Cancer 1996;74:S39–42.
- Patterson LH, McKeown SR, Ruparelia K, Double JA, Bibby MC, Cole S, et al. Enhancement of chemotherapy and radiotherapy of murine tumors by AQ4N, a bioreductively activated anti-tumor agent. Br J Cancer 2000;82:1984–90.
- Friery OP, Gallagher R, Murray MM, Hughes CM, Galligan ES, McIntyre IA, et al. Enhancement of the anti-tumor effect of cyclophosphamide by the bioreductive drugs AQ4N and tirapazamine. Br J Cancer 2000;82:1469–73.
- Gallagher R, Hughes CM, Murray MM, Friery OP, Patterson LH, Hirst DG, et al. The chemopotentiation of cisplatin by the novel bioreductive drug AQ4N. Br J Cancer 2001;85:625–9.
- Albertella MR, Loadman PM, Jones PH, Phillips RM, Rampling R, Burnet M, et al. Hypoxia-selective targeting by the bioreductive prodrug AQ4N in patients with solid tumors: results of a phase I study. Clin Cancer Res 2008;14:1096–104.
- Rischin D, Hicks RJ, Fisher R, Binns D, Corry J, Porceddu S, et al. Prognostic significance of [18F]-misonidazole positron emission tomography-detected tumor hypoxia in patients with advanced head and neck cancer randomly assigned to chemoradiation with or without tirapazamine: a substudy of Trans-Tasman Radiation Oncology Group study 98.02. J Clin Oncol 2006;24:2098–104.
- Papadopoulou MV, Bloomer WD. NLCQ-1 (NSC 709257): exploiting hypoxia with a weak DNA-intercalating bioreductive drug. Clin Cancer Res 2003;9:5714–20.
- Papadopoulou MV, Ji M, Rao MK, Bloomer WD. Reductive metabolism of the nitroimidazole-based hypoxia-selective cytotoxin NLCQ-1 (NSC 709257). Oncol Res 2003;14:21–9.
- Papadopoulou MV, Ji M, Bloomer WD. Schedule-dependent potentiation of chemotherapeutic drugs by the bioreductive compounds NLCQ-1 and tirapazamine against EMT6 tumors in mice. Cancer Chemother Pharmacol 2001;48:160–8.
- Papadopoulou MV, Ji M, Rao MK, Bloomer WD. 4-[3-(2-Nitro-1- imidazolyl)propylamino]-7-chloroquinoline hydrochloride (NLCQ-1), a novel bioreductive agent as radiosensitizer in vitro and in vivo: comparison with tirapazamine. Oncol Res 2001;12:325–33.
- Papadopoulou MV, Ji M, Bloomer WD. Synergistic enhancement of the antitumor effect of taxol by the bioreductive compound NLCQ-1, in vivo: comparison with tirapazamine. Oncol Res 2002;13:47–54.
- Papadopoulou MV, Ji M, Ji X, Bloomer WD. Therapeutic advantage from combining 5-fluorouracil with the hypoxia-selective cytotoxin NLCQ-1 in vivo; comparison with tirapazamine. Cancer Chemother Pharmacol 2002;50: 291–8.
- Papadopoulou MV, Ji X, Bloomer WD. Potentiation of alkylating agents by NLCQ-1 or TPZ in vitro and in vivo. J Exp Ther Oncol 2006;5:261–72.
- Wilson WR, Pullen SM, Degenkolbe A, Ferry DM, Helsby NA, Hicks KO, Atwell GJ, Yang S, Denny WA, Patterson AV. Water-soluble dinitrobenzamide mustard phosphate pre-prodrugs as hypoxic cytotoxins. Eur J Cancer. 2004;Suppl. 2(8):151.
- Patterson AV, Ferry DM, Edmunds SJ, Gu Y, Singleton RS, Patel K, et al. Mechanism of action and preclinical antitumor activity of the novel hypoxia-activated DNA cross-linking agent PR-104. Clin Cancer Res 2007;13:3922–32.
- Danson S, Johnson P, Ward T, Dawson M, Denneny O, Watson A. et al. Final results of a phase I clinical trial of the bioreductive drug RH1. J Clin Oncol 2007;25(18S):2514.
- Yan C, Kepa JK, Siegel D, Stratford IJ, Ross D. Dissecting the role of multiple reductases in bioactivation and cytotoxicity of the antitumor agent 2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1). Mol Pharmacol. 2008 Dec;74(6):1657–65. Epub 2008 Sep 15.
- Loadman PM, Phillips RM, Lim LE, Bibby MC. Pharmacological properties of a new aziridinylbenzoquinone, RH1 (2,5-diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone), in mice. Biochem Pharmacol 2000;59:831–7.
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89.
- Pastorekova S, Pastorek, J. Cancer-related carbonic anhydrase isozymes. In: Supuran CT, Scozzafava A, Conway J, eds. Carbonic Anhydrase—Its Inhibitors and Activators. Boca Raton, FL: CRC Press, 2004: 253–80.
- Hilvo M, Monti SM, De Simone G, Supuran CT, Parkkila S. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 2008;283:27799–809.
- Winum JY, Rami M, Scozzafava A, Montero JL, Supuran C. Carbonic anhydrase IX: a new druggable target for the design of antitumor agents. Med Res Rev 2008;28:445–63.
- Thiry A, Supuran CT, Masereel B, Dogné JM. Recent developments of carbonic anhydrase inhibitors as potential anticancer drugs. J Med Chem 2008;51:3051–6.
- Cecchi A, Supuran CT. Fluorescence- and spin-labeled carbonic anhydrase inhibitors. Curr Pharm Des 2008;14:699–707.
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229.
- Menchise V, De Simone G, Alterio V, Di Fiore A, Pedone C, Scozzafava A, et al. Carbonic anhydrase inhibitors: stacking with Phe131 determines active site binding region of inhibitors as exemplified by the X-ray crystal structure of a membrane-impermeant antitumor sulfonamide complexed with isozyme II. J Med Chem 2005;48:5721–7.
- Alterio V, Vitale RM, Monti SM, Pedone C, Scozzafava A, Cecchi A, et al. Carbonic anhydrase inhibitors: X-ray and molecular modeling study for the interaction of a fluorescent antitumor sulfonamide with isozyme II and IX. J Am Chem Soc 2006;128:8329–35.
- Abbate F, Casini A, Owa T, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: E7070, a sulfonamide anticancer agent, potently inhibits cytosolic isozymes I and II, and transmembrane, tumor-associated isozyme IX. Bioorg Med Chem Lett 2004;14:217–23.
- Garaj V, Pucetti L, Fasolis G, Winum J-Y, Montero J-L, Scozzafava A, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, and IX with sulfonamides incorporating 1,2,4-triazine moieties. Bioorg Med Chem Lett 2004;14:5427–33.
- De Simone G, Vitale RM, Di Fiore A, Pedone C, Scozzafava A, Montero JL, et al. Carbonic anhydrase inhibitors: hypoxia-activatable sulfonamides incorporating disulfide bonds that target the tumor-associated isoform IX. J Med Chem 2006;49:5544–51.
- Robertson N, Potter C, Harris AL. Role of carbonic anhydrase IX in human tumor cell growth, survival, and invasion. Cancer Res 2004;64:6160–5.
- Oosterwijk E. Carbonic anhydrase IX: historical and future perspectives. BJU Int 2008;101(Suppl 4):2–7.
- Lam JS, Pantuck AJ, Belldegrun AS, Figlin RA. G250: a carbonic anhydrase IX monoclonal antibody. Curr Oncol Rep 2005;7:109–15.
- Görlach A, Kietzmann T. Superoxide and derived reactive oxygen species in the regulation of hypoxia-inducible factors. Methods in Enzymology. 2007;435:421–46.
- Kietzmann T, Görlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression, Semin Cell Dev Biol. 2005;16:474–86.
- Toffoli S, Michiels C. <http://www.ncbi.nlm.nih.gov/pubmed/18445039?ordinalpos=10&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Intermittent hypoxia is a key regulator of cancer cell and endothelial cell interplay in tumours. FEBS J. 2008;275:2991–3002.
- Dewhirst MW, Cao Y, Moeller B. <http://www.ncbi.nlm.nih.gov/pubmed/18500244?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425–37.
- Nanduri J, Yuan G, Kumar GK, Semenza GL, Prabhakar NR. <http://www.ncbi.nlm.nih.gov/pubmed/18692603?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Transcriptional responses to intermittent hypoxia. Respir Physiol Neurobiol. 2008 164:277–81.
- Dewhirst MW. <http://www.ncbi.nlm.nih.gov/pubmed/17283112?ordinalpos=13&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Intermittent hypoxia furthers the rationale for hypoxia-inducible factor-1 targeting. Cancer Res. 2007;67:854–5.
- Martínez-Sánchez G, Giuliani A. <http://www.ncbi.nlm.nih.gov/pubmed/17550131?ordinalpos=39&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Cellular redox status regulates hypoxia inducible factor-1 activity. Role in tumour development. J Exp Clin Cancer Res. 2007;26:39–50.
- Bell EL, Chandel NS. <http://www.ncbi.nlm.nih.gov/pubmed/17705790?ordinalpos=13&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Mitochondrial oxygen sensing: regulation of hypoxia-inducible factor by mitochondrial generated reactive oxygen species. Essays Biochem. 2007;43:17–27.
- Cash TP, Pan Y, Simon MC. <http://www.ncbi.nlm.nih.gov/pubmed/17893032?ordinalpos=23&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Reactive oxygen species and cellular oxygen sensing. Free Radic Biol Med. 2007;43:1219–25.
- Guzy RD, Schumacker PT. <http://www.ncbi.nlm.nih.gov/pubmed/16857720?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–19.
- Wang J, Yi J. <http://www.ncbi.nlm.nih.gov/pubmed/18981733?ordinalpos=4&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Cancer cell killing via ROS: To increase or decrease, that is a question. Cancer Biol Ther. 2008;7:1875–84.
- Matsumoto K, Subramanian S, Murugesan R, Mitchell JB, Krishna MC. <http://www.ncbi.nlm.nih.gov/pubmed/17571957?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum> Spatially resolved biologic information from in vivo EPRI, OMRI, and MRI. Antioxid Redox Signal. 2007;9:1125–41.