
Abstract
A series of N-(5-methyl-isoxazol-3-yl/1,3,4-thiadiazol-2-yl)-4-(3-substitutedphenylureido) benzenesulfonamide derivatives has been designed, synthesized and screened for their in vitro human carbonic anhydrase (hCA; EC 4.2.1.1) inhibition potential. These newly synthesized sulfonamide compounds were assessed against isoforms hCA I, II, VII and XII, with acetazolamide (AAZ) as a reference compound. The majority of these compounds were found quite weak inhibitor against all tested isoforms. Compound 15 showed a modest inhibition potency against hCA I (Ki = 73.7 μM) and hCA VII (Ki = 85.8 μM). Compounds 19 and 25 exhibited hCA II inhibition with Ki values of 96.0 μM and 87.8 μM, respectively. The results of the present study suggest that, although the synthesized derivatives have weak inhibitory potential towards all investigated isoforms, some of them may serve as lead molecules for the further development of selective inhibitors incorporating secondary sulfonamide functionalities, a class of inhibitors for which the inhibition mechanism is poorly understood.
Introduction
The carbonic anhydrases (CAs; EC 4.2.1.1) are a large group of ubiquitous zinc containing enzymes, present in all mammals, including humanCitation1. CAs are encoded by six distinct gene families known as alpha (α), beta (β), gamma (γ), delta (δ) zeta (ζ) and eta (η) and all human CAs belongs to class alphaCitation2–4. These enzymes are well described by their different molecular features, oligomeric arrangement as well as kinetic propertiesCitation5. Till date, a total number of 15 different human carbonic anhydrases (hCA) isoforms have been discovered, among which CA I-III, CA VII and CA XIII are present in cytosol; CA IV, CA IX, CA XII and CA XIV are membrane bound; CA VA and CA VB are restricted to the mitochondrion and CA VI is secreted in milk and salivaCitation6. Functionally, CAs are primarily involved in catalyzing the rapid interconversion of CO2 and H2O to bicarbonate (HCO3-) and protons (H+) using a metal hydroxide nucleophilic mechanismCitation7. The hCAs play a pivotal role in a variety of physiological processes such as CO2/bicarbonate transportation, respiration, electrolyte secretion, gluoconeogensis, lipogenesis, ureagenesis, bone resorption, neuronal excitability and tumorigenicityCitation8. CAs have profound involvement in many pathological conditions like obesity, glaucoma, kidney dysfunction, osteoporosis, gastric ulcers, migraine, epilepsy and cancerCitation9. For example CA II deficiency is the major defect in disease conditions such as osteopetrosis, cerebral calcification and renal tubular acidosisCitation10. Another isoform, CA VII is implicated in generating neuronal excitation leading to the development of seizures as well as in the neuropathic pain controlCitation11. hCA II, IV and XII are druggable targets for anti-glaucoma agents while, hCA VII and hCA XIV are known drug targets for antiepileptic drugs. hCA IX and XII are anti-tumor drug targets.At present, CAs are attractive therapeutic targets to control these aforementioned diseases. The CAs are also essential for other living organism including pathogens.
Acetazolamide (AAZ) is considered as an excellent CA inhibitor (CAI) and is used for the treatment of glaucoma, epilepsy and altitude sicknessCitation12. Several other CAIs such as ethoxolamide (EZA), brinzolamide (BRZ), dorzolamide (DZA), methazolamide (MZA), and dichlorophenamide (DCP) are useful clinically as anti-glaucoma agents but, they are also used to treat various neurological as well as neuromuscular disorders such as epilepsy, genetic hemiplegic migraine and ataxia, tardive diskinesia, hypokalemic periodic paralysis, essential tremor and Parkinsons diseaseCitation13. Unfortunately, the presently used CAIs are non-selective to a particular CA isoform, which leads to the occurrence of peripheral side effects. Therefore, the development of novel CAIs having high potency and selectivity against specific isoform still represents a needful and challenging task to obtain newer compounds which could replace the “old ones”, in consequence avoiding side effects as well as improving therapeutic safety.
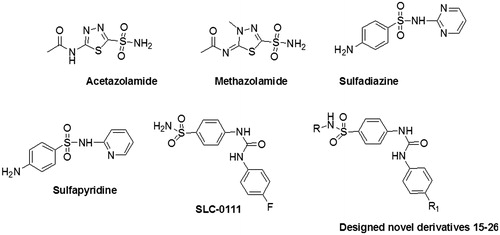
Sulfonamides containing molecules have presented a remarkable history in CA inhibition. The primary sulfonamide group (-SO2NH2) is present in these molecules which bind as anions to the Zn2+ ion in the enzyme active site with high affinity, and block catalysisCitation14. In the past, several research reports showed that the urea functional group along with sulfonamide also seemed to be beneficial to generate effective CA inhibitory activityCitation15. In the present study, we have used sulfamethoxazole/sulfamethiazole to develop potent CAIs and also phenyl urea moiety was also installed to this pharmacophore to develop new CAIs. showed some known CAIs and the molecular framework of our synthesized compound. The synthesized derivatives have been well characterized by using 1H NMR, 13C NMR and mass spectroscopy. The purity was analyzed by HPLC. The esterase activity was performed to study the potential of the newly synthesized compounds (15–26) towards hCA I, hCA II, hCA VII and hCA XII inhibition.
Figure 1. Sulfonamides CA inhibitors (acetazolamide, methazolamide and SLC-0111), sulfadrugs (sulfadiazine and sulfapyridine) and the designed novel compounds 15–26.
Materials and methods
Chemistry
All the chemicals and reagents were obtained from Sigma Aldrich (St. Louis, MO), Alfa Aesar (Massachusetts), S.D Fine Chemicals (India) and Merck (Darmstadt, Germany) and solvents for reaction medium were dried by standard methods. Analytical thin layer chromatography (TLC) was performed on commercially available silica gel (Kieselgel 60, F254) coated aluminum plates (Merck). Column chromatography purification was performed using silica gel Merck 100–200 mesh. Melting points were taken in open capillaries using model KSPII, KRUSS, (Germany). 1H NMR and 13C NMR were recorded on Jeol-400 MHz High Resolution NMR Spectrophotometer (JEOL USA, Inc., Peabody, MA). Chemical shifts in 1H NMR are reported in parts per million and coupling constant (J) in Hertz (Hz) using DMSO-d6 as solvent. The splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet. Mass spectra were recorded on an Agilent 6310 Ion trap LC/MS and the purity of final compounds was analyzed by using reverse phase HPLC (Shimadzu, Kyoto, Japan) with C-18 column.
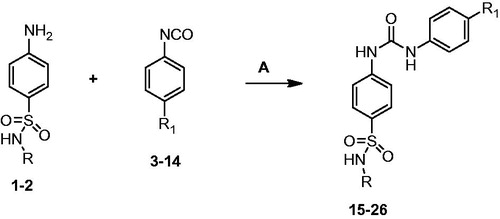
General procedure for the synthesis of novel N-(5-methyl-isoxazol-3-yl/1,3,4-thiadiazol-2-yl)-4–(3-substitutedphenylureido) benzenesulfonamides (15–26)
Sulfamethoxazole/sulfamethiazole (10 mmol) was fully dissolved in dried DMF and then subsequent isocyanate (10 mmol) was added dropwise to the reaction mixture and heated at 90–100 °C for 5–6 h. The appeared precipitate was filtered and washed with hot petroleum ether as well as water. The obtained crude products were purified by column chromatography using chloroform/methanol (98:02) as an eluent to furnish target compounds (15–26).The purity of the synthesized compounds was analyzed by HPLC using acetonitrile/methanol (98:02) as mobile phase ().
Scheme 1. Synthesis of N-(5-methyl-isoxazol-3-yl/1,3,4-thiadiazol-2-yl)-4-(3-substituted phenylureido)benzenesulfonamide derivatives. Reagent and conditions: A. Dried DMF reflux, 5–6 h.
N-(5-Methyl-isoxazol-3-yl)-4–(3-phenyl-ureido)-benzenesulfonamide (15)
White solid; yield 1.74 g; mp 240–242 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.28 (s, 3H, CH3), 6.13 (s, 1H, CH), 6.98 (t, 1H, Ar-H, J = 7.2 Hz), 7.27 (t, 2H, Ar-H, J = 7.6 Hz), 7.44 (d, 2H, Ar-H, J = 7.6 Hz), 7.63 (d, 2H, Ar-H, J = 9.1 Hz), 7.75 (d, 2H, Ar-H, J = 7.7 Hz), 8.82 (s, 1H, NH), 9.15 (s, 1H, NH), 11.29 (s, 1H, NH). 13C NMR (DMSO-d6): 12.08, 95.3, 117.7, 118.4, 122.3, 128.1, 128.8, 131.4, 139.1, 144.3, 152.1, 157.6, 170.2. LC–MS: m/e; 372 (M+). HPLC purity: 97.6%.
4-[3–(4-Fluoro-phenyl)-ureido]-N-(5-methyl-isoxazol-3-yl)-benzenesulfonamide (16)
White solid; yield 1.92 g; mp 228–230 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.28 (s, 3H, CH3), 6.12 (s, 1H, CH), 7.12 (t, 2H, Ar-H, J = 8.7 Hz), 7.43–7.47 (m, 2H, Ar-H), 7.61 (d, 2H, Ar-H, J = 9.1 Hz), 7.76 (d, 2H, Ar-H, J = 8.4 Hz), 8.85 (s, 1H, NH), 9.10 (s, 1H, NH), 11.28 (s, 1H, NH). 13C NMR (DMSO-d6): 12.07, 95.3, 115.2, 115.5, 117.7, 120.2, 120.3, 128.1, 131.5, 135.4, 144.3, 152.2, 156.4, 157.6, 158.7, 170.2. LC–MS: m/e; 391 (M + 1). HPLC purity: 97.1%.
4-[3–(4-Chloro-phenyl)-ureido]-N-(5-methyl-isoxazol-3-yl)-benzenesulfonamide (17)
White solid; yield 2.35 g; mp 238–240 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.28 (s, 3H, CH3), 6.12 (s, 1H, CH), 7.33 (d, 2H, Ar-H, J = 8.4 Hz), 7.47 (d, 2H, Ar-H, J= 8.4 Hz), 7.62 (d, 2H, Ar-H, J = 9.1 Hz), 7.75 (d, 2H, Ar-H, J = 8.4 Hz), 8.97 (s, 1H, NH), 9.20 (s, 1H, NH), 11.28 (s, 1H, NH). 13C NMR (DMSO-d6): 12.09, 95.4, 117.8, 120.0, 125.9, 128.1, 131.6, 138.2, 144.1, 152.1, 157.6, 170.2. LC–MS: m/e; 406 (M+). HPLC purity: 97.9%.
N-(5-Methyl-isoxazol-3-yl)-4-(3-p-tolyl-ureido)-benzenesulfonamide (18)
White solid; yield 1.49 g; mp 233–235 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.23 (s, 3H, CH3), 2.28 (s, 3H, CH3), 6.12 (s, 1H, CH), 7.08 (d, 2H, Ar-H, J = 7.6 Hz), 7.32 (d, 2H, Ar-H, J = 8.4 Hz), 7.61 (d, 2H, Ar-H, J = 9.1 Hz), 7.74 (d, 2H, Ar-H, J = 8.4 Hz), 8.71 (s, 1H, NH), 9.11 (s, 1H, NH), 11.20 (s, 1H, NH). 13C NMR (DMSO-d6): 12.0, 20.3, 95.3, 117.3, 118.5, 128.1, 129.2, 131.2, 131.3, 136.6, 144.4, 152.1, 157.6, 170.2. LC–MS: m/e; 386 (M+). HPLC purity: 99.4%.
4-[3–(4-Methoxy-phenyl)-ureido]-N-(5-methyl-isoxazol-3-yl)-benzenesulfonamide (19)
White solid; yield 1.84 g; mp 212–214 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.28 (s, 3H, CH3), 3.70 (s, 3H, CH3), 6.12 (s, 1H, CH), 6.86 (d, 2H, Ar-H, J = 8.4 Hz), 7.34 (d, 2H, Ar-H, J = 8.4 Hz), 7.61 (d, 2H, Ar-H, J = 8.4 Hz), 7.73 (d, 2H, Ar-H, J = 9.1 Hz), 8.63 (s, 1H, NH), 9.08 (s, 1H, NH), 11.21 (s, 1H, NH). 13C NMR (DMSO-d6): 12.0, 55.1, 95.3, 114.0, 117.5, 120.3, 128.1, 131.2, 132.1, 144.5, 152.3, 154.8, 157.6, 170.2. LC–MS: m/e; 402 (M+). HPLC purity: 99.7%.
4-(3-Benzyl-ureido)-N-(5-methyl-isoxazol-3-yl)-benzenesulfonamide (20)
White solid; yield 2.12 g; mp 218–220 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.27 (s, 3H, CH3), 4.29 (d, 2H, CH2, J = 6.1 Hz), 6.10 (s, 1H, CH), 6.82 (t, 1H, NH, J = 5.7 Hz), 7.21–7.33 (m, 5H, Ar-H), 7.57 (d, 2H, Ar-H, J = 9.1 Hz), 7.69 (d, 2H, Ar-H, J = 8.4 Hz), 9.08 (s, 1H, NH), 11.22 (s, 1H, NH). 13C NMR (DMSO-d6): 12.0, 42.7, 95.3, 117.1, 126.8, 127.1, 128.1, 128.3, 130.7, 139.9, 145.0, 154.7, 157.7, 170.2. LC–MS: m/e; 387 (M + 1). HPLC purity: 99.4%.
N-(5-methyl-[1,3,4]thiadiazol-2-yl)-4-(3-phenyl-ureido)-benzenesulfonamide (21)
White solid; yield 2.16 g; mp 226–228 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.45 (s, 3H, CH3), 6.98 (t, 1H, Ar-H, J = 7.6 Hz), 7.28 (t, 2H, Ar-H, J = 7.9 Hz), 7.44 (d, 2H, Ar-H, J = 8.4 Hz), 7.58 (d, 2H, Ar-H, J = 9.1 Hz), 7.68 (d, 2H, Ar-H, J = 9.1 Hz), 8.77 (s, 1H, NH), 9.09 (s, 1H, NH), 13.81 (s, 1H, NH). 13C NMR (DMSO-d6): 16.1, 117.6, 118.4, 122.2, 127.1, 128.8, 134.3, 139.2, 143.5, 152.2, 154.3, 167.7. LC–MS: m/e; 390 (M + 1). HPLC purity: 96.8%.
4-[3-(4-Fluoro-phenyl)-ureido]-N-(5-methyl-[1,3,4]thiadiazol-2-yl)-benzene sulfonamide (22)
White solid; yield 1.64 g; mp 252–254 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.45 (s, 3H, CH3), 7.10–7.14 (m, 2H, Ar-H), 7.43–7.47 (m, 2H, Ar-H), 7.58 (d, 2H, Ar-H, J = 8.3 Hz), 7.68 (d, 2H, Ar-H, J = 8.4 Hz), 8.81 (s, 1H, NH), 9.10 (s, 1H, NH), 13.82 (s, 1H, NH). 13C NMR (DMSO-d6): 16.1, 115.2, 115.5, 117.6, 120.2, 120.3, 127.0, 134.4, 135.6, 143.5, 152.3, 154.4, 156.4, 158.8, 167.7. LC–MS: m/e; 408 (M + 1). HPLC purity: 99.5%.
4-[3–(4-Chloro-phenyl)-ureido]-N-(5-methyl-[1,3,4]thiadiazol-2-yl)-benzene sulfonamide (23)
White solid; yield 2.0 g; mp 275–277 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.44 (s, 3H, CH3), 7.32 (d, 2H, Ar-H, J = 9.1 Hz), 7.48 (d, 2H, Ar-H, J = 9.1 Hz), 7.59 (d, 2H, Ar-H, J = 8.4 Hz), 7.69 (d, 2H, Ar-H, J = 9.1 Hz), 8.93 (s, 1H, NH), 9.14 (s, 1H, NH), 13.8 (s, 1H, NH).13C NMR (DMSO-d6): 16.1, 117.7, 120.0, 125.8, 127.1, 128.7, 134.5, 138.3, 143.3, 152.1, 154.4, 167.7. LC–MS: m/e; 423 (M+). HPLC purity: 98.6%.
N-(5-methyl-[1,3,4]thiadiazol-2-yl)-4-(3-p-tolyl-ureido)-benzenesulfonamide (24)
White solid; yield 1.75 g; mp 258–260 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.23 (s, 3H, CH3), 2.45 (s, 3H, CH3), 7.08 (d, 2H, Ar-H, J = 8.4 Hz), 7.32 (d, 2H, Ar-H, J = 8.4 Hz), 7.57 (d, 2H, Ar-H, J = 9.9 Hz), 7.67 (d, 2H, Ar-H, J = 9.9 Hz), 8.66 (s, 1H, NH), 9.05 (s, 1H, NH), 13.8 (s, 1H, NH).13C NMR (DMSO-d6): 16.09, 20.3, 117.5, 118.5, 127.0, 129.2, 131.1, 134.1, 136.6, 143.5, 152.2, 154.3, 167.7. LC–MS: m/e; 403 (M+). HPLC purity: 96.7%.
4-[3-(4-Methoxy-phenyl)-ureido]-N-(5-methyl-[1,3,4]thiadiazol-2-yl)-benzenesulfonamide (25)
White solid; yield 2.0 g; mp 238–240 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.48 (s, 3H, CH3), 3.70 (s, 3H, CH3), 6.86 (d, 2H, Ar-H, J = 9.1 Hz), 7.34 (d, 2H, Ar-H, J = 9.1 Hz), 7.57 (d, 2H, Ar-H, J = 8.4 Hz), 7.65 (d, 2H, Ar-H, J = 8.4 Hz), 8.58 (s, 1H, NH), 9.02 (s, 1H, NH), 13.8 (s, 1H, NH).13C NMR (DMSO-d6): 16.0, 55.1, 114.0, 117.4, 120.2, 127.0, 132.2, 134.0, 143.6, 152.3, 154.7, 167.7. LC–MS: m/e; 419 (M+). HPLC purity: 99.3%.
4-(3-Benzyl-ureido)-N-(5-methyl-[1,3,4]thiadiazol-2-yl)-benzenesulfonamide (26)
White solid; yield 2.2 g; mp 270–272 °C; 1H NMR (DMSO-d6,400 MHz): δ 2.44 (s, 3H, CH3), 4.28 (q, 2H, CH2, J = 6.0 Hz), 6.76 (t, 1H, NH, J = 6.1 Hz), 7.20–7.33 (m, 5H, Ar-H), 7.53 (d, 2H, Ar-H, J = 9.1 Hz), 7.62 (d, 2H, Ar-H, J = 8.4 Hz), 9.02 (s, 1H, NH), 13.8 (s, 1H, NH).13C NMR (DMSO-d6): 16.0, 42.7, 117.0, 126.8, 127.0, 127.1, 128.3, 133.5, 140.0, 144.2, 154.2, 154.8, 167.6. LC–MS: m/e; 404 (M + 1). HPLC purity: 99.3%.
Carbonic anhydrase inhibition assay
An applied photophysics stopped-flow instrument has been used for assaying the CA catalyzed CO2 hydration activityCitation16. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.4) as buffer, and 20 mM Na2SO4 for maintaining constant the ionic strength (this anion is not inhibitory and has a KI> 200 mM against these enzymes), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each measurement at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity, working with 10-fold decreasing inhibitor concentrations ranging between 0.1 nM and 10–100 μM (depending on the inhibitor potency, but at least five points at different inhibitor concentrations were employed for determining the inhibition constants). The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.1 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using the Cheng–Prusoff equation, and represent the mean from at least three different determinations. The human isoforms hCA I, II, VII and XII were recombinant enzymes produced as described earlier in our laboratoryCitation17–32.
Result and discussion
Chemistry
The synthetic route utilized for the preparation of N-(5-methyl-isoxazol-3-yl/1,3,4-thiadiazol-2-yl)-4–(3-substitutedphenylureido) benzenesulfonamide derivativesis depicted in .
An equimolar ratio of sulfamethoxazole/sulfamethiazole and substituted phenyl isocyantes were reacted in dry DMF to afford N-(5-methyl-isoxazol-3-yl/1,3,4-thiadiazol-2-yl)-4–(3-substituted phenylureido)benzenesulfonamide derivatives. All compounds were purified by using column chromatography using chloroform/methanol as eluent. The purity of the final compounds was analyzed by HPLC analysis using acetonitrile/methanol (98:02) as mobile phase. All compounds presented percent purity more than 95% and were fully characterized by NMR and mass spectroscopy.
We have used SLC-0111 as lead molecule for designing these compoundsCitation33,Citation34, as this derivative recently completed successfully the Phase I clinical trials as an anti-tumor agent. In addition, a range of secondary and tertiary sulfonamides has been investigated as CAIs recently, some of them showing effective and selective inhibition of several important isoformsCitation35–49. However, the inhibition mechanism with secondary and tertiary sulfonamides is unknown, as no X-ray crystal structures of such derivatives bound to the CA are available to date. Thus, the sulfonamides reported here incorporate in their molecule the urea fragment found in SLC-0111 and the secondary sulfonamide moiety present in sulfa drugs and several recently investigated CAIsCitation35–49.
In vitro carbonic anhydrase activity
All the synthesized compounds (15–26) were studied for the inhibition of four CA isozymes of human origin, i.e. hCA I, hCA II, hCA VII and hCA XII (). The following structure activity relationship (SAR) was obtained by analyzing CA inhibition data of :
In the isoxazole subseries (15–20) compound substituted phenyl ring (compound 15) at the terminal end has mild hCA inhibitory activity for both hCA I (Ki = 73.7 μM)and hCA VII (Ki = 85.8 μM) isoforms.
In this subseries, compound substituted with para-methoxy phenyl (compound 19) also exhibited low inhibition against hCA II (Ki = 96.0 μM). Moreover, this compound displayed a Ki value of >100 μM towards hCA I, VII and XII, thus seems to inactive for these isoforms. The remaining compounds in this subseries did not produce remarkable inhibitory potential against hCA I, II, VII and XII isoforms.
In thiadiazole subseries (21–26), compound 25 comprising para-methoxy phenyl ring at its terminal end has shown a modest inhibitory potential against hCA II with Ki value of 87.8 μM. This compound was inactive against hCA I, VII and XII (Ki= >100 μM).
Overall, the SAR study indicates that substitutions on the phenyl ring did not produce remarkable inhibitory potential against tested hCA isoforms except para-methoxy substitution. Therefore, further structural modifications are necessarily required to achieve more potent CAIs.
Table 1. hCA I, II, VII and XII inhibition with compounds 15–26, with AAZ as standard.
Table
Conclusions
A set of N-(5-methyl-isoxazol-3-yl/1,3,4-thiadiazol-2-yl)-4–(3-substitutedphenylureido) benzenesulfonamide derivatives were synthesized in a good yield and were characterized by using NMR and mass spectroscopy techniques. All compounds were subjected for their in-vitro CA inhibitory activity and only compound 15 showed CA inhibition against hCA I and hCA VII isoform. The selectivity ratio of compounds 19 and 25 for hCA II isoform makes them interesting leads for the development of potent and selective hCA II inhibitor.
Declaration of interest
Chandra Bhushan Mishra (DSK-PDF) and Shikha Kumari are thankful to the University Grant Commission for providing financial support. Author Amresh Prakash is thankful to SERB-YSS for financial support. Manisha Tiwari deeply acknowledges the financial support to carry out this research work. Work from Supuran laboratory was financed by two EU grants (Dynano and Metoxia).
Acknowledgements
The University Science Instrumentation Center (USIC), University of Delhi is acknowledged for providing NMR and Mass spectral characterization of the synthesized compounds.
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Del Pretea S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum-the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96
- Ceruso M, Antel S, Vullo D, et al. Inhibition studies of new ureido-substituted sulfonamides incorporating a GABA moiety against human carbonic anhydrase isoforms I-XIV. Bioorg Med Chem Lett 2014;22:6768–75
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–7
- Alterio V, Di Fiore A, D Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;8:4421–68
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–50
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem 2014;6:1149–65
- Shah GN, Bonapace G, Hu PY, et al. Carbonic anhydrase II deficiency syndrome (osteopetrosis with renal tubular acidosis and brain calcification): novel mutations in CA2 identified by direct sequencing expand the opportunity for genotype-phenotype correlation. Hum Mutat 2004;3:1–9
- Thiry A, Masereel B, Dogne JM, et al. Exploration of the binding mode of indanesulfonamides as selective inhibitors of human carbonic anhydrase type VII by targeting Lys 91. ChemMedChem 2007;2:1273–80
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO(2) capture. J Enzyme Inhib Med Chem 2013;28:229–30
- Supuran CT. Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;2:146–89
- Supuran CT, Winum JY. Designing carbonic anhydrase inhibitors for the treatment of breast cancer. Expert Opin Drug Discov 2015;10:591–7
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73
- Bonneau A, Maresca A, Winum JY, et al. Metronidazole-coumarin conjugates and 3-cyano-7-hydroxy-coumarin act as isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2013;28:397–401
- Sharma A, Tiwari M, Supuran CT. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J Enzyme Inhib Med Chem 2014;29:292–6
- Innocenti A, Casini A, Alcaro MC, et al. Carbonic anhydrase inhibitors: the first on-resin screening of a 4-sulfamoylphenylthiourea library. J Med Chem 2004;47:5224–9
- Casini A, Scozzafava A, Mincione F, et al. Carbonic anhydrase inhibitors: water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J Med Chem 2000;43:4884–92
- Mincione F, Starnotti M, Masini E, et al. Carbonic anhydrase inhibitors: design of thioureido sulfonamides with potent isozyme II and XII inhibitory properties and intraocular pressure lowering activity in a rabbit model of glaucoma. Bioorg Med ChemLett 2005;15:3821–7
- Supuran CT, Scozzafava A, Bogdan C, et al. Carbonic anhydrase inhibitors – Part 49: Synthesis of substituted ureido and thioureido derivatives of aromatic/heterocyclic sulfonamides with increased affinities for isozyme I. Eur J Med Chem 1998;33:83–93
- Avvaru BS, Wagner JM, Maresca A, et al. Carbonic anhydrase inhibitors. The X-ray crystal structure of human isoform II in adduct with an adamantyl analogue of acetazolamide resides in a less utilized binding pocket than most hydrophobic inhibitors. Bioorg Med ChemLett 2010;20:4376–81
- Wagner J, Avvaru BS, Robbins AH, et al. Coumarinyl-substituted sulfonamides strongly inhibit several human carbonic anhydrase isoforms: solution and crystallographic investigations. Bioorg Med Chem 2010;18:4873–8
- Carta F, Garaj V, Maresca A, et al. Sulfonamides incorporating 1,3,5-triazine moieties selectively and potently inhibit carbonic anhydrase transmembrane isoforms IX, XII and XIV over cytosolic isoforms I and II: Solution and X-ray crystallographic studies. Bioorg Med Chem 2011;19:3105–19
- Hen N, Bialer M, Yagen B, et al. Anticonvulsant 4-aminobenzenesulfonamide derivatives with branched-alkylamide moieties: X-ray crystallography and inhibition studies of human carbonic anhydrase isoforms I, II, VII, and XIV. J Med Chem 2011;54:3977–81
- Biswas S, Carta F, Scozzafava A, et al. Structural effect of phenyl ring compared to thiadiazole based adamantyl-sulfonamides on carbonic anhydrase inhibition. Bioorg Med Chem 2013;21:2314–18
- Di Fiore A, De Simone G, Menchise V, et al. Carbonic anhydrase inhibitors: X-ray crystal structure of a benzenesulfonamide strong CA II and CA IX inhibitor bearing a pentafluorophenylaminothioureido tail in complex with isozyme II. Bioorg Med Chem Lett 2005;15:1937–42
- Vitale RM, Alterio V, Innocenti A, et al. Carbonic anhydrase inhibitors. Comparison of aliphatic sulfamate/bis-sulfamate adducts with isozymes II and IX as a platform for designing tight-binding, more isoform-selective inhibitors. J Med Chem 2009;52:5990–8
- Temperini C, Cecchi A, Boyle NA, et al. Carbonic anhydrase inhibitors. Interaction of 2-N,N-dimethylamino-1,3,4-thiadiazole-5-methanesulfonamide with 12 mammalian isoforms: kinetic and X-ray crystallographic studies. Bioorg Med Chem Lett 2008;18:999–1005
- De Simone G, Di Fiore A, Menchise V, et al. Carbonic anhydrase inhibitors. Zonisamide is an effective inhibitor of the cytosolic isozyme II and mitochondrial isozyme V: solution and X-ray crystallographic studies. Bioorg Med Chem Lett 2005;15:2315–20
- Truppo E, Supuran CT, Sandomenico A, et al. Carbonic anhydrase VII is S-glutathionylated without loss of catalytic activity and affinity for sulfonamide inhibitors. Bioorg Med Chem Lett. 2012;22:1560–4
- Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–76
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902
- Scozzafava A, Carta F, Supuran CT. Secondary and tertiary sulfonamides: a patent review (2008 - 2012). Expert Opin Ther Pat 2013;23:203–13
- Alp C, Ozsoy S, Alp NA, et al. Sulfapyridine-like benzenesulfonamide derivatives as inhibitors of carbonic anhydrase isoenzymes I, II and VI. J Enzyme Inhib Med Chem 2012;27:818–24
- Ekinci D, Fidan I, Durdagi S, et al. Kinetic and in silico analysis of thiazolidin-based inhibitors of α-carbonic anhydrase isoenzymes. J Enzyme Inhib Med Chem 2013;28:370–4
- Abdel-Aziz AA, El-Azab AS, Ekinci D, et al. Investigation of arenesulfonyl-2-imidazolidinones as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:81–4
- Arslan M, Şentürk M, Fidan I, et al. Synthesis of 3,4-dihydroxypyrrolidine-2,5-dione and 3,5-dihydroxybenzoic acid derivatives and evaluation of the carbonic anhydrase I and II inhibition. J Enzyme Inhib Med Chem 2015;30:896–900
- Liu F, Martin-Mingot A, Lecornué F, et al. Carbonic Anhydrases inhibitory effects of new benzenesulfonamides synthesized by using superacid chemistry. J Enzyme Inhib Med Chem 2012;27:886–91
- Compain G, Martin-Mingot A, Maresca A, et al. Superacid synthesis of halogen containing N-substituted-4-aminobenzene sulfonamides: new selective tumor-associated carbonic anhydrase inhibitors. Bioorg Med Chem 2013;21:1555–63
- Métayer B, Mingot A, Vullo D, et al. New superacid synthesized (fluorinated) tertiary benzenesulfonamides acting as selective hCA IX inhibitors: toward a new mode of carbonic anhydrases inhibition by sulfonamides. Chem Commun 2013;49:6015–17
- Le Darz A, Mingot A, Bouazza F, et al. Fluorinated pyrrolidines and piperidines containing tertiary benzenesulfonamides: selective carbonic anhydrase II inhibitors. J Enzyme Inhib Med Chem 2015;30:737–45
- D'Ascenzio M, Carradori S, De Monte C, et al. Design, synthesis and evaluation of N-substituted saccharin derivatives as selective inhibitors of tumor-associated carbonic anhydrase XII. Bioorg Med Chem 2014;22:1821–31
- De Monte C, Carradori S, Secci D, et al. Cyclic tertiary sulfamates: selective inhibition of the tumor-associated carbonic anhydrases IX and XII by N- and O-substituted acesulfame derivatives. Eur J Med Chem 2014;84:240–6
- D'Ascenzio M, Carradori S, Secci D, et al. Selective inhibition of human carbonic anhydrases by novel amide derivatives of probenecid: synthesis, biological evaluation and molecular modelling studies. Bioorg Med Chem 2014;22:3982–8
- Carradori S, Mollica A, Ceruso M, et al. New amide derivatives of Probenecid as selective inhibitors of carbonic anhydrase IX and XII: biological evaluation and molecular modelling studies. Bioorg Med Chem 2015;23:2975–81
- Mollica A, Costante R, Akdemir A, et al. Exploring new Probenecid-based carbonic anhydrase inhibitors: synthesis, biological evaluation and docking studies. Bioorg Med Chem 2015;23:5311–18
- Gavernet L, Gonzalez Funes JL, Palestro PH, et al. Inhibition pattern of sulfamide-related compounds in binding to carbonic anhydrase isoforms I, II, VII, XII and XIV. Bioorg Med Chem 2013;21:1410–18