
Abstract
A series of 2-fluoro and 3-trifluoromethylphenylpiperazinylalkyl derivatives of 1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (4–21) were synthesized and evaluated for their serotonin (5-HT1A/5-HT7) receptor affinity and phosphodiesterase (PDE4B and PDE10A) inhibitor activity. The study enabled the identification of potent 5-HT1A, 5-HT7 and mixed 5-HT1A/5-HT7 receptor ligands with weak inhibitory potencies for PDE4B and PDE10A. The tests have been completed with the determination of lipophilicity and metabolic stability using micellar electrokinetic chromatography (MEKC) system and human liver microsomes (HLM) model. In preliminary pharmacological in vivo studies, selected compound 8-(5-(4-(2-fluorophenyl)piperazin-1-yl)pentyl)-1,3,7-trimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (9) behaved as a potential antidepressant in forced swim test (FST) in mice. Moreover, potency of antianxiety effects evoked by 9 (2.5 mg/kg) is greater than that of the reference anxiolytic drug, diazepam. Molecular modeling revealed that fluorinated arylpiperazinylalkyl derivatives of 1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione have major significance for the provision of lead compounds for antidepressant and/or anxiolytic application.
Introduction
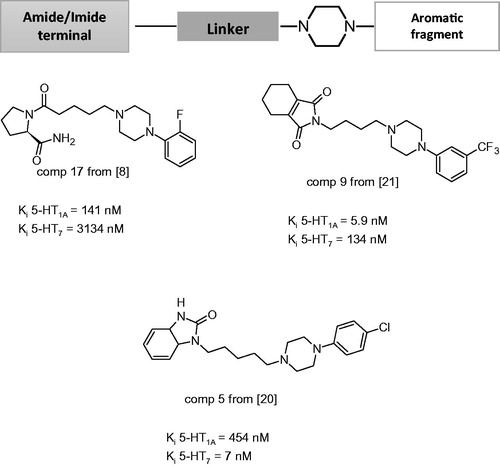
Over the past decade, we have witnessed unparalleled advances in our understanding of the basic biological processes that contribute to many human disorders, although a detailed understanding of the etiology of complex psychiatric disorders remains elusive. Psychiatric diseases are chronic and recurrent and have a complex etiology. Human genomics and biological studies have revealed an unprecedented number of promising molecular targets for neuropsychiatric disorders, including G-protein-coupled receptors and transporters, intracellular and synaptic proteins and microRNAs. Serotonin (5-HT) is one of the most attractive targets for medicinal chemists and the discovery of ligands with affinity for the family of 5-HT receptors (5-HTRs) is an area of intense research, because of the potential to find new therapeutic drugs, due to their involvement in numerous physiological and pathophysiological processes. Among the 14 5-HTRs identified, the 5-HT1A and 5-HT7 subtypes are the best studied due to effects of their full or partial agonists or antagonists on anxiety, depression and schizophreniaCitation1–6. A very important class of 5-HT receptors ligands are derivatives of 1,4-disubstituted arylpiperazine (). Such arylpiperazine derivative with long-chain substituents incorporated on the basic nitrogen of the phenylpiperazine ring – long-chain arylpiperazines (LCAPs) – are commonly studied classes of bioactive compoundsCitation7–24. Despite the enormous progress in central nervous system (CNS) drug discovery, particularly in the areas of mood disorders and schizophrenia, new drugs are still being sought. The main reasons behind the search for novel therapeutics are poor patient compliance, delayed onset of action and side effects, including emesis, sexual dysfunction, weight gain, diabetes mellitus or endocrinopathies. Roth et al. proposed that, for mood disorders and schizophrenia, attempting to develop more effective treatments by intentionally designing nonselective drugs that interact with several molecular targets would lead to new and more effective medications (“magic shotguns”)Citation25.
Figure 1. General structure of long-chain arylpiperazine (LCAPs) and their representatives of 5-HT1A and 5-HT7 receptor ligands.
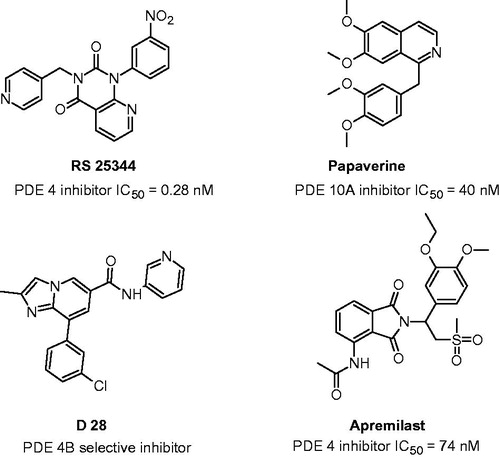
Cyclic nucleotide phosphodiesterases (PDEs) are a superfamily of enzymes that are involved in the regulation of the intracellular second messenger’s cyclic AMP (cAMP) and cyclic GMP (cGMP) by controlling their rates of hydrolysis. There are 11 different PDE families and each family typically has multiple isoforms and splice variants. The PDEs differ in their structures, distribution, modes of regulation and sensitivity to inhibitors. Since PDEs have been shown to play distinct roles in the processes of emotion and related learning and memory processes, selective PDE inhibitors, by preventing the breakdown of cAMP and/or cGMP, modulate mood and related cognitive activity. It is becoming increasingly clear that PDE inhibitors have therapeutic potential for the treatment of neuropsychiatric disorders involving disturbances of mood, emotion and cognitionCitation26. Moreover, PDE4 and PDE10 due to their presence in areas of the brain associated with the site of action of psychotropic drugs and their inhibitors have potential therapeutic relevance. In animal models, elevated intracellular cAMP level achieved by PDE inhibition have been shown to possess antidepressant-like effect. Moreover, antipsychotic effects can be reproduce by inhibition of the enzymes that degrade cAMP. Maxwell et al. used amphetamine-induced abnormalities in auditory event-related potentials to study the antipsychotic potential of nonreceptor based compounds such as rolipramCitation27,Citation28. The therapeutic potentials of PDE inhibitors have been explored in many areas; for instance, PDE4 inhibitors have been evaluated for the treatment of cognitive disorders, depression, and anxiety and PDE10 inhibitors for the treatment of schizophrenia and Huntington’s disease. Naturally occurring methylxanthines (theophylline, caffeine) were the first inhibitors of PDE to be discovered. To improve the potency and specificity for the inhibition of various PDEs, thousands of compounds with related structures have been synthesized. Today, selective inhibitors of PDE4 and PDE10 show a large of chemical diversity ().
Figure 2. Chemical structures and inhibitor activity (IC50) of inhibitors of PDE4 and PDE10.
For years, our attention has been focused on the development of LCAPs with the complex terminal part based on a purine moiety. Annulated derivatives of theophylline with an LCAP moiety demonstrate high affinity toward serotonin 5-HT1A and moderate affinity to 5-HT2A and 5-HT7 and the dopaminergic D2 receptor site. Preclinical results indicate that monosubstituted derivatives of LCAPs with an imidazo[2,1-f]purine-2,4-dione as the terminal part act as antipsychotics with additional antidepressant and anxiolytic propertiesCitation29–32. In view of this and our earlier chemical and pharmacological investigation, in the current study, we designed and synthesized a new series of 2-fluoro and 3-trifluoromethylphenylpiperazinyalkyl derivatives of 1H-imidazo[2,1-f]purine-2,4(3H,8H)-diones. The special properties of fluorine, such as its high electronegativity, small size and very low polarizability, impart a variety of properties important in medicinal chemistry. Fluoro or trifluoromethyl substitutions will generally have enhanced binding interactions, metabolic stability, changes in physicochemical properties and selective relativities toward of targeted molecules. Fluorinated drugs constitute the first (spiperone, haloperidol, droperidol) and second (risperidone, sertindole, paliperidone, iloperidone) generation of antipsychotic drugs and antidepressants (fluvoxamine, paroxetine, citalopram) from the selective serotonin reuptake inhibitors (SSRIs) group with extremely famous fluoxetine (Prozac®). Herein, we examined the effect of fluorine substitution on receptor affinity, selectivity, intrinsic activity for 5-HT1A and 5-HT7 receptor, and lipophilicity. Moreover, studies focused on the effect of the presence of annelated imidazole moiety at the 7, 8 position of purine system on PDE4B/PDE10A inhibition and metabolic stability. Finally, structure–activity relationships were analyzed and explained by molecular modeling.
Experimental protocols
Chemistry
Organic solvents (from Sigma-Aldrich and Chempur) were of reagent grade and were used without purification. All other reagents were from Sigma-Aldrich and Alfa Aesar. Thin-layer chromatography (TLC) was performed on Merck silica gel 60 F254 aluminum sheets (Merck, Darmstadt, Germany). Spots were detected by their absorption under UV light (λ = 254 nm). Analytical high-performance liquid chromatography (HPLC) was run on an Alliance Waters 2695 Separations Module equipped with a Chromolith SpeedROD RP 18.5 μm column (4.6 × 50 mm). Standard conditions were eluent system A (water/0.1% TFA), system B (acetonitrile/0.1% TFA). A flow rate of 5 mL/min and a gradient of (0–100)% B over 3 min were used. Detection was performed on a photodiode array (PDA) detector. Retention times (tR) are given in minutes. Nuclear magnetic resonance (NMR) spectra were measured in CDCl3 on a Varian Mercury-VX 300 spectrometer operating at 300.08 MHz (1H), 282 MHz (19F) and 75.46 MHz (13C). Sample concentrations were in the range from 10 mg/1 mL (1H and 19F) to 50 mg/mL for 13C, all spectra were acquired at 26 °C. The J values are in Hertz (Hz), and splitting patterns are designated as follows: s (singlet), d (doublet), t (triplet), m (multiplet). Mass spectrometry analyses: Samples were prepared in acetonitrile/water (10/90 v/v) mixture. The LC/MS system consisted of a Waters Acquity UPLC, coupled to Waters TQD mass spectrometer (electrospray ionization mode ESI-tandem quadrupole). All the analyses were carried out using an Acquity UPLC BEH C18, 50 × 2.1 mm reversed-phase column. A flow rate of 0.3 mL/min and a gradient of (5–5)% B over 5 min was used. Eluent A: water/0.1% HCO2H; eluent B: acetonitrile/0.1% HCO2H. Nitrogen was used for both nebulizing and drying gas. LC/MS data were obtained by scanning the first quadrupole in 0.5 s in a mass range from 100 to 700 m/z; eight scans were summed up to produce the final spectrum. Elemental analyses were found within ±0.4% of the theoretical values. Melting points (mp) were determined with a Büchi apparatus and are uncorrected. Column chromatography separations were carried out on column with Merck Kieselgel 60 (Darmstadt, Germany).
General procedure for the synthesis of phthalimide derivatives (VI–XI)
A mixture of 10 mmol of 1-(2-fluorophenyl)- or 1-(3-trifluoromethyl)phenyl)piperazine (I–II) and 10 mmol of appropriate 2-(bromoalkyl)-1H-isoindoline-1,3(2H)-dione (III–V), 30 mmol of potassium carbonate, catalytic amount of potassium iodide and 20 mL of acetonitrile was stirred at 82 °C temperature for 48 h. The reaction mixture evaporated under reduced pressure to obtain a residue, which was purified by column chromatography using dichloromethane/methanol (9/1) as an eluent.
General procedure for the deprotection of the phthalimide group (XII–XVI)
A mixtures of 1.2 mmol of appropriate compound VI–XI and 65% hydrazine monohydrate aqueous solution (15 mmol) in 99.8% ethanol were refluxed for 1 h. The reaction mixtures were cooled down and treated with an additional amount of 99.8% ethanol and concentrated HCl (1.3 mL). Then, the reaction mixtures were refluxed for 4 h and left overnight in a refrigerator. The precipitates were filtered off, and the solvents were evaporated. The residues were treated with n-hexane (20 mL) and NH3 (aqueous, 15 mL). The solutions were extracted with methylene chloride (3 × 15 mL), the combined extracts were dried over anhydrous Na2SO4, and the solvents were evaporated to give a products that were used without further purification (yield: 70–80%).
General procedure for 7-ketonyl derivatives of 8-bromo-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione (1–3)
2-(8-Bromo-1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-7H-purin-7-yl)acetaldehyde (1) was obtained according to the previously described procedureCitation29. 8-Bromo-1,3-dimethyl-7-(2-oxopropyl)-3,7-dihydro-1H-purine-2,6-dione (2) and 8-bromo-1,3-dimethyl-7-(2-oxo-2-phenylethyl)-3,7-dihydro-1H-purine-2,6-dione (3) were obtained as described below. 8-Bromo-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione (10 mmol), 1-chloropropan-2-one or 2-chloro-1-phenylethan-1-one (15 mmol), potassium carbonate (30 mmol) and acetonitrile (15 mL) were refluxed for 12 h. After this time, the acetonitrile was removed in vacuo and the resulting residues were dissolved in dichloromethane (20 mL). The organic layers were washed with water (2 × 20 mL) and dried over anhydrous Na2SO4, filtered and evaporated to dryness to give the crude products that were recrystallized from 96% ethanol (yield: 85–91%).
General procedure for the synthesis of 2-fluoro and 3-trifluoromethylphenylpiperazinyalkyl derivatives of 1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (4–21)
A mixture of 7-ketonyl derivatives of 8-bromo-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione 1–3 (5 mmol) with double amount of appropriate 2-fluoro or 3-trifluoromethylphenylpiperazinyalkylamine (10 mmol) was refluxed in 2-methoxyethanol (20 mL) for 12 h. The solvent was removed in vacuo, and the obtained residue was purified by flash column chromatography on silica gel using mixture of dichloromethane/methanol (9/1) or (9/1.2) as an eluent.
8-(3-(4-(2-Fluorophenyl)piperazin-1-yl)propyl)-1,3-dimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (4)
Yield: 62%, cream solid: m.p. 209–210 °C, tR = 0.831; 1H-NMR (300 MHz, CDCl3): δ (ppm): 2.07–2.16 (m, 2H, CH2CH2CH2), 2.46 (t, 2H, J = 7.3 Hz, CH2CH2CH2), 2.64–2.65 (m, 4H, N(CH2)2), 3.09–3.12 (m, 4H, (CH2)2N), 3.43 (s, 3H, N3–CH3), 3.59 (s, 3H, N1–CH3), 4.17 (t, 2H, J = 7.4 Hz, N8–CH2), 6.81–6.82 (d, 1H, J = 2.3 Hz, arom), 6.96–7.05 (m, 4H, arom), 7.39–7.40 (d, 1H, J = 2.3 Hz, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 26.21, 27.77, 30.07, 44.16, 46.23, 50.45, 50.49, 53.10, 54.43, 105.00, 107.39, 115.98, 116.25, 118.62, 118.86, 122.51, 124.46, 139.98, 147.43, 151.98, 154.04, 157.34. 19F NMR (282 MHz, CDCl3): δ (ppm): −123.19 (s, 1F). MS (EI) m/z: 440.38 [M +1]+. Anal. calcd for C22H26FN7O2: C, 60.12; H, 5.96; N, 22.31; found: C, 60.2; H, 6.04; N, 22.37.
8-(4-(4-(2-Fluorophenyl)piperazin-1-yl)butyl)-1,3-dimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (5)
Yield: 69%, cream solid: m.p. 144–145 °C, tR = 1.011; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.61–1.66 (m, 2H, CH2CH2CH2CH2), 1.92–2.02 (m, 2H, CH2CH2CH2CH2), 2.50 (t, 2H, J = 7.1 Hz, CH2CH2CH2CH2), 2.60–2.66 (m, 4H, N(CH2)2), 3.10–3.12 (m, 4H, (CH2)2N), 3.43 (s, 3H, N3–CH3), 3.59 (s, 3H, N1–CH3), 4.19 (t, 2H, J = 2.8 Hz, N8–CH2), 6.78–6.79 (d, 1H, J = 2.3 Hz, arom), 6.92–7.04 (m, 4H, arom), 7.40–7.41 (d, 1H, J = 3 Hz, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 23.30, 26.96, 27.52, 30.09, 46.11, 46.23, 50.38, 53.24, 57.62, 100.05, 107.60, 115.96, 116.24, 117.96, 118.85, 122.49, 124.42, 135.33, 139.98, 147.46, 151.98, 157.32, 168.14. 19F NMR (282 MHz, CDCl3): δ (ppm): −123.19 (s, 1F). MS (EI) m/z: 454.34 [M+ 1]+. Anal. calcd for C23H28FN7O2: C, 60.91; H, 6.22; N, 21.62; found: C, 61.2; H, 6.32; N, 21.37.
8-(5-(4-(2-Fluorophenyl)piperazin-1-yl)pentyl)-1,3-dimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (6)
Yield: 52%, Cream solid: m.p. 168–170 °C, tR = 1.145; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.40–1.43 (m, 2H, CH2CH2CH2CH2CH2), 1.62–1.66 (m, 2H, CH2CH2CH2CH2CH2), 1.89–1.97 (m, 2H, CH2CH2CH2CH2CH2), 2.45 (t, 2H, J = 2.8 Hz, CH2CH2CH2CH2CH2), 2.60–2.68 (m, 4H, N(CH2)2), 3.10–3.14 (m, 4H, (CH2)2N), 3.43 (s, 3H, N3–CH3), 3.60 (s, 3H, N1–CH3), 4.07 (t, 2H, J = 7.1 Hz, N8–CH2), 6.78–6.79 (d, 1H, J = 2.3 Hz, arom), 6.90–7.05 (m, 4H, arom), 7.40–7.41 (d, 1H, J = 3.1 Hz, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 24.32, 25.97, 27.75, 29.25, 30.07, 46.13, 50.23, 53.19, 58.11, 100.00, 107.52, 115.93, 116.21, 118.04, 118.89, 122.59, 124.42, 124.46, 139.94, 147.31, 151.95, 152.18, 154.01, 157.29. 19F NMR (282 MHz, CDCl3): δ (ppm): −123.19 (s, 1F). MS (EI) m/z: 468.43 [M + 1]+. Anal. calcd for C24H30FN7O2: C, 61.65; H, 6.47; N, 20.97; found: C, 61.42; H, 6.66; N, 21.07.
8-(3-(4-(2-Fluorophenyl)piperazin-1-yl)propyl)-1,3,7-trimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (7)
Yield: 72%, Cream solid: m.p. 160–161 °C, tR = 1.005; 1H-NMR (300 MHz, CDCl3): δ (ppm): 2.09–2.16 (m, 2H, CH2CH2CH2), 2.35 (s, 3H, CH3), 2.45 (t, 2H, J = 2.9 Hz, CH2CH2CH2), 2.63–2.65 (m, 4H, N(CH2)2), 3.09–3.12 (m, 4H, (CH2)2N), 3.42 (s, 3H, N3–CH3), 3.58 (s, 3H, N1–CH3), 4.10 (t, 2H, J = 7.4 Hz, N8–CH2), 6.92–7.05 (m, 4H, arom), 7.26 (s, 1H, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 10.32, 26.01, 27.74, 30.04, 41.17, 50.43, 50.48, 53.16, 54.67, 104.43, 115.98, 116.25, 118.83, 118.88, 122.57, 124.46, 127.13, 139.88, 147.71, 151.43, 152.01, 154.02, 157.32. 19F NMR (282 MHz, CDCl3): δ (ppm): −123.19 (s, 1F). MS (EI) m/z: 454.34 [M + 1]+. Anal. calcd for C23H28FN7O2: C, 60.91; H, 6.22; N, 21.62; found: C, 60.82; H, 6.41; N, 21.73.
8-(4-(4-(2-Fluorophenyl)piperazin-1-yl)butyl)-1,3,7-trimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (8)
Yield: 84%, Cream solid: m.p. 153–155 °C, tR = 1.155; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.55–1.66 (m, 2H, CH2CH2CH2CH2), 1.87–1.94 (m, 2H, CH2CH2CH2CH2), 2.33 (s, 3H, CH3), 2.53–2.68 (m, 6H, CH2CH2CH2CH2 + N(CH2)2), 3.10–3.12 (m, 4H, (CH2)2N), 3.43 (s, 3H, N3–CH3), 3.59 (s, 3H, N1–CH3), 4.05 (t, 2H, J = 3.1 Hz, N8–CH2), 6.93–7.05 (m, 4H, arom), 7.26 (s, 1H, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 10.41, 23.72, 24.53, 27.34, 27.74, 30.06, 43.17, 50.41, 53.25, 57.63, 58.31, 104.59, 115.96, 116.24, 118.88, 122.56, 124.42, 124.46, 126.98, 139.92, 147.77, 151.02, 151.43, 154.07. 19F NMR (282 MHz, CDCl3): δ (ppm): −123.19 (s, 1F). MS (EI) m/z: 468.43 [M+ 1]+. Anal. calcd for C24H30FN7O2: C, 61.65; H, 6.47; N, 20.97; found: C, 61.7; H, 6.32; N, 21.03.
8-(5-(4-(2-Fluorophenyl)piperazin-1-yl)pentyl)-1,3,7-trimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (9)
Yield: 92%, Cream solid: m.p. 143–145 °C, tR = 1.197; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.37–1.45 (m, 2H, CH2CH2CH2CH2CH2), 1.57–1.65 (m, 2H, CH2CH2CH2CH2CH2), 1.86–1.91 (m, 2H, CH2CH2CH2CH2CH2), 2.32 (s, 3H, CH3), 2.43 (t, 2H, J = 2.8 Hz, CH2CH2CH2CH2CH2), 2.60–2.66 (m, 4H, N(CH2)2), 3.10–3.13 (m, 4H, (CH2)2N), 3.42 (s, 3H, N3–CH3), 3.59 (s, 3H, N1–CH3), 4.01 (t, 2H, J = 7.1 Hz, N8–CH2), 6.91–7.16 (m, 4H, arom), 7.26 (s, 1H, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 5.67, 19.71, 21.40, 22.99, 24.52, 25.33, 38.50, 45.61, 48.51, 53.45, 95.12, 99.80, 111.19, 111.46, 114.13, 117.70, 111.81, 119.67, 122.07, 135.15, 143.01, 146.67, 147.25, 149.28, 152.57.19F NMR (282 MHz, CDCl3): δ (ppm): −123.19 (s, 1F). MS (EI) m/z: 482.39 [M + 1]+. Anal. calcd for C25H32FN7O2: C, 62.35; H, 6.7; N, 20.36; found: C, 62.42; H, 6.78; N, 20.37.
8-(3-(4-(2-Fluorophenyl)piperazin-1-yl)propyl)-1,3-dimethyl-7-phenyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (10)
Yield: 65%, Cream oil, tR = 1.232; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.18–1.42 (m, 2H, CH2CH2CH2), 1.94–2.04 (m, 2H, CH2CH2CH2), 2.53–2.73 (m, 4H, N(CH2)2), 2.96–3.12 (m, 4H, (CH2)2N), 3.32 (s, 3H, N3–CH3), 3.55 (s, 3H, N1–CH3), 3.63 (t, 2H, J = 4.1 Hz, N8–CH2), 6.50–6.55 (m, 2H, arom), 6.93–7.00 (m, 3H, arom), 7.35–7.58 (m, 4H, arom), 7.98–8.01 (d, J = 3.4 Hz, 1H, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 24.23, 25.92, 27.80, 29.69, 30.13, 42.36, 49.32, 50.37, 50.41, 52.99, 53.41, 54.78, 105.34, 115.90, 116.18, 118.80, 118.85, 122.40, 122.51, 124.40, 124.45, 128.26, 128.39, 151.74, 152.03, 154.06, 154.25, 157.32. 19F NMR (282 MHz, CDCl3): δ (ppm): −123.19 (s, 1F). MS (EI) m/z: 516.36 [M + 1]+. Anal. calcd for C28H30FN7O2: C, 65.23; H, 5.86; N, 19.02; found: C, 65.38; H, 6.08; N, 18.96.
8-(4-(4-(2-Fluorophenyl)piperazin-1-yl)butyl)-1,3-dimethyl-7-phenyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (11)
Yield: 75%, Cream solid: m.p. 133–134 °C, tR = 1.363; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.21–1.42 (m, 2H, CH2CH2CH2CH2), 1.70–1.92 (m, 2H, CH2CH2CH2CH2), 2.68–2.74 (m, 2H, CH2CH2CH2CH2), 2.86–2.92 (m, 4H, N(CH2)2), 3.08–3.16 (m, 4H, (CH2)2N), 3.32–3.39 (m, 4H, N3–CH3 + N8–CH2), 3.54–3.72 (m, 4H, N1–CH3 + N8–CH2), 6.80–7.05 (m, 5H, arom), 7.45–7.63 (m, 4H, arom), 8.04–8.06 (d, J = 8.0 Hz, 1H, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 23.30, 24.96, 27.52, 29.69, 30.90, 42.89, 49.26, 49.67, 57.68, 59.07, 60.90, 71.92, 115.98, 116.25, 118.92, 119.20, 122.78, 122.89, 124.45, 124.49, 127.38, 127.70, 128.08, 128.71, 128.87, 134.32, 149.19, 151.74, 154.19. 19F NMR (282 MHz, CDCl3): δ (ppm): −123.19 (s, 1F). MS (EI) m/z: 530.60 [M +1]+. Anal. calcd for C29H32FN7O2: C, 65.75; H, 6.09; N, 18.51; found: C, 65.73; H, 6.02; N, 18.53.
8-(5-(4-(2-Fluorophenyl)piperazin-1-yl)pentyl)-1,3-dimethyl-7-phenyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (12)
Yield: 77%, Cream oil, tR = 1.422; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.19–1.28 (m, 2H, CH2CH2CH2CH2CH2), 1.40–1.45 (m, 2H, CH2CH2CH2CH2CH2), 1.75–1.80 (m, 2H, CH2CH2CH2CH2CH2), 2.31 (t, 2H, J = 2.2 Hz, CH2CH2CH2CH2CH2), 2.57–2.63 (m, 4H, N(CH2)2), 3.07–3.14 (m, 4H, (CH2)2N), 3.44 (s, 3H, N3–CH3), 3.63 (s, 3H, N1–CH3), 4.11 (t, 2H, J = 3.4 Hz, N8–CH2), 6.91–7.05 (m, 5H, arom), 7.41–7.50 (m, 4H, arom), 8.04–8.03 (d, J = 5.0 Hz, 1H, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 23.30, 24.96, 27.52, 29.69, 30.90, 42.89, 49.26, 49.67, 52.99, 57.68, 59.07, 60.90, 71.92, 115.98, 116.25, 118.92, 119.20, 122.78, 122.89, 124.45, 124.49, 127.38, 127.70, 128.08, 128.71, 128.87, 134.32, 149.19, 151.74, 154.19. 19F NMR (282 MHz, CDCl3): δ (ppm): −123.14 (s, 1F). MS (EI) m/z: 544.55 [M+ 1]+. Anal. calcd for C30H34FN7O2: C, 66.28; H, 6.30; N, 18.04; found: C, 66.42; H, 6.06; N, 18.07.
1,3-Dimethyl-8-(3-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propyl)-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (13)
Yield: 78%, Cream solid: m.p. 110–112 °C, tR = 1.211; 1H-NMR (300 MHz, CDCl3): δ (ppm): 2.09–2.18 (m, 2H, CH2CH2CH2), 2.46 (t, 2H, J = 7.3 Hz, CH2CH2CH2), 2.64–2.65 (m, 4H, N(CH2)2), 3.24–3.27 (m, 4H, (CH2)2N), 3.43 (s, 3H, N3–CH3), 3.60 (s, 3H, N1–CH3), 4.18 (t, 2H, J = 3.3 Hz, N8–CH2), 6.82–6.83 (m, 2H, arom), 7.04–7.10 (m, 3H, arom), 7.40–7.41 (d, 1H, J = 2.3 Hz, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 26.96, 27.52, 30.09, 42.16, 46.75, 50.45, 50.49, 52.99, 54.43, 105.00, 107.39, 111.37, 115.09, 116.25, 117.22, 121.01, 122.51, 124.16, 129.53, 131.21, 151.28, 152.01, 154.04. 19F NMR (282 MHz, CDCl3): δ (ppm): −60.34 (s, 3F). MS (EI) m/z: 490.36 [M +1]+. Anal. calcd for C23H26F3N7O2: C, 56.44; H, 5.35; N, 20.03; found: C, 56.20; H, 5.30; N, 20.37.
1,3-Dimethyl-8-(4-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)butyl)-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (14)
Yield: 82%, Cream solid: m.p. 170–171 °C, tR = 1.254; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.48–1.53 (m, 2H, CH2CH2CH2CH2), 1.82–1.92 (m, 2H, CH2CH2CH2CH2), 2.40–2.54 (m, 6H, CH2CH2CH2CH2 + N(CH2)2), 3.10–3.17 (m, 4H, (CH2)2N), 3.34 (s, 3H, N3–CH3), 3.51 (s, 3H, N1–CH3), 4.04 (t, 2H, J = 3.1 Hz, N8–CH2), 6.79–6.80 (d, 1H, J = 2.3 Hz, arom), 6.91–7.01 (m, 2H, arom), 7.24–7.26 (m, 2H, arom), 7.40–7.41 (d, 1H, J = 3 Hz, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 26.96, 27.52, 29.69, 30.09, 42.16, 46.75, 50.45, 50.49, 52.99, 54.43, 105.00, 107.39, 112.22, 115.75 115.98, 117.92, 118.62, 121.01, 123.56, 129.53, 129.91, 151.28, 152.01, 154.04. 19F NMR (282 MHz, CDCl3): δ (ppm): −62.99 (s, 3F). MS (EI) m/z: 504.10 [M + 1]+. Anal. calcd for C24H28F3N7O2: C, 57.25; H, 5.61; N, 19.47; found: C, 57.02; H, 5.32; N, 19.37.
1,3-Dimethyl-8-(5-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)pentyl)-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (15)
Yield: 86%, Cream oil, tR = 1.301, 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.40–1.42 (m, 2H, CH2CH2CH2CH2CH2), 1.62–1.67 (m, 2H, CH2CH2CH2CH2CH2), 1.89–1.96 (m, 2H, CH2CH2CH2CH2CH2), 2.49 (t, 2H, J = 2.8 Hz, CH2CH2CH2CH2CH2), 2.70–2.73 (m, 4H, N(CH2)2), 3.26–3.28 (m, 4H, (CH2)2N), 3.43 (s, 3H, N3–CH3), 3.59 (s, 3H, N1–CH3), 4.06 (t, 2H, J = 7.1 Hz, N8–CH2), 6.78–6.79 (d, 1H, J = 2.3 Hz, arom), 7.05–7.09 (m, 4H, arom), 7.40–7.41 (d, 1H, J = 3.1 Hz, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 26.96, 27.52, 28.33, 29.69, 30.09, 42.16, 46.75, 50.45, 50.49, 52.99, 54.43, 105.00, 107.39, 111.97, 114.89, 116.25, 118.02, 121.01, 122.96, 124.56, 129.53, 131.42, 151.28, 152.01, 154.04. 19F NMR (282 MHz, CDCl3): δ (ppm): −63.14 (s, 3F). MS (EI) m/z: 518.19 [M +1]+. Anal. calcd for C25H30F3N7O2: C, 58.02; H, 5.84; N, 18.94; found: C, 58.22; H, 5.77; N, 18.97.
1,3,7-Trimethyl-8-(3-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propyl)-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (16)
Yield: 95%, Cream oil, tR = 1.172; 1H-NMR (300 MHz, CDCl3): δ (ppm): 2.11–2.13 (m, 2H, CH2CH2CH2), 2.34 (s, 3H, CH3), 2.47–2.49 (m, 2H, CH2CH2CH2), 2.64–2.65 (m, 4H, N(CH2)2), 3.23–3.26 (m, 4H, (CH2)2N), 3.43 (s, 3H, N3–CH3), 3.58 (s, 3H, N1–CH3), 4.10 (t, 2H, J = 3.3 Hz, N8–CH2), 7.03–7.16 (m, 4H, arom), 7.34–7.37 (m, 1H, C6H). 13C NMR (75 MHz, CDCl3) δ (ppm): 10.30, 26.96, 27.52, 28.33, 30.09, 42.16, 46.75, 50.45, 50.49, 52.99, 54.43, 105.00, 107.23, 112.07, 115.89, 118.32, 121.01, 123.36, 124.56, 129.53, 131.21, 151.28, 152.01, 154.04. 19F NMR (282 MHz, CDCl3): δ (ppm): −60.34 (s, 3F). MS (EI) m/z: 504.39 [M + 1]+. Anal. calcd for C24H28F3N7O2: C, 57.25; H, 5.61; N, 19.47; found: C, 57.20; H, 5.30; N, 19.37.
1,3,7-Trimethyl-8-(4-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)butyl)-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (17)
Yield: 87%, Cream solid: m.p. 188–190 °C, tR = 1.306; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.48–1.53 (m, 2H, CH2CH2CH2CH2), 1.76–1.78 (m, 2H, CH2CH2CH2CH2), 2.24–2.51 (m, 9H, CH3 + CH2CH2CH2CH2 + N(CH2)2), 3.10–3.13 (m, 4H, (CH2)2N), 3.30 (s, 3H, N3–CH3), 3.48 (s, 3H, N1–CH3), 3.94–3.98 (m, 2H, N8–CH2), 6.95–6.98 (m, 3H, arom), 7.13–7.25 (m, 2H, arom). 13C NMR (75 MHz, CDCl3) δ (ppm): 13.30, 26.96, 27.52, 28.33, 29.69, 30.09, 42.89, 49.26, 49.67, 57.68, 59.07, 71.92, 104.61, 107.33, 109.2, 111.77, 116.09, 118.12, 121.01, 123.36, 129.53, 131.21, 151.28, 152.01, 154.04. 19F NMR (282 MHz, CDCl3): δ (ppm): −63.12 (s, 3F). MS (EI) m/z: 519.12 [M +1]+. Anal. calcd for C25H30F3N7O2: C, 58.02; H, 5.84; N, 18.94; found: C, 58.22; H, 5.58; N, 19.07.
1,3,7-Trimethyl-8-(5-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)pentyl)-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (18)
Yield: 80%, Cream solid: m.p. 168–169 °C, tR = 1.340, 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.35–1.37 (m, 2H, CH2CH2CH2CH2CH2), 1.55–1.58 (m, 2H, CH2CH2CH2CH2CH2), 1.83–1.85 (m, 2H, CH2CH2CH2CH2CH2), 2.28–2.93 (m, 9H, CH3 + CH2CH2CH2CH2CH2 + N(CH2)2), 3.20–3.24 (m, 4H, (CH2)2N), 3.36 (s, 3H, N3–CH3), 3.54 (s, 3H, N1–CH3), 3.95–4.00 (m, 2H, N8–CH2), 6.99–7.07 (m, 3H, arom), 7.26–7.32 (m, 2H, arom). 13C NMR (75 MHz, CDCl3) δ (ppm): 10.20, 24.38, 25.91, 27.74, 29.25, 30.00, 43.21, 49.10, 52.84, 58.12, 99.83, 104.65, 112.13, 116.04, 118.76, 122.39, 126.00, 127.21, 129.51, 131.10, 131.51, 147.63, 151.08, 151.38, 151.90, 154.04. 19F NMR (282 MHz, CDCl3): δ (ppm): −63.14 (s, 3F). MS (EI) m/z: 532.15 [M + 1]+. Anal. calcd for C26H32F3N7O2: C, 58.75; H, 6.07; N, 18.44; found: C, 58.52; H, 6.10; N, 18.50.
1,3-Dimethyl-7-phenyl-8-(3-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propyl)-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (19)
Yield: 79%, Cream solid: m.p. 128–129 °C, tR = 1.501; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.82–2.02 (m, 2H, CH2CH2CH2), 2.34 (t, 2H, J = 7.3 Hz, CH2CH2CH2), 2.42–2.44 (m, 4H, N(CH2)2), 3.08–3.11 (m, 4H, (CH2)2N), 3.45 (s, 3H, N3–CH3), 3.63 (s, 3H, N1–CH3), 4.21 (t, 2H, J = 3.3 Hz, N8–CH2), 6.90–7.08 (m, 6H, arom), 7.33–7.49 (m, 4H, arom). 13C NMR (75 MHz, CDCl3) δ (ppm): 10.30, 25.92, 27.80, 29.69, 30.13, 42.36, 49.32, 50.37, 50.41, 52.99, 53.41, 54.78, 105.34, 112.07, 115.90, 116.18, 118.32, 121.01, 122.40, 124.40, 125.22, 128.26, 129.53, 131.21, 151.28, 152.01, 154.04, 154.25, 157.32. 19F NMR (282 MHz, CDCl3): δ (ppm): −60.34 (s, 3F). MS (EI) m/z: 566.49 [M + 1]+. Anal. calcd for C29H30F3N7O2: C, 61.58; H, 5.35; N, 17.34; found: C, 61.60; H, 5.33; N, 17.43.
1,3-Dimethyl-7-phenyl-8-(4-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)butyl)-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (20)
Yield: 81%, Cream oil; tR = 1.490; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.18–1.25 (m, 4H, CH2CH2CH2CH2), 1.42–1.52 (m, 6H, CH2CH2CH2CH2 + N(CH2)2), 3.39–3.64 (m, 10H, (CH2)2N + N3–CH3 + N1–CH3), 4.16 (t, 2H, J = 3.1 Hz, N8–CH2), 7.09–7.25 (m, 2H, arom), 7.43–7.52 (m, 8H, arom).
13C NMR (75 MHz, CDCl3) δ (ppm): 22.97, 24.96, 27.52, 29.96, 30.89, 42.15, 49.26, 49.77, 57.68, 59.17, 60.94, 72.02, 111.87, 115.91, 116.27, 119.12, 120.99, 122.87, 122.89, 124.54, 125.01, 128.08, 128.71, 128.87, 129.53, 131.01, 149.23, 151.18, 152.17, 154.01. 19F NMR (282 MHz, CDCl3): δ (ppm): −62.82 (s, 3F). MS (EI) m/z: 580.14 [M +1]+. Anal. calcd for C30H32F3N7O2: C, 62.17; H, 5.56; N, 16.92; found: C, 62.02; H, 5.32; N, 16.73.
1,3-Dimethyl-7-phenyl-8-(5-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)pentyl)-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (21)
Yield: 85%, Cream oil, tR = 1.540, 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.18–1.25 (m, 4H, CH2CH2CH2CH2CH2), 1.56–1.81 (m, 8H, CH2CH2CH2CH2CH2 + N(CH2)2), 3.36–3.63 (m, 10H, (CH2)2N + N3–CH3 + N1–CH3), 4.10–4.12 (m, 2H, N8–CH2), 7.10–7.25 (m, 2H, arom), 7.42–7.64 (m, 8H, arom). 13C NMR (75 MHz, CDCl3) δ (ppm): 23.33, 24.96, 27.52, 29.69, 30.90, 42.89, 49.26, 49.67, 52.99, 57.68, 59.07, 60.90, 71.92, 112.07, 115.89, 116.25, 118.32, 119.20, 121.01, 122.78, 122.89, 123.36, 128.08, 128.71, 128.87, 129.53, 131.21, 149.19, 151.28, 152.01, 154.04. 19F NMR (282 MHz, CDCl3): δ (ppm): −62.81 (s, 3F). MS (EI) m/z: 594.27 [M + 1]+. Anal. calcd for C31H34F3N7O2: C, 62.72; H, 5.77; N, 16.52; found: C, 62.61; H, 5.71; N, 16.67.
Radioligand binding assays
All of the compounds were tested in radioligand binding assays measuring their affinity for 5-HT1A and 5-HT7 receptors. The inhibition constants Ki were determined based on previously described protocolsCitation29,Citation32. One millimolar stock solutions of the compounds to be tested was prepared in DMSO. Serial dilutions of compounds were prepared in 96-well microplate in assay buffers using an automated pipetting system (epMotion 5070; Eppendorf). Radioligand binding was performed using cryopreserved membranes from cells stably expressing the relevant human receptor. Reagents and condition: 50 μL working solution of the tested compounds, 50 μL radioligand solution and 150 μL diluted membranes prepared in assay buffer were transferred to 96-well microplates. These were covered with sealing tape, mixed and incubated. The reaction was terminated by rapid filtration through UniFilter 96 GF/B filter microplate and 10 rapid washes with 200 μL 50 mM Tris buffer (4 °C, pH 7.4) were performed using vacuum manifold and 96-well pipettor. The UniFilter microplates were dried overnight at 37 °C in dry incubator. The UniFilter bottoms were sealed and 30 μL of liquid scintillator Betaplate Scint (PerkinElmer, Waltham, MA) was added to each well. The plates were allowed to equilibrate for 1 h, and then, radioactivity was counted in MicroBeta TriLux 1450 scintillation counter (PerkinElmer, Waltham, MA) at approximately 30% efficiency. Data were fitted to a one-site curve-fitting equation with Prism 5 (GraphPad Software Inc., La Jolla, CA) and Ki values were calculated using the Cheng–Prusoff equationCitation33. Each compound was tested in 10 concentrations from 1 × 10–5 M to 1 × 10–9 M (final concentration). All the assays were carried out in duplicates (n = 2). The inhibition constants Ki for D2 and 5-HT2A,6 receptors were determined based on previously described protocolsCitation29,Citation32.
The phosphodiesterase activity tests
Inhibition of PDE4B1 and PDE10A was measured using PDElight HTS cAMP phosphodiesterase assay kit (PDELightTM, Lonza) according to manufacturer’s recommendations. The percentage of inhibition was calculated to vehicle control (DMSO). Test and reference compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 mM and further diluted in assay buffer (10 mM Tris-HCl, 10 mM magnesium chloride and 0.05% Tween-20; pH 7.4) and eight concentrations were tested. Inhibition of PDE4B1 and PDE10A was measured using PDElight HTS cAMP phosphodiesterase assay kit (Lonza) according to the manufacturer’s recommendations. About 5 ng of human, recombinant PDE4B1 (Sigma-Aldrich, St. Louis, MO) or 10 ng PDE10A (BPS Biosciences) in assay buffer was incubated with reference (papaverine for PDE10A, 3-isobutyl-1-methylxanthine (IBMX) for PDE4B1) and tested compound for 20 min. After incubation, the cAMP substrate (final concentration 1.25 μM for PDE10A and 10 μM for PDE4B1) was added and incubated for 1 h. Then, PDELight AMP detection reagent was added and incubated 10 min. All reactions were carried out at 37 °C in white-walled, 384-well plate. Luminescence was measured in a multifunctional microplate reader (POLARstar Omega, BMG Labtech, Germany). All the assays were carried out in duplicates (n = 2).
Functional assays for 5-HT1A and 5-HT7 receptors
Test and reference compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 mM. Serial dilutions were prepared in 96-well microplate in assay buffers and eight concentrations were tested. All the assays were carried out in duplicates (n = 2). The EC50 values (concentration producing a half-maximal response) and IC50 values (concentration causing a half-maximal inhibition of the control agonist response) were determined by nonlinear regression analysis of the concentration response curves generated with mean replicate values using Hill equation curve fitting (GraphPad Prism 6.0 software; GraphPad Software Inc., La Jolla, CA). The log IC50 was used to obtain the antagonist equilibrium dissociation constant (Kb) by applying the Cheng–Prusoff EquationEquation (1)(1) :
(1)
where Kb – antagonist equilibrium dissociation constant, A – concentration of reference agonist in the assay, EC50A = EC50 value of reference agonist.
A cellular aeqorin-based functional assay was performed with recombinant CHO-K1 cells expressing mitochondrially targeted aequorin, human GPRC and the promiscuous G protein α16 for 5-HT1A. Assay was executed according to previously described protocolCitation34. After thawing, cells were transferred to (DMEM/HAM’s F12 with 0.1% protease-free BSA) and centrifuged. The cell pellet was resuspended in assay buffer and coelenterazine h was added at final concentration of 5 μM. The cell suspension was incubated at 16 °C, protected from light with constant agitation for 16 h and diluted with assay buffer to the concentration of 100 000 cells/ml. after 1 h of incubation, 50 μM of the cells suspension was dispensed using automatic injectors built into the radiometric and luminescence plate counter MicroBeta2 LumiJET (PerkinElmer, Waltham, MA) into white opaque 96-well microplate preloaded with test compounds. Immediate light emission generated following calcium mobilization was recordered for 30 s. In antagonist mode, after 25 min of incubation, the reference agonist was added to the above assay mix and light emission was recordered again. Final concentration of the reference agonist was to EC80 (100 nM for serotonin).
For the 5-HT7 receptor, adenylyl cyclase activity was monitored using cryopreserved CHO-K1 cells, expressing the human serotonin 5-HT7 receptor. Thawed cells were resuspended in stimulation buffer (HBSS, 5 mM HEPES, 0.5 IBMX and 0.1% BSA at pH 7.4) at 200 000 cells/mL. Ten microliters of cell suspension was added to 10 μL of tested compounds and loaded onto a white opaque half area 96-well microplate. An antagonist response experiment was performed with 10 nM serotonin as the reference agonist, and the agonist and antagonist were added simultaneously. Cell stimulation was performed for 60 min at room temperature. After incubation, cAMP measurements were taken with homogeneous time-resolved fluorescence energy transfer (TR-FRET) immunoassay using the LANCE Ultra cAMP kit (PerkinElmer, Waltham, MA). Ten microliters of EucAMP tracer working solution and 10 μL of ULight-anti-cAMP tracer working solution were added, mixed and incubated for 1 h. The TR-FRET signal was read on an EnVision microplate reader (PerkinElmer, Waltham, MA).
Determination of lipophilicity
Borate-phosphate buffer pH 7.0 with SDS (100 mM + 50 mM + 50 mM), SDS, 0.1 M of sodium hydroxide were purchased from Sigma-Aldrich (Poznań, Poland). Methanol (MeOH) HPLC purity. Reference drugs (purity > 99.5%) were purchased from different sources: barbital, phenobarbital (Polfa-Tarchomin, Poland), aminophenazone, caffeine (Polfa-Pabianice, Poland), chloramphenicol (Polfa-Kraków, Poland), phenytoin (Polfa-Warszawa, Poland), phenacetin (Azienda Chimica e Farmaceutica, Italy), benzocaine (Alfa Aesar, Germany), procaine (Amara Co., Poland). Trazodone was extracted from Trittico CR tablets (Aziende Chimiche Riunite Angelini Francesco (A.C.R.A.F.), Italy). The analyses were performed on Beckman P/ACE MDQ instrument (Beckman, Brea, CA) with DAD with UV detection at 220 nm. The electropherograms were recorded and analyzed with the 32 Karat Software version 8.0. Uncoated fused-silica capillaries (Beckman, Brea, CA; 39 cm distance to the detector, 50 μm internal diameter, 49 cm total length, 375 μm external diameter) were used. Background electrolyte, samples preparation and separation conditions were prepared according to methods of Bajda et.al.Citation35.
The logarithm of retention factor, log k, was calculated from the MEKC migration times according to the EquationEquation (2)(2)
Citation35:
(2)
where tR, tEOF and tMC are the migration time (min) of the solute, the EOF marker (MeOH) and the micelle marker (Sudan III), respectively. Estimated log p (cLog p) coefficients for the tested compounds were calculated from calibration curve obtained by linear regression of the log k and log p values of reference compounds according to EquationEquation (3)
(3) , where parameters b and a represent the slope and intercept of the curve, respectively:
(3)
Metabolism study
Human liver microsomes (HLMs, microsomes from liver, pooled, from human adult male and female), NADPH-regenerating system components and pentoxifylline were purchased from Sigma Aldrich (Poznań, Poland). Stock solutions of test compounds were prepared in ethanol. The final ethanol concentration in the incubation mixture was 0.38%. The incubation systems were composed of test compound (20 μM), HLMs (0.4 mg/mL), NADPH-regenerating system and potassium phosphate buffer (100 mM, pH 7.4). NADPH-regenerating system contained NADP, glucose-6-phosphate, glucose-6-phosphate dehydrogenase and potassium phosphate buffer (100 mM, pH 7.4). Firstly, the mixtures containing HLM, the test compound and a buffer were preincubated at 37 °C for 15 min before the addition of the NADPH-regenerating system. The resulting mixture was incubated for 15 min at 37 °C in a thermoblock. Next, pentoxifylline (20 μM, internal standard) was added, and the reaction was stopped by the addition of perchloric acid (69–72%, by volume). Proteins were sedimented by centrifugation. The supernatant was analyzed using UPLC/MS (Waters Corporation, Milford, MA) for qualitative analysis. The peak area ratios of analyte versus internal standard were used to calculate the % remaining at the end of 15 min of biotransformation. All the assays were carried out in duplicates (n = 2).
Pharmacology – general condition
The experiments were performed on male Albino Swiss mice (22–28 g). The animals were kept at a room temperature of 20 ± 1 °C and had free access to food (standard laboratory pellets) and tap water before the experiment. All the experiments were conducted in the light phase between 9 a.m. and 2 p.m. All the experimental procedures were approved by the Local Ethics Commission for Animal Experiments of Jagiellonian University in Cracow. Citalopram (Sigma-Aldrich, St. Louis, MO) was dissolved in distilled water. The investigated compound (9) and diazepam were suspended in a 1% aqueous solution of Tween 80 and was injected intraperitoneally (ip) 60 min before the test (citalopram 30 min before the test). Each experimental group consisted of six to ten animals, and all the animals were used only once.
Forced swim (porsolt) test in swiss albino mice
The experiment was carried out according to the method of Porsolt et al.Citation37. Mice were individually placed in a glass cylinder (25 cm high; 10 cm in diameter) containing 10 cm of water maintained at 23–25 °C and were left there for 6 min. A mouse was regarded as immobile when it remained floating on the water, making only small movements to keep its head above it. The total duration of immobility was recorded during the last 4 min of a 6-min test session.
Four-plate test in Swiss albino mice
The four-plate apparatus (BIOSEB) consists of a plastic cage (25 × 18 × 16 cm) floored by four identical rectangular metal plates (8 × 11 cm) separated from one another by a gap of 4 mm. The top of the cage is covered by a transparent Perspex lid that prevents escape behavior. The plates are connected to a device that can generate electric shocks. Following a 15-s habituation period, the animal’s motivation to explore a novel environment was suppressed by an electric foot shock (0.8 mA, 0.5 s) every time it moves from one plate to another during a 1-min test session. This action is referred to as a “punished crossing” and was followed by a 3-s shock interval, during which the animal can move across plates without receiving a shockCitation38. The number of punished crossings received by an animal was recorded during the 60-s period.
Spontaneous locomotor activity in mice
Locomotor activity was recorded with an Opto M3 multichannel activity monitor (MultiDevice Software v.1.3, Columbus Instruments). Swiss albino mice were individually placed in plastic cages (22 × 12 × 13 cm) for 30-min habituation period, and then, the ambulation was counted from 2 to 6 min or for 1 min 15 s, that is the time equal to the observation period in the forced swim test or the four-plate test, respectively. The cages were cleaned up with 70% ethanol after each mouse.
Statistics
All the data are presented as the mean ± SEM. The statistical significance of results was evaluated by a one-way analysis of variance (ANOVA) followed by Bonferroni’s comparison test. p < 0.05 was considered statistically significant.
Molecular modeling
The homology models of human 5-HT1A and 5-HT7 serotonin receptors adopted for docking in the present study were built on the template of β2 adrenergic receptor crystal structure (PDB ID: 2RH1). The homology models were generated using thoroughly tested methodCitation29 and were described in previously published papersCitation34,39–41. Sequence alignments between target receptors (UniProt database accession numbers P08908 and P34969, respectively) and the template were performed by hhsearch tool via GeneSilico MetaserverCitation42. The crude receptor models were obtained using SwissModelCitation43 and were validated by processing in Protein Preparation Wizard. For each receptor type, a set of bioactive compounds was selected for ligand-steered binding site optimization, which was performed using induced fit docking (IFD) workflowCitation44. The procedure resulted in a variety of comparative models, mirroring conformational plasticity of the protein. Ligand structures were optimized using LigPrep tool and were docked to the target models using Glide XP-docking procedure, setting flexible docking option, retaining ring conformations and nitrogen inversions as were selected in the previous step. H-bond constraints and centroid of a grid box (22 × 22 × 22 Å) for docking studies were located on Asp3.32. The remaining parameters were default. Phisicochemical (PSA) and ADME (QPPMDCK) properties were calculated with use of QikProp module. Glide, induced fit docking, LigPrep, Protein Preparation Wizard and QikProp were implemented in Small-Molecule Drug Discovery Suite (Schrödinger, Inc.), which was licensed for Jagiellonian University Medical College.
Results and discussion
Chemistry
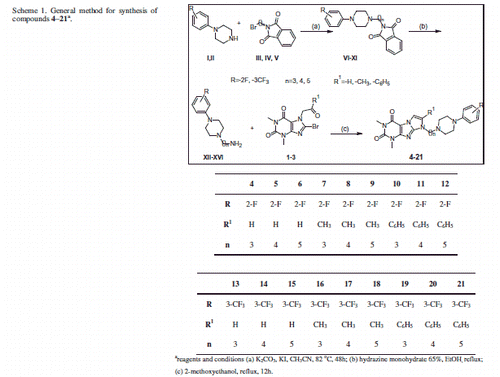
Following our previously developed route, the final derivatives of 1,3-dimethyl-(1H,8H)-imidazo[2,1-f]purine-2,4-dione (4–21) were achieved in via multistep synthesis. In order to carry out the process, two series of synthons were necessary: 7-ketonyl derivatives of 8-bromotheophylline (1–3) and N-(aminoalkyl)arylpiperazines (XII–XVI). Compound 1 was obtained from 8-bromotheophylline according to described earlier methodCitation29. Compounds 2 and 3 were obtained from 8-bromotheophylline in reaction of alkylation with α-chloro ketones in biphasic system, in the presence of potassium carbonate in acetonitrile. N-(aminoalkyl)arylpiperazines (XII–XVI) were obtained in two steps, according to the Gabriel methodCitation45, with minor modifications. First, 1-(2-fluorophenyl)- or 1-(3-trifluoromethyl)-phenyl)piperazine (I–II) were alkylated in biphasic system, using catalytic amount of potassium iodide with appropriate 2-(bromoalkyl)-1H-isoindoline-1,3(2H)-dione (III–V), in the presence of potassium carbonate in acetonitrile, to give the corresponding phthalimido derivatives. All intermediates were purified by column chromatography using dichloromethane/methanol (9/1) as an eluent. Next, phthalimido derivatives were hydrolyzed with hydrazine monohydrate to afford the N-(aminoalkyl)arylpiperazines (XII–XVI)Citation45. Final ligands 4–21 (Scheme 1) were synthesized as a result of cyclocondesation of 7-ketonyl derivatives of 8-bromotheophylline (1–3) with corresponding N-(aminoalkyl)arylpiperazines (XII–XVI), occurs by refluxing the in 2-methoxyethanol (Scheme 1). Note, in this condition, final compounds were obtained in good to moderate yields, depending on 7-ketonyl derivatives of 8-bromotheophylline used. All final compounds 4–21 in order to obtain analytic samples and further biological evaluation, were purified by column chromatography using a mixture of dichloromethane and methanol in various proportion as an eluent. The chemical structure of compounds 4–21 was verified by 1H, 13C and 19F NMR tests.
Scheme 1. General method for synthesis of compounds 4–21a.
Biological evaluation
In vitro examination of the newly synthesized compounds occurred in three steps. First, all of the compounds were tested in assays against cloned 5-HT1A and 5-HT7 receptors with standard competitive displacement assays and the inhibition constants Ki were determined. For structure–activity relationship studies, we examined the impact of three chemical elements: substitution at phenyl ring of LCAPs, the length of the linker between the purine core and LCAPs and substitution at the 7-position of the purine core.
All tested compounds possessed high affinity at 5-HT1ARs with some binding in the same range as serotonine (Ki from 0.1 to 33 nM) (), but the 3-CF3-phenyl-piperazinylalkyl derivatives of 1H-imidazo[2,1-f]purine-2,4(3H,8H)-diones were more active than their analogs with a 2-F moiety. For example, compound 7 exhibited the lowest affinity (Ki = 33 nM), and the affinity of compound 13 was more than six times higher than that of compound 4. The tetramethylene spacer (C4) between the purine core and LCAPs proved to be optimal for 5-HT1A sites and compounds with the highest affinity (one exception comp. 11) belong to this group. This optimal length of spacer leads to an absence of influence of the substituent at 7 position of the purine core for 5-HT1A affinities. In a group of 2-F analogs, it is easier to prove the unfavorable impact of the propyl (C3) alkyl chain than C4 preference. Moreover, the weaker influence of the 2-F substituent on 5-HT1AR affinity exposes the impact of a methyl moiety at the 7-position of imidazo[2,1-f]purine-2,4-dione (5 vs. 8, 8 vs. 11, 6 vs. 9, and 9 vs. 12). A spectrum of activities was observed for 5-HT7R, and tested compounds exhibited either a lack of affinity (4, 5, 13, 16), or high (comp. 12 Ki = 9 nM, comp. 21 Ki = 1 nM, comp. 18 and 20 Ki = 10 nM) or moderate affinity (comp. 6–11, 14, 17, Ki from 20–152 nM) (). The structure–activity relationship revealed that, in the case of this receptor, 3-CF3 derivatives in general bound with the highest affinity, and compound 21 with Ki = 1 nM belongs to this group. Due to the structural similarity between 5-HT1A and 5-HT7 receptors, many of the LCAPs described in the literature as 5-HT7R ligands derive from a structural modification of the terminal fragment present in 5-HT1AR ligands. Moreover, studies have revealed the critical role of the arylpiperazine substitution pattern in determining the receptor affinity. Volk et al.Citation46 found out that electron-donating substituents (–OCH3) were superior to electron-withdrawing (–Cl, –F) for binding to the 5-HT1A and 5-HT7 receptors. Our study confirmed that 2-F and 3-CF3 substituents at the phenyl ring of arylpiperazine were highly preferential for the 5-HT1AR over the 5-HT7R. The influence of substituents 2-F and 3-CF3 on 5-HT7 receptor affinity in the group of tested compounds appears to be equivocal, and a spectrum of activities was observed. It appears that, for LCAPs with a terminal fragment based on imidazo[2,1-f]purine-2,4-dione, the length of the aliphatic spacer (C5) is decisive for 5-HT7R affinity. This is in accordance with Leopoldo et al.Citation47, who found that, in a group of N-(1,2,3,4-tetrahydronaphthalen-1-yl)-4-arylpiperazinealkylamide derivatives, five methylene units separating the amide moiety and the piperazine ring were preferred for the affinity to 5-HT7Rs.
Table 1. The binding data and cLog p of compounds 4–21.
The second step of the in vitro study was the PDE inhibitory activity of targeted compounds using a bioluminescent detection system, based on the activity of PDEs which utilized second messenger cAMP. The AMP produced from the hydrolysis of cAMP was quantified using an AMP detection reagent that converts AMP directly to ATP. The therapeutic effect of theophylline (bronchodilatory, positive inotropic effect on heart, psychostimulant) has not been attributed to any single mechanism of action, but the inhibition of PDE was the most important. In order to identify which of the mechanisms involved in the central nervous system action of the LCAPs with imidazo[2,1-f]purine-2,4-dione as terminal fragment, PDE4B and PDE10A inhibitory activity was measured. It is noteworthy that, to assess the relative importance of receptor-dependent and/or receptor-independent mechanisms of action, the same range of concentrations as for receptor affinity were taken as a measure of PDE inhibitory activity (starting from 10−5 M). The investigated compounds had weak inhibitory potencies for PDE4B and PDE10A only at an initial concentration of 10−5 M, and no inhibition was detected at lower concentrations (, ). Moreover, the inhibitory effect of the tested compounds was compared with theophylline.
Figure 3. Inhibitor activity (%) against PDE10A for compound 9, theophylline and papaverine in concentration of 10–5 M and 10–5.5 M.
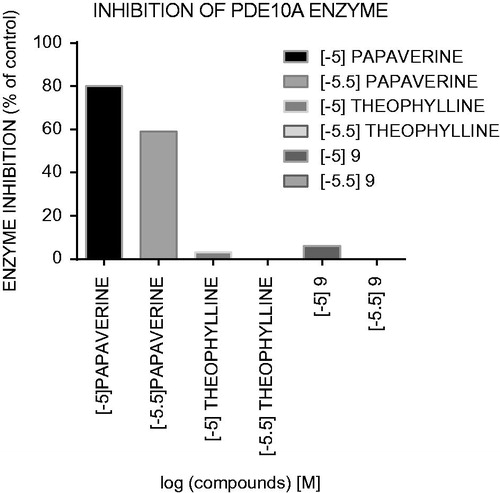
Table 2. Inhibition of PDE (%) of compounds 4–21.
On the basis of the binding affinity results, a series of compounds possessing affinity below 3 nM for 5-HT1AR (5, 8, 9, 12–21) and below 50 nM for 5-HT7 receptors (9, 12, 15, 17, 18, 20, 21) was selected for functional profile characterization. Intrinsic activity studies were performed using in vitro measures of receptor activation. No activation of any of the receptors was observed, and the compounds showed antagonist properties, especially the 5-HT1A (5, 9, 12, 14, 15, 17–21) ( and ). In case of 5-HT7R, there are significant differences between the binding and functional tests results for selected compounds. The reason for such a discrepancy is not clear, but one have to bear in mind, that the binding experiments are reflecting simply the competition at the level of a binding site which do not require intracellular signaling mechanisms (e.g. G-proteins or cAMP), whereas the functional tests require appropriate cellular machinery in addition to 5-HT7 receptor expression. In other words, affinity and lack of stimulation of particular signaling pathway is not equal to effective functional antagonism of the receptor. In fact, the functional tests provide additional level of characteristics for the tested compound and are more stringent than binding experiments and therefore constituting good tools to select compounds that are significantly active at 5-HT7 receptors.
Figure 4. Antagonist mode (Kb) of the selected compounds for 5-HT1A receptor.
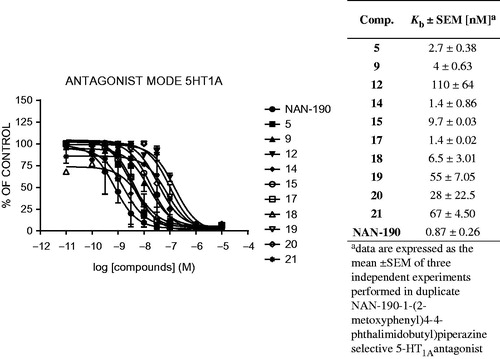
Figure 5. Antagonist mode (Kb) of selected compounds for the 5-HT7 receptor.
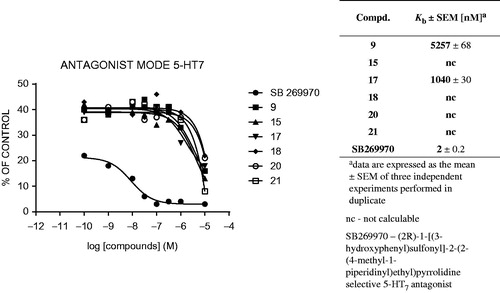
For 5-HT1A receptors, Paluchowska et al.Citation21 evaluated a series of 1-substituted 4-(4-arylpiperazin-1-yl)cyclohexane derivatives with different cyclic imide/amide termini, and their flexible tetramethylene analogs. Functional examination revealed that rigid ligands with 2-OCH3 groups in the aryl moiety and a cyclic imide system in the opposite terminal behaved like 5-HT1A receptor antagonists. On the other hand, unsubstituted, 3-Cl or 3-CF3 substituted derivatives, as well as those with a cyclic amide group in the terminal fragment, exhibited agonistic or partial agonistic activity. Our study revealed that for LCAPs with terminal fragments based on imidazo[2,1-f]purine-2,4-dione and a tetramethylene spacer, ligands with 3-CF3 group in the aryl moiety (14, 17, 20) showed 5-HT1A antagonist properties. Interestingly, in the group of ligands with a 2-F moiety and tetramethylene spacer, only compound 5 exhibited the ability to inhibit the 5-HT1A receptor. Elongation of the spacer from C4 to C5 for 2-F (9 and 12) and 3-CF3 (14 vs. 15, 17 vs. 18, 20 vs. 21) derivatives caused decreased in a value of Kb. The above observation indicates that not only the combination of structural features of arylpiperazinealkyl moiety found to be connected with the antagonistic properties of the ligands, but their overall effect also depends on the substitution in 7-position of terminal imidazo[2,1-f]purine-2,4-dione fragments.
Lipophilicity
In medicinal chemistry, numerous measured or calculated parameters have been used for the characterization of lipophilicity, but the logarithm of the n-octanol/water partition coefficient (log p) is still the generally accepted and primarily applied descriptor of lipophilicity in quantitative structure-activity relationship (QSAR) studies. The lipophilicity of a drug candidate can affect its both pharmacokinetic and pharmacodynamic properties. In particular, the ability of a molecule to cross the cell membrane depends on its partition coefficientCitation36. The MEKC system applied for estimation of lipophilicity uses micelles and closely mimics biological membranes; therefore, it is more suitable for physicochemical characterization of bioactive molecules. The “rule of five” defines the range of log p for good intestinal permeability and penetration to the brain. The estimated log p values for compounds 6, 8, 9, 11, 13 and 20 were in ranges defined by the “rule of five” (log p < 5), which indicates good intestinal permeability ().
Metabolic stability
Metabolic stability is a critical property for drug candidates, because it contributes to oral bioavailability and overall pharmacokinetic performanceCitation48. Metabolic stability is usually quantified by monitoring the disappearance over time of the parent compound in liver preparations. The disappearance of the parent compound was monitored by calculating the percentage of each compound remaining relative to 0 min and this was plotted against the incubation time. Currently, commercially available human liver microsomes (HLM) have been the most widely used in vitro models for metabolic stability studiesCitation49. Compounds 9 and 20 were selected for in vitro metabolic stability assays. It is noteworthy that this is the first report of the metabolic stability of LCAPs with terminal fragments based on imidazo[2,1-f]purine-2,4(3H,8H-dione). In terms of the conditions of our study, compounds 9 and 20 after 15 min of biotransformation, showed acceptable metabolic stability but 20 was slightly more susceptible to metabolism ().
Table 3. Metabolic stability screen of compounds 9 and 20 in human liver microsomes (HLM) and major metabolites characteristics.
Pharmacological in vivo evaluation
Finally, compound 9 was selected for further in vivo examination on the basis of the following. First, clinical and preclinical studies have shown that antidepressant-like and/or anxiolytic-like effects are mainly produced by serotonin receptor ligandsCitation50. Compound 9 (8-(5-(4-(2-fluorophenyl)piperazin-1-yl)pentyl)-1,3,7-trimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione) showed very high 5-HT1A receptor affinity (Ki = 2.5 nM) and high 5-HT7 receptor affinity (Ki = 30.8 nM). In our previous studies, a spectrum of receptor activities was observed for compounds with a substituent at the 7th position of the imidazo[2,1-f]purine-2,4-dione system. Some compounds were potent 5-HT1A and/or 5-HT7 receptor ligands with additional affinity for dopamine D2 receptors. Therefore, to test the feasibility of multireceptor activity, compound 9 was evaluated for affinity for D2 (Ki = 437 nM) and other serotonin subtype (5-HT2A,6 Ki > 1000 nM) according to the procedures described earlierCitation32,Citation34. Next, behavioral effects (antidepressant like) similar to those of selective serotonin reuptake inhibitors (SSRIs, e.g. fluoxetine) have been produced by antagonists of 5-HT1A and 5-HT7 receptorsCitation51. Moreover, an antagonist of 5-HT1A and/or 5-HT7 receptors induces an anxiolytic-like effect. Compound 9 showed 5-HT1A antagonistic properties exemplified by Kb = 4 nM. Our cumulative results prompted us to evaluate the activity of compound 9 in FST and four-plate tests in mice.
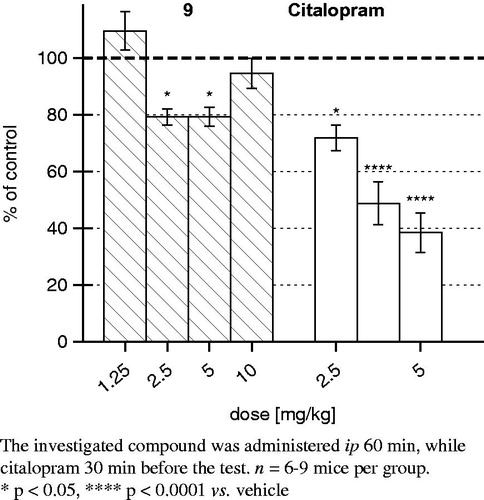
Compound 9 (1.25–5 mg/kg), as well as citalopram (1.25–5 mg/kg) used as a reference compound, revealed antidepressant-like properties. Compound 9 administered at two higher doses (2.5 and 5 mg/kg) significantly reduced the immobility time of mice in that test by 21%. The reference antidepressant citalopram shortened (by 28%, 51% and 61%, respectively) the immobility time of mice at all doses (). The additional dose (10 mg/kg) of compound 9 have been studied in FST and to check the type of dose–response curve. Compound 9 shows U-shaped dose–response relationship in the forced swim test. Some anxiolytics and antidepressants and newly synthesized compound (9) with different mechanisms of action can produce such U-shaped dose–response effects in animal models, for example Millan et al.Citation2, Wesołowska et al.Citation52.
Figure 6. The effect of the tested compound and 9 citalopram in the forced swim test (FST) in mice.
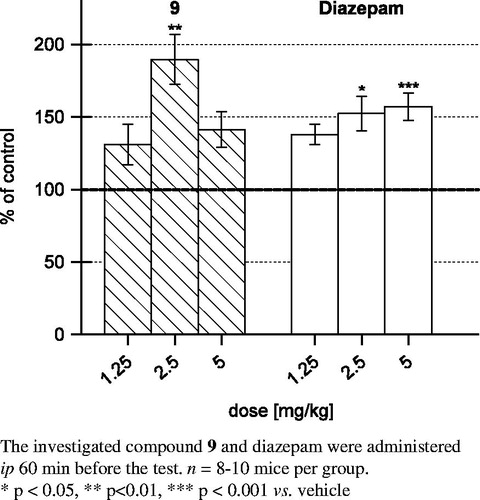
Compound 9 given at a dose of 2.5 mg/kg showed anxiolytic-like activity in the four-plate test, increasing by 90%, in a statistically significant manner, the number of punished crossings accepted by animals during the observation sessions. The reference anxiolytic drug, diazepam, administered at doses of 1.25, 2.5 and 5 mg/kg, significantly increased (by 38%, 52% and 57%, respectively) the number of punished crossings. It is worth noting that the potency of antianxiety effects evoked by 9 (2.5 mg/kg) is greater than that of the reference anxiolytic drug, diazepam, given at the same dose of 2.5 mg/kg. Compound 9 shows inverted U-shaped dose–response relationship in four-plate test. The effects of compound 9 and diazepam in the four-plate test are shown in .
Figure 7. The effect of the tested compound 9 and diazepam in the four-plate test in mice.
Moreover, the antidepressant-and/or anxiolytic-like properties of the investigated agents, citalopram and diazepam, seem to be specific, since when administered at effective doses they did not stimulate spontaneous locomotor activity in mice during the 4 min and 1 min 15 s sessions (i.e. at a time identical to the observation period in the FST and four-plate test, respectively) (data not shown).
Molecular modeling
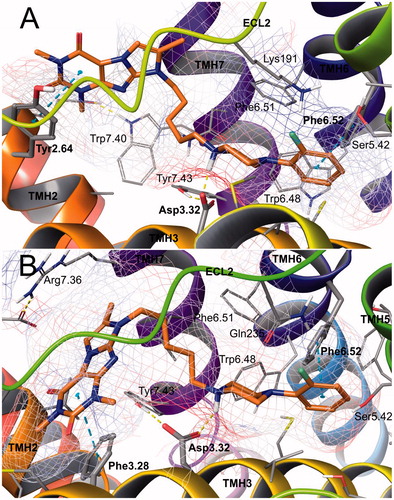
Docking experiments were performed for compound 9. The molecule was situated across the two cavities of the binding site: the first one constituted by the transmembrane helices (TMHs) 3–6 in both the receptor models, and the second one between TMHs 2 and 7 (5-HT1A receptor) or TMHs 3 and 7 (5-HT7 receptor). The anchoring interaction in both sites was a charge-reinforced hydrogen bond between the protonated nitrogen atom of the ligand and the carboxyl group of Asp3.32, as well as the CH-π interactions of the arylpiperazine and aromatic amino acid cluster in the deeper cavity, mainly Phe6.52 (). In the case of the 5-HT1A receptor, the electronegative 2-F group of the phenylpiperazine fragment was turned toward the electropositive receptor surface (blue color of the electrostatic potential grid), finding beneficial fluorine-bonding interaction with the side chain residues of Lys191 (the ɛ-amino group) from the second extracellular loop (ECL 2) (). The 5-HT7 receptor-binding site accepted the 2-fluorophenyl moiety in an analogous manner, exposing an electropositive surface of Gln235 (). The weak fluorine bonding interactions are considered to enrich the ligand–receptor contacts and therefore favorably adjust the ligand binding specificity. The imidazo[2,1-f]purine fragment of the molecule occupied the second cavity and established favorable, characteristic for each receptor type, aromatic interactions there. In the 5-HT1A receptor, it interacted by π-π stacking with phenyl ring of Tyr2.64 (). In the 5-HT7 receptor-binding site, the heterocyclic system formed essential π–π stacking with Phe3.28 ().
Figure 8. Binding modes of compound 9 in the site of 5-HT1A (A) and 5-HT7 (B) receptors. Amino acid residues engaged in ligand binding (within 4 Å from the ligand atoms) are displayed as sticks, whereas those forming H-bonds (dotted yellow lines) or π–π/CH-π stacking (dotted cyan lines) are represented as thick sticks. Van der Waals molecular surface of the receptor binding site is shown as grid, colored due to electrostatic potential. For the sake of clarity, ECL2 residues were hidden. TMH – transmembrane helix; ECL – extracellular loop.
Conclusion
In conclusion, the paper presents the synthesis of arylpiperazinylalkyl derivatives of 1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione with 2-fluoro and 3-trifluoromethyl moieties. The study allowed us to identify some potent 5-HT1A (5, 14, 17–21), 5-HT7 (17, 18, 20) and mixed 5-HT1A/5-HT7 (12, 15, 21) receptor ligands with weak inhibitory potencies for PDE4B and PDE10A. The study reveals that 5-HT1A and 5-HT7 receptor affinity benefits from the introduction of a fluorine moiety while introduction of imidazole ring into 7, 8 position of theophylline had no significant effect on inhibitory potencies for selected PDE. The tested compounds were in the ranges defined by the “rule of five” (log p < 5), which indicates good intestinal permeability and showed good metabolic stability. In preliminary pharmacological in vivo studies, selected 8-(5-(4-(2-fluorophenyl)piperazin-1-yl)pentyl)-1,3,7-trimethyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione (9) in the FST in mice behaved as a potential antidepressant. Moreover, potency of antianxiety effects evoked by 9 in the four-plate test (2.5 mg/kg) is greater than that of the reference anxiolytic drug, diazepam. The binding mode in the 5-HT1A and 5-HT7 receptors has been elucidated for the representative molecule by means of molecular docking, pointing out favorable interactions, characteristic for each receptor type. Studies might be beneficial to further investigation of the mechanism of CNS action of imidazo[2,1-f]purine-2,4(3H,8H)-dione derivatives.
Declaration of interest
This study was financially supported by the National Science Center Poland (NSC) funded grant based on the decision DEC-2012/07/B/NZ7/01173 and Funds of Statutory Activity UJ CM (K/ZDS/005533). The authors declare no conflict of interest.
Supplementary material available online
IENZ_1198902_Supplementary_Material.pdf
Download PDF (91.5 KB)References
- Millan MJ. Serotonin 5-HT2C receptors as a target for the treatment of depressive and anxious states: focus on novel therapeutic strategies. Therapie 2005;60:441–60
- Millan MJ, Brocco MD, ekeyne A. Anxiolytic properties of agomelatine, an antidepressant with melatoninergic and serotonergic properties: role of 5-HT2C receptor blockade. Psychopharmacology (Berl) 2005;77:448–58
- Wesołowska A. Potential role of the 5-HT6 receptor in depression and anxiety: an overview of preclinical data. Pharmacol Rep 2010;62:564–77
- Hedlund PB. The 5-HT7 receptor and disorders of the nervous system: an overview. Psychopharmacology (Berl) 2009;206:345–54
- Blier P, Ward NM. Is there a role for 5-HT1A agonists in the treatment of depression? Biol Psychiatry 2003;53:193–203
- Naughton M, Mulrooney JB, Leonard BE. A review of the role of serotonin receptors in psychiatric disorders. Hum Psychopharmacol 2000;15:397–415
- Canale V, Kurczab R, Partyka A et al. Towards novel 5-HT7 versus 5-HT1A receptor ligands among LCAPs with cyclic amino acid amide fragments: design, synthesis, and antidepressant properties. Part II. Eur J Med Chem 2015;92:202–11
- Canale V, Guzik P, Kurczab R et al. Solid-supported synthesis, molecular modeling, and biological activity of long-chain arylpiperazine derivatives with cyclic amino acid amide fragments as 5-HT7 and 5-HT1A receptor ligands. Eur J Med Chem 2014;78:10–22
- Medina RA, Vázquez-Villa H, Gómez-Tamayo JC, et al. The extracellular entrance provides selectivity to serotonin 5-HT7 receptor antagonists with antidepressant-like behavior in vivo. J Med Chem 2014;57:6879–84
- Cagnotto A, Salmona M, Casagni A, et al. Ros R. Targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors for developing effective antipsychotics: synthesis, biological characterization, and behavioral studies. J Med Chem 2014;57:9578–97
- Kowalski P, Mitka M, Jaśkowska J, et al. New arylpiperazines with flexible versus partly constrained linker as serotonin 5-HT(1A)/5-HT(7) receptor ligands. Arch Pharm (Weinheim) 2013;346:339–48
- Zajdel P, Marciniec K, Maślankiewicz A, et al. Antidepressant and antipsychotic activity of new quinoline- and isoquinolinesulfonamide analogs of aripiprazole targeting serotonin 5-HT1A/5-HT2A/5-HT7 and dopamine D2/D3 receptors. Eur J Med Chem 2013;60:42–50
- Lacivita E, De Giorgio P, Patarnello D, et al. Towards metabolically stable 5-HT7 receptor ligands: a study on 1-arylpiperazine derivatives and related isosters. Exp Brain Res 2013;230:569–82
- Zajdel P, Marciniec K, Maślankiewicz A, et al. Quinoline- and isoquinoline-sulfonamide derivatives of LCAP as potent CNS multi-receptor-5-HT1A/5-HT2A/5-HT7 and D2/D3/D4-agents: the synthesis and pharmacological evaluation. Bioorg Med Chem 2012;20:1545–56
- Lacivita E, Patarnello D, Stroth N et al. Investigations on the 1-(2-Biphenyl)piperazine motif: identification of new potent and selective ligands for the serotonin7 (5-HT7) receptor with agonist or antagonist action in vitro or ex vivo. J Med Chem 2012;55:6375–80
- Volk B, Gacsályi I, Pallagi K, et al. Optimization of (arylpiperazinylbutyl)oxindoles exhibiting selective 5-HT7 receptor antagonist activity. J Med Chem 2011;54:6657–69
- Hussainy RA, Verbeek J, van der Born D, et al. Design, synthesis, radiolabeling, and in vitro and in vivo evaluation of bridgehead iodinated analogues of N-{2-[4-(2-methoxyphenyl)piperazin-1-yl]ethyl}-N-(pyridin-2-yl)cyclohexanecarboxamide (WAY-100635) as potential SPECT ligands for the 5-HT1A receptor. J Med Chem 2011;54:3480–91
- Alonso D, Vázquez-Villa H, Gamo AM, et al. Development of fluorescent ligands for the human 5-HT1A receptor. ACS Med Chem Lett 2010;1:249–53
- Medina RA, Sallander J, Benhamu B, et al. Synthesis of new serotonin 5-HT7 receptor ligands. Determinants of 5-HT7/5-HT1A receptor selectivity. J Med Chem 2009;52:2384–92
- Siracusa MA, Salerno L, Modica MN, et al. Synthesis of new arylpiperazinylalkylthiobenzimidazole, benzothiazole, or benzoxazole derivatives as potent and selective 5-HT1A serotonin receptor ligands. J Med Chem 2008;51:4529–38
- Paluchowska MH, Bugno R, Tatarczyńska E, et al. The influence of modifications in imide fragment structure on 5-HT(1A) and 5-HT(7) receptor affinity and in vivo pharmacological properties of some new 1-(m-trifluoromethylphenyl)piperazines. Bioorg Med Chem 2007;15:7116–25
- Lepailleur A, Bureau R, Paillet-Loilier M, et al. Molecular modeling studies focused on 5-HT7 versus 5-HT1A selectivity. Discovery of novel phenylpyrrole derivatives with high affinity for 5-HT7 receptors. J Chem Inf Model 2005;45:1075–81
- Vermeulen ES, van Smeden M, Schmidt AW, et al. Novel 5-HT7 receptor inverse agonists. Synthesis and molecular modeling of arylpiperazine- and 1,2,3,4-tetrahydroisoquinoline-based arylsulfonamides. J Med Chem 2004;47:5451–66
- Perrone R, Berardi F, Colabufo NA, et al. Synthesis and structure-affinity relationships of 1-[omega-(4-aryl-1-piperazinyl)alkyl]-1-aryl ketones as 5-HT(7) receptor ligands. J Med Chem 2003;46:646–9
- Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov 2004;3:353–9
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci 2006;7:137–51
- Maxwell CR, Kanes SJ, Abel T, Siegel SJ. Phosphodiesterase inhibitors: a novel mechanism for receptor-independent antipsychotic medications. Neuroscience 2004;129:101–7
- Kanes SJ, Tokarczyk J, Siegel SJ, et al. Rolipram: a specific phosphodiesterase 4 inhibitor with potential antipsychotic activity. Neuroscience 2007;144:239–46
- Zagórska A, Pawłowski M, Siwek A, et al. Synthesis and pharmacological evaluation of novel tricyclic[2,1-f]theophylline derivatives. Arch Pharm (Weinheim) 2013;346:832–9
- Partyka A, Jarosz J, Wasik A, et al. Novel tricyclic[2,1-f]theophylline derivatives of LCAP with activity in mouse models of affective disorders. J Pharm Pharmacol 2014;66:1755–62
- Zagórska A, Jurczyk S, Pawłowski M, et al. Synthesis and preliminary pharmacological evaluation of imidazo[2,1-f]purine-2,4-dione derivatives. Eur J Med Chem 2009;44:4288–96
- Zagórska A, Kołaczkowski M, Bucki A, et al. Structure-activity relationships and molecular studies of novel arylpiperazinylalkyl purine-2,4-diones and purine-2,4,8-triones with antidepressant and anxiolytic activity. Eur J Med Chem 2015;97:142–54
- Cheng Y, Prusoff W. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 1973;22:3099–108
- Kołaczkowski M, Marcinkowska M, Bucki A, et al. Novel arylsulfonamide derivatives with 5-HT6/5-HT7 receptor antagonism targeting behavioral and psychological symptoms of dementia. J Med Chem 2014;57:4543–57
- Bajda M, Guła A, Więckowski K, Malawska B. Determination of lipophilicity of γ-butyrolactone derivatives with anticonvulsant and analgetic activity using micellar electrokinetic chromatography. Electrophoresis 2013;34:3079–85
- Waring MJ. Lipophilicity in drug discovery. Expert Opin Drug Discov 2010;5:235–48
- Porsolt RD, Bertin A, Blavet N, et al. Immobility induced by forced swimming in rats: effects of agents which modify central catecholamine and serotonin activity. Eur J Pharmacol 1979;57:201–10
- Aron C, Simon P, Larousse C, Boissier JR. Evaluation of a rapid technique for detecting minor tranquilizers. Neuropharmacology 1971;10:459–69
- Kołaczkowski M, Bucki A, Feder M, Pawłowski M. Ligand-optimized homology models of D1 and D2 dopamine receptors: application for virtual screening. J Chem Inf Model 2013;53:638–48
- Kowalski P, Jaśkowska J, Bojarski AJ, et al. Evaluation of 1-arylpiperazine derivative of hydroxybenzamides as 5-HT1A and 5-HT7 serotonin receptor ligands: an experimental and molecular modeling approach. J Heterocycl Chem 2011;48:192–8
- Chłoń-Rzepa G, Bucki A, Kołaczkowski M, et al. Arylpiperazinylalkyl aerivatives of 8-amino-1,3-dimethylpurine-2,6-dione as novel multitarget 5-HT/D receptor agents with potential antipsychotic activity. J Enzyme Inhib Med Chem 2015;25:1–15
- Kurowski MA, Bujnicki JM. Genesilico protein structure prediction meta-server. Nucleic Acids Res 2003;31:3305–7
- Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 2003;31:3381–5
- Sherman W, Day T, Jacobson MP, et al. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 2006;49:534–53
- Gabriel S. Ueber eine Darstellungsweise primärer amine aus den entsprechenden Halogenverbindungen. Chem Ber 1887;20:2224–36
- Volk B, Barkoczy J, Hegedus E, et al. Phenylpiperazinyl-butyl)oxindoles as selective 5-HT7 receptor antagonists. J Med Chem 2008;51:2522–32
- Leopoldo M, Lacivita E, Contino M, et al. Structure-activity relationship study on N-(1,2,3,4-tetrahydronaphthalen-1-yl)-4-aryl-1-piperazinehexanamides, a class of 5-HT7 receptor agents. 2. J Med Chem 2007;50:4214–21
- Di L, Kerns EH, Hong Y, et al. Optimization of a higher throughput microsomal stability screening assay for profiling drug discovery candidates. J Biomol Screen 2003;8:453–62
- Huang J, Si L, Fan Z, et al. In vitro metabolic stability and metabolite profiling of TJ0711 hydrochloride, a newly developed vasodilatory β-blocker, using a liquid chromatography-tandem mass spectrometry method. J Chromatogr B Analyt Technol Biomed Life Sci 2011;879:3386–92
- Carr GV, Lucki I. The role of serotonin receptor subtypes in treating depression: a review of animal studies. Psychopharmacology (Berl.) 2011;213:265–87
- Hitchcock SA, Pennington LD. Structure-brain exposure relationships. J Med Chem 2006;49:7559–83
- Wesołowska A, Nikiforuk A, Stachowicz K, Tatarczyńska E. Effect of the selective 5-HT7 receptor antagonist SB 269970 in animal models of anxiety and depression. Neuropharmacology 2006;51:578–86