
Abstract
α-Glucosidase inhibitors are described as the most effective in reducing post-prandial hyperglycaemia (PPHG) from all available anti-diabetic drugs used in the management of type 2 diabetes mellitus. As flavonoids are promising modulators of this enzyme’s activity, a panel of 44 flavonoids, organised in five groups, was screened for their inhibitory activity of α-glucosidase, based on in vitro structure–activity relationship studies. Inhibitory kinetic analysis and molecular docking calculations were also applied for selected compounds. A flavonoid with two catechol groups in A- and B-rings, together with a 3-OH group at C-ring, was the most active, presenting an IC50 much lower than the one found for the most widely prescribed α-glucosidase inhibitor, acarbose. The present work suggests that several of the studied flavonoids have the potential to be used as alternatives for the regulation of PPHG.
Graphical Abstract
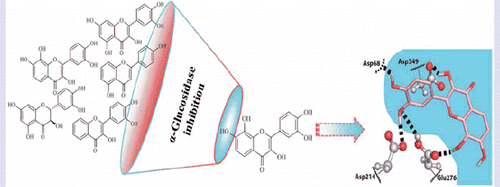
Introduction
Diabetes mellitus (DM) is one of the most significant public health concerns worldwide. According to the International Diabetes Federation, there are 415 million of adults with diabetes, probably reaching 642 million in 2040Citation1. DM is a multifactorial metabolic disorder, characterised by chronic hyperglycaemia and can be primarily classified as type 1 (insulin dependent DM) and type 2 (non-insulin dependent DM)Citation2. Type 2 DM is the most common form of DM, accounting for more than 90% of all diabetic patients, and results from the interaction between behavioural, environmental and genetic risk factorsCitation3,Citation4. Diabetic patients are more vulnerable to various forms of both short- and long-term complications, such an increased risk of common infections and cancer, increasing the morbidity and mortalityCitation4–6.
DM is characterised by absolute or relative deficiency in insulin secretion by pancreatic β-cells, increased insulin resistance and/or impairment of insulin action in target tissuesCitation7–10. Post-prandial hyperglycaemia (PPHG) is known as the plasma glucose value taken 1.5–2 h after a meal. The disequilibrium in the insulin regulatory system, culminates in high PPHG level (≥200 mg/dL) compared to individuals without type 2 DM (<140 mg/dL)Citation11. Prolonged hyperglycaemia has been associated with several macro- and micro-vascular complications, e.g. nephropathy, retinopathy and cardiovascular disordersCitation11–13. Therefore, the management of PPHG, in order to achieve blood glucose levels as close to normal as possible, is considered by the scientific community one of the major therapeutic strategies for type 2 DM treatmentCitation14.
An effective control of hyperglycaemia in type 2 DM includes the retarding, regulation and/or inhibition of carbohydrate hydrolysing enzymesCitation15. Among these, α-glucosidase, which is located in the brush border of the enterocytes of the jejunum, is the most important enzyme in carbohydrates digestionCitation16. It catalyses the hydrolysis of 1,4-α bonds of the unabsorbed oligo- and disaccharides, and converts them into monosaccharides, namely, glucose, which are absorbed in the upper jejunum, resulting in hyperglycaemiaCitation17,Citation18. Inhibitors of α-glucosidase can retard the decomposition and absorption of dietary carbohydrates by restricting the breakdown of linear or branched oligosaccharide units like α-limit dextrins, maltose and maltotriose to produce glucose, thereby preventing glucose absorption into blood stream and suppress the PPHGCitation19,Citation20.
The use of α-glucosidase inhibitors started in the 1970s and the first related therapeutic agents for diabetes were approved in the 1990sCitation18. Nowadays, several oral drugs are used such as voglibose, acarbose and miglitol to inhibit the α-glucosidase activityCitation20. However, their use has been associated with several adverse effects such as abdominal distention, flatulence, diarrhoea and pneumatosis cystoides intestinalisCitation11. Most of these side-effects occur when undigested carbohydrates are fermented by colonic bacteriaCitation21,Citation22. The commonly used α-glucosidase inhibitors were also reported to be associated with rare adverse hepatic eventsCitation23. Moreover, these drugs have low efficacy, with high IC50 valuesCitation24,Citation25. Thus, much effort has been undertaken to search for more effective and safer inhibitors.
Some flavonoids, such as morinCitation17, luteolinCitation26, baicaleinCitation27, kaempferolCitation28 and apigeninCitation24, have been shown to exhibit inhibitory effects against α-glucosidase enzymes. Nevertheless, the information found in the literature is disperse and variable, probably due to the use of different enzyme concentrations (0.3–1.7 U/mL)Citation8,Citation29,Citation30, from different origins (e.g. yeast and rat intestinal α-glucosidase)Citation18,Citation19,Citation31,Citation32, different substrates concentrations (1–20 mM)Citation19,Citation30,Citation33 and incubation time with the flavonoids (5 min–3 h)Citation8,Citation26,Citation28,Citation29,Citation34. The discrepancies between the studies bring difficulties to their comparison and consequently the conclusions about the flavonoids efficacy against α-glucosidase activity.
The present study aims to establish a structure–activity relationship of a panel of flavonoids (), divided in five groups, against α-glucosidase activity, covering a solid set of different structures. For this purpose, a microanalysis screening system was applied for testing the type of inhibition of the most active flavonoids from each group, and the positive control acarbose. Molecular docking calculations were also applied to complement the inhibitory activity studies and to predict the binding model of the selected flavonoids to the three-dimensional structure of α-glucosidase.
Figure 1. Chemical structures of the tested flavonoids.

The overall obtained results constitute an important step for the establishment of the adequate and ideal chemical structure of flavonoids to target α-glucosidase and consequently, to manage type 2 DM.
Materials and methods
Chemicals
The following reagents were purchased from Sigma-Aldrich Co. LLC (St. Louis, MO): α-glucosidase from Saccharomyces cerevisiae, p-nitrophenyl-α-D-glucopyranoside (pNPG), DMSO, NaHPO4, Na2HPO4, compounds D3 (baicalein), D4 (apigenin), D6 (kaempferol), D7 (luteolin), D8 (quercetin), D9 (morin), D16 (acacetin), D18 (rutin), E1 (naringenin), E2 (eriodictyol), E3 (taxifolin) and acarbose. Compounds A1, A2, A3, A4, A5, A6, B1, B2, B3, B4, C1, C2, C3, C4, C5, C6, C8, C9, D1 (chrysin), D2 (galangin) and D5 were obtained from Indofine Chemical Company, Inc. (Hillsborough, NJ). The flavonoids C7, C10, C11, C12, C13, D10, D11, D12, D13, D14 and D17 were synthesised as previously describedCitation35–37. For the enzymatic assays, flavonoids were dissolved in DMSO (the final concentration of DMSO in the reactional mixture was 4.76%).
In vitro inhibition assay for α-glucosidase activity
The α-glucosidase activity was measured using a method of Tadera et al.Citation31 slightly modified. The assay was carried out by monitoring the α-glucosidase-mediated transformation of the substrate pNPG into α-D-glucose and p-nitrophenol, at 405 nm. Briefly, in a 96-well plate, the enzyme (0.05 U/mL), dissolved in 100 mM phosphate buffer (pH 6.8), was pre-incubated at 37 °C for 5 min with the flavonoids (0–200 μM). In sequence, pNPG was added (600 μM) and incubated in the reaction mixture at 37 °C for 30 min. The enzymatic reaction was monitored spectrophotometrically, in a microplate reader (Synergy HT, BIO-TEK), by measuring the absorbance at 405 nm. The obtained values correspond to the slope measured between 5 and 20 min. The amount of DMSO used had no interference with the assay. Acarbose (0–2000 μM) was used as positive control. The obtained results represent at least three independent experiments.
Inhibitory kinetic analysis
The inhibitory kinetic analyses were performed for the most active flavonoids of each group (A–E): A5 (0–62.5 μM), B3 (0–50 μM), C7 (0–5 μM), D8 (quercetin) (0–12.5 μM), D10 (0–18.75 μM), E3 (taxifolin) (0–100 μM) and for the positive control, acarbose (0–2000 μM).
Briefly, the enzyme (0.05 U/mL), dissolved in 100 mM phosphate buffer (pH 6.8), was pre-incubated at 37 °C with the above mentioned flavonoids in a 96-well plate, for 5 min. In sequence, pNPG was added (300, 600 and 1200 μM) and incubated in the reaction mixture at 37 °C for 30 min.
The study of the inhibition type (competitive, uncompetitive, non-competitive or mixed) of the tested flavonoids was performed using the nonlinear regression Michaelis–Menten enzyme kinetics and the corresponding Lineweaver–Burk double reciprocal plots for each concentration of the inhibitor and substrate. The Ki values were calculated with GraphPad Software 6.0 by plotting the reciprocal of maximum velocity (1/Vmax) (y axis) against the flavonoid concentrations (x axis) ()Citation38–40.
The type of inhibition parameters were all calculated with GraphPad Software 6.0. The obtained results represent at least three independent experiments.
Molecular docking
Homology modelling
3D structures of Saccharomyces cerevisiae α-glucosidase (maltase, EC 3.2.1.20) are unavailable. There are, however, X-ray structures available for the isozyme isomaltase/α-methylglucosidase (EC 3.2.1.10). The isomaltase/α-methylglucosidase with the PDBCitation41 entry 3AJ7 has a high-resolution X-ray structure and a high sequence identity (72.51%) and sequence similarity score (0.54) with the Saccharomyces cerevisiae α-glucosidase MAL32 (UniProt entry P38158)Citation42.
We identified several differences between residues lining the binding pockets of α-glucosidase and isomaltase, such as the pairs Asp307/Glu204, Asp408/Glu411, Phe157/Tyr158, Thr215/Val216, Ala278/Gln279, Val303/Thr306 or Ala178/Cys179, respectively for α-glucosidase and isomaltase. Most of these differences involve very similar residues, but considering the differences in the size of the side chains (which can affect binding through stereochemical effects) and the high confidence entrusted by the extremely favourable sequence identity, we opted to build a high accuracy homology model of α-glucosidase using isomaltase as template.
Using MAL32 target sequence and the structure with the PDB ID 3AJ7 as template, we built the α-glucosidase homology model in SWISS-MODELCitation43 based on the target-template alignment using ProMod3. The resulting model has a Global Model Quality Estimation of 0.92 and a rmsd for 4452 atoms of 0.40 Å, which is excellent. Supplementary Figure S1 provides a summary of the homology modelling parameters and Supplementary Figure S2 the Ramachandran plot analysis of the homology model.
Docking protocol
The protonation states of the enzyme were predicted using the Protoss serverCitation44. Accordingly, His6, His97 and His251 were modelled in the charged form, Glu276 and Glu481 were modelled in the neutral form. All other residues were protonated according to their pKa at pH 7.
The substrate α-maltose was modelled inside the binding pocket and subsequently, to remove potential clashes and relax the structure of the model, Molecular Mechanics minimisation followed by classical MD simulations were done. This protocol was performed within the AMBER 12Citation45 suite using the ff99SBCitation46 force field for the protein and GLYCAM06Citation47 force field for maltose. The details for the energy minimisation and MD simulation are given in SI.
To validate the receptor for the docking protocol we extracted the last step from the MD simulation and re-docked the substrate α-maltose and also the inhibitor acarbose. Since the predicted binding modes, both for α-maltose and acarboseCitation48, behaved according to the expected catalytic binding mode and to the X-ray structure 3TOP, we proceeded to binding mode prediction of the most active flavonoids of each group (A–E).
AutoDock VinaCitation49 was used to generate binding poses using default parameters (see Supplementary Material for details), such as the exhaustiveness of the global search = 8 or the maximum number of output poses = 9. The search space was confined to a box of circa 15 × 20 × 17 centred on the site occupied by the second glucose in maltose. The receptor was treated as a rigid body and prepared using openbabelCitation50 for atom-typing and charge assignment using Gasteiger–Marsili chargesCitation51. Flavones atom-types and charges were attributed the same way.
Subsequently, the poses generated by AutoDock Vina were rescored using the scoring function ChemPLP, which is excellent for oligosaccharidesCitation52. We used the GOLD softwareCitation53 with the standard parameters for a “Rescoring Run” with local optimisation and selected the binding mode with the highest score.
Results
In vitro inhibition assay for α-glucosidase activity
The inhibitory effect of flavonoids against α-glucosidase activity is shown in . This effect was highly dependent on small variations of the flavonoids’ structures. According to the flavonoids’ structures, we divided the studied compounds into five groups (A–E).
Table 1. Structures and in vitro α-glucosidase inhibition by the studied flavonoids (IC50 μM, mean ± SEM).
Table 2. Ki values (mean ± SEM, μM) for the inhibition of yeast α-glucosidase by the selected flavonoids.
Flavonoids of A group
From all the flavones from A group, flavone A5 was noticeably the most effective, presenting an IC50 of 54 ± 3 µM. For the other tested compounds, it was not possible to achieve the IC50 value up to the concentrations of 100 µM (A6) or 200 µM (A1–A4). These results indicate that the presence of a 3-OH group in the C-ring is relevant for the inhibitory activity of flavonoids. Moreover, when comparing the flavonoid A5 and the flavonoid A6, we may conclude that the replacement of the 3′,4′-(OH)2, in the B-ring, by methoxy (–OMe) groups withdraws the inhibitory activity of compound A5.
Flavonoids of B group
From B group, flavone B1 was the less active. Comparing the activities of B1 (with a 5-OH in A-ring) and A1 (without substitutions) it is shown that the presence or absence of 5-OH in A-ring, per se, is indifferent for the inhibitory activity. However, the 5-OH substitution in A-ring, together with the 4′-OH (B3) or 3′,4′-(OH)2 (B4) in B-ring, increases the activity, comparing with the related compounds, from A group, which do not have 5-OH substitution.
Flavonoids of C group
C group has the most active flavonoid, C7 with an IC50 = 7.6 ± 0.4 µM, which is almost 80 times lower than the IC50 of the positive control, acarbose (IC50 = 607 ± 56 µM). Comparing the flavones C5 and C4, it is clear that the presence of –OH groups in the B-ring is more relevant in the 3′-position (C4, IC50 = 42 ± 4 µM) than in the 4’-position (C5, IC50 = 96 ± 10 µM). Flavone C1, with just one 7-OH group, did not achieve the IC50 value up to the maximum tested concentration (200 µM). However, adding another 3′-OH at the B-ring (C2) is enough to increase the inhibitory activity of the compound (IC50 = 53 ± 4 µM). In turn, increasing the complexity of the structure by the addition of a –OMe and/or a benzyloxy (–OBn) group (C8–C13) did not favour the activity.
Flavonoids of D group
In what concerns D group, the most effective compound was D8 (quercetin) with an IC50 = 15 ± 3 µM. The substitution in 3-position of the C-ring seems to be advantageous for the intended activity, since D7 (luteolin), with no substitution in 3-position, had a higher IC50 (46 ± 6 µM). Moreover, the relevance of the existence of a 3-OH substitution in the C-ring was evidenced by the low activity of D18 (rutin), which had a rutinose in this position. In turn, the close IC50 values of D8 (quercetin) and D10 (IC50 = 21 ± 2 μM) indicate that the presence of a 3-OH group or a 3-Cl atom in the C-ring is indifferent for α-glucosidase inhibition. The relevance of the presence of a catechol moiety in the B-ring is shown by the different IC50 values found for D7 (luteolin) (46 ± 6 µM), which have a catechol group in 3′,4′- positions, and the compounds D5 (apigenin) (IC50 = 82 ± 6 µM) and D4 (IC50 = 89 ± 3 µM), which had just one OH in the 4′- or 3′-positions, respectively. Similarly to the obtained results in C group, results of D group indicate that the methylation of the catechol arrangement in the B-ring (D15–D17) did not show any advantage for the studied activity.
Flavonoids of E group
Analysing the IC50 of flavonoids from E group, E3 (taxifolin) had the best inhibitory activity. Nonetheless, its IC50 was approximately 200 μM, the maximum tested concentration.
Inhibitory kinetics analysis
The type of inhibition of the most active flavonoids of each group (A–E), A5, B3, C7, D8 (quercetin), D10, E3 (taxifolin) and acarbose was deducted from the calculation of Km and maximum enzyme velocity (Vmax). These parameters were obtained by the nonlinear regression Michaelis–Menten enzyme kinetics and complemented with Lineweaver–Burk double reciprocal plots. As observed in , flavones A5 () and B3 () showed a mixed type inhibition. Concerning flavonoid C7, although the does not clearly differentiate between a mixed and a competitive inhibition type, the interception of the lines is close to the y axis, suggesting a competitive pattern. Flavonoid D8 (quercetin) () and the positive control acarbose () have shown a competitive inhibition type. Flavones D10 () and E3 (taxifolin) ( presented a non-competitive inhibition type, since the Km value remained constant and the Vmax value decreased with increasing concentrations of the flavonoids.
Figure 2. Lineweaver–Burk double reciprocal plots of α-glucosidase inhibition by A5 (A), B3 (B), C7 (C), D8 (quercetin) (D), D10 (E), E3 (taxifolin) (F) and acarbose (G).

The Ki values were calculated with GraphPad Software 6.0 by plotting the reciprocal of maximum velocity (1/Vmax) (y axis) against the flavonoid concentrations (x axis) ().
Binding mode of the selected inhibitors/flavonoids
We have investigated the binding poses of a selection of inhibitors within the reactive pocket of the active site of α-glucosidase [acarbose, A5, B3, C7, D8 (quercetin), D10 and E3 (taxifolin)]. It is important to realise that a few of them may bind in more than one sugar pocket, even predominantly in other sugar pockets that line the active site cavity (which were physiologically developed to bind the sugars of oligosaccharide chain tips), and in a lesser extent at the deeper, reactive pocket of the active site. However, for the purpose of drug discovery, the deep reactive pocket is by far the most relevant, because it is the one for which the drugs are always optimised to bind (an evidence of this is that all approved drugs for this target are described as competitive inhibitors). Therefore, it was the binding to this deep pocket that we explore in this section.
Acarbose
The predicted binding mode for acarbose in the homology model () positions the first glucose of acarbose in the exact same site as the first glucose of the α-maltose substrate, which was used to build and refine the model. Thus, the first glucose of acarbose is placed in the deep reactive pocket with 2,3,4-(OH)3 groups donating hydrogen bonds to Asp349 and Asp68. The oxygen atoms from these three hydroxyls accept hydrogen bonds from Arg212, His348 and Arg439. The single difference between acarbose first glucose and α-maltose is in the CH2OH group, since in α-maltose the C-4 has sp3 hybridisation while in acarbose the C-4 has sp2 hybridisation. Therefore, the α-maltose CH2OH group hydrogen bonds to both His111 and Asp214 while the same group in acarbose makes a single hydrogen bond with Asp68.
Figure 3. Predicted binding poses for acarbose (A) and flavones A5 (B), B1 and B3 (C), C7 (D), D8 (quercetin) (E), D10 (F) and E3 (taxifolin) (G). Hydrogen bonds are represented by dashed lines. (B) B1 is coloured salmon and B3 light blue. The residues that establish the most relevant interactions are also shown. Asp214 and Glu276 are the catalytic residues that participate in the hydrolysis reaction. Asp349 is also a conserved residue.

The nucleophilic Asp214 is placed below the acarbose C-1 carbon but does not accept any hydrogen bond from acarbose. Different to α-maltose binding, Glu276 donates one hydrogen bond to the N-glycosidic bond nitrogen and accepts one hydrogen bond from the 3-OH group of the second acarbose glucose.
To conclude, the first glucose, the N-glycosidic bond and the second glucose of acarbose, all occupy the same site as the substrate α-maltose and have a binding mode identical to α-maltose. This is compatible with a competitive mode of inhibition, which the Lineweaver-Burk double reciprocal plots of seem to corroborate.
Flavone A5
Flavones skeleton is quite different from the saccharides found in α-maltose and acarbose. Nevertheless, when we analyse the predicted binding poses for the most active compounds, we can observe similar interactions with binding pocket residues.
In the case of the most active compound of A group, A5, the catechol group in the B-ring and the OH substitution in the 3-position of the C ring, provide similar interactions to active site residues as found in α-maltose or acarbose (). The 3′,4′-(OH)2 B-ring substitutions donate hydrogen bonds to Asp214 and the neutral Glu276 donates one hydrogen bond to B-ring 3′-OH. The OH in the 3-position of the C-ring donates a hydrogen bond to the conserved Asp349.
The B-ring binds in the small crevice of the active site pocket, occupying the same site as the first glucose of acarbose and α-maltose. The C-ring occupies a similar site to α-maltose second glucose, in the entrance of the deep small pocket, while the A-ring is exposed to the larger binding pocket.
The Lineweaver–Burk double reciprocal plots () shows a mixed inhibition type. The binding mode described here for flavone A5 corresponds to the competitive inhibition mode. We directed the binding mode prediction to the active site, where the natural substrates bind the first two sugar rings and did not explore all possible modes of interaction of flavones with α-glucosidase, as the interactions with the active site are the most relevant for drug design. However, given the large size of the active site and knowing that many natural substrates are oligosaccharides, it is possible that the inhibitors bind to the sites where the oligosaccharides sugar rings (other than the first two) bind. Therefore, we cannot exclude that flavone A5 may bind with increased affinity to a different oligosaccharide site, which could interfere with substrate binding and explain the mixed inhibition type.
Flavone B3
When flavone B3 binds the reactive region of the active site it is predicted to bind the A- and C- rings close to the catalytic Glu276 and the conserved Asp349, establishing strong hydrogen bonds with them (). However, B3 does not insert into the deepest region of the catalytic pocket, formed by Asp349/Glu276/Asp214 triad (Asp214 is the nucleophilic residue), but instead in a larger, shallower region of the pocket bordered by hydrophobic residues (such as Phe157, Phe158, Phe177, Thr215 and Phe300) and polar residues (His245, His279, Glu304, Arg312 or Asp408). This binding mode is very similar to the one obtained for flavone B1 (). The additional 3′-OH group of flavone B3 might hydrogen bond His274, but it is not totally clear from our binding mode prediction.
Despite B3 binding outside the small crevice formed by the catalytic residues, the predicted mode of binding would still be more compatible with a competitive mode of inhibition. The reason is B3 C-ring binds exactly in the site of the α-maltose second glucose. The binding pose shown in will not allow for substrate binding, despite the fact that the data shown in seems to point to the opposite. As mentioned earlier, it is possible that the inhibitors bind to the sites where the oligosaccharides sugar rings (other than the first two) bind. These cases are less relevant than the one shown here for the actual purpose of drug discovery, but could allow for substrate binding even though interfering with the substrate pose.
Flavone C7
Comparing the inhibitory activity of C1 and C3 with C6, it seems that addition of –OH groups to the B-ring improves the inhibitory activity of group C flavonoids, with the 3′-OH substitution to have the greater impact on the inhibitory activity (). Flavone C7 has both of these substitutions, plus three more –OH substitutions, one on the C-ring (3-OH) and two on the A-ring (7,8-(OH)2).
The predicted binding mode for flavone C7 in the active site region places the B-ring deep in the small crevice formed by the catalytic residues. Glu276 hydrogen bonds to 3′-OH, and 3′-OH donates a hydrogen bond to Asp214. 4′-OH group is even deeper in the small pocket and hydrogen bonds Asp68.
The C- and A-rings bind in the entrance of the small crevice, with C-ring 3-OH substitution hydrogen bonding Asp349. The A- and C-rings are placed around Glu276, and 8-OH group donates a hydrogen bond to Glu276. The 7-OH substitution is exposed to the larger binding pocket and no specific interactions with 8-OH group can be identified.
Considering this binding mode, the strong hydrogen bond interactions with active site amino acids are consistent with a very favourable binding affinity to the active site, indicating a competitive type of inhibition.
Flavones D8 (quercetin) and D10
shows that flavone D8 (quercetin) () is a competitive inhibitor while the very similar D10 () is non-competitive. The predicted binding mode for flavones D8 (quercetin) and D10 brings the B-ring into the small reactive catalytic pocket formed by the Asp349/Glu276/Asp214 triad. The 3′-OH of flavone D8 (quercetin) makes two hydrogen bonds with Glu276, while D10, which is slightly rotated, accepts one hydrogen bond from Asp214 and donates another to Asp349 (). The 4′-OH of flavone D8 (quercetin) hydrogen bonds to Asp214 while 4′-OH from D10 hydrogen bonds to Asp68. The crucial difference between D8 (quercetin) and D10 seems to be the C-ring substitution. The 3-OH group in flavone D8 (quercetin) is able to hydrogen bond the conserved Asp349, while the hydrophobic 3-Cl-substitution in flavone D10 cannot provide any specific interaction. The lack of this interaction makes D10 to rotate and changes slightly the hydrogen bond pattern found for 3′,4′-(OH)2 substitution.
The extensive hydrogen bonding with the deep reactive pocket of flavone D8 (quercetin) is absolutely consistent with the competitive inhibition pattern seen in . D10, in contrast, loses the hydrogen bonds with one of the Asp349/Glu276/Asp214 triad members (namely with Asp214), weakening the interaction with the deep reactive pocket, also very well in line with the non-competitive pattern shown in . Therefore, extensive hydrogen bond with the Glu/Asp triad seems to be relevant to hold the inhibitors bound to the deep reactive pocket.
Flavanone E3 (taxifolin)
Flavanone E3 (taxifolin) contains 3′,4′-(OH)2 substitution in the B-ring, which were demonstrated to be important to drive the B-ring into the Glu276/Asp214/Asp349 triad pocketCitation32. However, the lack of C2 = C3 double bound makes the B-ring to move out of the plane of the A- and C-rings, destroying the overall planarity of the compounds, which has a major impact in the binding properties. The predicted binding pose of E3 (taxifolin) does not have the B-ring inserted in the small active site pocket. Instead, the entire flavanone moves to a larger, less deep pocket (). This result is consistent with the non-competitive mode of inhibition determined through the Lineweaver–Burk double reciprocal plots of . The B-ring binds a hydrophobic pocket formed by Phe157, Leu176, Phe177, Thr215, Leu218, Ala278 and Phe300, and with 3′-OH hydrogen bonding to Glu276. The 5-OH of A-ring hydrogen bonds to the backbone of Asp349 and 7-OH of the same ring hydrogen bonds to the Phe157 backbone.
Discussion
In this work, we explored a panel of flavonoids, covering a solid set of different structures, tested in the same model, under the same experimental conditions, which enabled us to draw relevant conclusions about the relationship structure versus activity.
Many of the studied flavonoids offer a promising alternative for the management of PPHG, presenting an IC50 ≤ 200 μM, much lower than the one found for acarbose (607 ± 56 μM), the positive control.
We started our study by testing a flavone without –OH groups (A1), which was not able to inhibit α-glucosidase. We also tested flavonoids A2, A3 and A4, and all of them showed low or no inhibitory activity. From A group, the most active flavonoid was A5, which has a 3-OH in the C-ring and presented an IC50 of 54 ± 3 µM. The replacement of the –OH groups of the catechol in B-ring by –OMe groups (A6) decreased the inhibitory activity of the flavonoid.
The importance of the –OH groups position was also highlighted when A5, with OH groups at 3-position of C-ring and at 3′- and 4′- positions of B-ring, was tested, because the obtained IC50 was lower than the one obtained for A5, with –OH groups at 5 position of A-ring and at 3′- and 4′- positions of B-ring.
We followed our rationale of increasing the complexity of the structure, by the addition of a catechol group in the A-ring. The most effective compound was C7, studied here for the first time, with an IC50 = 7.6 ± 0.4 µM. The presence of two catechol groups distributed in the A and B rings, together with a 3-OH group in the C-ring, seems to be the more favourable structure for the inhibition of the enzyme. In fact, and as it can be seen in , the other related compounds (C1–C6) that differ from C7 by the lack of an –OH group in some positions, have considerable weaker activities. Gao et al.Citation27 corroborate our results showing that B1 and C1 have no activity against α-glucosidase catalytic activity. In the present study, we observed that the addition of a catechol group to the B-ring in both compounds increased their activity, the –OH group in 5-position of the A-ring (B4) being better than in 7-position (C6) (IC50 = 66 ± 7 µM and IC50= 95 ± 7 µM, respectively). The obtained results also have shown that, as mentioned by Gao and co-workersCitation27, baicalein has a favourable structure for the intended effect, as observed in the present study for the same flavonoid [D3 (baicalein)], which presents an IC50 of 44 ± 3 μM. Nevertheless, and corroborating our results, the activity increased substantially with the hydroxylation at the B-ring. In addition, comparing the results obtained for C4 and C5 it is possible to infer that the presence of the –OH group at the 3′-position (C4) is more relevant than at the 4′-position (C5) of the B-ring, as C4 is more active than C5. In turn, with the basic structure studied by Gao et al.Citation54 (5,6,7-trihydroxyflavone), only the 4′-hydroxylation of the B-ring enhanced the inhibitory activity of the 5,6,7-trihydroxyflavone. We can also conclude from the obtained results that the addition of hydrophobic and bulky substituents in the flavonoid structure decreases its activity, as it was possible to see in flavonoids that possess –OMe and/or –OBn groups (C8–C13).
We also tested another group of flavonoids, based on chrysin (D1) structure, in the D group. Once again, our results are in agreement with Gao et al.Citation54, since chrysin had no inhibitory effect against α-glucosidase activity. Hydroxylation at 4′-position of the B-ring of chrysin (D1), giving apigenin (D5), increased the activity reaching an IC50 = 82 ± 6 µM. With this basic structure, the presence of an –OH group at 3′- (D4) or 4′- (D5) positions of the B-ring seems to be indifferent for the inhibitory activity.
It has been postulated that –OH groups in the B-ring are favourable for the inhibitory activity of the compoundsCitation16,Citation31,Citation32. Zeng et al.Citation24 compared the activity of morin and myricetin and concluded that myricetin, that has three –OH groups at the 3′-, 4′- and 5′-positions of the B-ring, was more potent than morin that has only two –OH groups at 2′- and 4′-positions. Comparing D6 (kaempferol) and D8 (quercetin) or D5 (apigenin) and D7 (luteolin), it is possible to conclude that the presence of the catechol group in the B-ring increased considerably the activity of the flavonoids. Additionally, our results indicate that D8 (quercetin) is a stronger inhibitor when compared with D9 (morin). These findings clearly demonstrate that the ortho –OH groups might be responsible for the higher inhibitory effect, which seems to promote the interaction between α-glucosidase and flavonoids, while the meta-position of –OH groups decreases the electron cloud density of band I resulting in a lower inhibitory activityCitation24,Citation32. Comparing the activity of D8 (quercetin) (IC50 = 15 ± 3 μM) and D7 (luteolin) (IC50 = 46 ± 6 μM), we can conclude that the presence of a 3-OH group in the C-ring increases the ability to inhibit α-glucosidase. This result is corroborated by Nicolle et al.Citation55 and XuCitation32 who suggested that the 3-OH in the C-ring may have the ability to make flavonoids bind into the binding pocket of yeast α-glucosidase properly to maintain a high inhibitory activity.
It is currently accepted that modification of any of the –OH groups in the A-ring diminishes the α-glucosidase inhibitory effects of flavonoidsCitation27,Citation56–58. From the D group analysis, it becomes clear that D3 (baicalein), which has –OH groups at 5-, 6- and 7-positions of A-ring, is more effective than D2 (galangin) and D1 (chrysin), with only –OH groups at 5- and 7-positions. These results are in agreement with Gao et al.Citation27, that studied parent compounds of baicalein and reported that removal of any –OH group from the 5-, 6- and 7-positions led to a dramatic loss of the inhibitory potency. Moreover, here we concluded that the 5-hydroxylation at the A-ring is important for the intended effect, since the IC50 of D6 (kaempferol) is almost three times lower than the IC50 of C5. Wang et al.Citation56 results validate this finding, as compounds that bear one acetyl group at the 5-position of the A-ring of chrysin almost induce the disappearance of the flavonoids’ inhibitory activity. It is known that hydrogen bonding is a crucial factor for the interactions between the enzyme and its substrates and for the conformation and orientation of the inhibitors in the active siteCitation8. Kumar et al.Citation57 showed that a 5-OH group of the A-ring acts as a hydrogen bond donor and consequently enhances the inhibitory activity of the flavonoid.
Our results illustrate that the presence of a 3-OH group in the C-ring is also relevant, evidenced by the lowest IC50 of D5 (apigenin) and D8 (quercetin) when compared with D6 (kaempferol) and D7 (luteolin), respectively. Accordingly, Silva et al.Citation25, also compared the effect of luteolin and quercetin and suggested that 3-hydroxylation of the C-ring increases the inhibitory activity against α-glucosidase. Furthermore, XuCitation32 gathered information about the interaction of flavonoids with α-glucosidase, analysed with Autodock, and reported that the 3′,4′-(OH)2 groups of the B-ring together with a 3-OH group in the C-ring are crucial for the inhibitory effect of flavonoids.
In the panel of flavonoids in this work, we tested a series of chlorinated flavonoids (D10–D14) for the first time. It has been postulated that under diabetic conditions, the inflammatory process is exacerbated and consequently there is an excessive production of reactive species, namely, HOClCitation59. It is currently known the reactivity of flavonoids with HOCl in order to form stable mono and dichlorinated productsCitation36. Our group has recently shown that chlorinated flavonoids were more efficient than their parent compounds in modulate human neutrophils’ activities, foreseeing a potential anti-inflammatory activityCitation36. These results raised our attention to these promising compounds, exploring here for the first time, their inhibitory effect against α-glucosidase. The chlorinated flavonoid D10 was one of the most active compounds tested in this work, presenting an IC50 similar to the one obtained for D8 (quercetin). Moreover, comparing compounds D10 and D8 (quercetin) it is possible to conclude that the substitution of a 3-OH group or a 3-Cl atom at the C-ring is almost indifferent for the inhibitory effect. In contrast, the results obtained with D18 (rutin), reveal that the addition of a larger group, in this case rutinose, at the 3-position of the C-ring, weakens the activity of the flavonoidCitation16.
Following the order of potency of the chlorinated flavonoids, it is possible to see that the second most effective compound was flavonoid D14, with –Cl atoms at 6 and 8 positions of the A-ring. However, when we have only one –Cl atom at 6 (D11) or 8 (D12) position of the A-ring, the inhibition of the enzyme decreased considerably.
We also tested a group of methoxylated compounds (D15–D17), and, corroborating the results obtained in A and C groups, the inhibitory effect on α-glucosidase was reduced by the introduction of an –OMe group, independently of the ring. According to the literature, the contribution of –OMe groups to the inhibitory effect of flavonoids depends on the basic structure of the flavonoid and/or with the type of substrate usedCitation60. Azuma et al.Citation60 indicated that the α-glucosidase inhibition was enhanced by the presence of 5-OMe groups at the A-ring, and at 3′-, 4′-positions of the B-ring, when compared with quercetin. In turn, Gao et al.Citation54 reported that, in contrast to –OH, the –OMe group substitution on the B-ring of 5,6,7-trihydroxyflavone was unfavourable for the activity.
It is known that the C2 = C3 double bond of flavonoids is crucial for their anti-inflammatory activityCitation37,Citation61,Citation62. Analysing E group and comparing the inhibitory effect with some flavonoids of D group, namely, D5 (apigenin) with E1 (naringenin), D7 (luteolin) with E2 (eriodictyol), D8 (quercetin) with E3 (taxifolin), it is clear that the lack of C2 = C3 double bond weakens the flavonoids activity. This finding is in agreement with other reports about this issueCitation31,Citation57. Xiao et al.Citation15 reported that the C2 = C3 double bond increases the π-conjugation of the bond linking the B and C rings, which favours near-planarity of the two rings. It is known that the molecules with near-planar structure easily enter the hydrophobic pockets in enzymes and subsequently can increase their inhibitory effect.
Once defined the effectiveness of the tested flavonoids, it was our aim to evaluate the type of α-glucosidase inhibition of the most active flavonoids from each group (A–E): A5, B3, C7, D8 (quercetin) and E3 (taxifolin). We also analysed the chlorinated flavonoid D10, since it was one of the most effective compounds, presenting an IC50 value similar to the one found for D8 (quercetin). Lineweaver–Burk double reciprocal plots were employed to investigate the inhibition kinetics of the tested compounds against α-glucosidase activity. As observed in , flavonoids A5 () and B3 () presented a mixed inhibition type (Km value increased and Vmax value decreased), which means that binding of the substrate or the inhibitor affects their enzyme binding affinity. In the mixed type inhibition, the inhibitor did not bind to the enzyme in the active site and both inhibitor and substrate can simultaneously be attached to the enzyme. However, the binding affinity for the substrate is decreased when the inhibitor is present. Concerning flavonoid C7, cannot clearly differentiate between a mixed and a competitive inhibition type. However, the interception of lines is close to the y axis. Moreover, the result obtained by docking calculations for flavone C7 suggested that the binding pose prediction was directed to the active site. Considering the in vitro and docking studies, the results indicate that C7 presents a competitive inhibition type. Flavonoid D8 (quercetin) () and acarbose () are also competitive inhibitors, since the Km value increased and the Vmax value remained constant. As shown in , flavonoids D10 () and E3 (taxifolin) () are non-competitive inhibitors (Km value remained constant and Vmax value decreased), which means that they can bind with equal affinity to both the free enzyme and the enzyme–substrate complex. In this inhibitory mechanism, the inhibitor binds to the enzyme at a site that is distinct from the substrate binding site (active site of the enzyme). A non-competitive inhibitor binds to an enzyme whether the substrate is at low or high concentrationsCitation63–67. To the best of our knowledge, this is the first report stating the type of inhibition for the mentioned flavonoids, except for D8 (quercetin), which was already reported as a mixed type inhibitorCitation16,Citation31,Citation32.
In what concerns acarbose, we verified a competitive inhibition type, as observed by the studies of Kim et al.Citation68 and Lopez et al.Citation69. Interestingly, Son et al.Citation70 referred that this compound has a mixed type of inhibition.
The Ki values can also be a useful tool to compare the activity of the compounds, being reflective of the binding affinity of the inhibitor for the enzyme. The tested flavonoids presented the following order of Ki values: C7 < D8 (quercetin) < A5 < D10 < B3 < E3 (taxifolin). These results show that flavonoid C7, followed by D8 (quercetin), exhibit the strongest affinity for the enzyme.
Flavones A5, D8 (quercetin) and C7 share very similar interactions with the active site and mimic interactions found between the first glucose of acarbose and the active site pocket. Moreover, the predicted mode of binding can rationalise the trends found across the series of studied flavonoids, namely the importance of 3-hydroxylation in the C-ring and the catechol group in the B-ring. The 3-OH substitution provides a hydrogen bond to Asp349, while the catechol in the B-ring hydrogen bonds Asp68, Asp214 and Glu276. Similarly, the inhibitor acarbose presents hydrogen bond interactions with the same active site amino acids.
Conclusions
Our results have shown that adequately substituted flavonoids are effective α-glucosidase inhibitors. The flavonoid structure, the position and number of OH groups are determinant factors for the intended effect. The most active flavonoids were C7 and D8 (quercetin), which indicates that hydoxylations at 5- and 7- or 8-positions of the A-ring, at 3′ and 4′-positions of the B-ring and at 3-position of the C-ring, as well as the C2 = C3 double bond in the C-ring, are critical for the inhibitory activity of flavonoids (). The results obtained in this study provide a series of potentially effective flavonoids that can be used as alternatives to the commonly administrated α-glucosidase inhibitors in the DM therapeutics.
Figure 4. Potential flavonoids ‘substitution pattern that contributes to increase the α-glucosidase inhibition.

Supporting information
Download PDF (850.9 KB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- International Diabetes Federation. IDF Diabetes Atlas, 2015. Available from: http://www.diabetesatlas.org/.
- Salsali A, Nathan M. A review of types 1 and 2 diabetes mellitus and their treatment with insulin. Am J Ther 2006;13:349–61.
- Wilke T, Boettger B, Berg B, et al. Epidemiology of urinary tract infections in type 2 diabetes mellitus patients: an analysis based on a large sample of 456,586 German T2DM patients. J Diabetes Complicat 2015;29:1015–23.
- Olokoba AB, Obateru OA, Olokoba LB. Type 2 diabetes mellitus: a review of current trends. Oman Med J 2012;27:269–73.
- Muller LM, Gorter KJ, Hak E, et al. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin Infect Dis 2005;41:281–8.
- Garg SK, Maurer H, Reed K, Selagamsetty R. Diabetes and cancer: two diseases with obesity as a common risk factor. Diabetes Obes Metab 2014;16:97–110.
- Taylor SI, Accili D, Imai Y. Insulin resistance or insulin deficiency. Which is the primary cause of NIDDM? Diabetes 1994;43:735–40.
- Cheng N, Yi WB, Wang QQ, et al. Synthesis and α-glucosidase inhibitory activity of chrysin, diosmetin, apigenin, and luteolin derivatives. Chin Chem Lett 2014;25:1094–8.
- Pereira DF, Cazarolli LH, Lavado C, et al. Effects of flavonoids on α-glucosidase activity: potential targets for glucose homeostasis. Nutrition 2011;27:1161–7.
- Kawser HM, Abdal DA, Han J, et al. Molecular mechanisms of the anti-obesity and anti-diabetic properties of flavonoids. Int J Mol Sci 2016;17:569–132.
- Sudhir R, Mohan V. Postprandial hyperglycemia in patients with type 2 diabetes mellitus. Treat Endocrinol 2002;1:105–16.
- Leiter LA, Ceriello A, Davidson JA, et al. Postprandial glucose regulation: new data and new implications. Clin Ther 2005;27(Suppl B):S42–S56.
- Ceriello A, Davidson J, Hanefeld M, et al. Postprandial hyperglycaemia and cardiovascular complications of diabetes: an update. Nutr Metab Cardiovasc Dis 2006;16:453–6.
- Ceriello A, Genovese S. Atherogenicity of postprandial hyperglycemia and lipotoxicity. Rev Endocr Metab Disord 2016;17:111–16.
- Xiao JB, Hogger P. Dietary polyphenols and type 2 diabetes: current insights and future perspectives. Curr Med Chem 2015;22:23–38.
- Li YQ, Zhou FC, Gao F, et al. Comparative evaluation of quercetin, isoquercetin and rutin as inhibitors of alpha-glucosidase. J Agric Food Chem 2009;57:11463–8.
- Zeng L, Zhang G, Liao Y, Gong D. Inhibitory mechanism of morin on α-glucosidase and its anti-glycation properties. Food Funct 2016;7:3953–63.
- Hakamata W, Kurihara M, Okuda H, et al. Design and screening strategies for alpha-glucosidase inhibitors based on enzymological information. Curr Top Med Chem 2009;9:3–12.
- Priscilla DH, Roy D, Suresh A, et al. Naringenin inhibits α-glucosidase activity: a promising strategy for the regulation of postprandial hyperglycemia in high fat diet fed streptozotocin induced diabetic rats. Chem Biol Interact 2014;210:77–85.
- Joshi SR, Standl E, Tong N, et al. Therapeutic potential of α-glucosidase inhibitors in type 2 diabetes mellitus: an evidence-based review. Expert Opin Pharmacother 2015;16:1959–81.
- Derosa G, Maffioli P. α-Glucosidase inhibitors and their use in clinical practice. Arch Med Sci 2012;8:899–906.
- Lebovitz HE. Alpha-glucosidase inhibitors. Endocrinol Metab Clin North Am 1997;26:539–51.
- Zhang L, Chen Q, Li L, et al. Alpha-glucosidase inhibitors and hepatotoxicity in type 2 diabetes: a systematic review and meta-analysis. Sci Rep 2016;6:32649–18.
- Zeng L, Zhang G, Lin S, Gong D. Inhibitory mechanism of apigenin on α-glucosidase and synergy analysis of flavonoids. J Agric Food Chem 2016;64:6939–49.
- Silva EL, Lobo JF, Vinther JM, et al. High-resolution α-glucosidase inhibition profiling combined with HPLC-HRMS-SPE-NMR for identification of antidiabetic compounds in Eremanthus crotonoides (Asteraceae). Molecules 2016;21:782.
- Yan J, Zhang G, Pan J, Wang Y. α-Glucosidase inhibition by luteolin: kinetics, interaction and molecular docking. Int J Biol Macromol 2014;64:213–23.
- Gao H, Nishioka T, Kawabata J, Kasai T. Structure-activity relationships for alpha-glucosidase inhibition of baicalein, 5,6,7-trihydroxyflavone: the effect of A-ring substitution. Biosci Biotechnol Biochem 2004;68:369–75.
- Na B, Nguyen PH, Zhao BT, et al. Protein tyrosine phosphatase 1B (PTP1B) inhibitory activity and glucosidase inhibitory activity of compounds isolated from Agrimonia pilosa. Pharm Biol 2016;54:474–80.
- Kim JS, Kwon CS, Son KH. Inhibition of alpha-glucosidase and amylase by luteolin, a flavonoid. Biosci Biotechnol Biochem 2000;64:2458–61.
- Kang S, Park JY, Choi H, Kim J. α-Glucosidase inhibitory activities of myricetin in animal models of diabetes mellitus. Food Sci Biotechnol 2015;24:1897–900.
- Tadera K, Minami Y, Takamatsu K, Matsuoka T. Inhibition of alpha-glucosidase and alpha-amylase by flavonoids. J Nutr Sci Vitaminol 2006;52:149–53.
- Xu H. Inhibition kinetics of flavonoids on yeast α-glucosidase merged with docking simulations. Protein Pept Lett 2010;17:1270–9.
- Abdullah NH, Salim F, Ahmad R. Chemical constituents of Malaysian U. cordata var. ferruginea and their in vitro α-glucosidase inhibitory activities. Molecules 2016;21:525.
- Meng Y, Su A, Yuan S, et al. Evaluation of total flavonoids, myricetin, and quercetin from Hovenia dulcis Thunb. as inhibitors of alpha-amylase and alpha-glucosidase. Plants Food Hum Nutr 2016;71:444–9.
- Sousa JLC, Proença C, Freitas M, et al. New polyhydroxylated flavon-3-ols and 3-hydroxy-2-styrylchromones: synthesis and ROS/RNS scavenging activities. Eur J Med Chem 2016;119:250–9.
- Freitas M, Ribeiro D, Tome SM, et al. Synthesis of chlorinated flavonoids with anti-inflammatory and pro-apoptotic activities in human neutrophils. Eur J Med Chem 2014;86:153–64.
- Ribeiro D, Freitas M, Tome SM, et al. Modulation of human neutrophils’ oxidative burst by flavonoids. Eur J Med Chem 2013;67:280–92.
- Das D. Biochemistry. 8th ed. Kolkata, India: Academic Publishers, 1993:89–111.
- Banerjee S. Inhibition of mackerel (Scomber scombrus) muscle lipoxygenase by green tea polyphenols. Food Res Int 2006;39:486–91.
- Nelson DL, Cox MM. Enzyme kinetics as an approach to understanding mechanism. In: Lehninger principles of biochemistry. 6th ed. New York: W. H. Freeman & Co.; 2014:P200–12.
- Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res 2000;28:235–42.
- Consortium TU. UniProt: a hub for protein information. Nucleic Acids Res 2015;43:D204–12.
- Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011;27:343–50.
- Bietz S, Urbaczek S, Schulz B, Rarey M. Protoss: a holistic approach to predict tautomers and protonation states in protein-ligand complexes. J Cheminform 2014;6:1–12.
- Case DA, Darden TA, Cheatham TE, et al. AMBER 12. University of California, San Francisco; 2012.
- Hornak V, Abel R, Okur A, et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006;65:712–25.
- Kirschner KN, Yongye AB, Tschampel SM, et al. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem 2008;29:622–55.
- Yamamoto K, Miyake H, Kusunoki M, Osaki S. Steric hindrance by 2 amino acid residues determines the substrate specificity of isomaltase from Saccharomyces cerevisiae. J Biosci Bioeng 2011;112:545–50.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61.
- O’Boyle NM, Morley C, Hutchison GR. Pybel: a Python wrapper for the OpenBabel cheminformatics toolkit. Chem Cent J 2008;2:1–7.
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity – a rapid access to atomic charges. Tetrahedron 1980;36:3219–28.
- Eldridge MD, Murray CW, Auton TR, et al. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des 1997;11:425–45.
- Jones G, Willett P, Glen RC, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–48.
- Gao H, Kawabata J. Importance of the B ring and its substitution on the α-glucosidase inhibitory activity of baicalein, 5,6,7-trihydroxyflavone. Biosci Biotechnol Biochem 2004;68:1858–64.
- Nicolle E, Souard F, Faure P, Boumendjel A. Flavonoids as promising lead compounds in type 2 diabetes mellitus: molecules of interest and structure-activity relationship. Curr Med Chem 2011;18:2661–72.
- Wang QQ, Cheng N, Yi WB, et al. Synthesis, nitric oxide release, and α-glucosidase inhibition of nitric oxide donating apigenin and chrysin derivatives. Bioorg Med Chem 2014;22:1515–21.
- Kumar V, Kumar S, Rani P. Pharmacophore modeling and 3D-QSAR studies on flavonoids as α-glucosidase inhibitors. Der Pharma Chem 2010;2:324–35.
- Gu C, Zhang H, Putri CY, Ng K. Evaluation of α-amylase and α-glucosidase inhibitory activity of flavonoids. Int J Food Nutr Sci 2015;2:1–6.
- Kaneto H, Katakami N, Matsuhisa M, Matsuoka TA. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat Inflamm 2010;2010:1–11.
- Azuma T, Kayano S, Matsumura Y, et al. Antimutagenic and α-glucosidase inhibitory effects of constituents from Kaempferia parviflora. Food Chem 2011;125:471–5.
- Ribeiro D, Freitas M, Lima JL, Fernandes E. Proinflammatory pathways: the modulation by flavonoids. Med Res Rev 2015;35:877–936.
- Ribeiro D, Freitas M, Tome SM, et al. Flavonoids inhibit COX-1 and COX-2 enzymes and cytokine/chemokine production in human whole blood. Inflammation 2015;38:858–70.
- Ault A. An introduction to enzyme kinetics. J Chem Educ 1974;51:381–6.
- Hüttl R, Frank N. Enzymatic kinetic determinations. In: Encyclopedia of analytical chemistry. New York: John Wiley & Sons, Ltd; 2006.
- Kakkar T, Pak Y, Mayersohn M. Evaluation of a minimal experimental design for determination of enzyme kinetic parameters and inhibition mechanism. J Pharmacol Exp Ther 2000;293:861–9.
- Ochs RS. Understanding Enzyme Inhibition. J Chem Educ 2000;77:1453–6.
- Waldrop GL. A qualitative approach to enzyme inhibition. Biochem Mol Biol Educ 2009;37:11–15.
- Kim M, Lee S, Lee H, et al. Comparative study of the inhibition of α-glucosidase, α-amylase, and cyclomaltodextrin glucanosyltransferase by acarbose, isoacarbose, and acarviosine–glucose. Arch Biochem Biophys 1999;371:277–83.
- Lopez D, Cherigo L, Spadafora C, et al. Phytochemical composition, antiparasitic and α-glucosidase inhibition activities from Pelliciera rhizophorae. Chem Cent J 2015;9:1–11.
- Son HU, Lee SH. Comparison of α-glucosidase inhibition by Cudrania tricuspidata according to harvesting time. Biomed Rep 2013;1:624–8.