
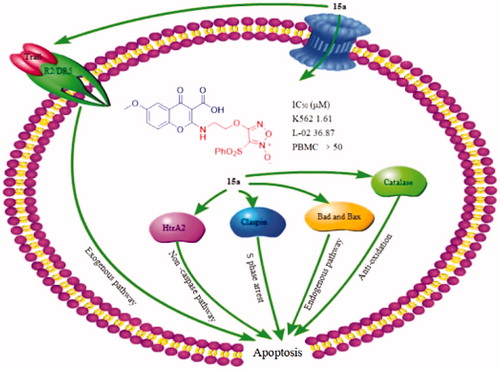
Abstract
A series of furoxan derivatives of chromone were prepared. The antiproliferative activities were tested against five cancer cell lines HepG2, MCF-7, HCT-116, B16, and K562, and two normal human cell lines L-02 and PBMCs. Among them, compound 15a exhibited the most potent antiproliferative activity. It was also found 15a produced more than 8 µM of NO at the peak time of 45 min by Griess assay. Generally, antiproliferative activity is positively related to NO release to some extent. Further in-depth studies on apoptosis-related mechanisms showed that 15a caused S-phase cell cycle arrest in a concentration-dependent manner and induced apoptosis significantly through mitochondria-related pathways. Human apoptosis protein array assay also demonstrated 15a increased the expression levels of pro-apoptotic Bax, Bad, HtrA2 and Trail R2/DR5. The expression of catalase and cell cycle blocker claspin were similarly up-regulated. In balance, 15a induced K562 cells death through both endogenous and exogenous pathways.
Graphical Abstract
1. Introduction
From ancient times, various natural products have been used as traditional medicines and are rich sources of bioactive compoundsCitation1,Citation2. Chromones (4H-chomen-4-one, 4H-1-benzopyran-4-one) are widely distributed oxygen-containing natural heterocyclic compounds from the plants of Polygonaceae, Umbelliferae, Sterculiaceae, Rhamnaceae, Liliaceae, Asteraceae etc., with a benzoylated γ-pyrone ring which is a part of the structure of flavonoid skeletonsCitation3–5. It is recognised as a privileged structure and a useful template for the design of novel compounds with potential pharmacological interest, particularly in the field of neurodegenerativeCitation6,Citation7, inflammatoryCitation8,Citation9, biocidalCitation10, immune-stimulatoryCitation11, infectious diseasesCitation12–14, as well as diabetesCitation15 and cancerCitation16–21. With respect to antitumor activity, chromones demonstrate toxicity against many kinds of tumour cells, including cervical epithelioid carcinoma, breast adenocarcinoma, hepatoma carcinoma, lung cancer, leukaemia, colon cancer and so on. The antiproliferative mechanisms involve cytotoxicity, anti-metastasis, anti-angiogenesis, chemoprevention, immunomodulation and so forthCitation22–27.
Chromone has emerged as one of the most important synthetic scaffolds for their antitumor activity. Chromone derivative LY294002 (2-morpholino-8-phenyl-4H-chromen-4-one, ) is a synthetic protein kinase inhibitor through the blockage of phosphatidylinositol-3-kinase (PI3K) signalling pathwayCitation26,Citation28,Citation29. Flavopiridol (2-(2-chlorophenyl)-5,7-dihydroxy-8-((3R,4S)-3-hydroxy-1-methylpiperidin-4-yl)-4H-chromen-4-one, ) is identified as the first cyclin-dependent kinase (CDK) inhibitor which blocks cell cycle progression and induces apoptotic cell death. It has entered Phase II clinical trialsCitation30,Citation31.
Figure 1. The chemical structures of reported chromone and furoxan derivatives. (A,B) Chromone derivatives; (C–E) furoxan-based NO donor derivatives.
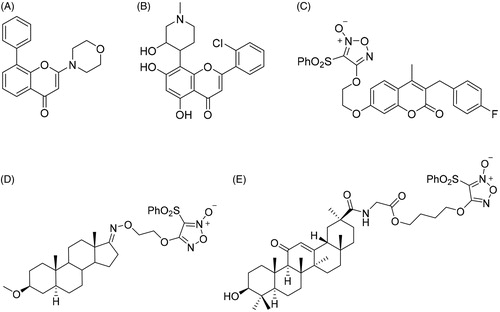
Nitric oxide (NO) acts as an important biological signalling molecule in a large variety of physiological processes, including neurotransmission, blood pressure regulation, defence mechanisms, smooth muscle relaxation and tumour growth inhibitionCitation32–34. Over the past decade, major advances of NO in cancer pathogenesis have been witnessed, suggesting an exciting future in the medical fieldCitation35,Citation36. Moreover, codelivery of NO with chemotherapeutic drugs enhances the suppression of tumour growthCitation37–40, and it is also discovered to enhance the efficacy of other treatments, such as photodynamic therapyCitation41–43, radiotherapyCitation44,Citation45 and ultrasound therapyCitation46,Citation47. However, NO has a half-life of only a few seconds in an aqueous environment, so NO release in specific targets is still a crucial challenge for antitumor therapyCitation48,Citation49. A wide range of NO donors have emerged as potential therapeutics to exploit the biological roles of NOCitation50–55. Some NO donors produce high levels of NO with a wide range of halflives in vitro and in vivo, and have been widely used in drug research, especially the type of furoxan ()Citation56–58.
In this work, 16 chromone derivatives (12a–d, 13a–d, 14a–d and 15a–d) with NO-releasing furoxan moiety were synthesised through different linkers. The linkers between the drug and NO donor influence NO releasing abilityCitation56. The antiproliferative activities against human tumour and normal cells were evaluated. Furthermore, in-depth apoptosis-related mechanisms of the most potent compound 15a, including cell cycle progression, induction of apoptosis, changes of mitochondrial membrane potential and the expression of apoptosis-related proteins, were also explored.
2. Experimental
2.1. Chemistry
All starting materials and solvents were purchased from commercial suppliers and used without purification, unless otherwise noted. 1H and 13C NMR spectra were measured on Bruker AV400 spectrometers with tetramethylsilane (TMS) as the internal standard. Chemical shifts were reported in δ (ppm). High-resolution mass spectra were obtained on HClASS XEVOG2XSQTof in the ESI mode (HR-ESI-MS). All the spectra were in Supplemental data.
2.1.1. General procedures for the synthesis of compounds 12a–d, 13a–d, 14a–d and 15a–d
A mixture of 10 or 11 (0.5 mmol) and HOBt (0.6 mmol) in anhydrous DMF (5 ml) was stirred at room temperature for 0.5 h. After the addition of 0.75 mmol 4-(2-aminoethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole-2-oxide (5a), 4-(3-aminopropoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole-2-oxide (5b), 4-((1-aminopropan-2-yl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole-2-oxide (5c) or 3-(phenyl-sulfonyl)-4-(2-(piperazin-1-yl)ethoxy)-1,2,5-oxadiazole-2-oxide (5d) and EDCI (0.75 mmol) to the solution, the mixture was further stirred at room temperature for 3 h. Then, poured into 20 ml of H2O and extracted with EtOAc (3 × 20 ml). The organic layers were combined, washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography eluting with dichloromethane/methanol system.
2.1.2 (2-(6-Methyl-4-oxo-4H-chromene-3-carboxamido)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (12a)
Yield: 52.6%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.45 (t, 1H, J = 6.1 Hz, –NH–), 9.05 (s, 1H, 3-ArH), 8.50 (s, 1H, 2′-ArH), 8.06–8.05 (d, 2H, J = 7.2 Hz, 3′,5′-ArH), 7.80 (s, 1H, 6′-ArH), 7.86–7.83 (t, 1H, J = 7.3 Hz, 4′-ArH), 7.77–7.74 (dd, 1H, J = 8.7, 1.9 Hz, 6-ArH), 7.72–7.68 (m, 2H, 5,8-ArH), 4.59–4.56 (t, 2H, J = 5.3 Hz, CH2–O), 3.83–3.79 (q, 2H, J = 11.0, 5.6 Hz, CH2–N–), 2.49 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 176.7, 163.3, 163.1, 159.3, 154.4, 137.7, 136.9, 136.8, 136.6, 130.4, 128.9, 125.0, 123.8, 119.0, 115.5, 111.1, 70.5, 37.8, 21.0; HRMS (ESI) m/z calcd for C21H16N3O8S [M−H]− 470.0658, found 470.0667.
2.1.3 4-(2-((3-Carboxy-6-methyl-4-oxo-4H-chromen-2-yl)amino)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (12b)
Yield: 47.6%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.76 (s, 1H, –COOH), 8.62 (s, 1H, –NH–), 8.00–7.98 (d, 2H, J = 7.6 Hz, 2′,6′-ArH), 7.85–7.81 (t, 1H, J = 7.5 Hz, 4′-ArH), 7.77 (s, 1H, 5-ArH), 7.66–7.62 (t, 2H, J = 8.2 Hz, 3′,5′-ArH), 7.51–7.48 (dd, 1H, J = 8.4, 1.9 Hz, 6-ArH), 7.24–7.22 (s, 1H, 8-ArH), 4.69–4.67 (t, 2H, J = 4.6 Hz, CH2-O), 4.08–4.06 (t, 2H, J = 4.5 Hz, CH2–O), 2.38 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 180.2, 164.1, 159.2, 153.0, 137.8, 136.6, 135.8, 133.8, 130.4, 128.8, 128.1, 126.0, 125.4, 117.3, 111.6, 96.8, 70.2, 49.2, 20.4; HRMS (ESI) m/z calcd for C22H18N3O8S [M−H]− 484.0815, found 484.0803.
2.1.4 4-(3-(6-Methyl-4-oxo-4H-chromene-3-carboxamido)propoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (12c)
Yield: 48.8%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.27–9.24 (t, 1H, J = 5.8 Hz, –NH–), 9.03 (s, 1H, 3-ArH), 8.06–8.04 (d, 2H, J = 7.4 Hz, 2′,6′-ArH), 7.94 (s, 1H, 8-ArH), 7.91–7.87 (t, 1H, J = 7.5 Hz, 4′-ArH), 7.77–7.75 (m, 2H, 3′,5′-ArH), 7.73–7.71 (dd, 1H, J = 6.4, 1.7 Hz, 6-ArH), 7.69–7.67 (d, 1H, J = 8.5 Hz, 5-ArH), 4.49–4.46 (t, 2H, J = 6.0 Hz, CH2–O), 3.51–3.46 (q, 2H, J = 12.7, 6.48 Hz, –CH2–), 2.46 (s, 3H, –CH3), 2.08–2.03 (m, 2H, CH2–N); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 176.7, 163.0, 162.8, 159.4, 154.4, 137.7, 136.7, 136.6, 130.5, 128.9, 125.1, 123.8, 118.9, 115.9, 111.0, 69.9, 35.7, 28.8, 21.0; HRMS (ESI) m/z calcd for C22H18N3O8S [M−H]− 484.0815, found 484.0803.
2.1.5 4-(3-((3-Carboxy-6-methyl-4-oxo-4H-chromen-2-yl)amino)propoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (12d)
Yield: 37.5%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.62 (s, 1H, –COOH), 8.60–8.43 (m, 1H, –NH–), 8.06–8.04 (d, 2H, J = 7.9 Hz, 2′,6′-ArH), 7.94–7.88 (m, 1H, 4′-ArH), 7.77–7.69 (m, 3H, 3′,5′,5-ArH), 7.47–7.45 (m, 1H, 8-ArH), 7.21–7.17 (t, 1H, J = 9.6 Hz, 6-ArH), 4.50–4.46 (m, 2H, CH2–O), 3.74–3.69 (q, 2H, J = 12.7, 6.2 Hz, –CH2–), 2.36 (s, 3H, –CH3), 2.20–2.17 (m, 2H, CH2–N); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.8, 163.3, 162.9, 161.4, 159.3, 152.8, 137.6, 136.6, 135.6, 130.5, 129.0, 125.3, 120.5, 117.2, 111.1, 96.4, 69.1, 47.6, 29.2, 20.8; HRMS (ESI) m/z calcd for C25H23N4O8S [M−H]− 539.1237, found 539.1229.
2.1.6 4-(2-(6-Methoxy-4-oxo-4H-chromene-3-carboxamido)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (13a)
Yield: 45.3%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.48–9.45 (t, 1H, J = 5.7 Hz –NH–), 9.05 (s, 1H, 3-ArH), 8.07–8.05 (d, 2H, J = 7.5 Hz, 2′,6′-ArH), 7.86–7.82 (t, 1H, J = 7.5 Hz, 4′-ArH), 7.79–7.76 (d, 1H, J = 8.9 Hz, 5-ArH), 7.71–7.67 (t, 2H, J = 8.0 Hz, 3′,5′-ArH), 7.55–7.53 (dd, 1H, J = 4.4, 2.9 Hz, 6-ArH), 7.51–7.50 (d, 1H, J = 3.1 Hz, 8-ArH), 4.59–4.56 (t, 2H, J = 5.1 Hz, CH2–O), 3.89 (s, 3H, –OCH3), 3.84–3.80 (q, 2H, J = 10.7, 5.4 Hz, CH2–N); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 176.4, 163.1, 159.3, 157.8, 150.9, 137.7, 136.5, 130.4, 128.8, 124.9, 124.7, 120.9, 114.9, 111.1, 105.7, 70.6, 56.4, 37.8; HRMS (ESI) m/z calcd for C21H16N3O9S [M−H]− 486.0607, found 486.0609.
2.1.7 4-(2-((3-Carboxy-6-methoxy-4-oxo-4H-chromen-2-yl)amino)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (13b)
Yield: 35.7%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.74–11.71 (t, 1H, J = 6.6 Hz, –COOH), 8.73–8.57 (q, 1H, –NH–), 8.01–7.96 (m, 1H, 2′,6′-ArH), 7.85–7.80 (m, 1H, 4′-ArH), 7.66–7.61 (m, 2H, 3′,5′-ArH), 7.41–7.38 (m, 1H, 5-ArH), 7.30–7.28 (m, 2H, 6,8-ArH), 4.69–4.68 (m, 2H, CH2–O), 4.09–4.08 (m, 2H, CH2–N), 3.83 (s, 3H, –OCH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.8, 177.5, 164.1, 159.1, 156.1, 155.9, 149.0, 137.7, 136.5, 130.4, 128.8, 122.3, 121.2, 118.8, 111.1, 107.6, 107.5, 70.1, 56.1, 40.6, 40.4, 40.2, 40.0, 39.8, 39.6, 39.4; HRMS (ESI) m/z calcd for C22H18N3O9S [M–H]− 500.0764, found 500.0761.
2.1.8 4-(3-(6-Methoxy-4-oxo-4H-chromene-3-carboxamido)propoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (13c)
Yield: 33.9%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.28–9.25 (t, 1H, J = 5.7 Hz, –NH–), 9.03 (s, 1H, 3-ArH), 8.05–8.04 (d, 2H, J = 7.5 Hz, 2′,6′-ArH), 7.90–7.87 (t, 1H, J = 7.5 Hz, 4′-ArH), 7.77 (s, 1H, 5-ArH), 7.74–7.73 (m, 2H, 3′,5′-ArH), 7.51–7.49 (m, 2H, 6, 8-ArH), 4.50–4.47 (t, 2H, J = 6.0 Hz, CH2–O), 3.89 (s, 3H, –OCH3), 3.51–3.47 (m, 2H, CH2–N), 2.10–2.03 (m, 2H, –CH2–); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 176.4, 162.8, 159.4, 157.7, 150.9, 137.6, 136.6, 130.5, 128.9, 124.9, 124.6, 120.8, 115.2, 111.0, 105.8, 69.9, 56.3, 35.8, 28.7; HRMS (ESI) m/z calcd for C22H18N3O9S [M−H]− 500.0764, found 500.0782.
2.1.9 4-(3-((3-Carboxy-6-methoxy-4-oxo-4H-chromen-2-yl)amino)propoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (13d)
Yield: 56.6%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.61 (s, 1H, –COOH), 8.50 (s, 1H, –NH–), 8.06–8.04 (d, 2H, J = 7.5 Hz, 2′,6′-ArH), 7.92–7.89 (t, 1H, J = 7.4 Hz, 4′-ArH), 7.78–7.74 (t, 2H, J = 7.8 Hz, 3′,5′-ArH), 7.35 (s, 1H, 5-ArH), 7.21 (s, 2H, 6,8-ArH), 4.48–4.45 (t, 2H, J = 5.7 Hz, CH2–O), 3.80 (s, 3H, –OCH3), 3.17–3.16 (d, 2H, J = 2.8 Hz, CH2–N), 2.18–2.15 (m, 2H, –CH2–); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.3, 155.9, 148.9, 137.6, 136.6, 130.5, 129.0, 126.0, 118.8, 111.1, 107.6, 96.4, 69.2, 56.1, 47.8, 29.2; HRMS (ESI) m/z calcd for C25H23N4O9S [M−H]− 555.1186, found 555.1173.
2.1.10 2-((1-(6-Methyl-4-oxo-4H-chromene-3-carboxamido)propan-2-yl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (14a)
Yield: 28.1%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.43–9.40 (t, 1H, J = 5.5 Hz, –NH–), 9.04–9.03 (d, 1H, J = 6.5 Hz, 3-ArH), 8.07–8.05 (d, 2H, J = 7.4 Hz, 2′,6′-ArH), 7.97–7.96 (d, 1H, J = 5.6 Hz, 8-ArH), 7.86–7.82 (t, 1H, J = 7.4 Hz, 4′-ArH), 7.76–7.74 (dd, 1H, J = 8.7 Hz, 6-ArH), 7.72–7.67 (m, 3H, 3′,5′, 5-ArH), 5.18–5.14 (m, 1H, –CH–), 3.86–3.80, 3.65–3.59 (m, 2H, –CH2–N–), 2.48 (s, 3H, ArH-CH3), 1.40–1.39 (d, 3H, J = 6.3 Hz, –CH–CH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 176.8, 163.3, 163.2, 158.8, 154.4, 137.6, 136.9, 136.5, 130.4, 128.9, 126.0, 125.0, 123.7, 119.0, 115.5, 78.5, 42.6, 21.0, 17.2; HRMS (ESI) m/z calcd for C21H16N3O9S [M–H]− 486.0607, found 486.0609.
2.1.11 4-((1-((3-Carboxy-6-methyl-4-oxo-4H-chromen-2-yl)amino)propan-2-yl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (14b)
Yield: 35.6%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.72 (s, 1H, –COOH), 8.58 (s, 1H, –NH–), 8.00–7.99 (d, 2H, J = 7.2 Hz, 2′,6′-ArH), 7.84–7.80 (t, 1H, J = 7.2 Hz, 4′-ArH), 7.74 (s, 1H, 5-ArH), 7.65–7.61 (t, 2H, J = 7.4 Hz, 3′,5′-ArH), 7.48–7.47 (d, 1H, J = 7.3 Hz, 8-ArH), 7.21–7.19 (d, 1H, J = 8.9 Hz, 6-ArH), 5.26–5.24 (m, 1H, CH–O), 4.11–4.00 (m, 3H, –CH2–), 2.37 (s, 3H, Ar–CH3), 1.40–1.39 (d, 3H, CH-CH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 180.6, 164.2, 163.7, 160.1, 153.6, 138.4, 137.4, 136.4, 134.4, 131.3, 129.8, 126.1, 121.3, 118.0, 111.9, 97.2, 69.9, 48.4, 30.0, 21.6; HRMS (ESI) m/z calcd for C22H18N3O9S [M–H]− 500.0764, found 500.0783.
2.1.12 4-(2-(4-(6-Methyl-4-oxo-4H-chromene-3-carbonyl)piperazin-1-yl)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (14c)
Yield: 29.7%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.51 (s, 1H, 3-ArH), 8.03–8.01 (d, 2H, J = 7.4 Hz, 2′,6′-ArH), 7.90–7.86 (t, 2H, J = 7.4 Hz, 4′,6-ArH), 7.76–7.72 (t, 2H, J = 8.0 Hz, 3′,5′-ArH), 7.69–7.66 (dd, 1H, J = 8.6, 1.9 Hz, 5-ArH), 7.62–7.60 (d, 1H, J = 8.6 Hz, 8-ArH), 4.54–4.51 (t, 2H, J = 5.0 Hz, –CH2–O), 3.58–3.56 (t, 2H, J = 4.9 Hz, CON–CH2), 3.27–3.24 (t, 2H, J = 5.4 Hz, CON–CH2), 2.81–2.79 (t, 2H, J = 5.1 Hz, –N–CH2–), 2.55–2.53 (t, 2H, J = 5.2 Hz, –CH2–N–C), 2.48–2.45 (t, 2H, J = 5.0 Hz, –CH2–N–C), 2.44 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 173.8, 162.5, 159.4, 156.5, 154.5, 137,8, 136.6, 136.2, 136.1, 130.5, 128.7, 127.8, 125.0, 123.8, 122.6, 118.9, 69.7, 55.9, 53.4, 52.8, 47.1, 41.9, 20.9; HRMS (ESI) m/z calcd for C22H18N3O9S [M–H]− 500.0764, found 500.0776.
2.1.13 4-(2-(4-(3-Carboxy-6-methyl-4-oxo-4H-chromen-2-yl)piperazin-1-yl)ethoxy)-3-(phenlsulfonyl)-1,2,5-oxadiazole 2-oxide (14d)
Yield: 27.6%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 14.1 (s, 1H, –COOH), 8.03 (m, 2H, 2′,6′-ArH), 7.92–7.89 (m, 1H, 4′-ArH), 7.86–7.83 (d, 1H, J = 9.0 Hz, 5-ArH), 7.79–7.75 (m, 3H, 3′,5′,8-ArH), 7.19–7.17 (dd, 1H, J = 8.3 Hz, 6-ArH), 4.56–4.54 (t, 1H, J = 5.1 Hz, CH–O), 3.54–3.52 (t, 3H, J = 4.6 Hz, CH–O, Ar–N–CH2), 3.17 (s, 2H, N–CH2), 2.86–2.84 (t, 2H, J = 5.2 Hz, N-CH2), 2.62–2.60 (t, 4H, J = 4.1 Hz, –CH2–N–CH2–), 2.25 (s, 3H, –CH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 191.2, 160.7, 154.3, 137.8, 136.6, 135.1, 130.6, 129.2, 128.7, 128.1, 127.8, 126.0, 120.2, 117.7, 111.0, 89.8, 70.3, 69.9, 55.7, 53.5, 49.1, 29.5, 20.6; HRMS (ESI) m/z calcd for C25H23N4O9S [M–H]− 555.1186, found 555.1171.
2.1.14 4-((1-(6-Methoxy-4-oxo-4H-chromene-3-carboxamido)propan-2-yl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (15a)
Yield: 29.5%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.45–9.42 (t, 1H, J = 11.6 Hz, –NH–), 9.04 (s, 1H, 3-ArH), 8.07–8.06 (d, 2H, J = 7.4 Hz, 2′,6′-ArH), 7.86–7.82 (t, 1H, J = 7.5 Hz, 4′-ArH), 7.79–7.77 (m, 1H, 5-ArH), 7.71–7.67 (t, 2H, J = 8.0 Hz, 3′,5′-ArH), 7.54–7.52 (q, 2H, J = 7.8, 3.2 Hz, 6,8-ArH), 5.18–5.14 (m, 1H, CH–O), 4.54–4.52 (m, 1H, –CH–N–), 3.90 (s, 3H, Ar–CH3), 3.66–3.59 (m, 1H, –CH–N–), 1.41–1.40 (d, 3H, CH–CH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 176.5, 163.2, 163.1, 158.8, 157.8, 150.9, 137.6, 136.5, 130.4, 128.9, 124.8, 120.9, 114.9, 111.1, 105.6, 78.5, 56.4, 42.6, 17.2; HRMS (ESI) m/z calcd for C21H16N3O10S [M–H]− 502.0556, found 502.0544.
2.1.15 4-((1-((3-Carboxy-6-methoxy-4-oxo-4H-chromen-2-yl)amino)propan-2-yl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (15b)
Yield: 22.1%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.7 (s, 1H, –COOH), 8.65 (s, 1H, –NH–), 8.00 (m, 2H, 2′,6′-ArH), 7.84–7.81 (t, 1H, J = 7.2 Hz, 4′-ArH), 7.65–7.63 (t, 2H, J = 7.5 Hz, 3′,5′-ArH), 7.37 (s, 1H, 5-ArH), 7.28 (s, 2H, 6,8-ArH), 5.28–5.21 (m, 1H, CH–O), 4.06–3.82 (m, 2H, –CH2–), 3.82 (s, 3H, Ar-CH3), 1.41–1.39 (d, 3H, CH–CH3); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 179.9 164.3, 163.2, 158.6, 155.9, 149.0, 137.6, 136.6, 130.3, 128.9, 128.1, 125.9, 122.5, 121.1, 118.9, 111.1, 107.4, 78.0, 70.2, 56.1, 29.5, 16.6; HRMS (ESI) m/z calcd for C22H18N3O10S [M–H]− 516.0713, found 516.0728.
2.1.16 4-(2-(4-(6-Methoxy-4-oxo-4H-chromene-3-carbonyl)piperazin-1-yl)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (15c)
Yield: 29.9%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.52 (s, 1H, 3-ArH), 8.04–8.01 (m, 2H, 2′,6′-ArH), 7.90–7.87 (t, 1H, J = 7.5 Hz, 4′-ArH), 7.77–7.73 (t, 2H, J = 8.1 Hz, 3′,5′-ArH), 7.69–7.67 (dd, 1H, J = 7.8, 1.9 Hz, 6-ArH), 7.46–7.43 (m, 2H, 5, 8-ArH), 4.55–4.52 (t, 2H, J = 10.1 Hz, CH2–O), 3.87 (s, 3H, –OCH3), 3.60–3.57 (t, 2H, J = 4.4 Hz, –CON–CH2), 3.28–3.26 (t, 2H, J = 7.5 Hz, –CON–CH2), 2.82–2.80 (t, 2H, J = 5.0 Hz, –N–CH2), 2.56–2.54 (t, 2H, J = 5.0 Hz, –CH2–N–C), 2.49–2.47 (t, 2H, J = 4.9 Hz, –CH2–N–C); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 173.5, 162.6, 159.4, 157.3, 156.4, 151.0, 137.8, 136.6, 130.5, 128.7, 124.9, 124.2, 121.9, 120.7, 110.9, 105.4, 69.7, 56.3, 55.9, 53.4, 52.8, 47.1, 41.9; HRMS (ESI) m/z calcd for C22H18N3O10S [M–H]− 516.0713, found 516.0714.
2.1.17 4-(2-(4-(3-Carboxy-6-methoxy-4-oxo-4H-chromen-2-yl)piperazin-1-yl)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (15d)
Yield: 37.8%. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 13.9 (s, 1H, -COOH), 8.04 (m, 2H, 2′,6′-ArH), 7.92–7.87 (m, 2H, 4′,5-ArH), 7.79–7.75 (m, 3H, 3′,5′,8-ArH), 7.03–7.00 (dd, 1H, J = 8.9, 3.0 Hz, 6-ArH), 4.56–4.54 (t, 2H, J = 5.0 Hz, CH2–O), 3.75 (s, 3H, –OCH3), 3.55 (s, 4H, Ar–N–(CH2)2), 2.86–2.84 (t, 2H, J = 5.0 Hz, –N–CH2), 2.62–2.60 (t, 4H, J = 4.6 Hz, –N–(CH2)2); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 190.8, 157.2, 156.9, 154.6, 151.5, 137.8, 136.6, 130.6, 128.7, 126.0, 121.6, 120.6, 120.4, 118.6, 113.0, 105.2, 89.8, 69.9, 60.4, 56.4, 56.2, 55.7, 29.5; HRMS (ESI) m/z calcd for C25H23N4O10S [M–H]− 571.1135, found 571.1122.
2.2. MTT assay
The colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to measure the cell viability of all of the above cell lines following treatment with target hybrids. Exponentially growing cells were added into 96-well plates at a concentration of 2000–4000 cells per well. Following attachment the cells were treated with varying concentrations (64, 16, 4, 1, 0.25, 0.0625 and 0.015625 µM) of target compounds in media and were kept for incubation for 72 h. MTT (20 µL, 5 mg/mL in PBS) was added to each well and the cells were incubated for another 3 h at 37 °C. Then the medium was removed, followed by the addition of 150 µL DMSO for each well. After that, the absorbance (OD) data of each well at 570 nm wavelength were measured by a Microplate Reader (BIO-RAD), and half inhibition rates (IC50) were calculatedCitation59,Citation60.
2.3. No releasing test
The levels of NO produced by each compound were determined by colorimetric assay using a nitrite colorimetric assay kit (Beyotime, China) according to the manufacturer’s instructions. Incubated in phosphate buffer (pH 7.4) containing 2% dimethyl sulfoxide and 10−5 M test compound (1 ml of 0.2 mM solution in 0.1 M phosphate buffer, pH 7.4) with freshly prepared L-cysteine (1 ml of 3.6 mM solution in 0.1 M phosphate buffer, pH 7.4) at 37 °C for 1, 2 and 3 h without air. After exposure to air for 10 min at 25 °C, aliquots of Griess reagent I (50 µL) and Griess reagent II (50 µL) were added to an equal volume (50 µL) of the incubation solution of each test compound. After 10 min, the absorbance was measured at 540 nm. The nitrite absorbance versus concentration curve was prepared using 1 M sodium nitrite solution under the same experimental conditions. The concentration of NO formed by a single test compound was calculated using different concentrations of nitrite as a standard.
2.4. Stability of 15a
Compound 15a was dissolved in culture medium to a final concentration of 100 µM from 50 mM stock solution in DMSO. The solutions were incubated at 37 °C. An aliquot (20 µL) of the incubation mixture was taken at different time points (0, 1, 2, 3, 4, 5, 6, 8, and 12 h). And the components were analysed by HPLC equipped with a C18 reverse phase column (Shimadzu, LC-6 AD) with the flow rate of 0.5 ml/min methanol-water (60: 40–100: 0) and detection at UV 230 nm.
2.5. Cell cycle study
The cells in the logarithmic growth phase were digested into six-well plates. The next day, cells were treated with different concentrations (0, 0.8, 1.6, 3.2 µM) of compound 15a. After 72 h, 5 × 105 cells were collected and fixed with a volume fraction of 70% ethanol for 2 h (or overnight), washed the fixative with PBS, added 100 µL RNase A in 37 °C water bath for 30 min, and finally 400 µL of PI stain was added, after 30 min under the dark at 4 °C, the cytometry was used to record the red fluorescence at the excitation wavelength of 488 nm, and the cell cycle distribution of the DNA content was evaluated.
2.6. Hoechst 333258 staining
K562 cells were incubated with different concentrations (0, 0.8, 1.6 and 3.2 µM) of 15a for a period of time, the medium containing the compound was removed, the cell smear was naturally dried, and immersed in 4% paraformaldehyde for 30 min or overnight to improve cell permeability, then soaked in PBS, washed for three times, added appropriate amount of Hoechst 33258 staining solution, fully covered, allowed to stand at room temperature for 10 min, finally immersed in PBS again, washed three times and sealed. Anti-fluorescence quenching after the liquid was sealed, the staining results were observed under a fluorescence microscope.
2.7. Analysis of cellular apoptosis
The cells were placed in six-well plates and incubated at 37 °C for 24 h, after which K562 cells were treated with different concentrations (0, 0.8, 1.6, 3.2 µM) of 15a for 24 h, washed with PBS, centrifuged, and collected. 500 µL of binding buffer suspension cells, 5 µL of Annexin V-FITC, and 5 µL of PI were mixed incubated at room temperature, protected from light, and reacted for 5–15 min, then analysed with flow cytometry instrument to detect cell apoptosis.
2.8. Cell mitochondrial membrane potential assay
K562 cells were cultured for 48 h in six-well plates with different concentrations of 15a (0, 0.8, 1.6 and 3.2 µM), then washed with PBS and stained with JC-1 in the dark at room temperature. Flow cytometry was used to measure the number of cells with collapsed mitochondrial membrane potential.
2.9. Quantitation of cellular proteins involved in apoptosis
The relative expression levels of 35 apoptosis-related proteins were evaluated using Human Apoptosis Array kit (R&D Systems, Abingdon, UK) in K562 cells. Proteins were extracted according to the manufacturer’s protocol from cells treated for 24 h with compound 15a (2 µM). The tool is fast, sensitive and economical, with 2.0 ml of array buffer added to each well, followed by array, The capture antibodies are retained in their specific locations, then incubated, transferred, diluted, added to Streptavidin-HRP, shaken. The platform shaker was incubated on the plate for 30 min, then the membrane was removed, 1 ml of the prepared Chemi Reagent Mix was evenly pipetted onto each membrane, incubated, the excess Chemi Reagent Mix was removed, and the membrane was placed in a self-developing film cartridge, exposed to X-ray film for 1–10 min. The positive signals seen on developed film can be quickly identified by placing the transparency overlay template on the array image and aligning it with the pairs of reference spots in three corners of each array. Creating templates, exporting files, averaging signals, finding backgrounds, comparing corresponding signals on the array to determine relative changes in apoptosis-related protein levels.
3. Results and discussion
3.1. Chemistry
The synthetic routine of target compounds is illustrated in Scheme 1. Compound 5 was synthesised in a three-step sequence according to the literatureCitation61, and then converted to various monophenylsulfonylfuroxans (5a–d) by the treatment with corresponding amino-substituted alcohol, ethanolamine, 3-aminopropanol, 1-aminopropan-2-ol and N-(2-hydroxyethyl)piperazine (Scheme 1).
Scheme 1. Synthesis of 5a–d, 12a–d, 13a–d, 14a–b and 15a–b. Reagents and conditions: (I) ClCH2COOH, NaOH (aq), reflux, 2 h; (II) 30% H2O2, AcOH, rt, 3 h; (III) fuming HNO3, 90 °C, 4 h; (IV) aminoalcohol, THF, NaH, 0 °C, 4 h; (V) POCl3, DMF, −10 °C, 15 h; (VI) H3NSO3, NaClO2, 0 °C, 12 h; (VII) 5a–d, HOBt, EDCI, DMF, rt, 3 h.
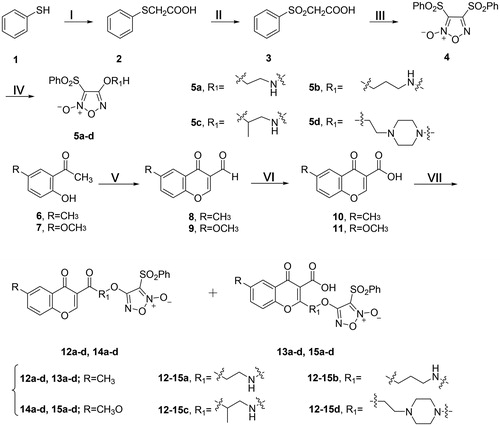
Compounds 8 and 9 were synthesised from 2-hydroxy-5-methoxyacetophenone (6) and 2-hydroxy-5-methylacetophenone (7), respectively, by using the Vilsmeier Haack reagent (POCl3 and DMF) at −10 °C. Then the aldehyde group was oxidised to carboxylic acid by sulphamic acid and sodium chloriteCitation62. Finally, the furoxan intermediates (5a–d) were reacted with the derivatives (10 and 11) of chromone to give the target compounds 12a–d plus 13a–d and 14a–d plus 15a–d, respectively (Scheme 1). The structures of all the derivatives were confirmed by 1H NMR, 13C NMR and high resolution mass spectrum (HR-MS).
3.2. Biological evaluation
3.2.1. Antiproliferative activity
Target chromone/furoxan hybrids (12a-d, 13a-d, 14a-d and 15a-d) were evaluated for their inhibitory effects against five different human cancer cell lines (hepatoma HepG2, breast carcinoma MCF-7, colorectal carcinoma HCT-116, melanoma B16 and chronic myeloid leukaemia K562), with the reference fluorouracil (5-Fu)Citation63–66. Meanwhile, the activities against the human normal hepatic L-02 cell line and peripheral blood mononuclear cells (PBMCs) were also evaluated since a potential anticancer drug candidate would be better to show selective cytotoxicity between malignant and normal cells.
As shown in , most target compounds displayed more potent inhibitory activities than corresponding chromone parent compound. For instance, the antiproliferative activities of the furoxan hybrids 12d, 14c, 14d and 15a showed IC50 values ranging from 1.61 to 15.92 µM against five cancer cell lines. Especially, they were more sensitive to K562 (IC50 1.61–4.94 µM) and HepG2 (IC50 4.86–7.91 µM) cells. Among 12a, 13a, 14a and 15a, 14a and 15a with a methoxy group at the 5-position of chromone displayed more potent antiproliferative activities against HepG2, HCT-116 and K562 cells than corresponding ones with methyl group. The results were in accord with previous literatures that bulky methoxy group was preferred for antiproliferative activityCitation67,Citation68. In addition, the presence of carboxyl groups also enhanced antiproliferaitve activity, which were more easily salified to increase water solubility. Furthermore, 12d and 14d with a nitrogen-containing heterocyclic ring showed stronger antiproliferative activities. Other compounds generally followed the above rules. The target derivatives were sensitive to K562 cell line, of which 15a displayed the most potent antiproliferative activity with an IC50 value of 1.61 µM.
Table 1. The antiproliferative effects of the target compounds against different human cancer and normal cell lines.
In addition, all the target compounds exhibited weak antiproliferative activities with IC50 values greater than 50 µM against PBMCs and above 28.90 µM against L-02 cells, which showed good selectivity between tumour and normal cells.
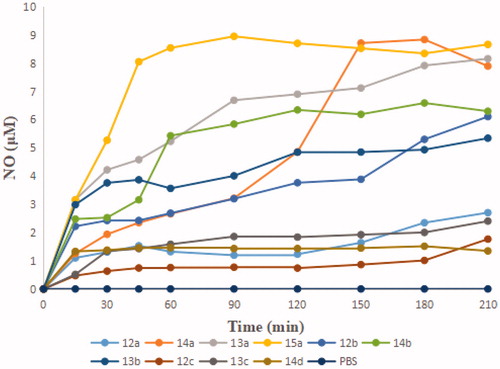
3.2.2. No releasing ability in vitro
The levels of NO released were tested by Griess assay. As shown in , the amounts were basically consistent with the potency of antiproliferative activity, such as 13a-b, 14b and 15a. In addition, the faster the amounts of NO released reached the peak, the stronger activity exhibited. 15a, which showed the strongest growth inhibitory activity, produced more than 8 µM of NO at the peak time of 45 min. Generally, the antiproliferative activity is somehow robust related to the amount of NO released.
Figure 2. The amounts of NO released by the target compounds 12a–d, 13a–d, 14a–d, and 15a–d.
3.2.3. Stability of 15a
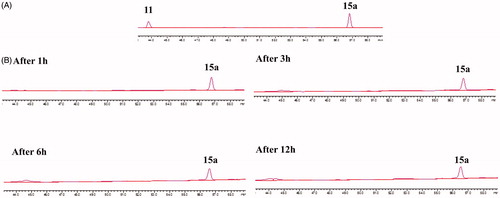
To test that if 15a was easy to hydrolyse, its stability was studied in cRPMI-1640 culture media supplemented with FBS under cell-free conditions. The results of HPLC analysis were summarised in . It was observed that 15a was relatively stable in cRPMI-1640 culture media within 12 h.
Figure 3. HPLC of (A) 11 and 15a in MeOH; (B) 15a in cell-free culture medium (cRPMI-1640) after incubation for 1, 3, 6, and 12 h.
3.2.4. Cell cycle analysis
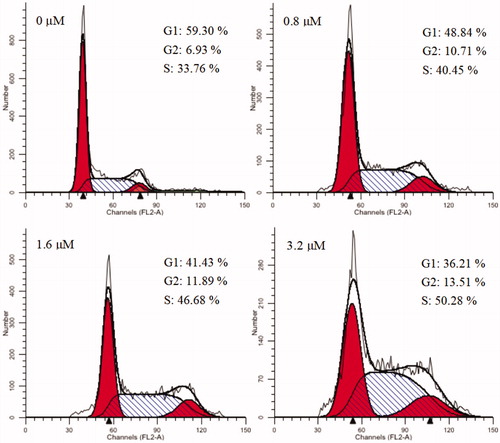
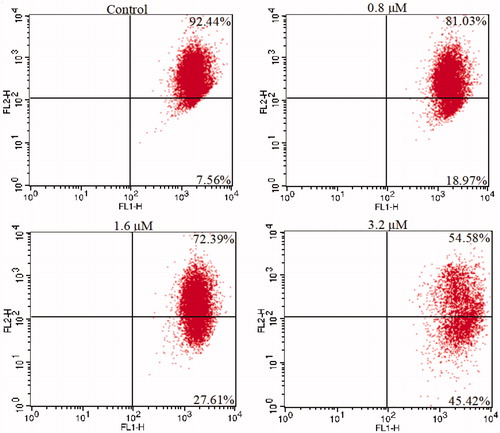
Cell cycle refers to the process that is experienced from the end of cell division to the end of the next cell division. Numerous anticancer molecules employ their impact via blocking cell cycle progression, inducing apoptosis, or the merged effects of bothCitation69–71. To verify the causal relation of cell proliferation inhibition and cell cycle arrest, K562 cells were treated with different concentrations of 15a (0, 0.8, 1.6 and 3.2 µM). Effects on cell cycle were determined by flow cytometry after propidium iodide (PI) staining. As shown in , cells in the S phase increased from 33.76% in the negative control group to 40.45, 46.68 and 50.28% in a concentration-dependent manner. These results revealed that 15a caused S phase arrest of K562 cells in a concentration-dependent manner.
Figure 4. Cell cycle analysis of 15a (0, 0.8, 1.6, and 3.2 μM) in K562 cells, cells were stained with PI and then cell cycle distribution was analysed by flow cytometry.
3.2.5. Morphological analysis by Hoechst 33258 staining
Cell apoptosis shows characteristic morphological changes, including cell shrinkage, chromatin condensation, apoptotic body formation, cytoskeletal disintegration, etc. Among them, the change of nucleus was the most significant one. Hoechst 33258, which stains the cell nucleus, is a membrane permeable dye with blue fluorescence. Live cells with uniformly light blue nuclei can be observed under fluorescence microscope after the treatment with Hoechst 33258. Apoptotic cells have bright blue nuclei on account of karyopyknosis and chromatin condensation; whereas, the nuclei of dead cells cannot be stainedCitation72.
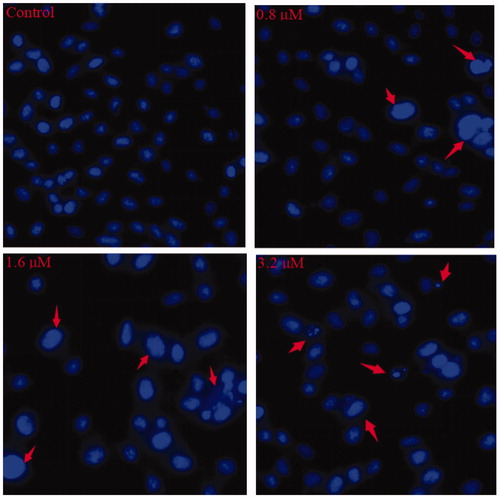
In , control cells showed no obvious morphological changes, while K562 cells exposed to 0.8 and 1.6 µM of 15a exhibited brightly blue fluorescence and revealed typical apoptotic morphology. After the treatment of 3.2 µM of 15a, the K562 cell membranes were ruptured and the nuclei were fragmented. These results strongly supported the pro-apoptotic effects of 15a.
Figure 5. Hoechst staining of 15a-treated K562 cells. The red arrows point to the cells with obvious morphological changes of apoptosis.
3.2.6. Cell apoptosis assay
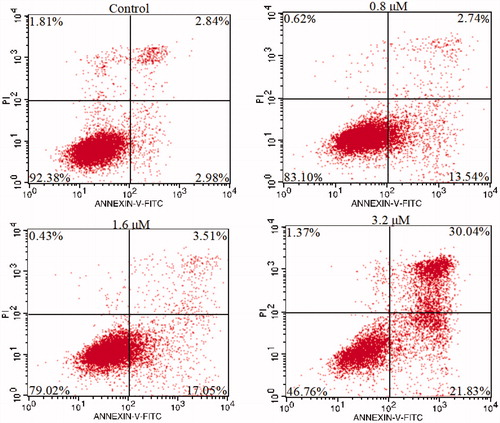
NO has been noted in cancer biology that is associated with cancer cell apoptosisCitation73. To further verify 15a induced apoptosis in K562 cells, K562 cells were treated with different concentrations (the same as the cell cycle test) of 15a for 72 h. Then, cells were harvested and stained with Annexin V-FITC and propidium iodide (PI), and the percentages of apoptotic cells were determined by flow cytometry. As shown in , the apoptotic rates of drug-treated cells were positively correlated with the concentrations. At concentrations of 0.8, 1.6 and 3.2 µM, the apoptotic rates were 16.28, 20.56 and 51.87%, respectively, compared with 5.82% in the negative control group, which confirmed that 15a induced apoptosis in K562 cells.
Figure 6. K562 cells were treated with 15a, stained with annexin V-FITC/PI and analysed by flow cytometry.
3.2.7. Mitochondria membrane potential analysis
Mitochondria are the main organelles that produce ATP and play important roles in the process of apoptosisCitation74–77. We further examined the mitochondrial membrane potentials of K562 cells treated with 15a to confirm its pro-apoptotic effects. K562 cells were treated with different concentrations of 15a for 48 h, then stained with the dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-caebocyanine (JC-1). The changes of mitochondrial membrane potentials were observed. As shown in , the depolarisation of mitochondria increased in a concentration-dependent fashion, indicating that 15a induced apoptosis through mitochondrial related pathways.
Figure 7. 15a induced mitochondrial depolarisation in K562 cells.
3.2.8. Effects on apoptosis-related protein
To further investigate the mechanism of action of 15a in K562 cells, the expression of related apoptotic proteins were tested using the Human Apoptotic Array Kit (). The apoptotic pathways of cells mainly include the mitochondrial pathway, the endoplasmic reticulum pathway and the death receptor pathway.
Figure 8. The effects exerted by 15a on the expression of apoptosis-related proteins using the Human Apoptosis Array kit in K562 cells.
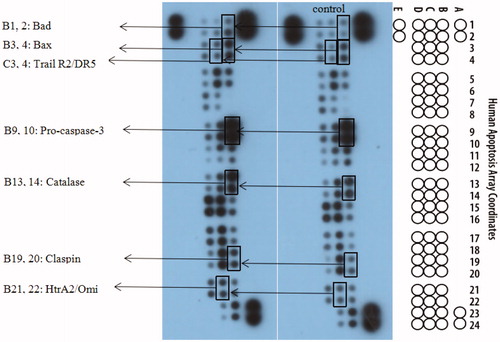
The Bcl-2 family is a major regulator of the mitochondrial apoptotic pathway. Bad and Bax are pro-apoptotic members that form homodimers with their family members, increasing mitochondrial permeability. Caspase-3 is activated by proteolytic processing of procaspase-3 and activates caspases chain, and finally causes apoptosisCitation78,Citation79. From experimental results (), increased expression of Bad and Bax was observed after the treatment with 15a compared to the control.
The death receptor pathway regulates apoptosis mainly through three pathways, including tumour necrosis factor receptor (TNFR), tumour necrosis factor-related apoptosis-inducing ligand (Trail), and factor associated suicide/ligand (Fas/FasL) pathways. The death ligand TRAIL binds to the receptor R2/DR5 on the cell surface and forms death-inducing signalling complex (DISC), thereby activates caspase 8, which in turn causes subsequent cascade reactionsCitation80,Citation81. The results also showed upregulated expression of Trail R2/DR5, which were the exogenous pathway results.
HtrA2 is a serine protease located in the mitochondria of eukaryotes. When cells are stimulated, in addition to inducing apoptosis through the mitochondrial pathway, Omi interacts with the antiapoptotic protein HAX-1 (HIS-associated protein X-1) via the caspases-independent pathway, causing HAX-1 degradation, thereby increasing the sensitivity of cells to apoptotic stimuli and inducing apoptosis. P53 also induces HtrA2 phosphorylation by regulating the actin cytoskeleton to counteract cell migration induced by Ras, thereby inhibiting tumour metastasisCitation82,Citation83. Catalase exists in all known animals and regulates the metabolism of reactive oxygen species in the body in a stable state, studies have shown that it inhibits NF-κB activation and promotes apoptosisCitation84,Citation85.
To ensure orderly progression of cell cycle, there are many quality control points known as cell cycle checkpoint proteins. Claspin, as a tumour suppressor, activates checkpoint kinase 1 (Chk1), sensitises cancer cell DNA during the S phase of the cell cycle, produces stress responses, and induces DNA damage and apoptosisCitation86–89. The expression of Claspin is increased to achieve the purpose of inhibiting the production of tumour cells.
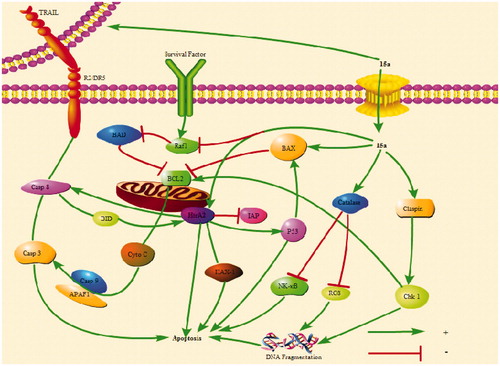
The above results indicated that 15a induced apoptosis of K562 cells by participating in both endogenous and exogenous pathways ().
Figure 9. 15a induced K562 cells death through multiple pathways. Bad and Bax participate in endogenous pathways. Trail R2/DR5 induces cell death through exogenous pathways. HtrA2 kills cells through non-caspase pathway. In addition, catalase promotes apoptosis by inhibiting NF-κB translocation and reactive oxygen content. Claspin indirectly induces DNA damage to kill cells.
4. Conclusion
In conclusion, 16 furoxan-based chromone derivatives were synthesised and tested for their antiproliferative activity. The results indicated that most of the target compounds exhibited stronger antiproliferative activity than the parent compound. 15a showed the most potent activity with an IC50 value of 1.61 µM against K562 cells. To further investigate its mechanism of action, the effects on cell cycle, morphological change, mitochondrial membrane potential and apoptosis-related proteins were evaluated. The results showed that 15a caused S phase arrest in K562 cells in a concentration-dependent manner. Cells treated with 15a showed obvious cell membrane rupture and nuclear fragmentation. The apoptotic rate of the treated cells was positively correlated with the concentration. Human apoptosis protein array assay also demonstrated 15a increased the expression levels of pro-apoptotic Bax, Bad, HtrA2, and Trail R2/DR5. The expression of Catalase and cell cycle blocker Claspin were similarly up-regulated. So, 15a induced K562 cells death through multiple pathways, including endogenous and exogenous pathways, as well as non caspase pathway. In addition, DNA damage indirectly killed cells. In brief, 15a as an antitumor drug candidate deserves further investigation.
Supplemental Material
Download PDF (3.5 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dutta S, Mahalanobish S, Saha S, et al. Natural products: an upcoming therapeutic approach to cancer. Food Chem Toxicol 2019;128:240–55.
- Fu YW, Luo JY, Qin JA, Yang MH. Screening techniques for the identification of bioactive compounds in natural products. J Pharm Biomed Anal 2019;168:189–200.
- Keri RS, Budagumpi S, Pai RK, et al. Chromones as a privileged scaffold in drug discovery: a review. Eur J Med Chem 2014;78:340–74.
- Rokaya MB, Münzbergová Z, Timsina B, et al. Don: a review of its botany, ethnobotany, phytochemistry and pharmacology. J Ethnopharmacol 2012;141:761–74.
- Duan YD, Jiang YY, Guo FX, et al. The antitumor activity of naturally occurring chromones: a review. Fitoterapia 2019;135:114–29.
- Dudek H, Datta SR, Franke TF, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997;275:661–5.
- Franaszek EC, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA 1999;96:8745–50.
- Medina EA, Morris IR, Berton MT. Phosphatidylinositol 3-kinase activation attenuates the TLR2-mediated macrophage proinflammatory cytokine response to Francisella tularensis live vaccine strain. J Immunol 2010;185:7562–72.
- Guo YQ, Yin T, Wang XM, et al. Traditional uses, phytochemistry, pharmacology and toxicology of the genus Cimicifuga: a review. J Ethnopharmacol 2017;209:264–82.
- Ruhs BN, Julia KCH, William PH, Tor SP. Developing environmentally benign and effective organic wood preservatives by understanding the biocidal and non-biocidal properties of extractives in naturally durable heartwood. Holzforschung 2008;62:264–9.
- Amira MG, Pierre DC, Marguerite T, et al. Anti-cancer and immunostimulatory activity of chromones and other constituents from Cassia petersiana. Z Naturforsch C 2007;62:331–8.
- Barre-Sinoussi F, Chermann JC, Rey F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983;220:868–71.
- Sriram D, Yogeeswari P, Dinakaran M, et al. Discovery of novel antitubercular 2,10-dihydro-4H-chromeno-[3,2-c]pyridin-3-yl derivatives. Eur J Med Chem 2010;45:120–3.
- Taylor WRJ, White NJ. Antimalarial drug toxicity: a review. Drug Safety 2004;27:25–61.
- Keane WF, Brenner BM. The risk of developing end-stage renal disease in patients with type 2 diabetes and nephropathy: the RENAAL study. Kidney Int 2003;63:1499–507.
- Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009;4:226–35.
- Yousuf I, Arjmand F, Tabassum SL, et al. Mechanistic insights into a novel chromone-appended Cu(II) anticancer drug entity: in vitro binding profile with DNA/RNA substrates and cytotoxic activity against MCF-7 and HepG2 cancer cells. Dalton Trans 2015;44:10330–42.
- Vlahos CJ, Matter WF, Hui KY, et al. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 1994;269:5241–8.
- Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 2000;60:1541–5.
- Samuels Y, Diaz LAJ, Schmidt-Kittler O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005;7:561–73.
- Gaspar A, Matos MJ, Garrido J, et al. Chromone: a valid scaffold in medicinal chemistry. Chem Rev 2014;114:4960–92.
- Klausmeyer P, Zhou Q, Scudiero DA, et al. Cytotoxic and HIF-1α inhibitory compounds from Crossosoma bigelovii. J Nat Prod 2009;72:805–12.
- Andrioli WJ, Conti R, Araujo MJ, et al. Mycoleptones A-C and polyketides from the endophyte Mycoleptodiscus indicus. J Nat Prod 2014;77:70–8.
- Asselin E, Mills GB, Tsang BK. XIAP regulates Akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer Res 2001;61:1862–8.
- Brognard J, Clark AS, Ni Y, et al. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res 2001;61:3986–97.
- Itoh N, Semba S, Ito M, et al. Phosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer 2002;94:3127–34.
- Valdameri G, Genoux-Bastide E, Peres B, et al. Substituted chromones as highly potent nontoxic inhibitors, specific for the breast cancer resistance protein. J Med Chem 2012;55:966–70.
- Ma YY, Wei SJ, Lin YC, et al. PIK3CA as an oncogene in cervical cancer. Oncogene 2000;19:2739–44.
- Shayesteh L, Lu Y, Kuo WL, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet 1999;21:99–102.
- Senderowicz AM, Sausville EA. Preclinical and clinical development of cyclin-dependent kinase modulators. J Natl Cancer Inst 2000;92:376–87.
- Kelland LR. Flavopiridol, the first cyclin-dependent kinase inhibitor to enter the clinic: current status. Expert Opin Investig Drugs 2000;9:2903–11.
- Song QL, Tan SW, Zhuang XT, et al. Nitric oxide releasing D-α-tocopheryl polyethylene glycol succinate for enhancing antitumor activity of doxorubicin. Mol Pharm 2014;11:4118–29.
- Bergendi L, Beneš L, Ďuračková Z, et al. Chemistry, physiology and pathology of free radicals. Life Sci 1999;65:1865–74.
- Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer 2006;6:521–34.
- Mocellin S, Bronte V, Nitti D. Nitric oxide, a double edged sword in cancer biology: searching for therapeutic opportunities. Med Res Rev 2007;27:317–52.
- Carpenter AW, Schoenfisch MH. Nitric oxide release: part II. Therapeutic applications. Chem Soc Rev 2012;41:3742–52.
- Ignarro LJ. Nitric oxide: biology and pathobiology. Cambridge, Massachusetts: Academic Press; 2000.
- Sang JR, Chen YC, Tao Y. Nitric oxide inhibits gastric cancer cell growth through the modulation of the Akt pathway. Mol Med Rep 2011;4:1163–7.
- De Luca A, Moroni N, Serafino A, et al. Treatment of doxorubicin-resistant MCF7/Dx cells with nitric oxide causes histone glutathionylation and reversal of drug resistance. Biochem J 2011;440:175–83.
- Chung MF, Liu HY, Lin KJ, et al. A pH-responsive carrier system that generates NO bubbles to trigger drug release and reverse P-glycoprotein-mediated multidrug resistance. Angew Chem Int Ed 2015;54:9890–3.
- Rapozzi V, Della Pietra E, Zorzet S, et al. Nitric oxide-mediated activity in anti-cancer photodynamic therapy. Nitric Oxide 2013;30:26–35.
- Xiang HJ, Deng Q, An L, et al. Tumor cell specific and lysosome-targeted delivery of nitric oxide for enhanced photodynamic therapy triggered by 808 nm near-infrared light. Chem Commun 2016;52:148–51.
- Huang L, Gao Z, Han G. Photoswitchable near-infraredemitting borondipyrromethene (BODIPY) nanoparticles. Part Part Syst Char 2017;34:1700223.
- De Ridder M, Verellen D, Verovski V, et al. Hypoxic tumor cell radiosensitization through nitric oxide. Nitric Oxide 2008;19:164–9.
- Fan W, Bu W, Zhang Z, et al. X-ray radiation-controlled NO-release for on-demand depth independent hypoxic radiosensitization. Angew Chem Int Ed 2015;54:14026–30.
- Liu J, Li C, Qu W, et al. Nitric oxide prodrugs and metallochemotherapeutics: JS-K and CB-3-100 enhance arsenic and cisplatin cytolethality by increasing cellular accumulation. Mol Cancer Ther 2004;3:709–14.
- Zhang K, Xu H, Jia Y, et al. Ultrasound-triggered nitric oxide release platform based on energy transformation for targeted inhibition of pancreatic tumor. ACS Nano 2016;10:10816–28.
- Chiueh CC. Neuroprotective properties of nitric oxide. Ann N Y Acad Sci 1999;890:301–11.
- Feng T, Wan J, Li P, et al. A novel NIR-controlled NO release of sodium nitroprusside-doped Prussian blue nanoparticle for synergistic tumor treatment. Biomaterials 2019;214:119213.
- Burgaud J-L, Ongini E, Soldato P. Nitric oxide-releasing drugs-a novel class of effective and safe therapeutic agents. Ann NY Acad Sci 2002;962:360–71.
- Chiueh CC, Hong JS, Leong SK. Nitric oxide: novel actions, deleterious effects, and clinical potential. Ann N Y Acad Sci 2002;402–14.
- Huerta S, Chilka S, Bonavida B. Nitric oxide donors: novel cancer therapeutics. Int J Oncol 2008;33:909–27.
- Serafim RAM, Primi MC, Trossini GHG, Ferreira EI. Nitric oxide: state of the art in drug design. Curr Med Chem 2012;19:386–405.
- Arrieta O, Blake M, de la Mata-Moya MD, et al. Phase II study. Concurrent chemotherapy and radiotherapy with nitroglycerin in locally advanced nonsmall cell lung cancer. Radiother Oncol 2014;111:311–15.
- Bonavida B, Baritaki S, Huerta-Yepez S, et al. Novel therapeutic applications of nitric oxide donors in cancer: roles in chemo- and immunosensitization to apoptosis and inhibition of metastases. Nitric Oxide 2008;19:152–57.
- Bonavida B, Garban H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol 2015;6:486–94.
- Lai Y, Shen L, Zhang Z, et al. Synthesis and biological evaluation of furoxan-based nitric oxide-releasing derivatives of glycyrrhetinic acid as anti-hepatocellular carcinoma agents. Bioorg Med Chem Lett 2010;20:6416–20.
- Aguirre G, Boiani M, Cerecetto H, et al. Furoxan derivatives as cytotoxic agents: preliminary in vivo antitumoral activity studies. Pharmazie 2006;61:54–59.
- Hu X, Jiao RW, Li HN, et al. Antiproliferative hydrogen sulfide releasing evodiamine derivatives and their apoptosis inducing properties. Eur J Med Chem 2018;151:376–88.
- Han T, Tian KT, Pan HQ, et al. Novel hybrids of brefeldin A and nitrogen mustards with improved antiproliferative selectivity: design, synthesis and antitumor biological evaluation. Eur J Med Chem 2018;150:53–63.
- Ling Y, Ye X, Zhang Z, et al. Novel nitric oxide-releasing derivatives of farnesylthiosalicylic acid: synthesis and evaluation of antihepatocellular carcinoma activity. J Med Chem 2011;54:3251–59.
- André F, Joana R, Tiago S, et al. Coumarin versus chromone monoamine oxidase B inhibitors: quo vadis? J Med Chem 2017;60:7206–12.
- Novak M, Žegura B, Baebler Š, et al. Influence of selected anti-cancer drugs on the induction of DNA double-strand breaks and changes in gene expression in human hepatoma HepG2 cells. Environ Sci Pollut R 2016;23:14751–61.
- Hussein EM, Al-Rooqi MM, Abd El-Galil SM, et al. Design, synthesis, and biological evaluation of novel N4-substituted sulfonamides: acetamides derivatives as dihydrofolate reductase (DHFR) inhibitors. BMC Chem 2019; 13(1):91.
- Kumar S, Lim SM, Ramasamy K, et al. Design, synthesis, antimicrobial and cytotoxicity study on human colorectal carcinoma cell line of new 4,4′-(1,4-phenylene) bis(pyrimidin-2-amine) derivatives. Chem Cent J 2018;12(1):73.
- Liu X, Chan SY, Chi-Lui Ho P. Comparison of the in vitro and in vivo effects of retinoids either alone or in combination with cisplatin and 5-fluorouracil on tumor development and metastasis of melanoma. Cancer Chemoth Pharm 2008;63:167–74.
- Anand P, Thomas SG, Kunnumakkara AB, et al. Biological activities of curcumin and its analogues (Congeners) made by man and mother nature. Biochem Pharmacol 2008;76:1590–611.
- Mortimer CG, Wells G, Wells JP, et al. Antitumor benzothiazoles. 26.1 2-(3,4-dimethoxyphenyl)-5-fluorobenzothiazole (GW 610, NSC 721648), a simple fluorinated 2-arylbenzothiazole, shows potent and selective inhibitory activity against lung, colon, and breast cancer cell lines. J Med Chem 2006;49:179–85.
- Chan KT, Meng FY, Li Q, et al. Cucurbitacin B induces apoptosis and S phase cell cycle arrest in BEL-7402 human hepatocellular carcinoma cells and is effective via oral administration. Cancer Lett 2010;294:118–24.
- Shen JK, Du HP, Yang M, et al. Casticin induces leukemic cell death through apoptosis and mitotic catastrophe. Ann Hematol 2009;88:743–52.
- Yang L, Wu S, Zhang Q, et al. 24-Dihydrocucurbitacin B induces G2/M cell-cycle arrest and mitochondria-dependent apoptosis in human breast cancer cells (Bcap37). Cancer Lett 2007;256:0–278.
- Li JF, Huang RZ, Yao GY, et al. Synthesis and biological evaluation of novel aniline-derived asiatic acid derivatives as potential anticancer agents. Eur J Med Chem 2014;86:175–88.
- Yasuda H. Solid tumor physiology and hypoxia-induced chemo/radio-resistance: novel strategy for cancer therapy: nitric oxide donor as a therapeutic enhancer. Nitric Oxide 2008;19:205–16.
- Susin SA, Zamzami N, Castedo M, et al. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95 and ceramide-induced apoptosis. J Exp Med 1997;186:25–37.
- Stefano S, Cristiana B, Jurek D, et al. Opposite role of changes in mitochondrial membrane potential in different apoptotic processes. FEBS Lett 2000;469:186–90.
- Liu X, Kim CN, Yang J, et al. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996;86:147–57.
- Susin SA, Zamzami N, Castedo M, et al. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med 1996;184:1331–41.
- Jeong SY, Gaume B, Lee YJ, et al. Bcl-x(L) sequesters its C-terminal membrane anchor in soluble, cytosolic homodimers. Embo J 2004;23:2146–55.
- Renault TT, Floros KV, Chipuk JE. BAK/BAX activation and cytochrome c release assays using isolated mitochondria. Methods 2013;61:146–55.
- Jo EB, Lee YS, Lee H, et al. Combination therapy with c-met inhibitor and TRAIL enhances apoptosis in dedifferentiated liposarcoma patient-derived cells. BMC Cancer 2019;19:496.
- Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol 1997;37:517–54.
- Goo HG, Rhim H, Kang S. HtrA2/Omi influences the stability of LON protease 1 and prohibitin, proteins involved in mitochondrial homeostasis. Exp Cell Res 2014;328:456–65.
- Yamauchi S, Hou YY, Guo AK, et al. p53-mediated activation of the mitochondrial protease HtrA2/Omi prevents cell invasion. J Cell Biol 2014;204:1191–207.
- Wang CY, Cusack JJC, Liu R, et al. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-κB. Nat Med 1999;5:412–17.
- Lüpertz R, Chovolou Y, Kampkötter A, et al. Catalase overexpression impairs TNF-alpha induced NF-kappaB activation and sensitizes MCF-7 cells against TNF-alpha. J Cell Biochem 2008;103:1497–511.
- Lin SY, Li K, Stewart GS, Elledge SJ. Human claspin works with BRCA1 to both positively and negatively regulate cell proliferation. Proc Natl Acad Sci USA 2004;101:6484–89.
- Lam MH, Liu Q, Elledge SJ, et al. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 2004;6:45–59.
- Azenha D, Lopes MC, Martins TC. Claspin functions in cell homeostasis-A link to cancer?. DNA Repair 2017;59:27–33.
- Tsimaratou K, Kletsas D, Kastrinakis NG, et al. Evaluation of claspin as a proliferation marker in human cancer and normal tissues. J Pathol 2007;211:331–39.