
Abstract
Bcr-AblT315I induced drug resistance remains a major challenge to chronic myelogenous leukemia (CML) treatment. Herein, we reported GZD856 as a novel orally bioavailable Bcr-AblT315I inhibitor, which strongly suppressed the kinase activities of both native Bcr-Abl and the T315I mutant with IC50 values of 19.9 and 15.4 nM, and potently inhibited proliferation of corresponding K562, Ba/F3WT and Ba/F3T315I cells with IC50 values of 2.2, 0.64 and 10.8 nM. Furthermore, GZD856 potently suppressed tumor growth in mouse bearing xenograft K562 and Ba/F3 cells expressing Bcr-AblT315I. Thus, GZD856 may serve as a promising lead for the development of Bcr-Abl inhibitors overcoming acquired imatinib resistance.
Graphical Abstract
Introduction
Chronic myelogenous leukemia (CML) is a hematological stem cell disorder caused by expansion of lymphocytic or myeloid blasts in the blood or bone marrow, and resulting from a t (9; 22) reciprocal translocation encoding the constitutively active Bcr-Abl tyrosine kinaseCitation1. Bcr-Abl, a 210-kDa non-receptor protein kinase that catalyzes the transfer of γ-phosphoryl group from ATP to the hydroxyl group of specific tyrosine residues in proteins, is a validated target for development of small molecule inhibitors to treat CMLCitation2. The first generation Bcr-Abl inhibitor imatinib (1, STI1571) has achieved significant clinical benefit and became the first-line drug for conventional treatment of CML ()Citation3–5. However, many patients developed emerging acquired resistance to imatinib with its widespread used in clinicCitation6. The primary mechanism of acquired imatinib resistance is the occurrence of point mutations in the Abl kinase domain, which effects the binding of imatinib in the ATP-binding siteCitation7. To date, more than 100 different point mutations have been identified in clinic.
Figure 1. (A) Chemical structures of FDA-approved Bcr-Abl inhibitors; (B) design of GZD856 as new Bcr-Abl inhibitor by scaffold hopping based on ponatinib.

To overcome imatinib resistance, several classes of second generation of Bcr-Abl inhibitors have been developedCitation8. Nilotinib (2)Citation9,Citation10, dasatinib (3)Citation11,Citation12 and bosutinib (4)Citation13 have been approved for the treatment of adults in all phases of CML with resistance to imatinib (). However, these second-generation inhibitors are not capable of inhibiting all the kinase mutants identified in patients, especially the most notably Bcr-AblT315I gatekeeper mutant that represents 15–20% of all clinical acquired resistancesCitation14. Thus, CML containing the T315I mutation remains a serious medical problem in clinic.
To address this unmet need, considerable efforts have been made to develop the third generation Bcr-Abl inhibitors which can override the T315I mutationCitation15–18, especially for AP24534 (5)Citation19,Citation20, GNF-7Citation21 and GZD824Citation22. Among them, only AP24534 (ponatinib, Iclusig®) has been approved for the treatment of resistant or intolerant CML and Ph+ ALL patients against imatinib, especially those harboring Bcr-AblT315I mutationCitation19,Citation20. However, FDA suspended the sale of ponatinib due to the increasing numbers of blood clots observed in ponatinib-treated patients after 10 monthsCitation23. Ponatinib was then reauthorized for sale a black-box warning and revised indication statement for concerns over risks of its usageCitation24.
As part of our continuous efforts to identify new molecules that could target Bcr-AblT315I mutantCitation22,Citation25, here we report the design, synthesis and biological evaluation of GZD856 (6)Citation26 as a potent Bcr-AblT315I inhibitor by substituting imidazo[1,2-b]pyridazine (five-fused-six membered ring) of ponatinib with pyrazole[1,5-a]pyrimidine (six-fused-five membered ring) by scaffold hopping strategies (), which also keep the hydrogen bond with the residue in kinase hinge region. GZD856 strongly suppressed the kinase activities of both native Bcr-Abl and the T315I mutant, and potently inhibited proliferation of corresponding K562, Ba/F3WT and Ba/F3T315I cells with low nM IC50s. It also displayed promising in vivo antitumor efficacy in mouse bearing xenograft K562 and Ba/F3 cells expressing Bcr-AblT315I.
Methodology
Computational study
The structure of Bcr-AblT315I protein was retrieved from the Protein Data Bank (PDB code: 3IK3), which was published by O'Hare et al.Citation20 The protein was processed using protein preparation wizard, which assigns bond orders and adds hydrogen and missing atoms. GZD856 was built by in LigPrep (LigPrep, version 2.5, Schrödinger, LLC, New York, NY, 2011) module using OPLS-2005 force field. Molecular docking was performed in Glide module (Glide, version 5.7, Schrödinger, LLC, New York, NY, 2011) with standard precision scoring function.
FRET-based Z′-lyte assay detecting peptide substrate phosphorylation
The effects of GZD856 on the kinase activity of Bcr-Abl and its mutants were assessed in 384-well plates using the FRET-based Z′-Lyte assay system according to manufacturer’s instructions (Invitrogen, Carlsbad, CA). Briefly, 10 µL per well reactions contained ATP concentration at 10 µM (for Bcr-Abl wildtype) or 5 µM (for T315I mutant), 2 µM Tyr2 peptide substrate in 50 mM HEPES (pH 7.5), 0.01% BRIJ-35, 10 mM MgCl2, 1 mM EGTA, 0.0247 µg/mL Bcr-Abl, and inhibitors as appropriate. The reaction was performed at room temperature for 2.0 h, and then 5 µL of development reagent was added for a further 2 h room temperature incubation followed by the addition of 5 µL of stop solution. Fluorescence signal ratio of 445 nm (coumarin)/520 nm (fluorescein) was examined on EnVision Multilabel Reader (Perkin-Elmer, Inc., Waltham, MA). The data were analyzed using Graphpad Prism5 (Graphpad Software, Inc., La Jolla, CA). The data were the mean values of three experiments.
Cells and reagents
K562 (Bcr-Abl fusion expression), U-937 and MOLT4 were purchased from ATCC and maintained as recommended by ATCC (Manassas, VA). Imatinib, dasatinib and nilotinib were purchased from Biocompounds Pharmaceutical Inc. (Shanghai, China). Ponatinib was synthesized by ourself. CCK-8 was purchased from Dojindo Molecular Technologies Inc. (Kumamoto, Japan). Dimethyl sulfoxide (DMSO) and Cremophor were purchased from Sigma-Aldrich (North Dorset, UK). Antibodies against Abl, p-Abl, Crkl, p-Crkl, STAT5 and p-STAT5, respectively, were all purchased from Cell Signaling Technology, Inc. (Danvers, MA).
Stably transformed Ba/F3 cells
The Ba/F3 cell lines stably Bcr-AblWT and Bcr-AblT315I mutant were self-established by following procedures similar to those described by von BubnoffCitation27. Briefly, wild-type Bcr-Abl p210 was cloned into pcDNA3.1(+) (Invitrogen, Carlsbad, CA). Point mutations were introduced to pcDNA3.1(+) Bcr-Abl using the QuickChange XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Ba/F3 cells were transfected with the constructs using Amaxa Cell Line Nucleofector Kit V (Lonza, Cologne, Germany) by electroporation. Stable lines were selected using Transfected Cells Cloning Kit (Stem Cell Technologies, Vancouver, Canada) with G418 (Merck, Whitehouse Station, NJ) and withdrawal of interleukin-3 (IL-3, R&D). Ba/F3 stable cell lines were verified by monitoring both DNA sequences through DNA sequencing and protein expression levels of the corresponding Bcr-Abl mutants through Western blotting analysis. Their responses to the imatinib, nilotinib and dasatinib were also hired for selecting the right clones. Parent Ba/F3 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and IL-3 (10 ng/mL), while all Bcr-Abl-transformed Ba/F3 stable cell lines were cultured in the similar medium except without IL-3.
Stably K562R (Q252H) cells
Imatinib-resistant K562 cells which expressed Bcr-Abl Q252H were self-established. Briefly, K562 cells were treated with a range of concentrations of imatinib (from 0.1 µM to 5 µM) over a 3 month period. Single clones were then selected and identified through DNA sequencing, and their response to imatinib, nilotinib and dasatinib were monitored as an internal reference.
Cellular antiproliferation assay using cell counting kit (CCK-8)
Cells in the logarithmic phase were plated in 96-well culture dishes (∼3000 cells/well). Twenty-four hours later, cells were treated with the corresponding compounds or vehicle control at the indicated concentration for 72 h. CCK-8 was added into the 96-well plates (10 µL/well) and incubated with the cells for 3 h. OD450 and OD650 were determined by a microplate reader. Absorbance rate (A) for each well was calculated as OD450–OD650. The cell viability rate for each well was calculated as V% = (As − Ac)/(Ab − Ac) × 100%, and the data were further analyzed using Graphpad Prism5 (Graphpad Software, Inc., La Jolla, CA) (As, absorbance rate of the test compound well; Ac, absorbance rate of the well without either cell or test compound; Ab, absorbance rate of the well with cell and vehicle control).
Western blot analysis
The Western blot analysis was carried out by following the protocol described previouslyCitation22. Briefly, after the indicated treatment, cell lysates were collected, dissolving cells in 1× SDS sample lysis buffer. After being sonicated and boiled, the supernatant of the cell lysate was used for Western blot analysis. Cell lysates were loaded to 8–12% SDS-PAGE and separated by electrophoresis. Separated proteins were then electrically transferred to a PVDF film. After being blocked with 1× TBS containing 0.1% Tween-20, and 5% nonfat milk, the film was incubated with corresponding primary antibody followed by HRP-conjugated secondary antibody. The protein lanes were visualized using ECL Western Blotting Detection Kit (GE Healthcare, Piscataway, NJ).
Animal studies
Male BALB/c or SCID nude mice were purchased from Vital River Laboratory Animal Technology Inc. (Beijing, China). All animal studies were approved by the Institutional Animal Use and Care Committee of Guangzhou Institute of Biomedicine and Health, Chinese Academy of Science.
Mice xenograft models
K562 and Bcr-AblT315I cells were re-suspended in normal saline (NS) solution (2.5 × 107 and 1 × 107 cell/mL respectively). A 0.2 mL amount of cell suspension was injected subcutaneously into the right flank of each mouse. Mice were randomly grouped based on the tumor volume when the mean tumor volume reached 100–200 mm3. GZD856 and imatinib were dissolved in a vehicle containing 1% DMSO, 22.5% Cremophor, 7.5% ethanol and 69% NS. Mice were treated for the 16 consecutive days once daily by oral gavage with GZD856 (10 mg/kg), imatinib (50 mg/kg) and vehicle, respectively. Tumor volume and body weight were monitored once every 2 days. Tumor volume was calculated as the L×W2/2 (L and W are the length and width of the tumor, respectively). Tumor volume data were analyzed with the one-way ANOVA method using software SPSS 17.0 (SPSS Inc., Chicago, IL).
Synthesis of GZD856
Reagents and solvents were obtained from commercial suppliers and used without further purification. Flash chromatography was performed using silica gel (300–400 mesh). All reactions were monitored by TLC, silica gel plates with fluorescence F254 were used and visualized with UV light. 1H and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer at 400 MHz and Bruker AV-500 spectrometer at 125 MHz, respectively (Bruker, Billerica, MA). Coupling constants (J) are expressed in hertz (Hz). Chemical shifts (δ) of NMR are reported in parts per million (ppm) units relative to internal control (TMS). The low or high resolution of ESI-MS was recorded on an Agilent 1200 HPLC-MSD mass spectrometer (Santa Clara, CA) or Applied Biosystems Q-STAR Elite ESI-LC-MS/MS mass spectrometer (Foster City, CA), respectively.
Methyl 3-ethynyl-4-methylbenzoate (9)
To a solution of methyl 3-iodo-4-methylbenzoate (7) (2.76 g, 10 mmol) and trimethylsilyl acetylene (0.98 g, 10 mmol) in acetonitrile (20 mL) were added Pd(dppf)Cl2 (0.07 g, 0.1 mmol), CuI (0.19 g, 1 mmol) and triethylamine (10 mL). The mixture was stirred at 80 °C for 2 h under argon atmosphere. The reaction mixture was filtered through a pad of Celite. The filtrate was concentrated, and the resulting residue was solved in CH3OH (25 mL) and treated with K2CO3 (9.8 g, 60 mmol) solution, and the mixture was stirred at room temperature for 3 h. The reaction solution was concentrated, and EtOAc and H2O were added to the residue. The organic layer was separated, and the aqueous layer was exacted with EtOAc (3 × 50 mL). The combined layers were dried over Na2SO4 and concentrated. The resulting crude was further purified by flash chromatography to give the product as a white solid (1.56 g, 90.0%). 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 1.2 Hz, 1H), 7.85 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.44 (d, J = 8.4 Hz, 1H), 4.49 (s, 1H), 3.84 (s, 3H), 2.44 (s, 3H). LC–MS (ESI): m/z 175 [M + H]+; 173 [M − H]−.
Methyl 4-methyl-3-(pyrazolo[1,5-a]pyrimidin-6-ylethynyl)-benzoate (11)
To a solution of 9 (385 mg, 2.2 mmol) in DMF (10 mL), 6-bromopyrazolo[1,5-a]pyrimidine (396 mg, 2 mmol), N,N-diisopropylethylamine (193 mg, 1.5 mmol), Pd(PPh3) Cl2 (7 mg, 0.01 mmol) and CuI (19 mg, 0.1 mmol) were placed in a vial with rubber septum. The mixture underwent three cycles of vacuum/filling with Ar. The mixture was stirred at 80 °C overnight and then quenched with H2O. EtOAc and more H2O were added for extraction. The combined organic layer was dried over Na2SO4, filtered, and concentrated, and the resulting residue was purified by chromatography, giving the title compound as a white solid (465 mg, 80.0%). 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H), 8.70 (s, 1H), 8.32 (s, 1H), 8.08 (s, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 6.82 (s, 1H), 3.86 (s, 3H), 2.56 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 165.4, 150.9, 146.5, 146.3, 145.3, 138.2, 132.2, 130.3, 129.5, 127.6, 122.0, 104.6, 97.3, 90.2, 87.8, 52.2, 20.5. LC–MS: m/z 292 [M + H]+.
4-Methyl-N-(4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-3-(pyrazolo[1,5-a]pyrimidin-6-ylethynyl)benzamide (6, GZD856)
A solution of methyl 4-methyl-3-(pyrazolo[1,5-a]pyrimidin-6-ylethynyl)-benzoate (145 mg, 0.5 mmol) and 4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)aniline (137 mg, 0.5 mmol) in anhydrous THF (10 mL) was added potassium tert-butoxide (336 mg, 3 mmol) in anhydrous THF (10 mL) slowly at −20 °C and stirred for 1 h. Then the reaction mixture was slowly warmed to room temperature and stirred for another 8 h. After evaporation of the solvent, the residue was re-dissolved in ethyl acetate. Then the organic phase was washed with brine, dried over anhydrous sodium sulfate, concentrated under reduced pressure. The residue was purified through silica gel column chromatography to the desired compound as yellow solid (200 mg, 75%). 1H NMR (400 MHz, DMSO-d6), 2.17 (s, 3H), 2.37 (m, 8H), 2.60 (s, 3H), 3.57 (s, 2H), 6.85 (d, J = 2.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.96 (dd, J = 8.0, 3.29 Hz, 1H), 8.06 (d, J = 2.00 Hz, 1H), 8.21 (dd, J = 4.2, 2.0 Hz, 2H), 8.34 (d, J = 2.0 Hz, 1H), 8.72 (d, J = 2.0 Hz, 1H), 9.58 (d, J = 2.00 Hz, 1H), 10.56 (s, 1H). 13C NMR (125 MHz, DMSO-d6) δ 165.5, 151.8, 147.4, 147.2, 144.8, 139.1, 139.0, 133.1, 132.9, 132.0, 131.7, 130.8, 129.4, 126.3, 124.4, 122.5, 118.2, 105.6, 98.2, 91.6, 88.4, 58.3, 55.5, 53.5, 46.5, 21.3. MS (ESI), m/z: 533 (M + H)+. HRMS (EI): calcd for C29H27F3N6O: 533.2198 [M + H]+; found: 533.2266.
Results and discussion
Synthetic route
GZD856 was readily prepared using palladium catalyzed Sonogashira reaction as the key step, which is illustrated in Scheme 1. Briefly, the commercial material methyl 3-iodo-4-methylbenzoate (7) was reacted with ethynyltrimethylsilane (8) under palladium catalysis to afford the Sonogashira coupling products, which was further deprotected to produce the alkyne 9. Then, coupling of alkyne 9 with 6-bromopyrazolo[1,5-a]pyrimidine (10) under the Sonogashira coupling conditions afforded the key intermediate 11. Ammonolysis of 11 with 4-((4-methylpiperazin-1-yl) methyl)-3-(trifluoromethyl) benzenamine (12) under basic condition yielded the desired GZD856 (6).
Scheme 1. Synthetic route of compound GZD856. Reagents and conditions: (a) i. Pd(dppf)2Cl2, CuI, Et3N, DMF, 80 °C; ii. K2CO3, MeOH, rt, 90%; (b) Pd(PPh3) Cl2, CuI, DIPEA, DMF, 80%; (c) t-BuOK, THF, −20 °C, 75%.

Kinase activities evaluation
The kinase inhibitory activities of GZD856 against Bcr-AblWT and Bcr-AblT315I were evaluated by using the well established FRET-based Z’-Lyte assayCitation22,Citation28. Imatinib and nilotinib were used as positive controls to validate the screening conditions. Under the screening conditions, imatinib and nolitinib potently inhibited the enzymatic activity of Bcr-AblWT with IC50 values of 98.2 and 43.5 nM, which were highly consistent to the reported dataCitation9,Citation29. As shown in , the designed GZD856 potently inhibited Bcr-AblWT and Bcr-AblT315I with IC50 values of 19.9 and 15.4 nM, respectively, which displayed similar activity to ponatinib (19), suggested that GZD856 can be a potent Bcr-Abl inhibitor overcoming acquired imatinib resistance. However, imatinib and nilotinib were both significantly less potent for Bcr-AblT315I mutation.
Table 1. Inhibitory activities of GZD856 against Bcr-AblWT and Bcr-AblT315I mutant.
Molecular docking studies
Molecular docking study was performed to investigate the binding mode of compound GZD856 with Bcr-AblT315I (PDB code: 3IK3) using Glide module (Glide, version 5.7 Schrödinger, LLC, New York, NY) with standard precision scoring function. GZD856 bound to the ATP binding site of the DFG-out conformation of Bcr-AblT315I (), which was similar to that of ponatinibCitation19,Citation20. The pyrazolo[1,5-a]pyrimidine of GZD856 formed an essential hydrogen bond with the NH of Met318 in the hinge region of Bcr-AblT315I. The amide formed two hydrogen bonds with Glu286 and Asp381, and the trifluoromethylphenyl group bound deeply into the hydrophobic pocket. The alkynyl linker made favorable van der Waals interactions with the gatekeeper Ile315 avoiding steric clash.
Figure 2. The predicted binding mode of GZD856 with Bcr-AblT315I. Hydrogen bonds are indicated by yellow hatched lines to key amino acids.

Cellular antiproliferation assay
The antiproliferative activity of GZD856 was also examined against a panel of leukemia cells with differing Bcr-Abl status (). Imatinib, nilotinib, ponatinib and taxol were utilized as positive controls to validate the screening conditions. As shown in , imatinib and nilotinib were both significantly less potent for K562R (Q252H) and Ba/F3T315I cells. GZD856 strongly suppressed the proliferation of K562, K562R (Q252H) and murine Ba/F3 cells ectopically expressing Bcr-AblWT and Bcr-AblT315I, with IC50 values of 2.2, 67.0, 0.64 and 10.8 nM, respectively, which were equivalent to the potency of ponatinib and much higher than the potency of imatinib and nilotinib (). As a confirmation of GZD856’s Bcr-Abl-selective effect, GZD856 was significantly less potent against the proliferation of MOLT4 and U937 leukemia cells (negative for Bcr-Abl expression), which are much selective than ponatinib. Further, GZD856 also displayed less potent antiproliferative activity against non-cancer cells HFL-1 (human embryonic lung fibroblasts) with IC50 value of 6.78 µMCitation26. The studies suggested that GZD856 could be a selective anti-cancer drug. However, the cytotoxic agent taxol inhibited the growth of all tested cells with similar IC50 values.
Table 2. GZD856 selectively and potently inhibited the proliferation of Bcr-Abl positive leukemia cells.
Western blot study
To further validate the cellular kinase inhibitory activity of GZD856, we examined its effects on the activation of Bcr-Abl and its downstream signals in Bcr-Abl positive K562 CML cells and stably transformed Ba/F3 cells expressing Bcr-AblWT and Bcr-AblT315I by Western blot analysis (. GZD856 efficiently suppressed the activation of both Bcr-Abl and downstream Crkl and STAT5 in dose-dependent manners in K562 CML cells. Similar inhibition was also observed in the Ba/F3 cells expressing Bcr-AblWT and Bcr-AblT315I. Not surprisingly, none of the three FDA approved drugs (imatinib, nilotinib, dasatinib) showed obviously suppression on the activation of Bcr-AblT315I Citation22.
Figure 3. GZD856 inhibits Bcr-Abl signaling in K562 and Ba/F3 stable cell lines expressing Bcr-AblWT and Bcr-AblT315I. Cells were treated with GZD856 at the indicated concentrations for 4.0 h, and whole cell lysates were then subjected to Western blot analyses. The results represent three independent experiments.

In vivo antitumor studies
Giving its highly promising antiproliferative activity in vitro and pharmacokinetic (PK) properties in vivo in ratsCitation26, GZD856 may possess good in vivo efficacy when orally administered. Thus, the in vivo antitumor effect of GZD856 was first evaluated in subcutaneous K562 xenograft model of human CML. Imatinib was used as a positive control at dose of 50 mg/kg/day to validate the animal models. The animals were dosed orally with GZD856 at 10 mg/kg/day. As shown in , GZD856 almost completely eradicated the tumor at doses of 10 mg/kg/day after 8 days of treatment, whereas imatinib only induced tumor stasis at a dose of 50 mg/kg/day. Notably, after the cessation of treatment (16 days of dosing), there was no sign of tumor recurrence in the following 7 days. Also, at the dose of 10 mg/kg/day, GZD856 was well tolerated with no mortality or significant body loss (<5% relative to vehicle-matched controls) during the treatment.
Figure 4. GZD856 potently suppressed tumor growth in K562 xenograft model of human CML. Mice bearing K562 xenografts were dosed orally once a day with GZD856 at 10 mg/kg dosages for 16 consecutive days. The data are representative of three independent experiments.

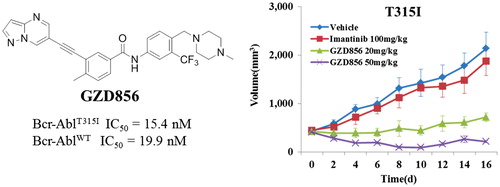
Similar to that of the K562 xenograft studies, the antitumor efficacy of GZD856 was further evaluated in a xenograft model using Ba/F3 cell expressing Bcr-AblT315I. The animals were administered GZD856 at doses of 20 and 50 mg/kg via oral gavage once daily for 16 days. Imatinib was again used as a reference compound and administered orally at dose of 100 mg/kg/day for 16 days. As shown in , GZD856 dose-dependently inhibited tumor growth in the xenograft model bearing Bcr-AblT315I. Although GZD856 did not show obvious inhibition of tumor growth at dose of 20 mg/kg/day, it induced almost about 90% tumor regression at a dose of 50 mg/kg/day after a 16 days consecutive treatment. In contrast, 100 mg/kg/day of imatinib failed to exhibit inhibition of tumor growth in the Bcr-AblT315I model.
Figure 5. GZD856 suppressed tumor growth in xenograft models of Ba/F3 cells expressing Bcr-AblT315I. Mice bearing xenograft Ba/F3 Bcr-AblT315I cells were orally dosed with GZD856 and imatinib at the indicated doses. Tumor sizes were monitored every 2 days (n = 10 for each group; error bar represents SE).

Conclusions
In summary, we have successfully discovered GZD856 as a new Bcr-Abl inhibitor which maintains significant inhibition against Bcr-Abl gatekeeper T315I mutant. GZD856 strongly suppressed both native Bcr-Abl and the T315I mutant with IC50 values of 19.9 and 15.4 nM, and potently inhibited proliferation of corresponding K562 and Ba/F3T315I cells with IC50 values of 2.2 and 10.8 nM. Western blot analysis further supported its kinase inhibition against Bcr-Abl and Bcr-AblT315I. In vivo efficacy studies in mouse xenograft models of human leukemia demonstrated that GZD856 potently suppresses growth of tumors driven by native Bcr-Abl and Bcr-AblT315I, which indicated that GZD856 is a promising anti-cancer lead for further development.
Disclosure statement
The authors report no declarations of interest.
Additional information
Funding
References
- Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973;243:290–3.
- Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 2009;113:1619–30.
- Capdeville R, Buchdunger E, Zimmermann J, et al. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov 2002;1:493–502.
- Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005;105:2640–53.
- Quintas-Cardama A, Kantarjian H, Cortes J. Imatinib and beyond-exploring the full potential of targeted therapy for CML. Nat Rev Clin Oncol 2009;6:535–43.
- Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006;355:2408–17.
- O’Hare T, Eide CA, Deininger MWN. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007;110:2242–9.
- Weisberg E, Manley PW, Cowan-Jacob SW, et al. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer 2007;7:345–56.
- Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005;7:129–41.
- Kantarjian HM, Giles F, Gattermann N, et al. Nilotinib (formerly AMN 107), a highly selective Bcr-Abl tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome positive chronic myelogenous leukemia inchronic phase following imatinib resistance and intolerance. Blood 2007;110:3540–6.
- Shah NP, Tran C, Lee FY, et al. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004;305:399–401.
- Quintas-Cardama A, Kantarjian H, Jones D, et al. Dasatinib (BMS-354825) is active in Philadelphia chromosome-positive chronic myelogenous leukemia after imatinib and nilotinib (AMN107) therapy failure. Blood 2007;109:497–9.
- Puttini M, Coluccia AM, Boschelli F, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl + neoplastic cells. Cancer Res 2006;66:11314–22.
- Modugno M. New resistance mechanisms for small molecule kinase inhibitors of Abl kinase. Drug Discov Today Technol 2014;11:5–10.
- Tanaka R, Kimura S. Abl tyrosine kinase inhibitors for overriding Bcr-Abl/T315I: from the second to third generation. Expert Rev Anticancer Ther 2008;8:1387–98.
- Noronha G, Cao J, Chow CP, et al. Inhibitors of ABL and the ABL-T315I mutation. Curr Top Med Chem 2008;8:905–21.
- Schenone S, Brullo C, Botta M. New opportunities to treat the T315I-Bcr-Abl mutant in chronic myeloid leukaemia: tyrosine kinase inhibitors and molecules that act by alternative mechanisms. Curr Med Chem 2010;17:1220–45.
- Lu XY, Cai Q, Ding K. Recent developments in the third generation inhibitors of Bcr-Abl for overriding T315I mutation. Curr Med Chem 2011;18:2146–57.
- Huang WS, Metcalf CA, Sundaramoorthi R, et al. Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methyl-piperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J Med Chem 2010;53:4701–19.
- O'Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009;16:401–12.
- Choi HG, Ren P, Adrian F, et al. A type-II kinase inhibitor capable of inhibiting the T315I ‘gatekeeper’ mutant of Bcr-Abl. J Med Chem 2010;53:5439–48.
- Ren XM, Pan XF, Zhang Z, et al. Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated breakpoint cluster region-abelson (Bcr-Abl) kinase and overcomes clinically acquired mutation-induced resistance against imatinib. J Med Chem 2013;56:879–94.
- “FDA asks manufacturer of the leukemia drug Iclusig (ponatinib) to suspend marketing and sales”. FDA Drug Safety Communication. U.S. Food and Drug Administration; 2013. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm373040.htm.
- Liu X, Kung A, Malinoski B, et al. Development of alkyne-containing pyrazolopyrimidines to overcome drug resistance of Bcr-Abl kinase. J Med Chem 2015;58:9228–37.
- Lu XY, Zhang Z, Ren XM, et al. Hybrid pyrimidine alkynyls inhibit the clinically resistance related Bcr-Abl(T315I) mutant. Bioorg Med Chem Lett 2015;25:3458–63.
- Zhang Z, Ren XM, Lu XY, et al. GZD856, a novel potent PDGFRα/β inhibitor, suppresses the growth and migration of lung cancer cells in vitro and in vivo. Cancer Lett 2016;375:172–8.
- von Bubnoff N, Veach DR, van der Kuip H, et al. A cell-based screen for resistance of Bcr-Abl-positive leukemia identifies the mutation pattern for PD166326, an alternative Abl kinase inhibitor. Blood 2005;105:1652–9.
- Jares-Erijman EA, Jovin TM. FRET imaging. Nat Biotechnol 2003;21:1387–95.
- Chan WW, Wise SC, Kaufman MD, et al. Conformational control inhibition of the Bcr-abl1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell 2011;19:556–68.