
Abstract
We evaluated the hCA (CA, EC 4.2.1.1) inhibitory activity of novel 4-(2-(2-substituted-thio-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamides (compounds 2–20) towards the isoforms I, II, IX, and XII. hCA Isoforms were effectively inhibited by most of new compounds comparable to those of AAZ. Compounds 2 and 4 showed interestingly efficient and selective antitumor (hCA IX and hCA XII) inhibitor activities (KIs; 40.7, 13.0, and 8.0, 10.8 nM, respectively). Compounds 4 and 5 showed selective hCA IX inhibitory activity over hCA I (SI; 95 and 24), hCA IX/hCA II (SI; 23 and 5.8) and selective hCA XII inhibitory activity over hCA I (SI; 70 and 44), hCA XII/hCA II, (SI; 17 and 10) respectively compared to AAZ. Compounds 12–17, and 19–20 showed selective inhibitory activity towards hCA IX over hCA I and hCA II, with selectivity ranges of 27–195 and 3.2–19, respectively, while compounds 12, 14–17, and 19 exhibited selective inhibition towards hCA XII over hCA I and hCA II, with selectivity ratios of 48–158 and 5.4–31 respectively, compared to AAZ. Molecular docking analysis was carried out to investigate the selective interactions among the most active derivatives, 17 and 20 and hCAs isoenzymes. Compounds 17 and 20, which are highly selective CA IX and XII inhibitors, exhibited excellent interaction within the putative binding site of both enzymes, comparable to the co-crystallized inhibitors.
Quinazoline-linked ethylbenzenesulfonamides inhibiting CA were synthesised.
The new molecules potently inhibited the hCA isoforms I, II, IV, and IX.
Compounds 4 and 5 were found to be selective hCA IX/hCA I and hCA IX/hCA II inhibitors.
Compounds 4 and 5 were found to be selective hCA XII/hCA I and hCA XII/hCA II inhibitors.
Compounds 12–17, 19, and 20 were found to be selective hCA IX/hCA I and hCA IX/hCA II inhibitors.
Compounds 12, 14–17, 19 were found to be selective hCA XII/hCA I and hCA XII/hCA II inhibitors.
Highlights
Compounds 4 and 5 are selective hCA IX and XII inhibitors over hCA I (selectivity ratios of 95, 23, and 24, 5.8, respectively) and hCA II (selectivity ratios of 70, 17, and 44, 10 respectively). Compounds 12–17, and 19–20 are selective hCA IX inhibitors over hCA I (selectivity ratios of 27-195) and hCA II (selectivity ratios of 3.2-19). Compounds 12, 14–17 and 19 are also selective hCA XII inhibitors over hCA I (selectivity ratios of 48-158) and hCA II (selectivity ratios of 5.4-31).
Graphical Abstract
1. Introduction
Carbonic anhydrases (CAs; EC 4.2.1.1) constitute the superfamily of metalloenzymes that catalyse the CO2 hydration and dehydration reactions. CAs are classified into eight genetically distinct families, named α-, β-, γ-, δ-, ζ-, η-, ɵ- and ι-CAsCitation1,Citation2. 15 α-class CA isozymes have been detected in humans, which are further classified into four different subsets on the basis of their subcellular localisation—CA I, II, III, VII, VIII, X, XI, XIIII are cytosolic proteins, CA IV is a glycosylphosphatidylinositol (GPI)-anchored protein, CA VA and VB are located in the mitochondrial matrix, CA VI is secreted, and CA IX, XII and XIV are trans-membrane isoformsCitation1–3. Human CAs (hCAs) are spread in the human body, and are implicated in a plethora of essential physiological processes. Therefore, the dysregulated expression and/or activity of the CAs can lead to various pathological conditionsCitation2. CA II is the most physiologically relevant CA isoform, implicated in various disorders including cerebral oedema, glaucoma (such as CA XII), and epilepsy. It is conversely off-target, as CA I, when targeting tumours where CA IX and XII are overexpressed and represent validated targets to combat the growth of both primary tumours and metastasisCitation4,Citation5. The high structural similarities between various CA isoforms necessitate high selectivity in the design of small-molecule anti-CA drugs for the treatment of diseases associated with CA dysregulation, to minimise the side effectsCitation3. Benzene sulphonamides are one of the best-known molecules clinically used as CA inhibitors. Additionally, “SLC-011 (), a benzenesulfonamide, is a selective CA IX/XII inhibitor currently being evaluated in a Phase I trial for the treatment of solid, metastatic tumors”Citation6–10. Sulphonamide derivatives are not only one of the most preferred CA inhibitor classesCitation9,Citation11–23, but also important COX-2 inhibitors and antitumor agentsCitation17,Citation19,Citation24–26. The quinazolinone scaffold is also used widely across medicinal chemistryCitation27–43. (6-Iodo or 7-flouro-2-merqapto-4-(3H)-quinazolinone3-yl)-benzenesulfonamides (A, ) have been shown to potently inhibit CA I, II, IX, and XIICitation44,Citation45. A number of 2-((3-benzyl-4-oxo-3,4-dihydroquinazolin-2-yl)thio)-N-(4-sulfamoylphenethyl)anildes (B, ) also showed potent inhibitory activity against different hCA isoformsCitation38. The 2-mercapto-4(3H)-quinazolinone derivatives containing ethylsulfonamide tail (C, ) showed strong inhibitory activity against different hCA isoforms with low-concentration inhibition constants
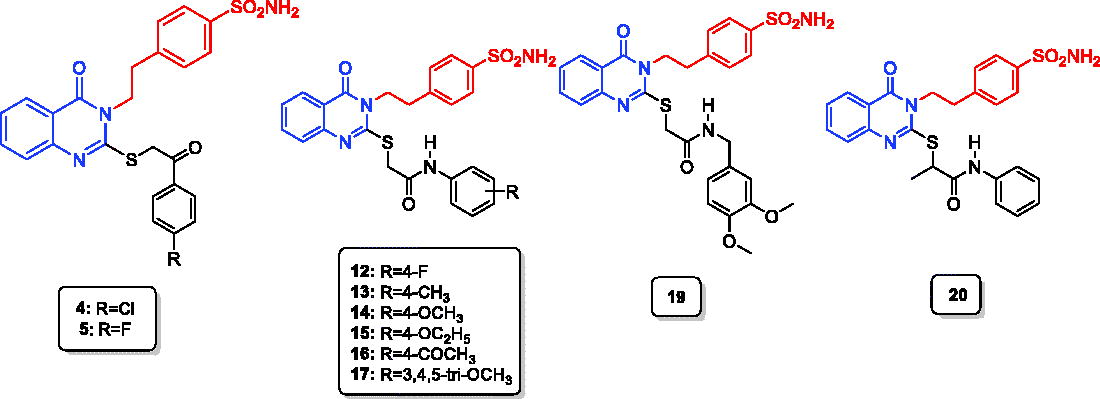
Figure 1. Structures of AAZ, SLC-0111, A–C, and the designed quinazoline derivatives (2–20) as CAIs.
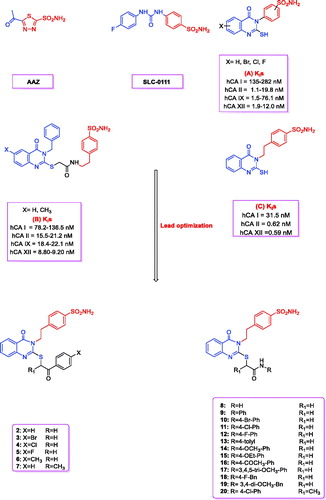
Here, we studied 2-mercaptoquinazolinone, (C, ) a slightly polar and non-selective hCA inhibitor. Because the sulfhydryl group has been reported to be associated with various metabolic and pharmacological problemsCitation46–49, we used a 2-mercaptoquinazolinone scaffold bearing an ethylsulfonamide head with alkylation of the thione group with a terminal lipophilic moiety, so that it can interact selectively with CA through both, hydrogen and hydrophobic interactions. Here, we synthesised various derivatives of 2-mercaptoquinazolinone (2–20, ) with different selectivity criteria for the hCA inhibitors, particularly for the tumor-associated hCA IX and hCA XII. The role of alkyl substituent in 2-mercaptoquinazolinone was computationally analysed and the conserved residues responsible for the target selectivity were identified.
2. Materials and methods
2.1. Chemistry
Melting points were recorded on a Barnstead 9100 electrothermal melting point apparatus (UK). IR spectra (KBr) were recorded on a FT-IR Perkin-Elmer spectrometer (Perkin Elmer Inc., MA). NMR (1H and 13C NMR) spectra were recorded with Bruker 700 MHz spectrometers (Zurich, Switzerland). Micro-analytical data (C, H, and N) were obtained using a Perkin-Elmer 240 analyser (Perkin Elmer Inc., MA) and agreed with the proposed structures within ±0.4% of the theoretical values. Mass spectra were recorded on a Varian TQ 320 GC/MS/MS mass spectrometer (Varian, Palo Alto, CA). Thione 1 and compounds 8–20 were prepared as described earlierCitation50,Citation51.
2.1.1. General procedure for synthesis of 4-(2-(2-((2-(4-substituted-phenyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamide (2–7)
A mixture of thione 1 (1 mmol, 361 mg) and potassium carbonate (3 mmol, 415 mg) in 6 ml acetone were stirred at room temperature for one hour. Appropriate phenacyl bromide (1 mmol) was added and the reaction mixture was stirred at room temperature for 9–12 h, filtered, and the crude solid was washed with water, dried and recrystallized from ethanol (1H & 13C NMR supplementary material).
4-(2-(4-Oxo-2-((2-oxo-2-phenylethyl)thio)quinazolin-3(4H)-yl)ethyl)benzenesulfonamide (2): m.p 246–247°; 94% yield; IR (KBr, cm−1) ν: 3284, 3237 (NH), 1665 (C=O), 1342, 1151 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 8.14 (t, 2H, J = 7.14 and 1.26 Hz), 8.04 (dd, 1H, J = 7.91 and 1.26 Hz), 7.82 (d, 2H, J = 8.26 Hz), 7.74 (t, 1H, J = 7.49 Hz), 7.66 (t, 1H, J = 16.71 and 6.96 Hz), 7.62 (t, 2H, J = 7.80 and 7.77 Hz), 7.52 (d, 2H, J = 8.26 Hz), 7.41 (t, 1H, J = 7.17 Hz), 7.37 (s, 2H), 6.98 (d, 1H, J = 8.12 Hz), 4.92 (s, 2H), 4.33 (t, 2H, J = 16.25 Hz), 3.14 (t, 2H, J = 16.20 Hz); 13C NMR (176 MHz, DMSO-d6): δ 194.04, 160.76, 156.10, 146.92, 143.11, 142.29, 136.90, 135.19, 134.01, 129.67, 129.29, 128.79, 126.92, 126.45, 125.87, 119.08, 45.69, 39.38, 33.67; Ms; m/z (479).
4-(2-(2-((2-(4-Bromophenyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamide (3): m.p 248–248°; 95% yield; IR (KBr, cm−1) ν: 3280, 3236 (NH), 1686 (C=O), 1340, 1153 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 8.07 (d, 2H, J = 8.26 Hz), 8.04 (d, 1H, J = 7.84 Hz), 8.85 (d, 2H, J = 8.19 Hz), 7.81 (d, 2H, J = 7.98 Hz), 7.68 (t, 1H, J = 7.63 Hz), 7.52 (d, 2H, J = 8.05 Hz), 7.41 (d, 1H, J = 7.45 Hz), 7.37 (s, 2H), 7.00 (d, 1H, J = 8.19 Hz), 4.89 (s, 2H), 4.32 (t, 2H, J = 16.05 Hz), 3.13 (t, 2H, J = 16.04 Hz); 13 C NMR (176 MHz, DMSO-d6): δ 193.41, 160.74, 156.02, 146.88, 143.12, 142.27, 135.9266, 135.26, 132.38, 130.81, 129.67, 128.10, 126.93, 126.55, 126.52, 126.45, 125.87, 119.08, 45.72, 39.27, 33.67; Ms; 558.0; Ms; (m/z; 557, M + 2; 559).
4-(2-(2-((2-(4-Chlorophenyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamide (4): m.p 250–251°; 93% yield; IR (KBr, cm−1) ν: 3281, 3239 (NH), 1684 (C=O), 1345, 1159 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 8.15 (d, 2H, J = 8.43 Hz), 8.04 (d, 1H, J = 7.85 Hz), 7.82 (d, 2H, J = 8.05 Hz), 7.70 (d, 2H, J = 8.40 Hz), 7.67 (d, 1H, J = 7.05 Hz), 7.52 (d, 2H, J = 8.05 Hz), 7.41 (t, 1H, J = 7.45 Hz), 7.37 (s, 2H), 6.99 (d, 1H, J = 8.19 Hz), 4.89 (s, 2H), 4.32 (t, 2H, J = 16.09 Hz), 3.13 (t, 2H, J = 16.06 Hz); 13 C NMR (176 MHz, DMSO-d6): δ 193.20, 160.74, 156.03, 146.89, 143.12, 142.28, 138.89, 135.60, 135.25, 130.73, 129.67, 129.43, 126.93, 126.53, 125.86, 119.08, 45.72, 39.28, 33.67; Ms; 514; Ms; (m/z; 513, M + 1; 514).
4-(2-(2-((2-(4-Fluorophenyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamide (5): m.p 249–250°; 92% yield; IR (KBr, cm−1) ν: 3278, 3238(NH), 1666 (C=O), 1342, 1152 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 8.23 (dd, 2H, J = 13.95 and 2.66 Hz), 8.04 (d, 1H, J = 7.84 Hz), 7 82 (d, 2H, J = 8.05 Hz), 7.68 (t, 1H, J = 7.63 Hz), 7.52 (d, 2H, J = 7.98 Hz), 7.46 (t, 2H, J = 8.71 Hz), 7.41 (t, 1H, J = 7.49 Hz), 7.37 (s, 2H), 6.99 (d, 1H, J = 8.12 Hz), 4.90 (s, 2H), 4.33 (t, 2H, J = 16.09 Hz), 3.14 (t, 2H, J = 16.06 Hz); 13 C NMR (176 MHz, DMSO-d6): δ 192.73, 166.36, 164.93, 160.7571, 156.07, 146.89, 143.12, 142.28, 135.24, 133.65, 133.64, 131.88, 131.83, 129.67, 126.92, 126.52, 125.87, 119.08, 116.41, 116.29, 45.70, 39.26, 33.67; Ms; m/z (497).
4-(2-(4-Oxo-2-((2-oxo-2-(p-tolyl)ethyl)thio)quinazolin-3(4H)-yl)ethyl)benzenesulfonamide (6): m.p 257–258°; 92% yield; IR (KBr, cm−1) ν: 3281, 3237 (NH), 1665 (C=O), 1339, 1150 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 8.04 (t, 3H, J = 7.12 and 4.90 Hz), 7.82 (d, 2H, J = 7.75 Hz), 7.68 (t, 1H, J = 7.59 Hz), 7.52 (d, 2H, J = 7.84 Hz), 7.42 (t, 3H, J = 7.84 and 10.71 Hz), 7.37 (s, 2H), 7.04 (d, 1H, J = 8.19 Hz), 4.90 (s, 2H), 4.33 (t, 2H, J = 15.79 Hz), 3.14 (t, 2H, J = 15.79 Hz), 2.44 (s, 3H); 13 C NMR (176 MHz, DMSO-d6): δ 193.40, 160.78, 156.12, 146.94, 144.46, 143.12, 142.30, 135.21, 134.33, 129.83, 129.67, 128.93, 126.91, 126.52, 125.95, 119.09, 45.65, 39.41, 33.67, 21.73; Ms; m/z (493).
4-(2-(4-Oxo-2-((1-oxo-1-phenylpropan-2-yl)thio)quinazolin-3(4H)-yl)ethyl)benzenesulfonamide (7): m.p 245–246°; 90% yield; IR (KBr, cm−1) ν: 3279, 3237 (NH), 1668 (C=O), 1347, 1154 (O=S=O); 1H NMR (700 MHz, DMSO-d6): δ 8.16 (d, 2H, J = 7.76 Hz), 8.29 (d, 1H, J = 7.85 Hz), 7.80 (d, 2H, J = 7.84 Hz), 7.73 (t, 1H, J = 7.31 Hz), 7.65–7.61 (m, 3H), 7.84 (d, 2H, J = 7.84 Hz), 7.39 (t, 1H, J = 7.52 Hz), 7.37 (s, 2H), 6.78 (d, 1H, J = 8.12 Hz), 5.75 (q, 1H, J = 7.16 Hz), 4.29–4.19 (m, 2H), 3.07 (t, 2H, t, J = 12.58 Hz), 1.57 (d, 3H, J = 7.19 Hz); 13 C NMR (176 MHz, DMSO-d6): δ 198.20, 160.67, 155.81, 146.87, 143.12, 142.23, 135.98, 135.15, 134.01, 129.69, 129.34, 128.95, 126.92, 126.57, 126.48, 125.46, 119.13, 46.23, 45.76, 33.60, 16.44; Ms; 493.00; Ms; m/z (493).
2.2. CA inhibition
The hCA I, II, IX, and XII isoenzyme inhibition assays were performed according to the reported method using the SX.18 MV-R stopped-flow instrument (Applied Photophysics, Oxford, UK)Citation52–54. All CA isoforms were recombinant isoforms obtained in-house, as reported earlierCitation55,Citation56.
2.3. Molecular docking method
The molecular docking protocol was conducted according to the reported methodsCitation28,Citation32,Citation33,Citation41–43,Citation57–64 using MOE 2008.10 from the Chemical Computing Group IncCitation65. The crystal structures of CA-IX (PDB ID: 5FL4) and CA-XII (PDB ID: 1JCZ) were obtained from the protein data bankCitation66,Citation67.
3. Results and discussion
3.1. Chemistry
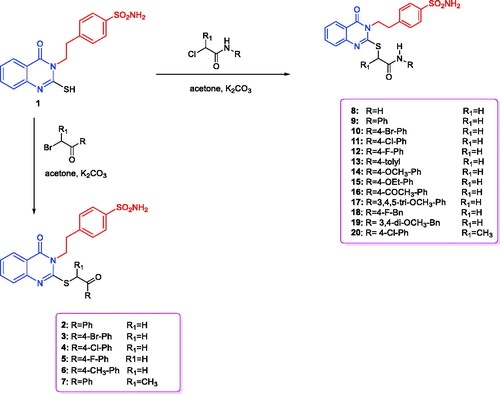
4-(2-(4-Oxo-2-thioxo-1,4-dihydroquinazolin-3(2H)-yl)ethyl)benzenesulfonamide (1) was obtained via the reaction of 4-(2-isothiocyanatoethyl)benzenesulfonamide, triethylamine and 2-aminobenzoic acid in boiling ethanolCitation50,Citation51 (Scheme 1). Stirring of compound 1 with potassium carbonate in acetone and different phenacyl bromides produced the corresponding 4-(2-(2-((2-(4-substituted-phenyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamides 2–7 with 90–95% yield. Various spectroscopic studies were conducted to validate the structures of the newly synthesised compounds, 2–7. The target compounds, 2–6, were validated by the diminishing of the thioamidic proton (NH–C=S) at 13.03 ppm and that of the thione moiety (NH–C=S) at 175.29 ppm, as well as by the presence of the phenacyl carbonyl group (SCH2COAr) at 194.04–192.73 ppm, with singlet peaks at 4.92–4.89 ppm and 39.41–39.26 ppm due to the phenacyl (SCH2COAr) moiety, in the 1H and 13C NMR spectra, respectively. Additionally, 4-(2-(4-oxo-2-((1-oxo-1-phenylpropan-2-yl)thio)quinazolin-3(4H)-yl)ethyl)benzenesulfonamide (7) was confirmed by presence of the carbonyl group of (S(CH)CH3COAr) at 198.20 ppm in the 13C NMR spectrum, as well as by the quartette (S(CH)CH3COAr) and doublet (S(CH)CH3COAr) peaks at 5.75 and 1.57 ppm respectively in the 1H NMR spectrum, together with the characteristic peaks (S(CH)CH3COAr) at 46.23 and (S(CH)CH3COAr) at 16.44 ppm in the 13C NMR spectrum. The ethylbenzenesulfonamide amino group (NH2) (in compounds 2–7) was long-established by the presence of a typical singlet peak at 7.37 ppm in the 1H NMR spectrum. The tails of aliphatic ethylbenzenesulfonamide moiety were fixed by triplet peaks at 4.33–4.32 and 3.15–3.12 ppm in the 1H NMR spectrum and distinctive peaks at 45.72–45.65 and 33.67 ppm in 13C NMR spectrum, respectively. 2-Substituted mercapto-4(3H)-quinazolinones (8–20) were prepared in 90–96% yield by mixing compound 1 and 2-chloro-N-substitutedamide in acetone at room temperature in the presence of potassium carbonateCitation51.
Scheme 1. Synthesis of the designed quinazoline derivatives (2–20).
3.2. CA inhibitory activity
The CA inhibitory activity of 4-(2-(2-(substituted-thio)-4(3H)-quinazolinon-3-yl)ethyl)benzenesulfonamides (compounds 2–20) towards hCA I, II, IV, and IX isoforms was measured and compared to acetazolamide (AAZ), a typical sulphonamide inhibitor. hCA I was effectively inhibited by compounds 2 and 4–13 with the inhibition-constant (KI) values ranging from 114.5–938.3 nM (AAZ: KI = 250.0 nM). Compounds 3 and 16 showed moderate activity with KI values of 1447.0 and 1697.0 nM, respectively, while compounds 14–15 and 17–20 showed weak activity with KI values ranging from 2048–5467 nM. Compounds 5, 8, 9, 11, 12, and 20 were verified to be effective hCA II inhibitors, with KI values of 25.4–95.4 nM (AAZ: KI = 12.0 nM). Compounds 2, 3, 4, 6, 7, 10, 14, and 16 showed modest hCA II inhibitory activity with KI values ranging between 116.2 and 266.1 nM, whereas compounds 13 and 15 showed a weak inhibitory activity with KI values of 304.6 and 1099.0 nM, respectively. Compounds 2–17 and 20 displayed potent hCA IX inhibitory activity with KI values ranging from 8.0 to 100.4 nM, which were greater than or nearly identical to that of AAZ (KI = 25.0 nM), whereas compounds 18 and 19 showed modest hCA IX inhibitory activity with KI values ranging between 256.4 and 145.1 nM, respectively. 4-(2-(2-(Substituted-thio)-4(3H)-quinazolinon-3-yl)ethyl)benzenesulfonamide derivatives 2, 4, 5, 8, 9, 11, 12, 13, 14, 16 and 17 showed potent hCA XII inhibitory activity with KI values of 2.4–49.1 nM compared to AAZ (KI = 5.7 nM), whereas compounds 3, 6, 7, 10, 15, 18, 19, and 20 exerted moderate hCA XII inhibitory activities with KI values of 59.7–113.4 nM (). On the other hand, the selectivity factor is critical goal to increase the value of the new synthesised compounds. New compounds, such as 2 and 4 showed characteristic effective and selective antitumor (hCA IX and hCA XII) carbonic anhydrase inhibitory activity with KI values (compound 2; 40.7 and 13.0 nM) and KI values (compound 4; 8.0, and 10.8 nM) compared with AAZ (KI values of 25 and 5.7 respectively). 4-(2-(2-((2-(4-Substituted-phenyl)-2-oxoethyl)thio)-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamides (compounds 2–7) showed high selectivity in the inhibition of hCA IX over hCA I and hCA II (in the range of 15.0–95.0 and 2.3–23.0, respectively), as well as selectivity in the inhibition of hCA XII over hCA I and hCA II (in the range of 5.5–70.0 and 2.5–17.0, respectively) ().
Table 1. Inhibition data of hCA isoforms hCA I, II, IX and XII for sulphonamides using AAZ as standard drug. 
Compounds 4 and 5 showed high selectivity in the inhibition of hCA IX over hCA I and hCA II, with selectivity ratios of 95.0 and 23.0, respectively for compound 4, and those of 24.0 and 5.8, respectively, for compound 5, compared with AAZ selectivity ratios of 10.0 and 0.5, respectively. Additionally, compounds 4 and 5 showed selective inhibition of hCA XII over hCA I and hCA II with selectivity ratios of 70.0 and 17.0, respectively, for compound 4, and 44.0 and 10.0, respectively, for compound 5, compared with AAZ selectivity ratios of 44.0 and 2.1, respectively. N-(substituted)-2-((4-oxo-3-(4-sulfamoylphenethyl)-3,4-dihydroquinazolin-2-yl)thio)amides (compounds 8–20) showed high selectivity in the inhibition of hCA IX over hCA I and hCA II, with selectivity ratios in the range 3.3–195.0 and 0.7–8.3 respectively, compared with AAZ selectivity ratios of 10.0 and 0.5 respectively, and that of hCA XII over hCA I and hCA II, respectively with selectivity ratios in the range 11.0–158.0 and 1.8–31.0, compared with AAZ selectivity ratios of 44.0 and 2.1 respectively. Compounds 12–17, and 19–20 showed selective inhibition of hCA IX over hCA I and hCA II, with selectivity ratios of 23.0–195.0 and 3.2–19.0, respectively, compared with AAZ selectivity ratios of 10.0 and 0.5. Additionally, Compounds 12, 14–17, and 19 showed selective inhibition of hCA XII over hCA I and hCA II, with selectivity ratios of 48.0–158.0 and 5.4–31.0, respectively, compared with AAZ selectivity ratios of 44.0 and 2.1 respectively.
3.3. Structure-activity relationship (SAR) analysis
Several synthesised quinazolinone derivatives (compounds 2–20) were potent inhibitors of the hCA isoforms.
3.3.1. SAR analysis of hCA I inhibition
SAR analysis of hCA I inhibition indicated revealed several key features. (1) 4–(2-(4-Oxo-2-((2-oxo-2-phenylethyl)thio)quinazolin-3(4H)-yl)ethyl)benzenesulfonamide (2), with a KI value of 592.7 nM, was more potent than 4-(2-(2-((1-(4-substituted-phenyl)-1-oxopropan-2-yl)thio)-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamides 3–4 and 4-(2-(4-oxo-2-((1-oxo-1-phenylpropan-2-yl)thio)quinazolin-3(4H)-yl)ethyl)benzenesulfonamide 7, with KI values of 758.7–1447 nM, but less potent than 4-(2-(2-((1-(4-flouro/4-methyl-phenyl)-1-oxopropan-2-yl)thio)-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamide 5 and 6 with KI values of 399.5–471.0 nM. (2) Unsubstituted-N-acetamide 8 (KI value = 114.5 nM) was more active than the corresponding N-phenylacetamide 9 (KI value = 459.7 nM. (3) Substitution of the phenyl ring of N-phenylacetamide 9 (KI value = 459.7 nM) resulted in substituted-N-phenylacetamides 10–17 and N-phenylpropanamide 20 with significantly decreased CA inhibitory activity (KI values = 548.6–3654 nM). (4) The hCA I inhibitory activity of N-(4-fluorophenyl)-2-((4-oxo-3-(4-sulfamoylphenethyl)-3,4-dihydroquinazolin-2-yl)thio)acetamide (12), with a KI value of 548.6 nM, was more stronger than the corresponding N-(4-fluorobenzyl)-2-((4-oxo-3-(4-sulfamoylphenethyl)-3,4-dihydroquinazolin-2-yl)thio)acetamide (18) KI of 2048 nM. (5) hCA I inhibition of N-acetamide 11, with a KI value of 726.4 nM, was more powerful than the corresponding N-propanamide 20 with a KI value of 3628 nM.
3.3.2. SAR analysis for hCA II inhibition
The SAR analysis for hCA II inhibition revealed several key features. (1) 4-(2-(2-((1-(4-Fluorophenyl/4-methylphenyl)-1-oxopropan-2-yl)thio)-4-oxoquinazolin-3(4H)-yl)ethyl)benzenesulfonamides 5 and 6 with KI values of 95.4–116.2 nM were more effective than unsubstituted phenyl and other substituted phenyl derivatives, such as compounds 2–4 and 7 with KI values of 140.8–202.6 nM. (2) hCA II inhibition of 2-((2-oxo-2-phenylethyl)thio)quinazolinone 2, with a KI value of 140.8 µM, was stronger than the corresponding 2-((1-oxo-1-phenylpropan-2-yl)thio)quinazolinone 7 with a KI value of 202.6 nM. (3) N-Phenylacetamide 9 with a KI value of 69.7 nM was less potent than unsubstituted-N-acetamide 8 (KI value = 25.4 nM). (4) Substitution of the phenyl ring of N-phenylacetamide 9 (KI; 69.7 nM) resulted in substituted-N-phenylacetamides 10–17 and N-phenylpropanamide 20 with considerably diminished CA II inhibitory activity (KI values of 75.4–684.2 nM); (5) The hCA II inhibitory effect of N-acetamide 11 (KI value = 92 nM) was less potent than the corresponding N-propanamide 20 (KI value = 75.4 nM). (6) The hCA II inhibitory activity of N-(4-fluorophenyl)acetamide 12, with a KI value of 87.6 nM, was stronger than the corresponding N-(4-fluorobenzyl)acetamide 18 (KI of 2048 nM).
3.3.3. SAR analysis of hCA IX inhibition
SAR analysis of hCA IX inhibition revealed several key factors. (1) The 2-((2-oxo-2-phenylethyl)thio)quinazolinone 2, with a KI value of 40.7 nM, was more potent than 2-((1-oxo-1-phenylpropan-2-yl)thio)quinazolinone 7 with KI value of 63.2 nM. (2) The induction of the activating group, such as the 4-methyl group on the phenyl ring of compound 2 (KI value = 40.7 nM) led to compound 6, with an increased hCA IX inhibitory activity (KI value = 25.1 nM). (3) The introduction of the deactivating group on phenyl ring of compound 2, such as the 4-bromo group, resulted in compound 3 with diminished hCA IX inhibition activity (KI value of 75.2 nM); in contrast, the introduction of 4-fluoro/4-chloro groups produced compounds 4–5 with boosted the inhibitory potency of the hCA IX (KI values of 8.0–16.5 nM). (4) N-propanamide 20, with a KI value of 18.6 nM, was powerful than the corresponding N-acetamide 11 with a KI value of 66.8 nM. (5) The introduction of activating/deactivating groups on the phenyl ring of compound 2 (KI value = 27.3 nM) yielded compounds 10–17 with reduced inhibitory activity (KI values = 35.9–100.4 nM), except for compounds 12 and 16, which had improved hCA IX inhibitory potency (KI values = 12.7–24.1 nM). (6) Substitution of the phenyl group of compound 12 (KI value = 12.7 nM) with a benzyl moiety resulted in compound 18, which had significantly reduced hCA IX inhibitory activity (KI value = 256.4 nM).
3.3.4. SAR analysis for hCA XII inhibition
SAR analysis for hCA XII inhibition revealed several key factors. (1) 2-((2-Oxo-2-phenylethyl)thio)quinazolinone 2, with a KI value of 13.0 nM, was more potent than 2-((1-oxo-1-phenylpropan-2-yl)thio)quinazolinone 7 with a KI value of 76.8 nM. (2) The introduction of a chloro/fluoro group at the phenyl ring, such as in compounds 4 and 5 (KI values = 9.1–10.8 nM), improved the hCA XII inhibition activity and was similar to that of compound 2 (KI value = 13.0 nM). (3) The unsubstituted N-acetamide, compound 8, (KI value = 2.4 nM) resulted in more powerful hCA XII inhibition than N-substituted amides, compounds 9–20, (KI values = 8.7–113.4 nM). (4) hCA XII inhibition of N-acetamide 11, with a KI value of 31.6 nM, was more powerful than that of the corresponding N-propanamide 20 with a KI value of 66.7 nM. (5) The substitution of the phenyl group of N-(4-fluorophenyl)acetamide 12 (KI value of 8.7 nM) with a benzyl moiety resulted in the N-(4-fluorobenzyl)acetamide 18, with sharply reduced CA inhibitory activity (KI value = 113.4 nM).
3.4. Molecular docking
3.4.1. Molecular docking of compounds 17 and 20 with CA IX and CA XII isoenzymes
To further investigate the interactions between the selected active compounds 17 and 20 with the hCAs targets, we performed docking simulations into the binding pockets of the hCA isoforms, IX and XII, using the MOE SuiteCitation65 (data are summarised in and ).
Figure 2. Docking modes of active compounds 17 and 20 in the binding pockets of CA isoenzyme IX (PDB 5FL4). Predicted binding mode of compounds 17 (2D and 3D in upper panel), and 20 (2D and 3D in lower panel) with the hCA-IX target.
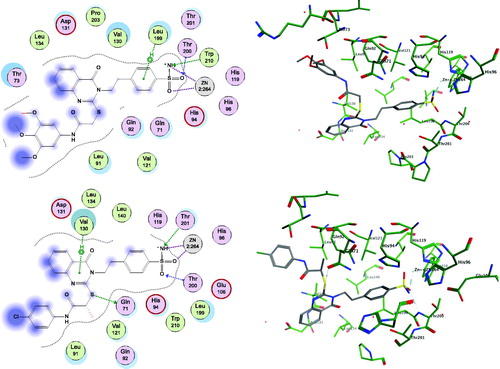
Figure 3. Docking modes of the active compounds 17 and 20 in the binding pockets of CA isoenzyme XII (PDB 1JCZ). Predicted binding mode of compounds 17 (2D and 3D in upper panel) and 20 (2D and 3D in lower panel) with hCA-XII target.
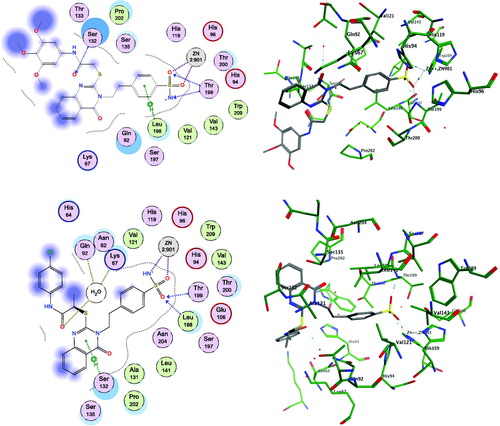
Both the compounds 17 and 20 were shown to directly interact with the zinc ion of CA IX and CA XII isoenzymes, via the sulphonamide anion of the active sites of both enzymes. However, the contributions of the quinazoline scaffold and the terminal bulky thioether fragments interaction are different, based on the CA isoform. In CA IX, the quinazoline ring of compound 20 interacts with the Gln71 residue through a stable hydrogen bond, and gets accommodated in the hydrophobic pocket lined by the Val121, Val130, Leu134, and Leu91 residues, thereby stabilising the binding (, lower panel). In addition, the terminal p-chlorobenzamide fragment formed a hydrophobic interaction with the Leu91 residue (, lower panel). In contrast, compound 17 was shown to bind similarly to the pocket of CA IX, except the unfavourable orientation of the quinazoline carbonyl moiety of compound 17 towards the hydrophobic pocket formed by Leu91 residue in CA IX (, upper panel). Also, the benzamide core showed a polar-nonpolar interaction with the Leu91 and Thr73 residues, as the bulky side chain causes steric hindrance, inducing conformational changes in the bulky thioether tail and the quinazoline groups (, upper panel). These differences in the binding of compounds 17 and 20 could be responsible for the observed differences in the KI values of the two compounds for CA IX.
Results also showed different interactions between CA XII and compounds 17 and 20 (). The carbonyl group on the quinazoline ring in compound 17 was stabilised by direct hydrogen bonding with the target residue Ser132 of CA XII (, upper panel). In addition, the Lys67 residue showed favourable hydrophobic binding to the quinazoline core of compound 17. The trimethoxybenzamide group of compound 17 was accommodated in the polar pocket of CA XII that included Ser132 and Thr133 residues (, upper panel). The placement of compound 20 within the CA XII pocket was not favoured, particularly because the quinazoline ring of compound 20 was trapped between the polar pocket of CA lined by the Ser135, Gln92, and Ser132 residues (, lower panel). Therefore, this interaction causes an energetically unfavourable change in the terminal benzamide and quinazoline scaffold of compound 20, which could be responsible for the decreased inhibitory activity of compound 20 (, lower panel).
3.4.2. Molecular orbital analyses
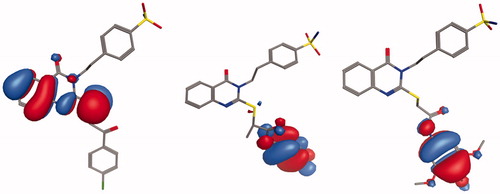
According to the frontier molecular orbital theory, HOMO and LUMO are the most important orbitals found in a molecule, as they can affect its biological activity, the molecular reactivity, the ionisation and the electron affinityCitation68–70. The molecular orbital analysis of the representative compounds 4, 17, and 20 () as an active and selective derivatives was done by exploring their structure-selectivity relationship. The electron transition from HOMO to LUMO occurs freely when the energy gap is small. The HOMO-LUMO energy gap for the compounds 4, 17, and 20 was calculated to be −0.3125, −0.2834, and −0.28949 eV, respectively. The negative energy values are indicative of a stable structure and confirm the eventual charge transfer interactions. The distributions and energy levels of the HOMO-LUMO orbitals computed for the above-mentioned compounds are represented in . HOMO and LUMO orbitals are mainly delocalised in the carbon and nitrogen of the quinazoline scaffolds and the sulphur ether atoms in the active compound 4. While they are mainly delocalised in the S-linker of the benzamide moiety, ring substituents in the compounds 17 and 20 reverse their interactions with the enzyme isoforms. These results indicate that the affinity of the selective compounds for the CA IX and CA XII binding sites could be because of the involvement of the thioether moiety, and that the quinazoline moiety could mostly provide the structural basis and the lipophilic function, contributing strongly to their selectivity. In addition, the low HOMO-LOMO energy gap suggests that the molecules have high stability and are in their lowest energy conformation.
Figure 4. Molecular orbital spatial distribution and localisation for the HOMO and LUMO of three representative compounds, 4 (left panel), 17 (middle panel), and 20 (right panel).
4. Conclusion
The CA inhibitory activity of 4-(2-(2-(substituted-thio)-4(3H)-quinazolinon-3-yl)ethyl)benzenesulfonamides (compounds 2–20) towards the hCA I, II, IV, and IX isoforms was assessed and compared with acetazolamide (AAZ), a typical sulphonamide inhibitor. Of the different hCA isoforms, hCA I was effectively inhibited by the compounds 2 and 4–13, with inhibition constant (KI) values in the range of 114.5–938.3 nM (AAZ: KI value of 250.0 nM), while compounds 3 and 14–20 showed moderate to weak CA inhibitory activity with KI values of 1447.0–5467 nM. Compounds 5, 8, 9, 11, 12, and 20 were revealed to be effective hCA II inhibitors, with KI values of 25.4–95.4 nM (AAZ: KI value of 12.0 nM). Compounds 2, 3, 4, 6, 7, 10, 13, 14, 15, and 16 showed modest to weak hCA II inhibitory activity with KI values ranging between 116.2 and 1099.0 nM. Compounds 2–17 and 20 displayed potent hCA IX inhibitory activity with KI values ranging from 8.0 to 100.4 nM compared to AAZ (KI value of 25.0 nM), whereas compounds 18 and 19 showed modest hCA IX inhibitory activity with KI values ranging between 256.4 and 145.1 nM, respectively. Ethylbenzenesulfonamide derivatives, 2, 4, 5, 8, 9, 11, 12, 13, 14, 16, and 17 showed potent hCA XII inhibitory activities with KI values of 2.4–49.1 nM compared to AAZ (KI value of 5.7 nM), whereas compounds 3, 6, 7, 10, 15, 18, 19, and 20 showed moderate hCA XII inhibitory activities with KI values of 59.7–113.4 nM. Compounds 2 and 4 showed characteristic effective and selective antitumor (hCA IX and hCA XII) carbonic anhydrase inhibitory activity with KI values (compound 2; 40.7 and 13.0 nM) and KI values (compound 4; 8.0, 10.8 nM). Compounds 2–7 showed high selectivity ratios for the inhibition of hCA IX over hCA I (15.0–95.0) and hCA IX over hCA II (2.3–23.0), while selectivity ratios of hCA XII over hCA I (5.5–70.0) and hCA XII over hCA II (1.4–17.0). Compounds 4 and 5 displayed selective inhibitory activity towards hCA IX over hCA I with selectivity ratios of 95.0 and 24.0 respectively, and hCA IX over hCA II with selectivity ratios of 23.0 and 5.8 respectively, as well as, selective inhibitory activity for hCA XII over hCA I and hCA XII over hCA II (selectivity ratios of 70.0, 44.0 and 17.0, 10.0, respectively). Compounds 12–17, and 19–20 exhibited selective inhibitory activities towards hCA IX over hCA I and hCA IX over hCA II (selectivity ratios of 23.0–195.0 and 3.2–19.0, respectively). In addition, compounds 8, 12, 14–17, and 19 showed selective inhibitory activity towards hCA XII over hCA I and hCA XII over hCA II (selectivity ratios of 48.0–158.0 and 5.4–31.0, respectively). Docking study of the selective derivatives, compounds 17 and 20, with the hCAs revealed consistent interactions, particularly selectivity-oriented hydrophobic and aromatic interactions through the S-alkyl substituent.
Supplemental Material
Download PDF (470.8 KB)Acknowledgements
The authors thank the Deanship of Scientific Research and RSSU at King Saud University for their technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019;14:1175–97.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Alterio V, Fiore AD, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Nocentini A, Supuran CT. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: a patent review (2008–2018). Expert Opin Therap Patents 2018;28:729–40.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60.
- Supuran CT. Carbonic anhydrases. Bioorg Med Chem 2013;21:1377–8.
- Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors: synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406.
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors: synthesis of sulfonamides incorporating DTPA tails and of their zinc complexes with powerful topical antiglaucoma properties. Bioorg Med Chem Lett 2001;11:575–82.
- Scozzafava A, Menabuoni L, Mincione F, et al. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem 1999;42:2641–50.
- Sheldrick GM. A short history of SHELX. Acta Crystallogr 2008;64:112–22.
- Abdel-Aziz AA, El-Azab AS, Abu El-Enin MA, et al. Synthesis of novel isoindoline-1,3-dione-based oximes and benzenesulfonamide hydrazones as selective inhibitors of the tumor-associated carbonic anhydrase IX. Bioorg Chem 2018;80:706–13.
- Abdel-Aziz AA, El-Azab AS, Ekinci D, et al. Investigation of arenesulfonyl-2-imidazolidinones as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:81–4.
- Angeli A, Abdel-Aziz AA, Nocentini A, et al. Synthesis and carbonic anhydrase inhibition of polycyclic imides incorporating N-benzenesulfonamide moieties. Bioorg Med Chem 2017;25:5373–9.
- Mohamed MA, Abdel-Aziz AA, Sakr HM, et al. Synthesis and human/bacterial carbonic anhydrase inhibition with a series of sulfonamides incorporating phthalimido moieties. Bioorg Med Chem 2017;25:2524–9.
- Abdel-Aziz AA, Angeli A, El-Azab AS, et al. Synthesis and biological evaluation of cyclic imides incorporating benzenesulfonamide moieties as carbonic anhydrase I, II, IV and IX inhibitors. Bioorg Med Chem 2017;25:1666–71.
- Abdel-Aziz AA, El-Azab AS, Ceruso M, Supuran CT. Carbonic anhydrase inhibitory activity of sulfonamides and carboxylic acids incorporating cyclic imide scaffolds. Bioorg Med Chem Lett 2014;24:5185–9.
- Alaa A-M, El-Azab AS, El-Subbagh HI, et al. Design, synthesis, single-crystal and preliminary antitumor activity of novel arenesulfonylimidazolidin-2-ones. Bioorg Med Chem Lett 2012;22:2008–14.
- El-Azab AS, Abdel-Aziz AA, Ayyad RR, et al. Inhibition of carbonic anhydrase isoforms I, II, IV, VII and XII with carboxylates and sulfonamides incorporating phthalimide/phthalic anhydride scaffolds. Bioorg Med Chem 2016;24:20–5.
- Abdel-Aziz AA, Angeli A, El-Azab AS, et al. Synthesis and anti-inflammatory activity of sulfonamides and carboxylates incorporating trimellitimides: dual cyclooxygenase/carbonic anhydrase inhibitory actions. Bioorg Chem 2019;84:260–8.
- Abdel-Aziz AA, El-Azab AS, Ghiaty AH, et al. 4-Substituted benzenesulfonamides featuring cyclic imides moieties exhibit potent and isoform-selective carbonic anhydrase II/IX inhibition. Bioorg Chem 2019;83:198–204.
- Abdel-Aziz AA, El-Azab AS, Bua S, et al. Design, synthesis, and carbonic anhydrase inhibition activity of benzenesulfonamide-linked novel pyrazoline derivatives. Bioorg Chem 2019;87:425–31.
- El-Azab AS, Abdel-Aziz AA, Bua S, et al. Synthesis and comparative carbonic anhydrase inhibition of new Schiff’s bases incorporating benzenesulfonamide, methanesulfonamide, and methylsulfonylbenzene scaffolds. Bioorg Chem 2019;92:103225.
- El-Azab AS, Abdel-Aziz AA, Bua S, et al. New anthranilic acid-incorporating N-benzenesulfonamidophthalimides as potent inhibitors of carbonic anhydrases I, II, IX, and XII: synthesis, in vitro testing, and in silico assessment. Eur J Med Chem 2019;181:111573.
- Abdel-Aziz AA, El-Azab AS, Abou-Zeid LA, et al. Synthesis, anti-inflammatory, analgesic and COX-1/2 inhibition activities of anilides based on 5,5-diphenylimidazolidine-2,4-dione scaffold: molecular docking studies. Eur J Med Chem 2016;115:121–31.
- Abdel-Aziz AA, El-Azab AS, Alanazi AM, et al. Synthesis and potential antitumor activity of 7-(4-substituted piperazin-1-yl)-4-oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies. J Enzyme Inhib Med Chem 2016;31:796–809.
- Al-Suwaidan IA, Alanazi AM, El-Azab AS, et al. Molecular design, synthesis and biological evaluation of cyclic imides bearing benzenesulfonamide fragment as potential COX-2 inhibitors. Part 2. Bioorg Med Chem Lett 2013;23:2601–5.
- Abdel-Aziz AA, Abou-Zeid LA, ElTahir KE, et al. Design, synthesis of 2,3-disubstitued 4(3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies. Bioorg Med Chem 2016;24:3818–28.
- Abdel-Aziz AA, Abou-Zeid LA, ElTahir KEH, et al. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibitory activities and molecular docking studies of substituted 2-mercapto-4(3H)-quinazolinones. Eur J Med Chem 2016;121:410–21.
- Alanazi AM, Abdel-Aziz AA-M, Al-Suwaidan IA, et al. Design, synthesis and biological evaluation of some novel substituted quinazolines as antitumor agents. Eur J Med Chem 2014;79:446–54.
- Alanazi AM, Al-Suwaidan IA, Abdel-Aziz AA-M, et al. Design, synthesis and biological evaluation of some novel substituted 2-mercapto-3-phenethylquinazolines as antitumor agents. Med Chem Res 2013;22:5566–77.
- Al-Obaid AM, Abdel-Hamide SG, El-Kashef HA, et al. Substituted quinazolines, part 3. synthesis, in vitro antitumor activity and molecular modeling study of certain 2-thieno-4(3H)-quinazolinone analogs. Eur J Med Chem 2009;44:2379–91.
- Al-Suwaidan IA, Abdel-Aziz AA, Shawer TZ, et al. Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4(3H)quinazolinone analogues. J Enzyme Inhib Med Chem 2016;31:78–89.
- Al-Suwaidan AM, Alanazi AA, Abdel-Aziz MA, Mohamed AS. Design, synthesis and biological evaluation of 2-mercapto-3-phenethylquinazoline bearing anilide fragments as potential antitumor agents: molecular docking study. Bioorg Med Chem Lett 2013;23:3935–41.
- El-Azab AS, Abdel-Hamide SG, Sayed-Ahmed MM, et al. Novel 4 (3H)-quinazolinone analogs: synthesis and anticonvulsant activity. Med Chem Res 2013;22:2815–27.
- El-Azab AS, Al-Omar MA, Abdel-Aziz AA, et al. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: molecular docking study. Eur J Med Chem 2010;45:4188–98.
- El-Azab AS, Eltahir KE. Synthesis and anticonvulsant evaluation of some new 2,3,8-trisubstituted-4(3H)-quinazoline derivatives. Bioorg Med Chem Lett 2012;22:327–33.
- El-Azab AS, Abdel-Aziz A-M, Ng SW, Tiekink ER. 6-Methyl-3-phenyl-2-sulfanylidene-1,2,3,4-tetrahydroquinazolin-4-one. Acta Crystallogr 2012;68:o862–o862.
- El-Azab AS, Abdel-Aziz AA, Bua S, et al. Synthesis of benzensulfonamides linked to quinazoline scaffolds as novel carbonic anhydrase inhibitors. Bioorg Chem 2019;87:78–90.
- El-Azab AS, ElTahir KE, Attia SM. Synthesis and anticonvulsant evaluation of some novel 4 (3H)-quinazolinones. Monatshefte Für Chemie-Chemical Monthly 2011;142:837–48.
- Mohamed MA, Ayyad RR, Shawer TZ, et al. Synthesis and antitumor evaluation of trimethoxyanilides based on 4(3H)-quinazolinone scaffolds. Eur J Med Chem 2016;112:106–13.
- Alanazi AM, Abdel-Aziz AA, Shawer TZ, et al. Synthesis, antitumor and antimicrobial activity of some new 6-methyl-3-phenyl-4(3H)-quinazolinone analogues: in silico studies. J Enzyme Inhib Med Chem 2016;31:721–35.
- El-Azab AS, Al-Dhfyan A, Abdel-Aziz AA, et al. Synthesis, anticancer and apoptosis-inducing activities of quinazoline-isatin conjugates: epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies. J Enzyme Inhib Med Chem 2017;32:935–44.
- El-Azab AS, Abdel-Aziz AA, Ghabbour HA, Al-Gendy MA. Synthesis, in vitro antitumour activity, and molecular docking study of novel 2-substituted mercapto-3-(3,4,5-trimethoxybenzyl)-4(3H)-quinazolinone analogues. J Enzyme Inhib Med Chem 2017;32:1229–39.
- Bozdag M, Alafeefy AM, Vullo D, et al. Benzenesulfonamides incorporating bulky aromatic/heterocyclic tails with potent carbonic anhydrase inhibitory activity. Bioorg Med Chem 2015;23:7751–64.
- Bozdag M, Alafeefy AM, Altamimi AM, et al. Synthesis of new 3-(2-mercapto-4-oxo-4H-quinazolin-3-yl)-benzenesulfonamides with strong inhibition properties against the tumor associated carbonic anhydrases IX and XII. Bioorg Med Chem 2017;25:2782–8.
- Kitamura K, Aihara M, Osawa J, et al. Sulfhydryl drug-induced eruption: a clinical and histological study. J Dermatol 1990;17:44–51.
- Wolf R, Brenner S. An active amide group in the molecule of drugs that induce pemphigus: a casual or causal relationship? Dermatology (Basel, Switzerland) 1994;189:1–4.
- Hospers GA, Eisenhauer EA, de Vries EG. The sulfhydryl containing compounds WR-2721 and glutathione as radio- and chemoprotective agents. A review, indications for use and prospects. Br J Cancer 1999;80:629–38.
- Epstein DL, Grant WM. Carbonic anhydrase inhibitor side effects: serum chemical analysis. Arch Ophthalmol 1977;95:1378–82.
- Bozdag M, Alafeefy AM, Carta F, et al. Synthesis 4-[2-(2-mercapto-4-oxo-4H-quinazolin-3-yl)-ethyl]-benzenesulfonamides with subnanomolar carbonic anhydrase II and XII inhibitory properties. Bioorg Med Chem 2016;24:4100–7.
- Alkahtani HM, Abdalla AN, Obaidullah AJ, et al. Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: dual inhibitors of EGFR/HER2. Bioorg Chem 2019;95:103461.
- Akurathi V, Dubois L, Lieuwes NG, et al. Synthesis and biological evaluation of a 99mTc-labelled sulfonamide conjugate for in vivo visualization of carbonic anhydrase IX expression in tumor hypoxia. Nuclear Med Biol 2010;37:557–64.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73.
- Nocentini A, Trallori E, Singh S, et al. 4-hydroxy-3-nitro-5-ureido-benzenesulfonamides selectively target the tumor-associated carbonic anhydrase isoforms ix and xii showing hypoxia-enhanced antiproliferative profiles. J Med Chem 2018;61:10860–74.
- Nocentini A, Gratteri P, Supuran CT. Phosphorus versus sulfur: discovery of benzenephosphonamidates as versatile sulfonamide-mimic chemotypes acting as carbonic anhydrase inhibitors. Chemistry 2019;25:1188–92.
- Nocentini A, Lucidi A, Perut F, et al. Supuran, alpha, gamma-diketocarboxylic acids and their esters act as carbonic anhydrase ix and xii selective inhibitors. ACS Med Chem Lett 2019;10:661–5.
- El-Husseiny WM, El-Sayed MA, Abdel-Aziz NI, et al. Structural alterations based on naproxen scaffold: Synthesis, evaluation of antitumor activity and COX-2 inhibition, and molecular docking. Eur J Med Chem 2018;158:134–43.
- Abdel-Sayed MA, Bayomi SM, El-Sherbeny MA, et al. Synthesis, anti-inflammatory, analgesic, COX-1/2 inhibition activities and molecular docking study of pyrazoline derivatives. Bioorg Med Chem 2016;24:2032–42.
- Alanazi AM, El-Azab AS, Al-Suwaidan IA, et al. Structure-based design of phthalimide derivatives as potential cyclooxygenase-2 (COX-2) inhibitors: anti-inflammatory and analgesic activities. Eur J Med Chem 2015;92:115–23.
- Al-Suwaidan IA, Abdel-Aziz NI, El-Azab AS, et al. Antitumor evaluation and molecular docking study of substituted 2-benzylidenebutane-1,3-dione, 2-hydrazonobutane-1,3-dione and trifluoromethyl-1H-pyrazole analogues. J Enzyme Inhib Med Chem 2015;30:679–87.
- El-Sayed MA, Abdel-Aziz NI, Abdel-Aziz AA, et al. Synthesis, biological evaluation and molecular modeling study of pyrazole and pyrazoline derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Part 2. Bioorg Med Chem 2012;20:3306–16.
- El-Azab AS, Abdel-Aziz AA, Ahmed HEA, et al. Exploring structure-activity relationship of S-substituted 2-mercaptoquinazolin-4(3H)-one including 4-ethylbenzenesulfonamides as human carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2020;35:598–609.
- Abdel-Aziz AA, El-Azab AS, AlSaif NA, et al. Synthesis, anti-inflammatory, cytotoxic, and COX-1/2 inhibitory activities of cyclic imides bearing 3-benzenesulfonamide, oxime, and beta-phenylalanine scaffolds: a molecular docking study. J Enzyme Inhib Med Chem 2020;35:610–21.
- El-Azab AS, Mary YS, Panicker CY, et al. DFT and experimental (FT-IR and FT-Raman) investigation of vibrational spectroscopy and molecular docking studies of 2-(4-oxo-3-phenethyl-3, 4-dihydroquinazolin-2-ylthio)-N-(3, 4, 5-trimethoxyphenyl) acetamide. J Mol Struct 2016;1113:133–45.
- Q. M. Molecular Operating Environment (MOE) Chemical Computing Group, Canada. 2012. Available from: http://www.chemcomp.com [last accessed on 30 Feb 2013].
- Leitans J, Kazaks A, Balode A, et al. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem 2015;58:9004–9.
- Whittington DA, Waheed A, Ulmasov B, et al. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci USA 2001;98:9545–50.
- Clare BWA. Frontier orbital energies in quantitative structure–activity relationships: a comparison of quantum chemical methods. J Theor Chim Acta 1994;87:415–30.
- Clare BW. The relationship of charge transfer complexes to frontier orbital energies in QSAR. J Mol Struct THEOCHEM 1995;331:63–78.
- Heaton CA, Miller AK, Powell RL. Predicting the reactivity of fluorinated compounds with copper using semi-empirical calculations. J Fluorine Chem 2001;107:1–3.