
Abstract
Sulphamate and sulphamide derivatives have been largely investigated as carbonic anhydrase inhibitors (CAIs) by means of different experimental techniques. However, the structural determinants responsible for their different binding mode to the enzyme active site were not clearly defined so far. In this paper, we report the X-ray crystal structure of hCA II in complex with a sulphamate inhibitor incorporating a nitroimidazole moiety. The comparison with the structure of hCA II in complex with its sulphamide analogue revealed that the two inhibitors adopt a completely different binding mode within the hCA II active site. Starting from these results, we performed a theoretical study on sulphamate and sulphamide derivatives, demonstrating that electrostatic interactions with residues within the enzyme active site play a key role in determining their binding conformation. These findings open new perspectives in the design of effective CAIs using the sulphamate and sulphamide zinc binding groups as lead compounds.
Graphical Abstract
Introduction
Carbonic anhydrases (CAs; EC: 4.2.1.1) are a family of metalloenzymes present in all kingdoms of life that catalyse the interconversion of carbon dioxide and bicarbonateCitation1. Based on their structural features, they are grouped into seven different classes, namely α-, β-, γ-, δ-, ζ-, η- and θ-CAs. α-CAs are predominantly expressed in vertebrates, bacteria, algae and cytoplasm of green plants, β-CAs in bacteria, algae and chloroplasts, γ-CAs in archaea and some bacteria, δ- and ζ-CAs in some marine diatoms, η-CAs only in the protozoan parasite Plasmodium spp., whereas the recently discovered θ-class has been so far found only into the marine diatom Phaeodactylum tricornutumCitation1–8. Humans encode 12 catalytically active α-CA isozymes, which differ in molecular features, oligomeric arrangement, kinetic properties and cellular localisation, with isoforms I, II, III, VII and XIII localised in the cytosol, CA IV, IX, XII and XIV associated with the cell membrane, CA VA and VB confined in mitochondria, and CA VI secreted in saliva and milkCitation1. All catalytically active human (h) CAs contain in the active site a Zn2+ ion essential for catalysis; this ion is coordinated by three conserved histidine residues (His94, His96 and His119) and a water molecule/hydroxide ionCitation1. hCAs participate in several physiological processes, among which pH homeostasis, CO2 and HCO3− transport, cell differentiation and proliferation, respiration, bone resorption, neurotransmission, ureagenesis, gluconeogenesis, lipogenesis, and fertilisationCitation9,Citation10. Abnormal levels and/or activities of these enzymes have been often associated with different human diseases, such as glaucoma, epilepsy, high-altitude sickness, as well as cancerCitation11. For these reasons, hCAs represent an important target for the design of inhibitors or activators with biomedical applicationsCitation11,Citation12.
The most studied carbonic anhydrase inhibitors (CAIs) are sulphonamide derivatives (R-SO2NH2), which are able to bind in a tetrahedral geometry the active site zinc ion, substituting the water molecule/hydroxide ion present in the native enzymeCitation1. These molecules have been largely investigated, due to their capability to strongly bind to the hCA active site, with many such agents in clinical useCitation11,Citation13; however, the occurrence of various undesired side effects due to the lack of selectivity against the different CA isoforms strongly limits their use as drugsCitation1,Citation11. Therefore, other CAI classes with different zinc-binding groups (ZBGs) have been developed over the years, with sulphamates (R-O-SO2NH2) and sulphamides (R-NH-SO2NH2) among the most important ones. These compounds differ from sulphonamides for the additional presence of an electron withdrawing group, an oxygen atom in the case of sulphamatesCitation14 and an NH group in the case of sulphamidesCitation15. As observed for sulphonamides, also sulphamates and sulphamides exert their inhibitory action through coordination to zinc ion and consequent substitution of the water molecule/hydroxide ionCitation1. Plenty of studies has been reported showing that many sulphamates possess effective inhibitory properties against all known human isoformsCitation1,Citation11,Citation16–19, with some derivatives, such as the sugar sulphamate topiramate (compound 1 in ), successfully used for the treatment of a variety of diseases such as epilepsy, migraine, and obesityCitation20,Citation21. Although the sulphamide group was initially considered not particularly suitable for obtaining potent CAIsCitation22, several compounds containing a primary sulphamide moiety have also been proved to possess a high CA inhibition activityCitation1,Citation11,Citation19,Citation23. As an example, compound JNJ-26990990 (2) (see ), which presents excellent anticonvulsant activity and can be potentially used in the treatment of multiple forms of epilepsy, is also a nanomolar inhibitor of several CA isoformsCitation24,Citation25.
Figure 1. Structural formulas of topiramate (1), JNJ-26990990 (2), 2-methyl-5-nitro-imidazole-sulphamate (3), 2-methyl-5-nitro-imidazole-sulphamide (4) and the topiramate sulphamide analogue (5). hCA II inhibition constants for compounds 3 and 4 are also reportedCitation26.

We recently reported the synthesis of a series of sulphonamide/sulphamide/sulphamate derivatives incorporating nitroimidazole moietiesCitation26. Inhibition studies against isoforms I, II, IX, and XII showed that these compounds, in particular, the sulphamate/sulphamide derivatives 3 and 4 (), are good CAIs, with KI values in the nanomolar range. Moreover, compound 4 was demonstrated to inhibit in vitro the hypoxia-induced extracellular acidosis in two cell lines overexpressing CA IX and to enhance in vivo, in co-treatment with doxorubicin, sensitisation towards radiotherapy and chemotherapy of CA IX containing tumoursCitation26. The X-ray crystal structure of the hCA II/4 adduct was also reported, highlighting the principal interactions responsible for the binding of the inhibitor to the enzyme active siteCitation26.
Within a research project aimed at understanding at the atomic level, the inhibition properties of sulphamate/sulphamide CAIs, here we report the X-ray crystal structure of the hCA II/3 adduct and compare it with the previously obtained hCA II/4 structure. Surprisingly, even if the two inhibitors differ for only one atom (see ), they adopt a completely different binding mode within the CA II active site. Binding free energy calculations have been used to rationalise this result.
Materials and methods
Crystallisation, X-ray data collection, and refinement
Crystals of the hCA II/3 complex were prepared by soaking hCA II 100K crystals (obtained using the hanging drop vapour diffusion technique) for 1 h in the crystallisation solution (1.3 M sodium citrate, 100 mM Tris-HCl, pH 8.5) saturated with the inhibitor. Prior to X-ray data collection, crystals of the complex were transferred from the drops to a cryoprotectant solution prepared by the addition of 20% glycerol to the precipitant solution and then flash-cooled to 100K in a nitrogen stream. A complete dataset was collected at 1.80 Å resolution from a single crystal, at 100 K, with a copper rotating anode generator developed by Rigaku and equipped with Rigaku Saturn CCD detector.
Diffraction data were indexed, integrated and scaled using the HKL2000 software packageCitation27. A total of 107,169 reflections were measured and reduced to 22,183 unique reflections. Crystal parameters and relevant X-ray data collection statistics can be found in . Initial phases were calculated using hCA II crystallised in the P21 space group (PDB code 1CA2)Citation28 as starting model after deletion of non-protein atoms. An initial round of rigid body refinement followed by simulated annealing and individual B-factor refinement was performed using the programme Crystallography and NMR system (CNS)Citation29,Citation30. Model visualisation and rebuilding were performed using the graphics programme OCitation31. After an initial refinement, limited to the enzyme structure, a model for the inhibitor was easily built and introduced into the atomic coordinates set for further refinement. Crystallographic refinement was carried out against 95% of the measured data. The remaining 5% of the observed data, which was randomly selected, was used for Rfree calculations to monitor the progress of refinement. Restraints on inhibitor bond angles and distances were taken from the Cambridge Structural DatabaseCitation32, whereas standard restraints were used on protein bond angles and distances throughout refinement. Water molecules were built into peaks >3σ in |Fo| − |Fc| maps that demonstrated appropriate hydrogen-bonding geometry. Several alternate cycles of refinement and manual model building were performed to reduce the Rwork and Rfree to the final values of 0.157 and 0.195, respectively. Relevant refinement statistics can be found in . The refined model contained 2055 protein atoms, 237 waters, and one inhibitor molecule. Coordinates and structure factors have been deposited with the Protein Data Bank (accession code 5O07).
Table 1. Data collection and refinement statistics. Values in parentheses refer to the highest resolution shell (1.86–1.80 Å).
Computational study
Systems preparation
Complex_O and complex_N models were built from the hCA II/3 and hCA II/4Citation26 crystallographic structures, by replacing the 2-methyl-5-nitro-imidazole moiety of the two inhibitors with a methyl group. The third model, namely complex_NO, was obtained by substituting the N2 atom of complex_N with an oxygen atom. Hydrogen atoms were added to all the models and their positions were energy minimised by 500 steps of Conjugate Gradient using the Discover module of InsightII package (Insight2000, Accelrys, San Diego, CA).
The partial atomic charges for ligands and zinc ion were obtained by quantum mechanical (QM) calculations (B3LYP/6–31 G*) using the Gaussian09 softwareCitation33 via the Restrained ElectroStatic Potential (RESP) fitting procedure as implemented in the PyRED serverCitation34,Citation35. The charges calculations were performed on model systems including the ligand, the zinc ion and the side chains of the three coordinating histidine residues. Since literature data suggest that the sulphamate and sulphamide groups, similarly to sulphonamidesCitation36,Citation37, bind the zinc ion in a deprotonated form, the total charge for ligands was set at −1 e. A charge of 1.5 e was obtained for the zinc ion, whereas a high negative charge was derived for the deprotonated nitrogen atom N1 (∼ −1.7 e) in all the three ligands. A complete list of the partial charges computed for the ligands atoms is reported in . The General AMBER force fieldCitation38, and the AMBERff14SB force fieldCitation39 were used for the ligands and proteins, respectively. Van der Waals parameters for the Zn2+ ion were adopted from the work of Li et al.Citation40 (σ = 1.271; ɛ (kcal/mol) = 0.00330286).
Table 2. Partial atomic charges (e) computed for the three ligands in complex_O, complex_N and complex_NO, respectively. Charges were calculated via the RESP fitting procedure as implemented in the PyRED server using Gaussian09 software.
Binding free energy calculations
The binding free energies (ΔGbind in kcal/mol) were calculated using the Molecular Mechanics/Generalised Born Surface Area (MM/GBSA) methodCitation41,Citation42 implemented in AmberTools14Citation43. Moreover, to identify the key protein residues responsible for the ligands binding process, the binding free energy was decomposed on a per-residue basis.
For each complex, the binding free energy of MM/GBSA was estimated as follows:
where ΔGbind is the binding free energy and Gcomplex, Gprotein and Gligand are the free energies of complex, protein, and ligand, respectively. The energies were estimated as shown below:
If ligands have similar structures and binding modes, it is acceptable to exclude the entropy contribution (–TΔS) in practiseCitation42,Citation44,Citation45. Then the binding free energy is evaluated byCitation46:
where ΔEgas, the complete gas phase force field energy, is the molecular mechanics (MM) part ΔEMM, including van der Waals (ΔEvdW) and electrostatic (ΔEelec) contributions; ΔGsol is the solvation free energy, and is the sum of electrostatic (ΔGGB) and non-polar (ΔGSA) interactions. The electrostatic solvation free energy (ΔGGB) is evaluated via Generalised Born implicit solvation modelCitation47, and the non-polar solvation free energy (ΔGSA) is estimated by the Linear Combination of Pairwise Overlaps (LCPO) methodCitation48.
Results and discussion
Crystal structure of hCA II in complex with compound 3 was determined at 1.80 Å resolution, revealing a clear electron density for the inhibitor molecule in the enzyme active site (). The model was refined with CNSCitation29,Citation30, giving final Rwork and Rfree values of 15.7% and 19.5%, respectively. The average B factors were 12.1 Å2 for the protein, 23.2 Å2 for the solvent and 16.0 Å2 for the inhibitor molecule. Data collection and refinement statistics are shown in .
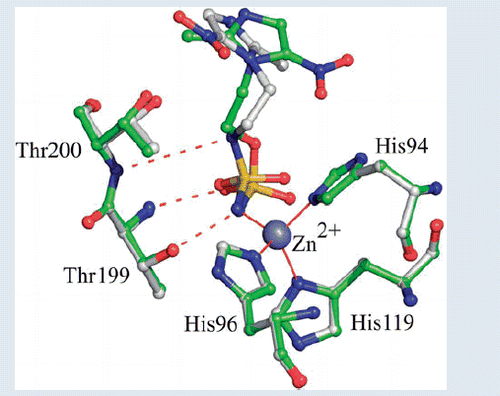
Figure 2. Active site region in the hCAII/3 complex. Hydrogen bonds, active site Zn2+ coordination and residues establishing van der Waals interactions (distance <4.0 Å) with the inhibitor are reported. Sigma-A weighted |2Fo-Fc| simulated annealing omit map (at 1.0 sigma) relative to the inhibitor molecule is also shown.

The binding of the inhibitor to hCA II did not generate major changes in the protein structure as proved by the low value of the r.m.s.d. calculated by superimposing the Cα atoms in the adduct and the non-inhibited enzyme (0.3 Å). Similarly to what previously observed for other hCA II/sulphamate complexes solved so farCitation49–65, compound 1 interacts directly with the zinc ion of the active site, with its sulphamate nitrogen atom N1 (for atom numbering see ) displacing the water molecule/hydroxide ion, which in the not-inhibited enzyme occupies the fourth coordination position. Additional hydrogen bonds between the sulphamate moiety and residues within the enzyme active site contribute to stabilise the binding. In detail, the sulphamate nitrogen atom N1 donates a hydrogen bond to the Thr199OG1 atom, whereas one of the two sulphamate sp2 oxygens accepts another hydrogen bond from the main chain nitrogen of the same residue (). No other polar interactions were observed between the inhibitor and enzyme residues, but a large number of van der Waals contacts were present, with the imidazole ring being located in the middle of the active site cavity and the nitro group being oriented towards the hydrophilic region of it ()Citation66.
To compare the binding mode of compounds 3 and 4 to the hCA II active site, the crystallographic structures of the hCA II/3 and hCA II/4 adducts were superimposed showing that the two inhibitors adopt a completely different binding mode to the enzyme (). Main differences were observed in the orientation of the imidazole rings, which were rotated of about 140° in the two complexes (). Because of the different orientation, inhibitor 4 established a higher number of favourable interactions with active site residues (), thus explaining its higher affinity for the enzyme (see KI values in ). Since compounds 3 and 4 differ only for one atom (O3 instead of N2) in their ZBG (see ), the structural basis of the different orientation of the imidazole rings in the active site cavity should be searched in the interactions that this atom can establish with neighbouring residues within the active site cavity. In the hCA II/4 complex, the nitrogen atom N2 is at 3.2 Å from the Thr200OG1 atom; this distance being compatible with the formation of a weak hydrogen bond interaction. On the contrary, in the hCA II/3 complex, the distance between the sulphamate oxygen O3 and the Thr200OG1 atom becomes of 4.7 Å. This slide away causes the rearrangement of the imidazole ring within the active site and the loss of the hydrogen bond interactions between the nitroimidazole moiety and residues His64 and Thr200.
Figure 3. (A) Structural superposition between hCA II/3 (green) and hCA II/4 (white, PDB code 4MO8)Citation26. (B) Active site region in the hCAII/4 complex. Hydrogen bonds, active site Zn2+ coordination and residues establishing van der Waals interactions (distance <4.0 Å) with the inhibitor are reported.

To understand if the different position assumed by N2 and O3 atoms in the enzyme active site was associated to a peculiarity of the two complexes under investigation, or to a more general behaviour of sulphamate and sulphamide derivatives, a comparative analysis of all hCA II/sulphamate and hCA II/sulphamide structures available in the PDB was undertakenCitation25,Citation26,Citation49–65,Citation67–71. Surprisingly, the analysis of all these structures revealed that, independently of the nature of the moiety attached to the ZBG, the distance between the Thr200OG1 atom and the sulphamide nitrogen N2 in hCA II/sulphamide complexes was generally shorter than the corresponding distance between the sulphamate oxygen O3 and the same enzyme atom in hCA II/sulphamate complexes (see and ). Moreover, in most of the hCA II/sulphamide adducts, such a distance is compatible with the formation of an H-bond, the situation not observed in the case of enzyme/sulphamate complexes.
Table 3. Distances between Thr200OG1 atom and the sulphamide N2 atom in hCA II/sulphamide complexes. Only sulphamides of the type R-NH-SO2NH2 were considered.
Table 4. Distances between Thr200OG1 atom and the sulphamate O3 atom in hCA II/sulphamate complexes.
To understand why the sulphamate oxygen O3 atom was always pushed away from the Thr200OG1 atom with respect to the corresponding atom in sulphamides, binding free energy calculations were carried out. At this aim, the MM/GBSA method, which allows obtaining a per-residue decomposition of the binding free energy, was utilised. To make results independent on the nature of the moiety attached to the ZBG, simplified models of sulphamate/sulphamide derivatives were used. In particular, three model systems, hereafter indicated as complex_O, complex_N and complex_NO, were built. The first two models were obtained starting from the hCA II/3 and hCA II/4 crystallographic structures and replacing the 2-methyl-5-nitro-imidazole moiety of the two inhibitors with a methyl group. The third model was obtained by substituting the N2 atom of complex_N with an oxygen atom. It is important to highlight that, whereas complex_O and complex_N represent a simplified version of the hCA II/sulphamate and hCA II/sulphamide crystal structures, complex_NO corresponds to a hypothetical hCA II/sulphamate adduct, where the oxygen atom O3 is forced to assume the same position occupied by N2 in hCA II/sulphamide complexes. Before calculations, hydrogen atoms, which were not visible in the crystallographic structures, were added to the models and their positions were energy minimised using the Discover module of InsightII package. It is worth of note that in all the protonated complexes, in agreement with what observed in the neutronic structure of hCA II crystallised at pH 7.5 (PDB code 4Q49)Citation72, the hydrogen bound to the Thr200OG1 atom was oriented towards Pro201O atom, in a direction opposite to the position of the ligand (). Consequently, the Thr200OG1 atom can act only as a hydrogen bond acceptor when interacting with the ligand. Accordingly, in complex_N Thr200OG1 atom establishes a hydrogen bond interaction with the N2 atom of the ligand (), which is a hydrogen bond donor. On the contrary, in complex_O and complex_NO, it cannot form such interaction with O3 atom, since the O3 atom can act only as hydrogen bond acceptor ().
Figure 4. Detail of the active site in the model systems Complex_N (A), Complex_O (B) and Complex_NO (C). In all three cases the ligand, the zinc ion, the three coordinating histidines, Glu106, and enzyme residues giving a major contribution to ligand binding are shown. Only polar hydrogens are shown. Hydrogen bonds are highlighted with red dotted lines, while the distances between O3 and Thr200OG1 are indicated with black arrows.

reports results of MM/GBSA calculations, which allowed the identification of all the enzyme residues, beyond the zinc ion, giving a stabilising contribution to the binding of the ligands. Interestingly, in all three model systems four residues, namely Val143, Leu198, Thr199 and Thr200, were identified as major contributors to the binding. Among these, Val143, Leu198, and Thr199 contribute in a similar way in all complexes, whereas Thr200 provides a different contribution to binding free energy in each model, thus confirming the critical role, suggested by crystallographic studies, played by this residue for sulphamate/sulphamide binding. In particular, this residue interacts more favourably with ligand in the case of complex_N, showing the lowest value of total binding free energy (ΔGbind-Thr200 = −3.164 Kcal/mol), whereas it interacts less favourably with ligand in complex_NO with a total binding free energy value of −1.290 Kcal/mol. These data can be explained by looking at the individual energy components of ΔGbind-Thr200 reported in . Major differences are observed in the contribution of the electrostatic term (ΔEelec). This term has always a positive value, indicating in all three complexes the presence of unfavourable charge interactions between Thr200 and ligand. Such unfavourable interactions can be mainly ascribed to the repulsion between the partial charges on the backbone nitrogen atom of Thr200 and the N1 atom of the three ligands. Although these atoms are quite far apart in all models (4.5 Å), the energetic calculations probably overestimate their charge repulsion due to the very high negative charge on N1 atom obtained through QM methods (see “Materials and Methods” section). However, since the distance between the backbone nitrogen atom of Thr200 and the N1 atom of the ligand is the same in all three systems, the extent of this repulsive interaction can be considered the same in all of them. Thus, additional contributions have to be considered to explain the observed differences in the electrostatic term. A detailed inspection of the three model systems reveals the presence, in the case of the complex_NO, of additional repulsive interactions between the negative partial charges on O3 and Thr200OG1 atoms, which are at a relatively close distance (3.2 Å) (), leading to the highest value of ΔEelec (2.673 Kcal/mol). In complex_O, where the distance between O3 and Thr200OG1 atoms is larger (4.7 Å) (), this repulsive electrostatic contribution is significantly reduced ( ΔEelec = 1.076 Kcal/mol), thus giving a justification for the preferential binding of sulphamate in this conformation, as observed in crystallographic studies. Finally, in complex_N, ΔEelec is further reduced (0.526 Kcal/mol) due to the stabilising contribution of the N3-Thr200OG1 hydrogen bond ().
Table 5. Per-residue binding energy decomposition (given in kcal/mol), calculated by the MM/GBSA method for complex_N, complex_NO and complex_O. Only residues contributing more than −1.0 kcal/mol to the binding are reported.
Table 6. Individual energy components (kcal/mol) of ΔGbind-Thr200 calculated by the MM/GBSA method for complex_N, complex_NO and complex_O.
In conclusion, energetic calculations showed that in the crystallographic structures of hCA II/sulphamate adducts the O3 sulphamate oxygen atom prefers to be placed in a position more distant from the Thr200OG1 atom with respect to the corresponding N2 atom in hCA II/sulphamide complexes, in order to reduce unfavourable electrostatic interactions.
Conclusions
Sulphamates and sulphamides derivatives have been largely investigated as CAIsCitation1,Citation14,Citation15 by means of different experimental techniques. However, the structural determinants responsible for their different binding mode to the enzyme active site were not clearly defined so far. In this paper, we report a combined crystallographic and theoretical study on these compounds, demonstrating that electrostatic interactions with residues within the enzyme active site play a key role in determining the binding conformation of these molecules. Due to these interactions, molecules that differ only for one atom, as in the case of compounds 3 and 4, can assume a completely different orientation within the CA active site. A similar situation was observed also in the case of topiramate 1 and its sulphamide analogue 5 (see ). Indeed, also in this case, a single atom substitution creates differences in the arrangement of the organic scaffold with the CA II active site, and consequently in KI values against the enzymeCitation69. These findings open new important perspectives in the field of CAI drug design. Indeed, as mentioned in the ‘Introduction’ section, in the past sulphamide derivatives were considered not particularly suitable for obtaining potent CAIs, mainly due to lower acidity of the sulphamide group with respect to sulphamate one and to the lower tendency to form the anionic form required for CA inhibitionCitation22. The study here reported demonstrates that other factors can play a key role in determining the affinity of sulphamide/sulphamate derivatives for the CA active site and that, as observed for compounds 3 and 4, these factors can also lead to a higher affinity of sulphamide derivatives with respect to the corresponding sulphamates for CAs.
Acknowledgements
We thank Maurizio Amendola and Giosuè Sorrentino for their skilful technical assistance with X-ray measurements.
Disclosure statement
The authors report no declaration of interest.
References
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum: the eta-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96.
- De Simone G, Di Fiore A, Capasso C, Supuran CT. The zinc coordination pattern in the eta-carbonic anhydrase from Plasmodium falciparum is different from all other carbonic anhydrase genetic families. Bioorg Med Chem Lett 2015;25:1385–9.
- Kikutani S, Nakajima K, Nagasato C, et al. Thylakoid luminal theta-carbonic anhydrase critical for growth and photosynthesis in the marine diatom Phaeodactylum tricornutum. Proc Natl Acad Sci USA 2016;113:9828–33.
- Xu Y, Feng L, Jeffrey PD, et al. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008;452:56–61.
- Smith KS, Jakubzick C, Whittam TS, Ferry JG. Carbonic anhydrase is an ancient enzyme widespread in prokaryotes. Proc Natl Acad Sci USA 1999;96:15184–9.
- Alterio V, Monti SM, Simone G. Thermal-stable carbonic anhydrases: a structural overview. In: Frost SC, McKenna R, eds. Carbonic anhydrase: mechanism, regulation, links to disease, and industrial applications. Amsterdam, The Netherlands: Springer; 2014:387–404.
- Alterio V, Langella E, De Simone G, Monti SM. Cadmium-containing carbonic anhydrase CDCA1 in marine diatom Thalassiosira weissflogii. Mar Drugs 2015;13:1688–97.
- Parkkila S. An overview of the distribution and function of carbonic anhydrase in mammals. EXS 2000;90:79–93.
- Supuran CT. Carbonic anhydrases – an overview. Curr Pharm Des 2008;14:603–14.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Supuran CT, De Simone G, eds. Carbonic anhydrases as biocatalysts: from theory to medical and industrial applications. Amsterdam, The Netherlands: Elsevier; 2015.
- Supuran CT, Scozzafava A, Conway J, eds. Carbonic anhydrase: its inhibitors and activators. Boca Raton, FL: CRC Press; 2004.
- Winum JY, Scozzafava A, Montero JL, Supuran CT. Sulfamates and their therapeutic potential. Med Res Rev 2005;25:186–228.
- Winum JY, Scozzafava A, Montero JL, Supuran CT. Therapeutic potential of sulfamides as enzyme inhibitors. Med Res Rev 2006;26:767–92.
- De Monte C, Carradori S, Secci D, et al. Cyclic tertiary sulfamates: selective inhibition of the tumor-associated carbonic anhydrases IX and XII by N- and O-substituted acesulfame derivatives. Eur J Med Chem 2014;84:240–6.
- Carradori S, Secci D, De Monte C, et al. A novel library of saccharin and acesulfame derivatives as potent and selective inhibitors of carbonic anhydrase IX and XII isoforms. Bioorg Med Chem 2016;24:1095–105.
- Winum JY, Colinas PA, Supuran CT. Glycosidic carbonic anhydrase IX inhibitors: a sweet approach against cancer. Bioorg Med Chem 2013;21:1419–26.
- Krishnamurthy VM, Kaufman GK, Urbach AR, et al. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem Rev 2008;108:946–1051.
- De Simone G, Supuran CT. Antiobesity carbonic anhydrase inhibitors. Curr Top Med Chem 2007;7:879–84.
- van Passel L, Arif H, Hirsch LJ. Topiramate for the treatment of epilepsy and other nervous system disorders. Expert Rev Neurother 2006;6:19–31.
- Maryanoff BE, McComsey DF, Costanzo MJ, et al. Comparison of sulfamate and sulfamide groups for the inhibition of carbonic anhydrase-II by using topiramate as a structural platform. J Med Chem 2005;48:1941–7.
- Gavernet L, Gonzalez Funes JL, Palestro PH, et al. Inhibition pattern of sulfamide-related compounds in binding to carbonic anhydrase isoforms I, II, VII, XII and XIV. Bioorg Med Chem 2013;21:1410–8.
- Parker MH, Smith-Swintosky VL, McComsey DF, et al. Novel, broad-spectrum anticonvulsants containing a sulfamide group: advancement of N-((benzo[b]thien-3-yl)methyl)sulfamide (JNJ-26990990) into human clinical studies. J Med Chem 2009;52:7528–36.
- Di Fiore A, De Simone G, Alterio V, et al. The anticonvulsant sulfamide JNJ-26990990 and its S,S-dioxide analog strongly inhibit carbonic anhydrases: solution and X-ray crystallographic studies. Org Biomol Chem 2016;14:4853–8.
- Rami M, Dubois L, Parvathaneni NK, et al. Hypoxia-targeting carbonic anhydrase IX inhibitors by a new series of nitroimidazole-sulfonamides/sulfamides/sulfamates. J Med Chem 2013;56:8512–20.
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997;276:307–26.
- Eriksson AE, Jones TA, Liljas A. Refined structure of human carbonic anhydrase II at 2.0 A resolution. Proteins 1988;4:274–82.
- Brünger AT, Adams PD, Clore GM, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 1998;54:905–21.
- Brünger AT. Version 1.2 of the crystallography and NMR system. Nat Protoc 2007;2:2728–33.
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst A 1991;47:110–19.
- Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B 2002;58:380–8.
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision A.1. 2009.
- Vanquelef E, Simon S, Marquant G, et al. R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res 2011;39:W511–17.
- Wang F, Becker J-P, Cieplak P, Dupradeau F-Y. R.E.D. Python: Object oriented programming for Amber force fields, Université de Picardie-Jules Verne, Sanford Burnham Prebys Medical Discovery Institute, 2013.
- King RW, Burgen AS. Sulphonamide complexes of human carbonic anhydrases. Ultraviolet difference spectroscopy. Biochim Biophys Acta 1970;207:278–85.
- Taylor PW, King RW, Burgen AS. Influence of pH on the kinetics of complex formation between aromatic sulfonamides and human carbonic anhydrase. Biochemistry 1970;9:3894–902.
- Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem 2004;25:1157–74.
- Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 2015;11:3696–713.
- Li P, Merz KM Jr. Taking into account the ion-induced dipole interaction in the nonbonded model of ions. J Chem Theory Comput 2014;10:289–97.
- Tsui V, Case DA. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 2000;56:275–91.
- Kollman PA, Massova I, Reyes C, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 2000;33:889–97.
- Case DA, Cerutti DS, Cheatham I, et al. AMBER 14. San Francisco: University of California; 2014.
- Wang J, Morin P, Wang W, Kollman PA. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J Am Chem Soc 2001;123:5221–30.
- Langella E, D’Ambrosio K, D’Ascenzio M, et al. A combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes in the active site of carbonic anhydrases. Chemistry 2016;22:97–100.
- Homeyer N, Gohlke H. Free energy calculations by the molecular mechanics Poisson–Boltzmann surface area method. Mol Inform 2012;31:114–22.
- Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004;55:383–94.
- Weiser J, Shenkin PS, Still WC. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J Comp Chem 1999;20:217–30.
- Recacha R, Costanzo MJ, Maryanoff BE, Chattopadhyay D. Crystal structure of human carbonic anhydrase II complexed with an anti-convulsant sugar sulphamate. Biochem J 2002;361:437–41.
- Lopez M, Paul B, Hofmann A, et al. S-glycosyl primary sulfonamides – a new structural class for selective inhibition of cancer-associated carbonic anhydrases. J Med Chem 2009;52:6421–32.
- Lopez M, Vu H, Wang CK, et al. Promiscuity of carbonic anhydrase II. Unexpected ester hydrolysis of carbohydrate-based sulfamate inhibitors. J Am Chem Soc 2011;133:18452–62.
- Mahon BP, Lomelino CL, Ladwig J, et al. Mapping selective inhibition of the cancer-related carbonic anhydrase IX using structure–activity relationships of glucosyl-based sulfamates. J Med Chem 2015;58:6630–8.
- Vitale RM, Alterio V, Innocenti A, et al. Carbonic anhydrase inhibitors. Comparison of aliphatic sulfamate/bis-sulfamate adducts with isozymes II and IX as a platform for designing tight-binding, more isoform-selective inhibitors. J Med Chem 2009;52:5990–8.
- Lloyd MD, Pederick RL, Natesh R, et al. Crystal structure of human carbonic anhydrase II at 1.95 A resolution in complex with 667-coumate, a novel anti-cancer agent. Biochem J 2005;385:715–20.
- Lloyd MD, Thiyagarajan N, Ho YT, et al. First crystal structures of human carbonic anhydrase II in complex with dual aromatase-steroid sulfatase inhibitors. Biochemistry 2005;44:6858–66.
- Leese MP, Leblond B, Smith A, et al. 2-substituted estradiol bis-sulfamates, multitargeted antitumor agents: synthesis, in vitro SAR, protein crystallography, and in vivo activity. J Med Chem 2006;49:7683–96.
- Woo LW, Jackson T, Putey A, et al. Highly potent first examples of dual aromatase-steroid sulfatase inhibitors based on a biphenyl template. J Med Chem 2010;53:2155–70.
- Cozier GE, Leese MP, Lloyd MD, et al. Structures of human carbonic anhydrase II/inhibitor complexes reveal a second binding site for steroidal and nonsteroidal inhibitors. Biochemistry 2010;49:3464–76.
- Woo LW, Fischer DS, Sharland CM, et al. Anticancer steroid sulfatase inhibitors: synthesis of a potent fluorinated second-generation agent, in vitro and in vivo activities, molecular modeling, and protein crystallography. Mol Cancer Ther 2008;7:2435–44.
- Temperini C, Innocenti A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Interaction of the antitumor sulfamate EMD 486019 with twelve mammalian carbonic anhydrase isoforms: kinetic and X-ray crystallographic studies. Bioorg Med Chem Lett 2008;18:4282–6.
- Sippel K, Stander A, Tu C, et al. Characterization of carbonic anhydrase isozyme specific inhibition by sulfamated 2-ethylestra compounds. Lett Drug Design Discov 2008;8:678–84.
- Moeker J, Mahon BP, Bornaghi LF, et al. Structural insights into carbonic anhydrase IX isoform specificity of carbohydrate-based sulfamates. J Med Chem 2014;57:8635–45.
- Mahon BP, Lomelino CL, Salguero AL, et al. Observed surface lysine acetylation of human carbonic anhydrase II expressed in Escherichia coli. Protein Sci 2015;24:1800–7.
- Leese MP, Jourdan F, Kimberley MR, et al. Chimeric microtubule disruptors. Chem Commun (Camb) 2010;46:2907–9.
- Leese MP, Jourdan FL, Gaukroger K, et al. Structure-activity relationships of C-17 cyano-substituted estratrienes as anticancer agents. J Med Chem 2008;51:1295–308.
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810.
- De Simone G, Pizika G, Monti SM, et al. Hydrophobic substituents of the phenylmethylsulfamide moiety can be used for the development of new selective carbonic anhydrase inhibitors. Biomed Res Int 2014;2014:523210.
- Brynda J, Mader P, Šicha V, et al. Carborane-based carbonic anhydrase inhibitors. Angew Chem Int Ed Engl 2013;52:13760–3.
- Winum JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;49:7024–31.
- Di Fiore A, Monti SM, Innocenti A, et al. Carbonic anhydrase inhibitors: crystallographic and solution binding studies for the interaction of a boron-containing aromatic sulfamide with mammalian isoforms I-XV. Bioorg Med Chem Lett 2010;20:3601–5.
- Mader P, Pecina A, Cigler P, et al. Carborane-based carbonic anhydrase inhibitors: insight into CAII/CAIX specificity from a high-resolution crystal structure, modeling, and quantum chemical calculations. Biomed Res Int 2014;2014:389869.
- Michalczyk R, Unkefer CJ, Bacik JP, et al. Joint neutron crystallographic and NMR solution studies of Tyr residue ionization and hydrogen bonding: implications for enzyme-mediated proton transfer. Proc Natl Acad Sci USA 2015;112:5673–8.