
Abstract
Inorganic anions inhibit the metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1) generally by coordinating to the active site metal ion. Cyanate was reported as a non-coordinating CA inhibitor but those erroneous results were subsequently corrected by another group. We review the anion CA inhibitors (CAIs) in the more general context of drug design studies and the discovery of a large number of inhibitor classes and inhibition mechanisms, including zinc binders (sulphonamides and isosteres, dithiocabamates and isosteres, thiols, selenols, benzoxaboroles, ninhydrins, etc.); inhibitors anchoring to the zinc-coordinated water molecule (phenols, polyamines, sulfocoumarins, thioxocoumarins, catechols); CAIs occluding the entrance to the active site (coumarins and derivatives, lacosamide), as well as compounds that bind outside the active site. All these new chemotypes integrated with a general procedure for obtaining isoform-selective compounds (the tail approach) has resulted, through the guidance of rigorous X-ray crystallography experiments, in the development of highly selective CAIs for all human CA isoforms with many pharmacological applications.
1. Introduction
Carbonic anhydrase (CA, EC 4.2.1.1) was discovered almost a century ago by Meldrum and RoughtonCitation1 who described its efficient catalysis for the reversible hydration of blood CO2 to bicarbonate and protons. Subsequently, such a process was shown to be relevant physiologically for many systems in organisms throughout the phylogenetic treeCitation2,Citation3. Relevant discoveries on the catalytic and inhibition mechanisms of this enzyme were thereafter mainly done by a Swedish scientist, in the period from the 60 s to the 80 s, S. Lindskog, who proposed the catalytic mechanism that is still valid todayCitation4, i.e. the enzyme contains a nucleophilic zinc hydroxide species that can hydrate CO2 in a two-step, “ping-pong” process, as follows: (i) the attack of CO2 by the zinc hydroxide bound within the active site of the enzyme resulting in formation of bicarbonate, which is released into solution, and (ii) regeneration of the zinc hydroxide nucleophilic species by a proton transfer reaction from a zinc-coordinated water molecule to the reaction medium, which is the rate-determining step of the catalytic cycleCitation4. The first investigation of anion CA inhibitors (CAIs) were also performed by Lindskog on both native and cobalt(II)-substituted enzymesCitation5, demonstrating by means of electronic spectroscopic techniques (using the cobalt-enzyme) that such inhibitors coordinate to the catalytic metal ion in a tetrahedral or trigonal-bipyramidal geometryCitation3–5. The first crystallographic studies of a CA isoform were also reported by researchers from Sweden (Lindskog’s and Nyman’s groups)Citation6, whereas the first complete structure, that of human (h) isoform hCA II was published in 1972Citation7.
Sulphonamides were reported to be CAIs in 1940 by Mann and KeilinCitation8 who also reported some side effects that were observed with the newly discovered (in 1940) antibacterial drug sulphanilamide (4-amino-benzenesulfonamide). Krebs on the other hand published the first comprehensive structure-activity relationship (SAR) study of sulphonamide inhibitorsCitation9 and together with Roughton, one of the co-discoverers of the enzyme, proposed CA as “a tool in studying the mechanism of reactions involving H2CO3, CO2 or HCO3−”Citation10, which is role that these enzymes still have today after thousands of valuable studies that have been performed worldwideCitation11–15. This is probably due primarily to the fact that CAs are drug targets for a multitude of disorders, which are aspects that will be discussed later in this review.
Nowadays, eight CA genetic families are known in organisms from across the phylogenetic tree, more precisely α-, β-, γ-, δ-, ζ-, η-, θ- and ι-CAsCitation16–23. Many representatives from these classes have been fully characterised and X-ray crystallographic structures are now known for several (and in some cases many) members of the α-, β-, γ-, ζ- and θ- classesCitation16–23, which constitutes an excellent example of convergent evolution at the molecular level. However, the structures of δ-, η- and ι-CAs have not yet been reported to date.
Importantly, the progress in X-ray crystallography over the last few decades has revealed the elusive binding modes of CO2 and bicarbonate, which are the two main substrates of the enzymeCitation24,Citation25 (. Indeed, as seen in for the α-class hCA II, CO2 is bound in a hydrophobic pocket that is lined with residues Val121, Vals143, Trp209 and Leu198 in close proximity (but not directly coordinated) to the zinc ion, whereas bicarbonate is coordinated to the metal ion in a monodentate fashion. Highly relevant studies from the groups of McKenna and Djinovic-CarugoCitation24 based on high pressure and cryo-crystallographic data demonstrated that the substrate binding modes are probably similar in all CA classes. Such a conclusion was further supported by crystallographic structures of β-CAs by McKenna’s groupCitation25, in which the binding mode of CO2 to a β-CA expressed in the bacterium Pseudomonas aeruginosa (psCA3) was reported.
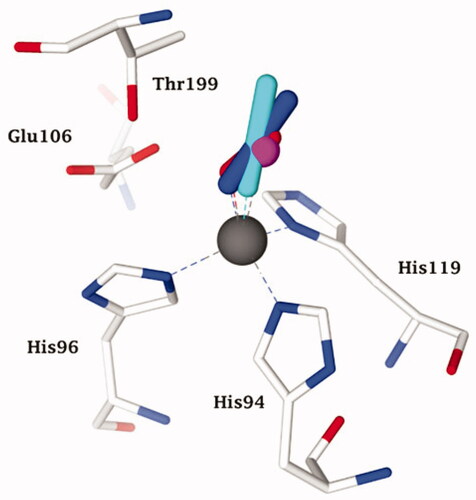
Figure 1. Active site view of the superimposed hCA II adducts with its main substrates: CO2 (green, pdb 2VVA) and bicarbonate (pink, pdb 2VVB) Citation24. The zinc ion is shown as a grey sphere with its three coordinated protein ligands (green/red), His94, 96 and 119. Amino acid residues involved in the binding of the substrates/inhibitors, at the bottom of the active site, are also shown.
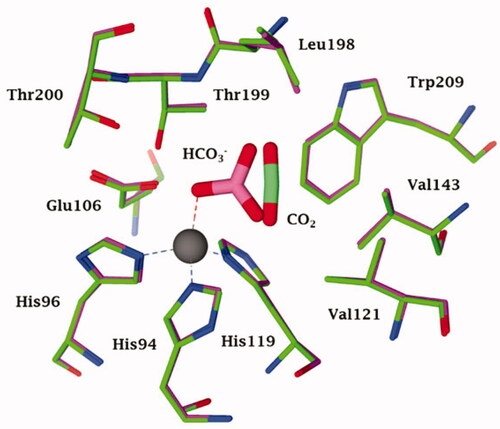
Figure 2. Representation of the superimposed X-ray crystallographic data for hCA II adducts with cyanate (cyan, pdb 4E5QCitation31) bromide (magenta, pdb 1RAZCitation32) azide (blue, pdb 1RAYCitation32) O2 (presumably as superoxide anion or radical anion; red, pdb 5EOICitation33) with a tetrahedrally coordinated active site zinc ion (grey sphere). The key active site zinc ligands (His94, 96 and 119) and “gate-keeping” residues (Thr199, Glu106) are also shown.
2. Anion inhibitors
As mentioned above, LindskogCitation3–5 reported some of the first anion CA inhibition studies, considering the well-known fact that many (in)organic anions have a high affinity for complexing transition metal ions in solution or within the active sites of metalloenzymesCitation26–30. Such pioneering studies in this field, before the X-ray crystallographic techniques became widely available (i.e. around 1995), used various biophysical techniques, such as circular dichroism, electron paramagnetic spectroscopy (EPR), electronic spectroscopy (UV/Vis), and NMR techniques together with metal-substituted CAs (incorporating Co(II), Cu(II), Mn(II), Ni(II), Cd(II) and other transition metal ions which have the desired spectral/paramagnetic properties, of which Zn2+, a d10 metal ion, is devoid). This allowed the investigation of the interactions of CA with various anions such as cyanide, cyanate, thiocyanate, azide, halides, carboxylates, hydrogen sulphide, and more complex anions (ferrocyanide, ferricyanide, etc.)Citation27–30. Such interesting and valuable studies were mainly reported by the groups of ColemanCitation29,Citation30 and Bertini-ScozzafavaCitation27,Citation28, which reinforced the earlier conclusionsCitation3–5 that the anion CAIs coordinate to the metal ion in the enzyme active site in a tetrahedral or more rarely, a trigonal bipyramidal geometryCitation26–30, as shown schematically in for some anion inhibitors, again using hCA II as model enzyme and high-resolution X-ray crystallographyCitation31–33.
Figure 3. Representation of the superimposed hCA II adducted to cyanate (cyan, pdb 4E5QCitation31) bicarbonate (pink, pdb 2VVBCitation24) urea (as anion) (white, pdb 1BV3 [39]), and trithiocarbonate (light blue, pdb 3K7KCitation40) The zinc ion (grey sphere), its protein ligands (His94, 96 and 119) and gate-keeping residues (Thr199, Glu106) are also shown.
![Figure 3. Representation of the superimposed hCA II adducted to cyanate (cyan, pdb 4E5QCitation31) bicarbonate (pink, pdb 2VVBCitation24) urea (as anion) (white, pdb 1BV3 [39]), and trithiocarbonate (light blue, pdb 3K7KCitation40) The zinc ion (grey sphere), its protein ligands (His94, 96 and 119) and gate-keeping residues (Thr199, Glu106) are also shown.](/cms/asset/6911aa26-9fda-4687-825d-fb54f97c3ed6/ienz_a_1882453_f0003_c.jpg)
Clearly based on the data in , all these anion inhibitors from significantly different chemical classes (e.g. halides, azide, cyanate and even oxygen, presumably bound as anion or radical anionCitation33) act as monodentate ligands to the zinc ion, which is in its usual tetrahedral geometry, with three additional ligands (residues His94, His96 and His119). All these (and other) anions have their closest atom to zinc at a distance of about 2 Å, and most of these anions show a rather low affinity (usually millimolar to micromolarCitation26) for this CA isoform. Thus, the zinc remains tetrahedrally coordinated upon inhibition by displacement of a water molecule ligand (or hydroxide) by many different anions. However, in 1993, the results of an X-ray crystallography study performed by the Liljas' group showed thatCitation34 cyanate and cyanide (well known “metal poisons”) do not inhibit CAs by this metal binding mechanism but instead are bound at a non-coordinating distance, presumably in the same location that the substrate CO2 binds (n.b. the CO2 binding mode was not precisely known at that time). Liljas presented these results in 1992 at an international bioinorganic chemistry conference in Florence prior to their publication in the following yearCitation34, in which many scientists active in the CA field participated. This discovery was considered by many to be a turning point in our understanding of CA inhibition mechanisms. However, these surprising findings presented by Liljas were difficult to rationalise, especially considering the fact that Bertini and Scozzafava’s groups, who continued the early research lines pioneered by Lindskog and Coleman, mentioned aboveCitation5,Citation29, presented highly convincing evidence that cyanate and cyanide coordinate to the metal ion (zinc or cobalt) in the CA active site based on data from electronic spectroscopy, EPR and NMR methodsCitation27,Citation28. One of us (CTS) proposed at the 1992 meeting that cyanate and presumably also cyanide might instead be hydrated by the CA enzyme given that these anions have a shape that is rather similar to that of the substrate CO2, hypothesis which has been validated in an interesting computational study by Merz and Banci’s groupsCitation35. Although the eventual experimental validation that CAs can hydrate cyanate and cyanide was more arduous than initially thought, ultimately Maren’s group and one of our groups (CTS) demonstrated that this was definitively the caseCitation36. Maren’s laboratory was highly skilled in measuring inhibition constants of anions, including halides, cyanate, cyanide, etcCitation37,Citation38. as well as sulphonamides, considering that he was a co-discoverer of several sulphonamide CAIs that have been used clinically for decades as well as the developer of two assay methods based on the CO2 hydration reactionCitation2. Maren’s group measured the liberation of ammonia from carbamate formed by CA-catalyzed cyanate hydrolysisCitation36. Although the ammonia formation was measured, the amount of ammonia was near the limits of detection for the method that was used and no clear-cut conclusions could be drawn. However, we proposed that cyanate once hydrolysed to carbamate within the CA active site may act as a suicide inhibitor, remaining strongly coordinated to the zinc ionCitation36, which might also explain the uncommon crystallographic results reported in ref.Citation34 The controversy was resolved years later by McKenna’s groupCitation31 who reported a high-resolution X-ray crystal structure of hCA II with cyanate bound (. Cyanate is directly coordinated to the metal ion just like most other anion inhibitors that have been investigatedCitation31. Although this definitive data was published in 2014, which should have “put the debate to rest”, the egregiously incorrect conclusion from the Liljas paper of 1993Citation34 has not yet been rectified by a retraction or correction. Furthermore, the protein database files of the two adducts to hCA II with cyanate and cyanide from Liljas’s group have not been deposited in the Protein Database.
However, this research line ultimately produced some interesting results. Cyanamide (H2N-CN or NH = C=NH, as possible tautomers) is another molecule with a similar linear shape with respect to the sp hybridised carbon atom as cyanate and CO2. Briganti et al.Citation39 investigated the role of cyanamide as either a substrate or inhibitor for CAs by using kinetic, electronic spectroscopic and X-ray crystallographic techniques. Again, and to the great surprise, it has been possible to demonstrate that cyanamide is initially acting as a weak inhibitor, which can then undergo a hydration reaction within the enzyme active site resulting in the formation of urea (more precisely the ureate anion), which can remain coordinated to the metal ion, as shown in , in which other structurally-related anions, such as cyanateCitation31, carbonateCitation24 and trithiocarbonateCitation40 are also shown superimposed.
Figure 4. Representation of the binding of A) O2− to a Zn2+/Cu2+ hCA II (pdb 5EOICitation33) and B) nitrite to Cu2+ substituted Cu2-hCA II (pdb 6PDVCitation43) Distances of the closest atom of the anion to the metal ion are: Zn-O (in the O2 adduct) of 1.88 Å; Cu-O (nitrite adduct) of 2.14 Å.
Cyanamide (unlike cyanate) was thus proven to be a new CA substrate, which is not that surprising considering that it is isosteric and isoelectronic with CO2Citation39. However, the most interesting result was probably that urea could not be displaced from the inhibited enzyme even by using highly effective, low nanomolar sulphonamide inhibitors, which demonstrated that ureate is probably one of the most effective CAIs known to dateCitation39. It should also be noted that urea (neutral molecule) is a very ineffective inhibitor (no KI was measured up to 100 mM) probably because it cannot be readily deprotonated in aqueous solutions for enzyme measurement assaysCitation39. Thus, only the deprotonated form, which is formed in situ through the suicide inhibition effects of cyanamide acts as a CAI, which is in fact the situation we expected (but could not initially confirm) for cyanate. As seen in , cyanate, carbonate and trithiocarbonate (all zinc binders) do not completely overlap in the X-ray crystal structures but they bind in a similar fashion, being monodentate ligands of the Zn(II) ion, which is in its stable tetrahedral geometryCitation24,Citation31,Citation40. The same geometry was observed also for the ureate adduct () and it was also bound in a monodentate fashion like many other small anions that have been reported to date. However, the ureate is tilted and has a very different orientation towards the gate-keeping residues Glu106-Thr199Citation39. In a subsequent studyCitation41, by using cryo-crystallography, it has been possible to “freeze” and observe the formation of the intermediates in the CA-catalyzed cyanamide hydration reaction, which revealed a new proposed reaction mechanism for this hydrolytic process.
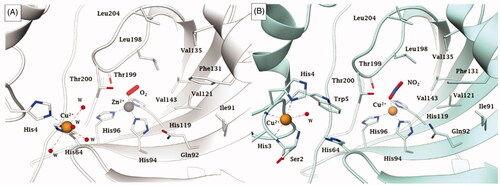
Apart the active site metal ion, hCA II has an additional metal binding site, at its entranceCitation42. Copper(II) binds rather efficiently in this additional site, being coordinated by His64, His4 and several water moleculesCitation33,Citation42. This form of the enzyme, with Zn(II) at its active site and Cu(II) at the secondary site is referred to as Zn/Cu-hCA II, in contrast to the enzyme which has zinc substituted by copper also within the catalytic centre (Cu2-hCA II)Citation43. Recently, McKenna’s group proposed that the Cu2-hCA II also has nitrite reductase activity, with nitrite being initially coordinated (as a bidentate ligand) to the active site copper ion (). In contrast, as mentioned also above, the Zn/Cu-hCA II was observed with an O2 molecule coordinated to zinc (), which most probably is a superoxide anion (or anion radical), as suggested by computational techniquesCitation33. A major overall conclusion is that all serious studies performed to date on hCA II demonstrate that anions are coordinated to the metal ion in the enzyme active site.
3. Sulphonamides and other zinc binders, the tail approach
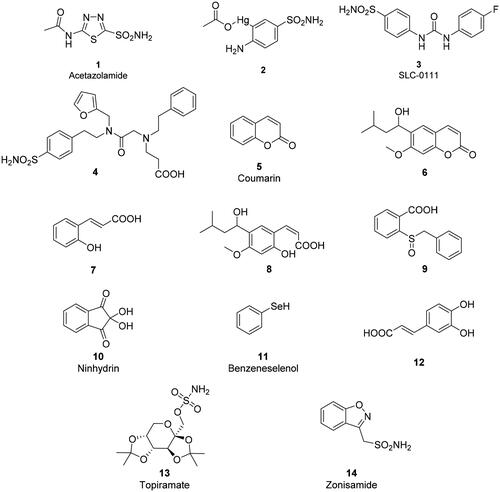
Primary sulphonamidesCitation44–46 were known for decadesCitation8–10 to potently inhibit CAs by binding as RSO2NH- anions to the Zn(II) ion within the enzyme active site, as shown by using various spectroscopic techniquesCitation5,Citation12,Citation13,Citation30 as well as a crystallographic study from Liljas’ group, in which acetazolamide 1, and 3-acetoxymercuri-4-aminobenzenesulfonamide 2 (), and the anion thiocyanate, were crystallised in complexes with hCA IICitation47. As for the anions mentioned in the preceding paragraphs, the sulphonamide inhibitors substituted the water/hydroxide ion acting as the fourth ligand to the tetrahedrally coordinated zinc site in a monodentate coordination with the metal ion through the deprotonated nitrogen atom of the sulphonamide moiety, resulting in a favourable hydrogen bond with Thr199, a conserved residue in most α-CAs. In contrast, thiocyanate binding was reported to result in a trigonal-bipyramidal coordination of the zinc centre because SCN- added to the coordination sphere of the non-inhibited enzymeCitation47. Since then, hundreds of sulphonamides were crystallised in complexes with various CA isoforms and in all of them the sulphonamide zinc-binding group (ZBG) participates in the same interactions as mentioned above for acetazolamide (reviewed in refs.Citation14,Citation48).
Although acetazolamide and other first generation sulphonamide CAIs (such as methazolamide, ethoxzolamide, dichlorophenamide, etc.; refer to refs.Citation11,Citation12,Citation14 for their structures) are potent, low nanomolar CAIs, they are non-selective inhibitorsCitation11, strongly inhibiting most hCA isoforms that are known to date (12 catalytically active isoforms that have been reported, hCA I-VA, VB, VI, VII, IX, XII-XIVCitation11,Citation12,Citation14) As a consequence, the first- and even second-generation inhibitors used clinically resulted in a range of documented side effects connected to the inhibition of off-target isoformsCitation48–57.
For such reasons, designing isoform-selective CAIs became the main issue in the revival of the CA field in the mid to late 90 s for obtaining compounds with an improved safety profile and efficacy compared to the inhibitors available at that timeCitation58–60. The tail approach has thus emerged in that period as an innovative modality for developing CA-selective inhibitorsCitation61. The idea is very simple: the attachment of moieties that may induce the desired physico-chemical properties (e.g. enhanced hydro- or liposolubility) to scaffolds of simple aromatic/heterocyclic sulphonamides (e.g. sulphanilamide, metanilamide, orthanilamide, and their derivatives; 5-amino-1,3,4-thiadiazole-2-sulphonamide, 4-methyl-5-imino-1,3,4-thiadiazoline-2-sulphonamide, and many other simple sulphonamides etc.) might lead to interactions with the external part of the CA active site, and not only with residues near the zinc catalytic centre that are the most highly conserved in all hCA isoforms. Indeed, the region at the entrance of the active site cavity is the most variable between the different human isoformsCitation61. Detailed kinetic studies were thereafter performed on over 10,000 different sulphonamide derivativesCitation62–71, which were synthesised and investigated in our laboratories and an additional 15,000 derivatives were received from academic groups and pharmaceutical companies in collaboration with the Florence group. Crystallography in complex with various isoforms was done on several hundred such derivatives. This type of research demonstrated that the hypothesis was correct and that it is possible to design sulphonamide CAIs that are selective for each isoform of interest by tailoring the dimensions (length, bulkiness, etc.) as well as the chemical nature of the various tailsCitation61–71. This research led to the discovery of many new sulphonamide, sulfamate and sulfamide classes, including the ureido-benzene-sulphonamidesCitation70,Citation72 as well as derivatives possessing more than one tail attached to the scaffold, such as dual-tailed or tri-tailed CAIsCitation73,Citation74. Of the many compounds discovered in this way includes SLC-0111, compound 3, which is a CA IX/XII-selective inhibitor that is now in clinical trials as an antitumor/antimetastatic agent (see discussion later in the text). Another notable example is the three-tailed sulphonamide 4 ().Citation71 Both SLC-0111 and sulphonamide 4 strongly inhibit and bind to hCA IX ()Citation71,74.
Figure 5. Active site view of A) hCA IX catalytic domain adducted to SLC-0111 (magenta)Citation74 and B) hCA II adducted to the three tailed inhibitor 4 (in green).Citation71 H-bonds are represented as black dashed lines. The Zn(II) ion (grey sphere) and some important amino acid residues involved in the binding of inhibitors are shown.
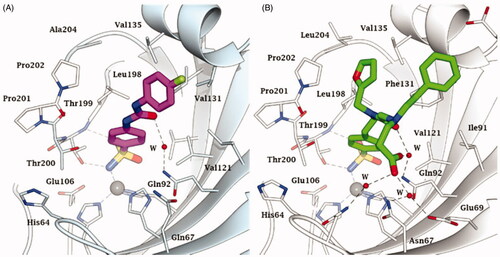
Figure 6. Historical overview on the discovery of the main chemotypes with CA inhibitory activity.
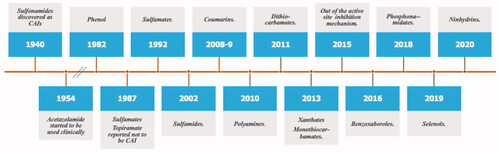
The tail approach was virtually the only strategy used to obtain CAIs from the moment it was disclosed, and it has been adopted by research groups all over the worldCitation71,Citation75–91. For space reasons only a limited number of such research was cited here, as the field was recently reviewedCitation14,Citation48. The advantage of the tail approach over other strategies to obtain isoform-selective CAIs are the facility of the syntheses, the versatility and possibility to introduce a variety of tails/scaffolds with different chemical properties, and thus to generate chemical diversity. As a consequence, these procedures were extended to many other classes of CAIs, not only to the sulphonamides and their isosteres (sulfamides/sulfamates, which are not discussed here in details; see refs.Citation14,Citation48 for recent reviews). These new chemotypes will be discussed in the next paragraph, together with some new CA inhibition mechanisms which emerged more recently.
4. Other chemotypes with CA inhibitory power
By comparing the sulphur-based, carbon-basedCitation45 and phosphorus-based ZBGs, a range of novel classes of non-sulphonamide CAIs which interact with the zinc ion were reported, including phosphonamidatesCitation91, dithiocarbamatesCitation92 and their derivatives (monothiocarbamatesCitation93, xanthates and trithiocarbonatesCitation94) selenolsCitation95,Citation96, carboxylatesCitation97, hydroxamatesCitation98, benzoxaborolesCitation99–101, and carbamatesCitation102. In addition, other classes of inhibitors that have been developed which exhibit a different inhibition mechanism compared to the zinc binders discussed above. It should be noted that many of the new chemotypes bind further away from the metal ion, interacting both with the hydrophobic and hydrophilic halves of the CA active siteCitation14,Citation48. These new inhibition mechanisms include:
CAIs that anchor to the zinc-coordinated water moleculeCitation103,Citation104. Polyamines, such as spermine and its derivativesCitation103, the simple phenolCitation104 and many of its derivativesCitation105–108 together with polyphenolsCitation109 inhibit CAs in this way. This inhibition mechanism is also shared by the sulfocoumarins and their derivatives, as demonstrated by Zalubovskis groupCitation110–113 using kinetic and crystallographic studies. These classes of highly innovative CAIs were developed by the Riga groupCitation110–113 starting from coumarins and their derivatives as a class of inhibitors with a totally new inhibition mechanism, which has been unravelled by some of usCitation114–124. Thioxocoumarins are the only exception, as they bind similar to the phenols, anchoring with their exocyclic sulphur atom to the zinc coordinated water moleculeCitation114.
Coumarins and many of their derivatives such as mono- and dithio-, as well as seleno/tellurium-coumarins but also many other structurally related compoundsCitation115–124 were shown to possess a very particular inhibition mechanism. This was demonstrated for the simple coumarin 5 and one of its derivative (compound 6), a natural product isolated from the Australian plant Leionema ellipticumCitation115,Citation116. The lactone ring of the coumarin is hydrolysed by the esterase activity of CA, leading to the formation of cis- or trans-2-hydroxycinnamic acid derivatives, of types 7 and 8 (), which thereafter bind far from the zinc ion, at the entrance of the active site cavity (the region which is more variable among the different human isoforms, as mentioned above). As a consequence, coumarins and their derivatives behave as pro-drug inhibitors, but also show a very high selectivity for many CA isoforms, depending on the substitution pattern on their bicyclic ring systemCitation115–124. Many representatives belonging to this family of CAIs in fact possess extremely good selectivity ratios for inhibiting isoforms of interest (e.g. CA IX and XII) versus highly distributed, “house-keeping” off-target isoforms, such as CA I and IICitation115–124.
Probably one of the strangest CA inhibition mechanism is the one reported by De Simone’s and Carradori’s group for a benzoic acid derivative (compound 9) which binds outside the active site cavity, blocking the His64 residue (the proton shuttle residue) in its out conformationCitation125. Again, X-ray crystallography was crucial for demonstrating this highly interesting CA inhibition mechanism.
An overview of the main new chemotypes acting as CAIs discovered up until now is shown in , in which the “fresh” discovery of the ninhydrins is also mentionedCitation126. Indeed, ninhydrins, such as compound 10, possess a gem-diol stable functionality which has been shown by means of kinetic and computational techniques to coordinate to the metal ion from the enzyme active siteCitation126. Furthermore, shows the X-ray crystal structures of some of the new chemotypes detected in the last period, such as the selenol 11Citation95 (), unsubstituted ninhydrin 10Citation126 () and a catechol derivative 12Citation127 () which has been recently reported to bind to hCA II by a new inhibition mechanism. Like many phenols, the catechol 12 is anchored to the zinc-coordinated water molecule, but being a diphenol, the second OH moiety additionally binds to the “deep water” molecule in the active site. As mentioned above, selenol 11 and the ninhydrin 10 () directly coordinate to the zinc ion in the enzyme active site (. Many of these inhibition mechanisms were thoroughly reviewed in ref.Citation128.
Figure 7. Active site view of hCA II complexed to A) selenol 11 (pdb 6hX5) Citation96, B) ninhydrin 10 (predicted in silico)Citation127 and C) catechol derivative 12 (pdb 6YRI) Citation128. H-bonds are represented as black dashed lines. The Zn(II) ion (grey sphere) and residues involved in its coordination and some active site residues near the binding of inhibitors are shown in CPK colours. Water molecules are shown as red spheres.
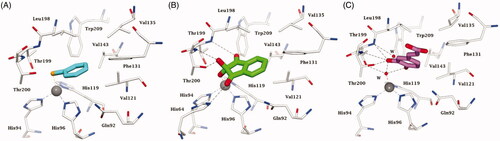
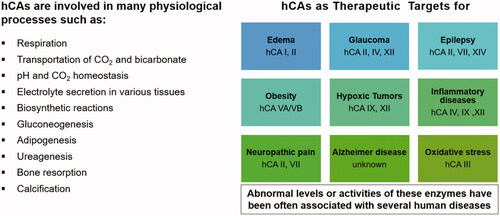
Figure 8. hCAs as drug targetsCitation11,Citation14,Citation51–57. From edoema and glaucoma, to obesity, neuropathic pain, hypoxic cancers, cerebrovascular diseases and oxidative stress, many isoforms are involved in diverse pathologies for which isoform-selective inhibitors showed a relevant potential to be translated to clinical entities.
5. New applications of the CAIs
For the last 65 years the CAIs were used as diureticsCitation2,Citation51,Citation53,Citation55 and anti-glaucoma agentsCitation2,Citation56–59, and they still have a firm place for the management of these conditionsCitation53,Citation57. However, in the last decade, several other highly relevant applications emerged, probably due to the availability of much more isoform-selective inhibitors. They include the use of CAIs such as acetazolamide 1, topiramate 13 and zonisamide 14 as antiepilepticsCitation129,Citation130 and anti-obesity agentsCitation54,Citation131,Citation132, targeting diverse isoforms (CA II and VII for the antiepileptic use, and CA VA/VB as antiobesity agents) Citation129–132. In fact, as seen from , most of the CA isoforms, alone (or in combinations of few of them) possess therapeutic/pharmacological applications.
Chart 1. Structure of the compounds 1–14 discussed in this review.
In fact, nowadays the CAIs were proposed to be used or are already used for the management of the following pathologies, in addition to the classical ones mentioned above: (i) treatment of idiopathic intracranial hypertension, by targeting hCA I and II but presumably also brain/cerebrospinal fluid isoformsCitation133; (ii) cerebral ischemia/diabetic cerebrovascular pathologies (the targeted isoforms seem to be hCA IX and XII, possibly also hCA VA)Citation134; (iii) neuropathic pain, by targeting the bran/peripheral nervous system isoforms hCA II and VIICitation135,Citation136; (iv) inflammation/arthritis, by targeting hCA IX/XII and possibly hCA IVCitation137–140; (v) hypoxic tumours, by targeting the two isoforms discovered to be associated with tumours, hCA IXCitation141 and hCA XIICitation142. This is in fact the field in which the highest number of studies have been published in the last decadesCitation143–155. Most of the proof-of-concept studies which validated the various isoforms for the management of such widespread diseases were conducted either in Florence or by collaborations with well-known groups worldwide, which used the compounds developed in Florence for these studies. Highly important for the success of these investigations was the fact that the X-ray crystal structures of almost all hCA isoforms (except hCA VB, X and XI) were published in the last 15 yearsCitation156–161. This allowed for highly focussed drug design campaigns in order to obtain isoform-selective compounds for all these enzymes, and again using the tail approachCitation61 and a variety of newly discovered chemotypes, discussed above, were essential for the success of these efforts.
6. CA activators and their applications
The carbonic anhydrase activators (CAAs) have been the focus of significant controversy for a long time up until 1997 when the first X-ray crystal structure of a complex of an activator (histamine) bound to hCA II was reported and the activation mechanism was revealed in molecular level detailCitation162. The activator, which was observed to be directly bound within the enzyme active site, promotes the formation of an enzyme – activator complex, in which the proton shuttling moieties present in the activator participate in the rate-determining step of the catalytic cycle, i.e. the transfer of a proton from the zinc-coordinated water to the external reaction medium, similar to the natural proton shuttle, which is residue His64 (in many CA isoforms), as mentioned aboveCitation162,Citation163. In such enzyme-activator complexes, the proton transfer is intramolecular, being more efficient compared to the intermolecular transfer to buffer molecules, which are not bound within the enzyme cavityCitation162,Citation163. Many X-ray crystal structures with amines and amino acid activators were reported thereafter, in addition to histamineCitation162, including L- and D-His bound to hCA II and hCA I, L- and D-Phe, D-Trp, L-adrenaline and pyridinium derivatives of histamine bound to hCA II, which confirmed this general CA activation mechanism outlined aboveCitation164–167. The thirteen catalytically active mammalian CAs (e.g. CA I-VA, VB, VI, VII, IX, XII-XV) were also investigated for their interaction with a rather large library of amino acids and aminesCitation168,Citation169, whereas several drug design studies have also been performedCitation170–174. Why are these studies relevant? In the last few years it has been demonstrated that CAAs of the amino acid type (e.g. D-Phe) can enhance memory and learning, which is antagonised by the simultaneous administration of a CAI of the sulphonamide type (e.g. acetazolamide) Citation175. A recent study showed that administration of the D-Phe to rats rapidly activated the extracellular signal-regulated kinase (ERK) pathways, which are involved in critical steps of memory formation, both in the cortex and the hippocampus, two brain areas crucially involved in memory processingCitation175,Citation176. Even more recently, the same groups (including the Florence team) demonstrated that CAAs play a crucial role in extinction of contextual fear memoryCitation177, opening the way towards the validation of CAs as new targets for improving cognition, but also in therapeutic areas, such as phobias, obsessive-compulsive disorder, generalised anxiety, and post-traumatic stress disorders, for which few efficient therapies are available to dateCitation175–177.
7. Discussion and conclusions
The drug design landscape for modulators of CA activity, both inhibitors and activators, appears to be a dynamic and healthy field, in which many relevant advances have been achieved in the last several decades through continuous contributions from many research groups, as outlined above. However, recently an event disturbed this situation. In an egregious polemic from Jonsson and LiljasCitation178, the measurements of some CA inhibition data were queried, in addition to some other aspects that will be discussed later in this article. This action was based on some typographical errors found in Supplementary Information from 5 papers from our group (among the > 1600 that have been published). We have replied to the above-mentioned articleCitation178 in an Editorial published in the same journalCitation179 as well as two additional review articlesCitation153,Citation180. Jonsson and Liljas replied to our editorialCitation179 with a new comment published recentlyCitation181. Furthermore, Liljas “promoted” their initial articleCitation178 by writing a denigrating and arrogant letter to the president of the University of Florence about one of us (CTS), stating that he is, “bothered about how long his [Supuran’s] poorly performed science could continue without serious criticism”, and “I followed Supuran's crystallographic investigations with horror”, but no technical comments regarding our presumed crystallographic errors were mentioned in this letter or in their papers, although Liljas himself is a crystallographer. We worked in this field in collaboration with well-established crystallographers from several laboratories in Italy, Germany, USA, Australia and Singapore over the yearsCitation178. Liljas continued, “the measurements are so poorly done that an undergraduate student reporting these in his works should have failed” and “we tried to publish our manuscript in the journals where Supuran has published his work but failed. This covers a good number of journals”, meaning probably that serious journals did not take into consideration his poorly formulated polemic. Liljas ends his letter to the rector: “The work by Supuran that we criticise needs to be repeated. There may be really excellent inhibitors among the hundreds of compounds that he has studied, but his analysis does not give a trustworthy account. It is important that the field and his co-workers become fully aware of the quality of his analyses and to know that much has to be done again”. In other words, this scientist who published completely erroneous conclusions based on crystallography data as shown here for the cyanate and cyanide examplesCitation34 and elsewhere by othersCitation31 and who has not withdrawn or corrected these published results after 27 years, finds it insufficient to publish his conceptually erroneous polemicCitation178, but also wants to defame our group to our institution. We have replied to the technical issues raised by Jonsson and Liljas, as mentioned above. In their comment on our replyCitation180 they indicated they were “happy by our prompt response” and that some clarifications were achievedCitation181, however now their criticism relating to the measurement of inhibition constants was extended to the entire medicinal chemistry community, not only to our group, stating that the “original article was not intended to raise specific concerns regarding just one research group but rather was intended to raise concerns regarding medicinal chemistry publications in general, and we are glad that Prof. Supuran agrees that there is room for improvement!” CTS hardly agrees with anything from their analysis. Such a strong and prejudicial opinion from someone without experience in the medicinal chemistry field is disappointing. Notably, the “best practice” examples for the measurement of KIs that Jonsson and Liljas provide in their replyCitation181 are the highly controversial articles from Johnson & Johnson regarding topiramate 13Citation182–185. Indeed, topiramate was initially reported to not be a CAICitation182, then a millimolar inhibitorCitation183, then a micromolar inhibitorCitation184 until finally it was established to be a sub-micromolar inhibitorCitation185 by the same group of Johnson & Johnson who initially discovered the compound. We have reported the X-ray crystal structure of this important drug bound to hCA II and demonstrated that it is a low nanomolar inhibitor for many CA isoformsCitation186. There are also other unusual comments from Jonsson and LiljasCitation181 regarding the native mass spectrometry measurements. Presumably, the main issue with this method is that it can be used to measure dissociation constants that are in very good agreement with the stopped flow method used in Florence, in addition to other methods such as isothermal titration calorimetry (ITC) and differential scanning calorimetry (DSC)Citation187,Citation188.
As noted above, there are no original contributions in the literature from Jonsson and/or Liljas regarding the measurement of inhibition constants (for CAs or any other enzyme) nor for any drug design studies targeting CAs or other druggable proteins. Jonsson is a biochemist who published several papers in the CA field and Liljas was a crystallographer (now retired for some time), who, as we mentioned earlier, contributed some important X-ray crystallographic studies in the 70 s and 80 s on CAs and then some questionable CA data in the 90 s (as discussed above). It should be noted that there is no analysis at all of our crystallographic work in the two papers by Jonsson and LiljasCitation178,Citation181 or in the letter to the rector, from more than 250 papers that we have published in that field, as mentioned here and elsewhereCitation12,Citation14. However, the polemic by Jonsson and LiljasCitation178 has been supported by another scientist, Matulis, based on his recent reference to this workCitation189.
Matulis entered the CA field in 2005 when he published his first paper with the Johnson & Johnson groupCitation190, on a fluorescent thermal shift assay (FTSA) for measuring CA inhibition. In this method, the extent of protein unfolding is monitored as a function to temperature when heated, alone and in the presence of inhibitors, providing a way to measure the affinity of inhibitors as well as some thermodynamic parameters. This method requires high concentrations of enzyme (around 5–10 µM), it uses a very high concentration (50 µM) of a fluorescent dye, ANS (8-anilino-1-naphthalene-sulphonate) as well as a 50 mM phosphate bufferCitation190. We and others have demonstrated that sulphonates can be strong inhibitors of the CAsCitation110, and phosphate is also an anion CA inhibitorCitation26. Just recently one of us (AA) retested phosphate as an inhibitor of hCA I and II at a concentration of 10 nM to 100 mM by the stopped-flow assay. At 50 mM, an inhibition of 13.0% of the activity of hCA I and 19.1% of the activity of hCA II was measured (unpublished results from our laboratories). Thus, the competition of the buffer, dye and inhibitor for binding to the metal ion (or elsewhere in the CA active site), coupled with the large concentrations of enzyme that is required results in this method providing information of limited usefulness regarding CA activity. FTSA was however extensively used in the subsequent Matulis papers, in combination with ITC, which as mentioned by Linkuviene et al. is one of the “most robust techniques” for measuring interactions between proteins and small molecule ligands in drug designCitation191. ITC also uses high concentrations of enzymes in the assay system (6–20 µM), and as mentioned in the Matulis papers the buffer is again 50 mM phosphate plus 100 mM NaCl. In addition to phosphate, chloride is also a CAI that binds to the metal ion active siteCitation26. Chloride is even more inhibitory than phosphate, as documented earlier by many groups. The KI of this anion is 6 mM against hCA I, 200 mM against hCA II, 1.8 mM against hCA VII, 138 mM against mCA XIII (m = murine enzyme), 33 mM against hCA IX and 0.8 mM against hCA XIVCitation192. Thus, it is unclear how robust the inhibition data is in these ITC studies when such high concentrations of CA anion inhibitors are being used.
In the subsequent papers from Matulis’ groupCitation193–198, all of which were inspired by the tail approach (see above), reported by us, a combination of several CA inhibition assays were performed, including the FTSA methodCitation190 and the ITC assay mentioned above, in addition to the stopped flow assay, which members of the Matulis group learned in 2014 in Florence at the request of Matulis (and we accepted them and taught them the CO2 hydrase assay developed originally by KhalifahCitation199 which has been used in Florence since 2001). Notably, the stopped flow assay uses nanomolar concentrations of CA in the system and buffers such as Hepes or Tris at 10–20 mM (which do not interfere with the metal ion), phenol red (at a concentration of 0.2 mM as an indicator) as well as low concentrations of salts to maintain a constant ionic strength (typically 20 mM NaClO4, which is not inhibitory to most CAsCitation26,Citation192) Needless to say, all the methods used by Matulis gave very different results, and it is often difficult to understand which of them should be considered for evaluating the inhibitors from his laboratory.
Thus, the next step of Matulis’ research was to investigate the so called “intrinsic” parameters for characterising the binding of inhibitors to various CA isoforms (there is a series of 24 papers in which “intrinsic” is the keyword, repeated with great insistence, but we will consider only the one in which the method is presented in detailCitation200) The main issue with the measurements of CA inhibition from all other laboratories around the world and in all historical periods, is according to Matulis that “many protein-ligand binding reactions are linked to protonation/deprotonation reactions or various conformational changes”, which were not considered by other researchers. N.b., conformational changes are not considered in this “intrinsic” parameter model either. Thus, the measurement of the energetic contributions for enzyme-inhibitor complex formation is considered to be correct only by considering additional contributions such as protonation/deprotonation reactions of the ligand and the protein. In this way, the observed parameter (e.g. KI, ΔH, ΔG, etc.) must be “corrected” to obtain the “intrinsic” value (KIin) by dividing the observed parameter (KIobs) by the product of the fraction of deprotonated sulphonamide (fsulf) and the fraction of enzyme in the protonated form (fCA)Citation201. In other words, KIin = KIobs/(fsulf x fCA). The theoretical considerations for this “correction” of the observed KI values are based on an old but correct work from Khalifah’s groupCitation202 in which the binding of imidazole and related compounds to hCA I was investigated. Based on this work Matulis proposes the “intrinsic” parameter model considering that “only the deprotonated form of the sulphonamide binds to the CA active site, and that, the Zn-coordinated hydroxide must be protonated before it can be replaced by the amino group of the sulphonamide”. He cites the above-mentioned Khalifah paperCitation202 (based on data for imidazole, which binds in neutral form to the enzyme and does not interact with the metal ion or the water/hydroxide coordinated to the metal ionCitation202) to justify his model for sulphonamides, which is erroneous for several reasons which we will detail here:
Apart the deprotonation of the sulphonamide moiety of the inhibitor and the protonation of the zinc hydroxide species of the enzyme, no hydrogen-bonding or hydrophobic interactions are considered at all in the “intrinsic” model. The Khalifah hypothesisCitation202, which is outdated, was appropriate for investigating imidazole and related azoles as CAIs but was never applied to sulphonamides. In fact, in a very important study from Klebe and Cavalli’s groupsCitation203 it has been demonstrated by using ITC, X-ray crystallography and molecular dynamics (MD) simulations that hydrophobicity is the key factor for the binding kinetics of sulphonamide inhibitors to this enzyme. In fact, by combining the three techniques mentioned above, Klebe and Cavalli’s groupsCitation203 showed that the protein-sulphonamide association rate (kon) dramatically increases with the hydrophobicity of the inhibitor, thus demonstrating the existence of a pre-binding stage in which the enzyme-inhibitor adduct is stabilised by the hydrophobic half of the CA active siteCitation204. Such hydrophobic interactions are the only ones occurring in the so called “F conformation” of the sulphonamide inhibitor adduct, which in its neutral form (RSO2NH2) makes a hydrogen bond to the zinc-coordinated water, which is similar to the phenols or polyamines binding to the enzyme by the zinc-anchoring mechanism discussed aboveCitation128. Then the “S state” emerges, which is characterised by geometries that allow hydrogen bonding of the still neutral SO2NH2 moiety of the inhibitor with Thr199, Thr200, and finally the Zn2+-bound hydroxyl ion (not to zinc-water as in the Matulis model). Thus, the sulphonamide approaches hCA II in its neutral state (not deprotonated as in the Matulis model), binds to the hydrophobic patch, and in the end interacts with the Zn2+ ion, being deprotonated nearby it, before finally coordinating to the zinc but only if the distance between the terminal NH and the zinc ion is within about 2.50 − 2.75 Å, i.e. the final thermodynamic minimumCitation203. These highly relevant data, which are in total contrast with the entire Matulis “intrinsic” theory, were thereafter validated by interesting work from the groups of KlebeCitation205,Citation206 and WhitesidesCitation207–209, and are supported by our analysis of the hydrophobicity of the CA active siteCitation204 as well as the report of several classes of inhibitors which anchor to zinc-bound hydroxide/water moleculeCitation128. Whitesides’ group elegantly demonstrated using ITC, crystallography and MD that the differences in binding between homologous sulphonamide ligands stem from changes in the number and organisation of active site localised water molecules rather than (or perhaps in addition to) the release of structured water from the apposed hydrophobic surfacesCitation207. Again, the hydrophobic effects, not at all considered in the Matulis intrinsic model, are the main players in the inhibition process. In a subsequent studyCitation210 again from the very famous laboratory of Whitesides, it was demonstrated that hydrophobicity is essential also for the binding of anion inhibitors (never investigated by Matulis). It was shown that the binding of the Hofmeister anions is determined by where, and how strongly, they associate with concavities on the surfaces of proteins and how, upon binding, they alter the structure of water within those hydrophobic concavities. It should be stressed here again that in the “intrinsic” method of Matulis, the measurements are done in 50 mM phosphate buffer and 100 mM NaCl, and that these anions are both inhibitory to CAsCitation26,Citation192, as detailed above, because they bind the zinc ion sometimes with a rather relevant affinity, but this phenomenon is never considered properly, and leads to erroneous measurements (apart from the conceptually wrong model used, see above and below).
Matulis considers that sulphonamides bind to the zinc ion in the CA active site with the sulphonamide group in the deprotonated, negatively charged form, to the water bound form (the so-called acidic form) of the enzyme, which as shown above, is not correct. Although noting that the association rates of sulphonamides to CA are slow, he considers this as “unexpected” considering that association is driven by a strong electrostatic interaction between the negatively charged sulphonamide and the positively charged Zn2+. Again, only electrostatic and acid-base equilibria are considered and not any hydrogen bonding and/or hydrophobic contacts, which are as important (or maybe even more important) than the electrostatic interactions in this context. This leads to the discovery of supposedly “kinetic – selective CAIs” Citation211. For decades, the kinetics of association (kon) and dissociation (koff) of the sulphonamide inhibitors to hCA II were well knownCitation212,Citation213. Maren and his group reported the kinetic values for more than 25 sulphonamides directly by measurement of association rate constants (kon) and equilibrium constants (KI), which were used to measure the dissociation rate constants, koffCitation212,Citation213. The values for kon ranged from 0.003 to 31 x 106 L−1 M−1, whereas the koff range was exceedingly low, 0.01–0.05 s−1. Thus, the inhibitory activity was entirely influenced by the association rate kon as also detailed by the groups of Klebe, Cavalli and WhitesidesCitation203–210, which depends primarily on hydrophobic effects. That is, another major error in the analysis of Matulis and colleagues is the lack of considering the hydrophobic effects when analysing the kinetics of association of the sulphonamide inhibitor within the active site. By analysing such low dissociation rates, they reported several ligands to be “kinetic-selective” against certain CA isozymesCitation211. Such a proposal is completely at odds with the well-established selectivity of CAIs for 15 CA isoforms from vertebrate organisms that is based on extensive thermodynamic measurements by groups from all over the world. Will the inhibitors reach the thermodynamic equilibrium, or will they “hurry up” to selectively inhibit some isoforms for which the association rates are faster, and in this way provide novel therapeutic applications, as suggestedCitation211? We leave the answer to this question to the reader.
Matulis suggested that the observed experimental inhibition constants should not be directly translated into a structure − activity relationship as done by all medicinal chemists, instead proposing that the observed gain in inhibitor potency might be attributed to an increase in pKa for the ionisable pharmacophore (presumably deprotonation of the sulphonamide moiety, the only pharmacophore that he has explored) and a corresponding “jump” in the observed affinity instead of to an improvement in ligand-protein interactions. Based on such a model, the explanation he gave for the difference in inhibition between CF3SO2NH2 (a very potent CAI, KI of 2 nM against hCA II, as measured by Maren’s group and confirmed by othersCitation213) and CH3SO2NH2 (a very weak CAI, KI of 0.1 mM against hCA II, again data from Maren and confirmed by other groupsCitation213) is dubious. Matulis predicts a 6-fold difference in the KI values between the two sulphonamides based on this theory, whereas the actual measured difference is 5 orders of magnitude. Thus, according to Matulis, only the quantification of intrinsic parameters may allow for the correct understanding of the inhibition mechanism. However, this method has another major issue: it works only with aromatic/heterocyclic primary sulphonamides (the data on aliphatic sulphonamides is illustrated above). All other inhibitor classes (and even a cyclic sulphonamide such as saccharinCitation214) cannot be measured with this approach (personal communication from Matulis to CTS). In fact, there are no studies from his group on other types of inhibitors apart primary sulphonamides. As showed here, there is a large variety of other chemotypes with potent CA inhibitory action, and their inhibition effects can be measured and compared well with the sulphonamides by using the stopped-flow CO2 hydrase assay queried by Liljas and referred to by Matulis. Note that other groups (Klebe, Whitesides, etc.) which use ITC and other techniques, have not queried the measurements done in our laboratory. So, what is the real problem?
Although we collaborated in the past with Matulis, his behaviour towards some of us should be discussed here. In 2013 at a conference organised by De Simone and one of us (CTS) in Naples, Italy, Matulis was invited as speaker and presented the ITC inhibition method. CTS asked whether the high concentration of sulphonate dye and the sulphonamide inhibitor will all compete for the same binding site and asked whether these effects were considered? There was not an adequate reply from Matulis. Later, during that conference, another author of this paper (RZ) was approached by Matulis who asked him to send him all his compounds that had been tested in Florence, so that he can check if the inhibition data were correct. RZ was shocked by the request and did not send the compounds to his group in Vilnius. The next year, in 2014, as already mentioned here, Matulis asked CTS to host some of his group members in Florence in order to learn the stopped flow assay. CTS agreed, and two students from Matulis’ group spent several months in Florence and learned the assay. In early 2015 after their stopped flow assay was working in Vilnius, Matulis wrote an e-mail to RZ asking him to no longer collaborate with the Florence group because “they do the stopped-flow measurements in an incorrect manner”. RZ replied to Matulis copying CTS and indicated his refusal to collaborate. Only at that point, considering this unethical behaviour, CTS decided to stop any interaction with this colleague.
Returning to the Jonsson and Liljas polemicCitation178, although these two colleagues have not made contributions to anticancer drugs or drug design, they questioned the drug candidate, SLC-0111, which is in Phase Ib/II clinical trials for the treatment of advanced, metastatic solid tumoursCitation215. Jonsson and Liljas state that “it is doubtful that SLC-0111 has the required KI to advance to clinical trials” and that the “commercial interests of pharmaceutical companies and patients may be hurt” Citation178, although the paper in which we describe the compoundCitation72 is not correctly cited and there are no new data on this compound in their commentary. SLC-0111 was reported by the Supuran groupCitation72 in efforts that spanned many years during which thousands of compounds were synthesised, assayed and characterised by X-ray crystallography and other biophysical techniques. SLC-0111 belongs to a class of ureidobenzene sulphonamides which were also assessed for their “druggability” criteria, including ADME (Absorption, Distribution, Metabolism, Excretion). Extensive in vitro and in vivo analysis in appropriate cancer models were then performed in Florence and other laboratories. Although the targets of this compound, CA IX/CA XII, are highly expressed within the hypoxic niches of solid tumours that represents potentially a minor portion of the total tumour cell population, these hypoxic cells have the properties for self-renewalCitation216–218, migration/invasionCitation219–221, survival in acidic tumour microenvironmentCitation222–227, and significantly contribute to resistance to chemo-, radio-, and immune-therapiesCitation228–236. The combination of SLC-0111 and other CAIs with chemotherapy agentsCitation237–239, immunotherapyCitation240–242 and radiotherapyCitation243,Citation244 has been investigated, including SLC-0111, in combination with proton pump inhibitorsCitation236, antimetabolitesCitation237, cisplatinCitation238, APE1-Ref-1 inhibitorsCitation239 and histone deacetylase inhibitorsCitation240, which resulted in excellent synergistic antitumor/antimetastatic effects. Such studies showed a synergistic effect between the CAI and the second antitumor agent, but also the lack of endothelial toxicityCitation241, which are highly important and desirable features for a sustained therapeutic response. The extensive studies reported in these and other papers, provided solid positive pre-clinical data to warrant the initiation of Phase 1 clinical trials in 2014, of which a Phase 1 safety trial with SLC-0111 (as a monotherapeutic agent), has been completedCitation215 and a Phase 1 b trial is currently underway to evaluate SLC-0111 in combination with gemcitabine in metastatic pancreatic cancer patients whose tumours are CA IX positive (ClinicalTrials.gov Identifier: NCT03450018).
Other groups also used SLC-0111 in various biomedical studies in which selective inhibition of some CA isoforms was needed. These include the effects on prostate cancer cells of SLC-0111 alone or in combination with daunorubicinCitation242, radiobiological effects of CA IX inhibition in human breast cancer cellsCitation243, microvascular endothelial cell pH regulationCitation244, glycolysis and migration suppression in pulmonary microvascular endothelial cellsCitation245,Citation246, involvement of CA isoforms in mitochondrial biogenesis and in the control of lactate production in human Sertoli cellsCitation246. Very recently, SLC-0111 was combined with 3-O-acetylbetulin for the treatment of CA IX-positive tumours by Vordermark’s group, again with excellent synergistic effectsCitation247. Dedhar’s group recently reported the interconnection between CA IX inhibition and the amino acid and acid/base transporters in hypoxic tumoursCitation248 as well as the possibility to overcome the adaptive resistance to KRAS and MEK inhibitors by using such anticancer agents in combination with SLC-0111Citation249. All these studies confirmed the usefulness of this clinical candidate, SLC-0111, in tumours and other biomedical conditions. Although Jonsson and Liljas presumably read our replyCitation180, in their reply to our reply they continue that they, “would have liked to see a properly reported inhibition study of this compound to make it obvious that it indeed is a good enough inhibitor to enter Phase I/Phase II studies”Citation181.
Thus, the CA field seems to be now in a typical ancient Greek tragedy situation: we have the Deus ex machina (the couple Jonsson and Liljas) who decide who is doing good and who is doing bad experiments (without however having any documented expertise in enzyme inhibition measurements) and then we have the Computantis (accountant, Matulis) who needs to check the measurements from other laboratories, and asking (in the rare occasions when he acted as reviewer to our papers) for a huge amount of supporting information data that are without any usefulness to anybody. Is this dogmatism or science?
Disclosure statement
All authors declare no conflict of interest except FC and CTS who are among the discoverers of SC-0111.
References
- Meldrum NU, Roughton FJ. Carbonic anhydrase. Its preparation and properties. J Physiol 1933;80:113–42.
- Maren TH. Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev 1967;47:595–781.
- Lindskog S, Malmstrom BG. Metal binding and catalytic activity in bovine carbonic anhydrase. J Biol Chem 1962;237:1129–37.
- Lindskog S, Coleman JE. The catalytic mechanism of carbonic anhydrase. Proc Natl Acad Sci USA 1973;70:2505–8.
- Thorslund A, Lindskog S. Studies of the esterase activity and the anion inhibition of bovine zinc and cobalt carbonic anhydrases. Eur J Biochem 1967;3:117–23.
- Strandberg B, Tilander B, Fridborg K, et al. The crystallization and x-ray investigation of one form of human carbonic anhydrase. J Mol Biol 1962;5:583–4.
- Liljas A, Kannan KK, Bergstén PC, et al. Crystal structure of human carbonic anhydrase C. Nat New Biol 1972;235:131–7.
- Keilin D, Mann T. Carbonic anhydrase. Purification and nature of the enzyme. Biochem J 1940;34:1163–76.
- Krebs HA. Inhibition of carbonic anhydrase by sulphonamides. Biochem J 1948;43:525–8.
- Krebs HA, Roughton FJ. Carbonic anhydrase as a tool in studying the mechanism of reactions involving H(2)CO(3), CO(2) or HCO(3)'. Biochem J 1948;43:550–5.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81.
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68.
- Krishnamurthy VM, Kaufman GK, Urbach AR, et al. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem Rev 2008;108:946–1051.
- Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev 2020;40:2485–565.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- (a) Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. (b) Supuran CT. Carbonic anhydrases and metabolism. Metabolites 2018;8:25.
- (a) Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32. (b) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. (c) Bertucci A, Moya A, Tambutté S, et al. Carbonic anhydrases in anthozoan corals-a review. Bioorg Med Chem 2013;21:1437–50.
- (a) Kikutani S, Nakajima K, Nagasato C, et al. Thylakoid luminal theta-carbonic anhydrase critical for growth and photosynthesis in the marine diatom Phaeodactylum tricornutum. Proc Natl Acad Sci USA 2016;113:9828–33. (b) Jin S, Vullo D, Bua S, et al. Structural and biochemical characterization of novel carbonic anhydrases from Phaeodactylum tricornutum. Acta Crystallogr D Struct Biol 2020;76:676–86. (c) Del Prete S, Bua S, Supuran CT, Capasso C. Escherichia coli γ-carbonic anhydrase: characterisation and effects of simple aromatic/heterocyclic sulphonamide inhibitors. J Enzyme Inhib Med Chem 2020;35:1545–54. (d) Kaur J, Cao X, Abutaleb NS, et al. Optimization of acetazolamide-based scaffold as potent inhibitors of vancomycin-resistant enterococcus. J Med Chem 2020;63:9540–62.
- (a) Xu Y, Feng L, Jeffrey PD, et al. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008;452:56–61. (b) Cox EH, McLendon GL, Morel FM, et al. The active site structure of Thalassiosira weissflogii carbonic anhydrase 1. Biochemistry 2000;39:12128–30. (c) Alterio V, Langella E, Viparelli F, et al. Structural and inhibition insights into carbonic anhydrase CDCA1 from the marine diatom Thalassiosira weissflogii. Biochimie 2012;94:1232–41.
- (a) Capasso C, Supuran CT. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin Ther Targets 2015;19:1689–704. (b) Supuran CT, Capasso C. The eta-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin Ther Targets 2015;19:551–63. (c) Supuran CT, Capasso C. Antibacterial carbonic anhydrase inhibitors: an update on the recent literature. Expert Opin Ther Pat 2020;30:963–82. (d) Angeli A, Pinteala M, Maier SS, et al. Inhibition of α-, β-, γ-, δ-, ζ- and η-class carbonic anhydrases from bacteria, fungi, algae, diatoms and protozoans with famotidine. J Enzyme Inhib Med Chem 2019;34:644–50.
- (a) Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. Isme J 2019;13:2094–106. (b) Del Prete S, Nocentini A, Supuran CT, Capasso C. Bacterial ι-carbonic anhydrase: a new active class of carbonic anhydrase identified in the genome of the Gram-negative bacterium Burkholderia territorii. J Enzyme Inhib Med Chem 2020;35:1060–8. (c) Petreni A, De Luca V, Scaloni A, et al. Anion inhibition studies of the Zn(II)-bound ι-carbonic anhydrase from the Gram-negative bacterium Burkholderia territorii. J Enzyme Inhib Med Chem 2021;36:372–6. (d) De Luca V, Petreni A, Nocentini A, et al. Effect of sulfonamides and their structurally related derivatives on the activity of ι -carbonic anhydrase from Burkholderia territorii. Int J Mol Sci 2021;22:571.
- (a) Tripp BC, Bell CB, 3rd, Cruz F, et al. A role for iron in an ancient carbonic anhydrase. J Biol Chem 2004;279:6683–7. (b) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal beta-class (Cab) and gamma-class (Cam) carbonic anhydrases. Curr Top Med Chem 2007;7:901–8.
- (a) Del Prete S, Vullo D, Fisher GM, et al. Discovery of a new family of carbonic anhydrases in the malaria pathogen Plasmodium falciparum-the η-carbonic anhydrases. Bioorg Med Chem Lett 2014;24:4389–96. (b) De Simone G, Di Fiore A, Capasso C, Supuran CT. The zinc coordination pattern in the η-carbonic anhydrase from Plasmodium falciparum is different from all other carbonic anhydrase genetic families. Bioorg Med Chem Lett 2015;25:1385–9.
- (a) Domsic JF, Avvaru BS, Kim CU, et al. Entrapment of carbon dioxide in the active site of carbonic anhydrase II. J Biol Chem 2008;283:30766–71. (b) Sjöblom B, Polentarutti M, Djinovic-Carugo K. Structural study of X-ray induced activation of carbonic anhydrase. Proc Natl Acad Sci USA 2009;106:10609–13.
- Aggarwal M, Chua TK, Pinard MA, et al. Carbon dioxide "trapped" in a β-carbonic anhydrase. Biochemistry 2015;54:6631–8.
- De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29.
- (a) Bertini I, Luchinat C, Scozzafava A. Binding affinity of bicarboxylate ions for cobalt (II) bovine carbonic anhydrase. Bioinorg Chem 1978;9:93–100. (b) Bertini I, Luchinat C, Scozzafava A. Interaction of cobalt-bovine carbonic anhydrase with the acetate ion. Biochim Biophys Acta 1976;452:239–44.
- (a) Bertini I, Luchinat C, Pierattelli R, Vila AJ. The interaction of acetate and formate with cobalt carbonic anhydrase. An NMR study. Eur J Biochem 1992;208:607–15. (b) Bertini I, Luchinat C, Scozzafava A. A 31P NMR study of phosphate in presence of cobalt(II)- and copper(II)- substituted bovine carbonic anhydrase B. FEBS Lett 1978;93:251–4.
- (a) Taylor JS, Coleman JE. Electron spin resonance of 63Cu and 65Cu carbonic anhydrases. Resolution of nitrogen ligand superhyperfine structure. J Biol Chem 1973;248:749–55. (b) Coleman JE, Coleman RV. Magnetic circular dichroism of Co (II) carbonic anhydrase. J Biol Chem 1972;247:4718–28.
- Coleman JE. Mechanism of action of carbonic anhydrase. Substrate, sulfonamide, and anion binding. J Biol Chem 1967;242:5212–9.
- West D, Pinard MA, Tu C, et al. Human carbonic anhydrase II-cyanate inhibitor complex: putting the debate to rest. Acta Crystallogr F Struct Biol Commun 2014;70:1324–7.
- Jönsson BM, Håkansson K, Liljas A. The structure of human carbonic anhydrase II in complex with bromide and azide. FEBS Lett 1993;322:186–90.
- Ferraroni M, Gaspari R, Scozzafava A, et al. Dioxygen, an unexpected carbonic anhydrase ligand. J Enzyme Inhib Med Chem 2018;33:999–1005.
- Lindahl M, Svensson LA, Liljas A. Metal poison inhibition of carbonic anhydrase. Proteins 1993;15:177–82.
- Peng Z, Merz KM, Jr, Banci L. Binding of cyanide, cyanate, and thiocyanate to human carbonic anhydrase II. Proteins 1993;17:203–16.
- Supuran CT, Conroy CW, Maren TH. Is cyanate a carbonic anhydrase substrate? Proteins 1997;27:272–8.
- Maren TH. Kinetics, equilibrium and inhibition in the Hansson histochemical procedure for carbonic anhydrase: a validation of the method. Histochem J 1980;12:183–90.
- Maren TH. Inhibition by anions of human red cell carbonic anhydrase B: physiological and biochemical implications. Science 1976;194:746–7.
- Briganti F, Mangani S, Scozzafava A, et al. Carbonic anhydrase catalyzes cyanamide hydration to urea: is it mimicking the physiological reaction? J Biol Inorg Chem 1999;4:528–36.
- Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. X-ray crystal studies of the carbonic anhydrase II-trithiocarbonate adduct-an inhibitor mimicking the sulfonamide and urea binding to the enzyme. Bioorg Med Chem Lett 2010;20:474–8.
- Guerri A, Briganti F, Scozzafava A, et al. Mechanism of cyanamide hydration catalyzed by carbonic anhydrase II suggested by cryogenic X-ray diffraction. Biochemistry 2000;39:12391–7.
- Nettles WL, Song H, Farquhar ER, et al. Characterization of the Copper(II) Binding Sites in Human Carbonic Anhydrase II. Inorg Chem 2015;54:5671–80.
- Andring JT, Kim CU, McKenna R. Structure and mechanism of copper-carbonic anhydrase II: a nitrite reductase. IUCrJ 2020;7:287–93.
- Carta F, Scozzafava A, Supuran CT. Sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2012;22:747–58.
- Supuran CT. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem 2018;33:485–95.
- Scozzafava A, Carta F, Supuran CT. Secondary and tertiary sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2013;23:203–13.
- Eriksson AE, Kylsten PM, Jones TA, Liljas A. Crystallographic studies of inhibitor binding sites in human carbonic anhydrase II: a pentacoordinated binding of the SCN- ion to the zinc at high pH. Proteins 1988;4:283–93.
- Supuran CT. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin Drug Discov 2020;15:671–86.
- Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites 2017;7:48.
- Supuran CT. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88.
- Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin Ther Pat 2013;23:681–91.
- Nocentini A, Supuran CT. Advances in the structural annotation of human carbonic anhydrases and impact on future drug discovery. Expert Opin Drug Discov 2019;14:1175–97.
- Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12.
- Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35.
- Supuran CT. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat 2018;28:713–21.
- Supuran CT, Altamimi ASA, Carta F. Carbonic anhydrase inhibition and the management of glaucoma: a literature and patent review 2013-2019. Expert Opin Ther Pat 2019;29:781–92.
- Supuran CT. The management of glaucoma and macular degeneration. Expert Opin Ther Pat 2019;29:745–7.
- Wistrand PJ. Carbonic anhydrase research: a clinical perspective, past and future. EXS 2000;90:597–609.
- Maren TH. Sulfonamides and secretion of aqueous humor. J Exp Zool 1997;279:490–7.
- Supuran CT, Nicolae A, Popescu A. Carbonic anhydrase inhibitors. Part 35. Synthesis of Schiff bases derived from sulfanilamide and aromatic aldehydes: the first inhibitors with equally high affinity towards cytosolic and membrane-bound isozymes. Eur J Med Chem 1996;31:431–8.
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem 1999;42:2641–50.
- D'Ascenzio M, Secci D, Carradori S, et al. 1,3-Dipolar Cycloaddition, HPLC Enantioseparation, and Docking Studies of Saccharin/Isoxazole and Saccharin/Isoxazoline Derivatives as Selective Carbonic Anhydrase IX and XII Inhibitors. J Med Chem 2020;63:2470–88.
- Güzel O, Temperini C, Innocenti A, et al. Carbonic anhydrase inhibitors. Interaction of 2-(hydrazinocarbonyl)-3-phenyl-1H-indole-5-sulfonamide with 12 mammalian isoforms: kinetic and X-ray crystallographic studies. Bioorg Med Chem Lett 2008;18:152–8.
- Biswas S, Aggarwal M, Güzel Ö, et al. Conformational variability of different sulfonamide inhibitors with thienyl-acetamido moieties attributes to differential binding in the active site of cytosolic human carbonic anhydrase isoforms. Bioorg Med Chem 2011;19:3732–8.
- Wagner J, Avvaru BS, Robbins AH, et al. Coumarinyl-substituted sulfonamides strongly inhibit several human carbonic anhydrase isoforms: solution and crystallographic investigations. Bioorg Med Chem 2010;18:4873–8.
- Avvaru BS, Wagner JM, Maresca A, et al. Carbonic anhydrase inhibitors. The X-ray crystal structure of human isoform II in adduct with an adamantyl analogue of acetazolamide resides in a less utilized binding pocket than most hydrophobic inhibitors. Bioorg Med Chem Lett 2010;20:4376–81.
- Carta F, Garaj V, Maresca A, et al. Sulfonamides incorporating 1,3,5-triazine moieties selectively and potently inhibit carbonic anhydrase transmembrane isoforms IX, XII and XIV over cytosolic isoforms I and II: solution and X-ray crystallographic studies. Bioorg Med Chem 2011;19:3105–19.
- Carta F, Birkmann A, Pfaff T, et al. Lead development of thiazolylsulfonamides with carbonic anhydrase inhibitory action. J Med Chem 2017;60:3154–64.
- Alterio V, Vitale RM, Monti SM, et al. Carbonic anhydrase inhibitors: X-ray and molecular modeling study for the interaction of a fluorescent antitumor sulfonamide with isozyme II and IX. J Am Chem Soc 2006;128:8329–35.
- Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3.
- Mboge MY, Mahon BP, Lamas N, et al. Structure activity study of carbonic anhydrase IX: selective inhibition with ureido-substituted benzenesulfonamides. Eur J Med Chem 2017;132:184–91.
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902.
- Tanpure RP, Ren B, Peat TS, et al. Carbonic anhydrase inhibitors with dual-tail moieties to match the hydrophobic and hydrophilic halves of the carbonic anhydrase active site. J Med Chem 2015;58:1494–501.
- Bonardi A, Nocentini A, Bua S, et al. Sulfonamide inhibitors of human carbonic anhydrases designed through a three-tails approach: improving ligand/isoform matching and selectivity of action. J Med Chem 2020;63:7422–44.
- Gieling RG, Babur M, Mamnani L, et al. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J Med Chem 2012;55:5591–600.
- Bozdag M, Carta F, Ceruso M, et al. Discovery of 4-hydroxy-3-(3-(phenylureido)benzenesulfonamides as SLC-0111 analogues for the treatment of hypoxic tumors overexpressing carbonic anhydrase IX. J Med Chem 2018;61:6328–38.
- Akocak S, Lolak N, Bua S, et al. Synthesis and biological evaluation of novel N,N'-diaryl cyanoguanidines acting as potent and selective carbonic anhydrase II inhibitors . Bioorg Chem 2018;77:245–51.
- Lolak N, Akocak S, Bua S, et al. Design and synthesis of novel 1,3-diaryltriazene-substituted sulfonamides as potent and selective carbonic anhydrase II inhibitors. Bioorg Chem 2018;77:542–7.
- Lomelino CL, Mahon BP, Carta F, et al. Kinetic and X-ray crystallographic investigations on carbonic anhydrase isoforms I, II, IX and XII of a thioureido analog of SLC-0111. Bioorg Med Chem 2016;24:976–81.
- Angeli A, Tanini D, Peat TS, et al. Discovery of new selenoureido analogues of 4-(4-Fluorophenylureido)benzenesulfonamide as Carbonic Anhydrase Inhibitors. ACS Med Chem Lett 2017;8:963–8.
- Eldehna WM, Abo-Ashour MF, Berrino E, et al. SLC-0111 enaminone analogs, 3/4-(3-aryl-3-oxopropenyl) aminobenzenesulfonamides, as novel selective subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform IX. Bioorg Chem 2019;83:549–58.
- Congiu C, Onnis V, Deplano A, et al. Synthesis of sulfonamides incorporating piperazinyl-ureido moieties and their carbonic anhydrase I, II, IX and XII inhibitory activity. Bioorg Med Chem Lett 2015;25:3850–3.
- Moi D, Nocentini A, Deplano A, et al. Appliance of the piperidinyl-hydrazidoureido linker to benzenesulfonamide compounds: synthesis, in vitro and in silico evaluation of potent carbonic anhydrase II, IX and XII inhibitors. Bioorg Chem 2020;98:103728.
- Abo-Ashour MF, Eldehna WM, Nocentini A, et al. Novel synthesized SLC-0111 thiazole and thiadiazole analogues: determination of their carbonic anhydrase inhibitory activity and molecular modeling studies. Bioorg Chem 2019;87:794–802.
- Alkhaldi AAM, Al-Sanea MM, Nocentini A, et al. 3-Methylthiazolo[3,2-a]benzimidazole-benzenesulfonamide conjugates as novel carbonic anhydrase inhibitors endowed with anticancer activity: design, synthesis, biological and molecular modeling studies. Eur J Med Chem 2020;207:112745.
- Iikuni S, Ono M, Watanabe H, et al. Cancer radiotheranostics targeting carbonic anhydrase-IX with 111In- and 90Y-labeled ureidosulfonamide scaffold for SPECT imaging and radionuclide-based therapy. Theranostics 2018;8:2992–3006.
- Eldehna WM, Fares M, Ceruso M, et al. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII. Eur J Med Chem 2016;110:259–66.
- Eldehna WM, Abo-Ashour MF, Nocentini A, et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur J Med Chem 2019;162:147–60.
- Carta F, Vullo D, Osman SM, et al. Synthesis and carbonic anhydrase inhibition of a series of SLC-0111 analogs. Bioorg Med Chem 2017;25:2569–76.
- Akgul O, Singh S, Andring JT, et al. Handling drug-target selectivity: a study on ureido containing Carbonic Anhydrase inhibitors. Eur J Med Chem 2020;113035.DOI:https://doi.org/10.1016/j.ejmech.2020.113035
- Nocentini A, Gratteri P, Supuran CT. Phosphorus versus sulfur: discovery of benzenephosphonamidates as versatile sulfonamide-mimic chemotypes acting as carbonic anhydrase inhibitors. Chemistry 2019;25:1188–92.
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun (Camb) 2012;48:1868–70.
- Vullo D, Durante M, Di Leva FS, et al. Monothiocarbamates strongly inhibit carbonic anhydrases in vitro and possess intraocular pressure lowering activity in an animal model of glaucoma. J Med Chem 2016;59:5857–67.
- Carta F, Akdemir A, Scozzafava A, et al. Xanthates and trithiocarbonates strongly inhibit carbonic anhydrases and show antiglaucoma effects in vivo. J Med Chem 2013;56:4691–700.
- Angeli A, Tanini D, Nocentini A, et al. Selenols: a new class of carbonic anhydrase inhibitors. Chem Commun (Camb) 2019;55:648–51.
- Tanini D, Capperucci A, Ferraroni M, et al. Direct and straightforward access to substituted alkyl selenols as novel carbonic anhydrase inhibitors. Eur J Med Chem 2020;185:111811.
- Langella E, D'Ambrosio K, D'Ascenzio M, et al. A Combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes in the active site of carbonic anhydrases. Chemistry 2016;22:97–100.
- Di Fiore A, Maresca A, Supuran CT, et al. Hydroxamate represents a versatile zinc binding group for the development of new carbonic anhydrase inhibitors. Chem Commun (Camb) 2012;48:8838–40.
- Alterio V, Cadoni R, Esposito D, et al. Benzoxaborole as a new chemotype for carbonic anhydrase inhibition. Chem Commun (Camb) 2016;52:11983–6.
- Langella E, Alterio V, D'Ambrosio K, et al. Exploring benzoxaborole derivatives as carbonic anhydrase inhibitors: a structural and computational analysis reveals their conformational variability as a tool to increase enzyme selectivity. J Enzyme Inhib Med Chem 2019;34:1498–505.
- Nocentini A, Supuran CT, Winum JY. Benzoxaborole compounds for therapeutic uses: a patent review (2010–2018). Expert Opin Ther Pat 2018;28:493–504.
- De Simone G, Angeli A, Bozdag M, et al. Inhibition of carbonic anhydrases by a substrate analog: benzyl carbamate directly coordinates the catalytic zinc ion mimicking bicarbonate binding. Chem Commun (Camb) 2018;54:10312–5.
- Carta F, Temperini C, Innocenti A, et al. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–22.
- Nair SK, Ludwig PA, Christianson DW. Two-site binding of phenol in the active site of human carbonic anhydrase II: structural implications for substrate association. J Am Chem Soc 1994;116:3659–60.
- Innocenti A, Vullo D, Scozzafava A, et al. Carbonic anhydrase inhibitors: interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg Med Chem Lett 2008;18:1583–7.
- Innocenti A, Vullo D, Scozzafava A, et al. Carbonic anhydrase inhibitors: inhibition of mammalian isoforms I-XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg Med Chem 2008;16:7424–8.
- (a) Bayram E, Senturk M, Kufrevioglu OI, et al. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–5. (b) Innocenti A, Gülçin I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I-XV. Bioorg Med Chem Lett 2010;20:5050–3.
- Nocentini A, Bonardi A, Gratteri P, et al. Steroids interfere with human carbonic anhydrase activity by using alternative binding mechanisms. J Enzyme Inhib Med Chem 2018;33:1453–9.
- Karioti A, Carta F, Supuran CT. Phenols and polyphenols as carbonic anhydrase inhibitors. Molecules 2016;21:1649.
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300.
- Grandane A, Tanc M, Žalubovskis R, Supuran CT. Synthesis of 6-aryl-substituted sulfocoumarins and investigation of their carbonic anhydrase inhibitory action. Bioorg Med Chem 2015;23:1430–6.
- Pustenko A, Stepanovs D, Žalubovskis R, et al. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:767–75.
- Grandane A, Nocentini A, Werner T, et al. Benzoxepinones: a new isoform-selective class of tumor associated carbonic anhydrase inhibitors. Bioorg Med Chem 2020;28:115496.
- Ferraroni M, Carta F, Scozzafava A, et al. Thioxocoumarins show an alternative carbonic anhydrase inhibition mechanism compared to coumarins. J Med Chem 2016;59:462–73.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- Temperini C, Innocenti A, Scozzafava A, et al. The coumarin-binding site in carbonic anhydrase accommodates structurally diverse inhibitors: the antiepileptic lacosamide as an example and lead molecule for novel classes of carbonic anhydrase inhibitors. J Med Chem 2010;53:850–4.
- Supuran CT. Coumarin carbonic anhydrase inhibitors from natural sources. J Enzyme Inhib Med Chem 2020;35:1462–70.
- Touisni N, Maresca A, McDonald PC, et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7.
- Davis RA, Vullo D, Maresca A, et al. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg Med Chem 2013;21:1539–43.
- De Luca L, Mancuso F, Ferro S, et al. Inhibitory effects and structural insights for a novel series of coumarin-based compounds that selectively target human CA IX and CA XII carbonic anhydrases. Eur J Med Chem 2018;143:276–82.
- Maresca A, Scozzafava A, Supuran CT. 7,8-disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8.
- Maresca A, Supuran CT. Coumarins incorporating hydroxy- and chloro-moieties selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II. Bioorg Med Chem Lett 2010;20:4511–4.
- Bonneau A, Maresca A, Winum JY, Supuran CT. Metronidazole-coumarin conjugates and 3-cyano-7-hydroxy-coumarin act as isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2013;28:397–401.
- D'Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Commun (Camb) 2015;51:302–5.
- Bouzina A, Berredjem M, Nocentini A, et al. Ninhydrins inhibit carbonic anhydrases directly binding to the metal ion. Eur J Med Chem 2021;209:112875.
- D'Ambrosio K, Carradori S, Cesa S, et al. Catechols: a new class of carbonic anhydrase inhibitors. Chem Commun (Camb) 2020;56:13033–6.
- (a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. (b) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72.
- (a) Aggarwal M, Kondeti B, McKenna R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:717–24. (b) Thiry A, Dogné JM, Supuran CT, Masereel B. Anticonvulsant sulfonamides/sulfamates/sulfamides with carbonic anhydrase inhibitory activity: drug design and mechanism of action. Curr Pharm Des 2008;14:661–71.
- (a) Hen N, Bialer M, Yagen B, et al. Anticonvulsant 4-aminobenzenesulfonamide derivatives with branched-alkylamide moieties: X-ray crystallography and inhibition studies of human carbonic anhydrase isoforms I, II, VII, and XIV. J Med Chem 2011;54:3977–81. (b) Thiry A, Dogné JM, Supuran CT, Masereel B. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr Top Med Chem 2007;7:855–64.
- (a) Muñoz W, Lamm A, Poppers D, Lamm S. Acetazolamide promotes decreased consumption of carbonated drinks and weight loss. Oxf Med Case Reports 2018;2018:omy081. (b) Bejaoui M, Pantazi E, De Luca V, et al. Acetazolamide protects steatotic liver grafts against cold ischemia reperfusion injury. J Pharmacol Exp Ther 2015;355:191–8. (c) Supuran CT. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert Opin Emerg Drugs 2012;17:11–5. (d) Supuran CT, Di Fiore A, De Simone G. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert Opin Emerg Drugs 2008;13:383–92.
- (a) Costa G, Carta F, Ambrosio FA, et al. A computer-assisted discovery of novel potential anti-obesity compounds as selective carbonic anhydrase VA inhibitors. Eur J Med Chem 2019;181:111565. (b) Costa G, Gidaro MC, Vullo D, et al. al. Active components of essential oils as anti-obesity potential drugs investigated by in silico techniques. J Agric Food Chem 2016;64:5295–300. (c) Arechederra RL, Waheed A, Sly WS, et al. al. Effect of sulfonamides as carbonic anhydrase VA and VB inhibitors on mitochondrial metabolic energy conversion. Bioorg Med Chem 2013;21:1544–8.
- (a) Supuran CT. For the treatment of idiopathic intracranial hypertension. Expert Rev Neurother. 2015;15:851–856; (b) Supuran CT. Drug interaction considerations in the therapeutic use of carbonic anhydrase inhibitors. Expert Opin Drug Metab Toxicol 2016;12:423–31.
- a)Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. Enzyme Inhib Med Chem 2016;31:894–9. (b) Angeli A, di Cesare Mannelli L, Trallori E, et al. Design, synthesis, and X-ray of selenides as new class of agents for prevention of diabetic cerebrovascular pathology. ACS Med Chem Lett 2018;9:462–7.
- (a) Carta F, Di Cesare Mannelli L, Pinard M, et al. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg Med Chem 2015;23:1828–40. (b) Vullo D, Voipio J, Innocenti A, et al. Carbonic anhydrase inhibitors. Inhibition of the human cytosolic isozyme VII with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett 2005;15:971–6.
- Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother 2016;16:961–8.
- Margheri F, Ceruso M, Carta F, et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J Enzyme Inhib Med Chem 2016;31:60–3.
- (a) Bua S, Di Cesare Mannelli L, Vullo D, et al. Design and synthesis of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J Med Chem 2017;60:1159–70. (b) Bua S, Lucarini L, Micheli L, et al. Bioisosteric development of multitarget nonsteroidal anti-inflammatory drug-carbonic anhydrases inhibitor hybrids for the management of rheumatoid arthritis. J Med Chem 2020;63:2325–42. (c) Akgul O, Di Cesare Mannelli L, Vullo D, et al. Discovery of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the management of rheumatoid arthritis. J Med Chem 2018;61:4961–77.
- (a) Micheli L, Bozdag M, Akgul O, et al. Pain relieving effect of-NSAIDs-CAIs hybrid molecules: systemic and intra-articular treatments against rheumatoid arthritis. Int J Mol Sci 2019;20:1923. (b) Lucarini L, Durante M, Sgambellone S, et al. Effects of new NSAID-CAI hybrid compounds in inflammation and lung fibrosis. Biomolecules 2020;10:1307.
- Berrino E, Milazzo L, Micheli L, et al. Synthesis and evaluation of carbonic anhydrase inhibitors with carbon monoxide releasing properties for the management of rheumatoid arthritis. J Med Chem 2019;62:7233–49.
- Pastorek J, Pastoreková S, Callebaut I, et al. Cloning and characterization of MN, a human tumor-associated protein with a domain homologous to carbonic anhydrase and a putative helix-loop-helix DNA binding segment. Oncogene 1994;9:2877–88.
- Türeci O, Sahin U, Vollmar E, et al. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc Natl Acad Sci USA 1998;95:7608–13.
- Svastová E, Hulíková A, Rafajová M, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004;577:439–45.
- Cecchi A, Hulikova A, Pastorek J, et al. Carbonic anhydrase inhibitors. Design of fluorescent sulfonamides as probes of tumor-associated carbonic anhydrase IX that inhibit isozyme IX-mediated acidification of hypoxic tumors. J Med Chem 2005;48:4834–41.
- Ahlskog JK, Dumelin CE, Trüssel S, et al. In vivo targeting of tumor-associated carbonic anhydrases using acetazolamide derivatives. Bioorg Med Chem Lett 2009;19:4851–6.
- Buller F, Steiner M, Frey K, et al. Selection of carbonic anhydrase IX inhibitors from one million DNA-encoded compounds. ACS Chem Biol 2011;6:336–44.
- Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–76.
- Krall N, Pretto F, Decurtins W, et al. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5.
- Ebbesen P, Pettersen EO, Gorr TA, et al. Taking advantage of tumor cell adaptations to hypoxia for developing new tumor markers and treatment strategies. J Enzyme Inhib Med Chem 2009;24: 1–39.
- Pettersen EO, Ebbesen P, Gieling RG, et al. Targeting tumour hypoxia to prevent cancer metastasis. From biology, biosensing and technology to drug development: the METOXIA consortium. J Enzyme Inhib Med Chem 2015;30:689–721.
- Supuran CT, Alterio V, Di Fiore A, et al. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev 2018;38:1799–836.
- McDonald PC, Winum JY, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012;3:84–97.
- Angeli A, Carta F, Nocentini A, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites 2020;10:412.
- Burianova V, Kalinin S, Supuran CT, Krasavin M. Radiotracers for positron emission tomography (PET) targeting tumour-associated carbonic anhydrase isoforms. Eur J Med Chem 2020;2021:113046.
- Andring JT, Fouch M, Akocak S, et al. Structural basis of nanomolar inhibition of tumor-associated carbonic anhydrase IX: X-ray crystallographic and inhibition study of lipophilic inhibitors with acetazolamide backbone. J Med Chem 2020;63:13064–75.
- Alterio V, Hilvo M, Di Fiore A, et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc Natl Acad Sci USA 2009;106:16233–8.
- Whittington DA, Waheed A, Ulmasov B, et al. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci USA 2001;98:9545–50.
- Di Fiore A, Truppo E, Supuran CT, et al. Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorg Med Chem Lett 2010;20:5023–6.
- Di Fiore A, Monti SM, Hilvo M, et al. Crystal structure of human carbonic anhydrase XIII and its complex with the inhibitor acetazolamide. Proteins 2009;74:164–75.
- Alterio V, Pan P, Parkkila S, et al. The structural comparison between membrane-associated human carbonic anhydrases provides insights into drug design of selective inhibitors. Biopolymers 2014;101:769–78.
- Pilka ES, Kochan G, Oppermann U, Yue WW. Crystal structure of the secretory isozyme of mammalian carbonic anhydrases CA VI: implications for biological assembly and inhibitor development. Biochem Biophys Res Commun 2012;419:485–9.
- Briganti F, Mangani S, Orioli P, et al. Carbonic anhydrase activators: X-ray crystallographic and spectroscopic investigations for the interaction of isozymes I and II with histamine. Biochemistry 1997;36:10384–92.
- (a) Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73. (b) Supuran CT, Balaban AT. Carbonic anhydrase activators. Part 8. pKa–activation relationship in a series of amino acid derivatives activators of isozyme II. Rev Roum Chim 1994;39:107–13. (c) Duda D, Tu C, Qian M, et al. Structural and kinetic analysis of the chemical rescue of the proton transfer function of carbonic anhydrase II. Biochemistry 2001;40:1741–8.
- Temperini C, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase activators. Activation of isozymes I, II, IV, VA, VII, and XIV with l- and d-histidine and crystallographic analysis of their adducts with isoform II: engineering proton-transfer processes within the active site of an enzyme. Chemistry 2006;12:7057–66.
- (a) Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase activation and the drug design. Curr Pharm Des 2008;14:708–15. (b) Temperini C, Innocenti A, Scozzafava A, et al. Carbonic anhydrase activators: L-Adrenaline plugs the active site entrance of isozyme II, activating better isoforms I, IV, VA, VII, and XIV. Bioorg Med Chem Lett 2007;17:628–35.
- (a) Temperini C, Scozzafava A, Vullo D, et al. Carbonic anhydrase activators. Activation of isoforms I, II, IV, VA, VII, and XIV with L- and D-phenylalanine and crystallographic analysis of their adducts with isozyme II: stereospecific recognition within the active site of an enzyme and its consequences for the drug design. J Med Chem 2006;49:3019–27. (b) Temperini C, Innocenti A, Scozzafava A, et al. Carbonic anhydrase activators: kinetic and x-ray crystallographic study for the interaction of D- and L-tryptophan with the mammalian isoforms I-XIV. Bioorg Med Chem 2008;16:8373–837.
- (a) Dave K, Ilies MA, Scozzafava A, et al. An inhibitor-like binding mode of a carbonic anhydrase activator within the active site of isoform II. Bioorg Med Chem Lett 2011;21:2764–8. (b) Bhatt A, Mondal UK, Supuran CT, et al. crystal structure of carbonic anhydrase II in complex with an activating ligand: implications in neuronal function. Mol Neurobiol 2018;55:7431–7.
- (a) Vullo D, Innocenti A, Nishimori I, et al. Carbonic anhydrase activators: activation of the human isoforms VII (cytosolic) and XIV (transmembrane) with amino acids and amines. Bioorg Med Chem Lett 2007;17:4107–12. (b) Pastorekova S, Vullo D, Nishimori I, et al. Carbonic anhydrase activators: activation of the human tumor-associated isozymes IX and XII with amino acids and amines. Bioorg Med Chem 2008;16:3530–6.
- Vullo D, Nishimori I, Innocenti A, et al. Carbonic anhydrase activators: an activation study of the human mitochondrial isoforms VA and VB with amino acids and amines. Bioorg Med Chem Lett 2007;17:1336–40.
- a). Saada MC, Vullo D, Montero JL, et al. Carbonic anhydrase I and II activation with mono- and dihalogenated histamine derivatives. Bioorg Med Chem Lett 2011;21:4884–7. (b) Saada MC, Vullo D, Montero JL, et al. Mono- and di-halogenated histamine, histidine and carnosine derivatives are potent carbonic anhydrase I, II, VII, XII and XIV activators. Bioorg Med Chem 2014;22:4752–8. (c) Vistoli G, Aldini G, Fumagalli L, et al. Activation effects of carnosine- and histidine-containing dipeptides on human carbonic anhydrases: a comprehensive study. Int J Mol Sci 2020;21:1761.
- (a) Supuran CT, Dinculescu A, Balaban AT. Carbonic anhydrase activators. Part 5. CA II activation by 2,4,6-trisubstituted pyridinium cations with 1-(ω-aminoalkyl) side chains. Rev Roum Chim 1993;38:343–9. (b) Draghici B, Vullo D, Akocak S, et al. Ethylene bis-imidazoles are highly potent and selective activators for isozymes VA and VII of carbonic anhydrase, with a potential nootropic effect. Chem Commun (Camb) 2014;50:5980–3.
- (a) Akocak S, Lolak N, Bua S, et al. Activation of human α-carbonic anhydrase isoforms I, II, IV and VII with bis-histamine schiff bases and bis-spinaceamine substituted derivatives. J Enzyme Inhib Med Chem 2019;34:1193–8. (b) Akocak S, Lolak N, Bua S, et al. α-Carbonic anhydrases are strongly activated by spinaceamine derivatives. Bioorg Med Chem 2019;27:800–4.
- a). Chiaramonte N, Maach S, Biliotti C, et al. Synthesis and carbonic anhydrase activating properties of a series of 2-amino-imidazolines structurally related to clonidine. J Enzyme Inhib Med Chem 2020;35:1003–10. (b) Angeli A, Vaiano F, Mari F, et al. Psychoactive substances belonging to the amphetamine class potently activate brain carbonic anhydrase isoforms VA, VB, VII, and XII. J Enzyme Inhib Med Chem 2017;32:1253–9.
- (a) Zhang Y, Supuran CT, Barboiu M. Exponential activation of carbonic anhydrase by encapsulation in dynameric host matrices with chiral discrimination. Chemistry 2018;24:715–20. (b) Le Duc Y, Licsandru E, Vullo D, et al. Carbonic anhydrases activation with 3-amino-1H-1,2,4-triazole-1-carboxamides: discovery of subnanomolar isoform II activators. Bioorg Med Chem 2017;25:1681–6. (c) Licsandru E, Tanc M, Kocsis I, et al. A class of carbonic anhydrase I - selective activators. J Enzyme Inhib Med Chem 2017;32:37–46. (d) Zhang Y, Legrand YM, Petit E, et al. Dynamic encapsulation and activation of carbonic anhydrase in multivalent dynameric host matrices. Chem Commun (Camb) 2016;52:4053–5. (e) Abdelrahim MY, Tanc M, Winum JY, et al. Dominant behaviours in the expression of human carbonic anhydrase hCA I activity. Chem Commun (Camb) 2014;50:8043–6.
- Canto de Souza L, Provensi G, Vullo D, et al. Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology 2017;118:148–56.
- Blandina P, Provensi G, Passsani MB, et al. Carbonic anhydrase modulation of emotional memory. Implications for the treatment of cognitive disorders. J Enzyme Inhib Med Chem 2020;35:1206–14.
- Schmidt SD, Costa A, Rani B, et al. The role of carbonic anhydrases in extinction of contextual fear memory. Proc Natl Acad Sci USA 2020;117:16000–8.
- Jonsson BH, Liljas A. Perspectives on the classical enzyme carbonic anhydrase and the search for inhibitors. Biophys J 2020;119:1275–80.
- Angeli A, Carta F, Nocentini A, et al. Response to perspectives on the classical enzyme carbonic anhydrase and the search for inhibitors. Biophys J 2021;120:178–81.
- Supuran CT. Experimental carbonic anhydrase inhibitors for the treatment of hypoxic tumors. J Exp Pharmacol 2020;12:603–17.
- Jonsson BH, Liljas A. Comments to the editor due to the response by the supuran group to our article. Biophys J 2021;120:182–3.
- Maryanoff BE, Nortey SO, Gardocki JF, et al. Anticonvulsant O-alkyl sulfamates. 2,3:4,5-Bis-O-(1-methylethylidene)-beta-D-fructopyranose sulfamate and related compounds. J Med Chem 1987;30:880–7.
- Dodgson SJ, Shank RP, Maryanoff BE. Topiramate as an inhibitor of carbonic anhydrase isoenzymes. Epilepsia 2000;41:35–9.
- Klinger AL, McComsey DF, Smith-Swintosky V, et al. Inhibition of carbonic anhydrase-II by sulfamate and sulfamide groups: an investigation involving direct thermodynamic binding measurements. J Med Chem 2006;49:3496–500.
- Shank RP, Smith-Swintosky VL, Maryanoff BE. Carbonic anhydrase inhibition. Insight into the characteristics of zonisamide, topiramate, and the sulfamide cognate of topiramate. J Enzyme Inhib Med Chem 2008;23:271–6.
- Casini A, Antel J, Abbate F, et al. Carbonic anhydrase inhibitors: SAR and X-ray crystallographic study for the interaction of sugar sulfamates/sulfamides with isozymes I, II and IV. Bioorg Med Chem Lett 2003;13:841–5.
- Nguyen GTH, Tran TN, Podgorski MN, et al. Nanoscale ion emitters in native mass spectrometry for measuring ligand-protein binding affinities. ACS Cent Sci 2019;5:308–18.
- (a) Nguyen GTH, Nocentini A, Angeli A, et al. Perfluoroalkyl substances of significant environmental concern can strongly inhibit human carbonic anhydrase isozymes. Anal Chem 2020;92:4614–22. (b) Nguyen GTH, Leung WY, Tran TN, et al. Mechanism for the binding of netropsin to hairpin DNA revealed using nanoscale ion emitters in native mass spectrometry. Anal Chem 2020;92:1130–7.
- Matulis D. Structural details of the enzymatic catalysis of carbonic anhydrase II via a mutation of valine to isoleucine. IUCrJ 2020;7:953–4.
- Matulis D, Kranz JK, Salemme FR, Todd MJ. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 2005;44:5258–66.
- Linkuvienė V, Krainer G, Chen WY, Matulis D. Isothermal titration calorimetry for drug design: precision of the enthalpy and binding constant measurements and comparison of the instruments. Anal Biochem 2016;515:61–4.
- (a) Vullo D, Ruusuvuori E, Kaila K, et al. Carbonic anhydrase inhibitors: inhibition of the cytosolic human isozyme VII with anions. Bioorg Med Chem Lett 2006;16:3139–43. (b) Vullo D, Franchi M, Gallori E, et al. Carbonic anhydrase inhibitors. inhibition of cytosolic isozymes I and II and transmembrane, cancer-associated isozyme IX with anions. J Enzyme Inhib Med Chem 2003;18:403–6. (c) Innocenti A, Vullo D, Pastorek J, et al. Carbonic anhydrase inhibitors. Inhibition of transmembrane isozymes XII (cancer-associated) and XIV with anions. Bioorg Med Chem Lett 2007;17:1532–7. (d) Innocenti A, Lehtonen JM, Parkkila S, et al. Carbonic anhydrase inhibitors. Inhibition of the newly isolated murine isozyme XIII with anions. Bioorg Med Chem Lett 2004;14:5435–9.
- (a) Sūdžius J, Baranauskienė L, Golovenko D, et al. 4-[N-(substituted 4-pyrimidinyl)amino]benzenesulfonamides as inhibitors of carbonic anhydrase isozymes I, II, VII, and XIII. Bioorg Med Chem 2010;18:7413–21. (b) Čapkauskaitė E, Zubrienė A, Baranauskienė L, et al. Design of [(2-pyrimidinylthio)acetyl]benzenesulfonamides as inhibitors of human carbonic anhydrases. Eur J Med Chem 2012;51:259–70.
- Zubrienė A, Čapkauskaitė E, Gylytė J, et al. Benzenesulfonamides with benzimidazole moieties as inhibitors of carbonic anhydrases I, II, VII, XII and XIII. J Enzyme Inhib Med Chem 2014;29:124–31.
- Dudutienė V, Matulienė J, Smirnov A, et al. Discovery and characterization of novel selective inhibitors of carbonic anhydrase IX. J Med Chem 2014;57:9435–46.
- Dudutienė V, Zubrienė A, Smirnov A, et al. Functionalization of fluorinated benzenesulfonamides and their inhibitory properties toward carbonic anhydrases. ChemMedChem 2015;10:662–87.
- Zakšauskas A, Čapkauskaitė E, Jezepčikas L, et al. Design of two-tail compounds with rotationally fixed benzenesulfonamide ring as inhibitors of carbonic anhydrases. Eur J Med Chem 2018;156:61–78.
- Zakšauskas A, Čapkauskaitė E, Jezepčikas L, et al. Halogenated and di-substituted benzenesulfonamides as selective inhibitors of carbonic anhydrase isoforms. Eur J Med Chem 2020;185:111825.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. J Bio. Chem 1971;246:2561–73.
- Paketurytė V, Zubrienė A, Ladbury JE, Matulis D. Intrinsic thermodynamics of protein-ligand binding by isothermal titration calorimetry as aid to drug design. Methods Mol Biol. 2019;1964:61–74.
- Kišonaitė M, Zubrienė A, Capkauskaitė E, et al. Intrinsic thermodynamics and structure correlation of benzenesulfonamides with a pyrimidine moiety binding to carbonic anhydrases I, II, VII, XII, and XIII. PLoS One 2014;9:e114106.
- (a) Khalifah RG, Zhang F, Parr JS, Rowe ES. Thermodynamics of binding of the CO2-competitive inhibitor imidazole and related compounds to human carbonic anhydrase I: an isothermal titration calorimetry approach to studying weak binding by displacement with strong inhibitors. Biochemistry 1993;32:3058–66. (b) An H, Tu C, Duda D, et al. Chemical rescue in catalysis by human carbonic anhydrases II and III. Biochemistry 2002;41:3235–42.
- Gaspari R, Rechlin C, Heine A, et al. Kinetic and structural insights into the mechanism of binding of sulfonamides to human carbonic anhydrase by computational and experimental studies. J Med Chem 2016;59:4245–56.
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810.
- Glöckner S, Ngo K, Sager CP, et al. Conformational changes in alkyl chains determine the thermodynamic and kinetic binding profiles of carbonic anhydrase inhibitors. ACS Chem Biol 2020;15:675–85.
- Glöckner S, Ngo K, Wagner B, et al. The influence of varying fluorination patterns on the thermodynamics and kinetics of benzenesulfonamide binding to human carbonic anhydrase II. Biomolecules 2020;10:509.
- Snyder PW, Mecinovic J, Moustakas DT, et al. Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proc Natl Acad Sci USA 2011;108:17889–94.
- Fox JM, Kang K, Sastry M, et al. Water-restructuring mutations can reverse the thermodynamic signature of ligand binding to human carbonic anhydrase. Angew Chem Int Ed Engl 2017;56:3833–7.
- Breiten B, Lockett MR, Sherman W, et al. Water networks contribute to enthalpy/entropy compensation in protein-ligand binding. J Am Chem Soc 2013;135:15579–84.
- Fox JM, Kang K, Sherman W, et al. Interactions between Hofmeister anions and the binding pocket of a protein. J Am Chem Soc 2015;137:3859–66.
- Talibov VO, Linkuvienė V, Matulis D, Danielson UH. Kinetically selective inhibitors of human carbonic anhydrase isozymes I, II, VII, IX, XII, and XIII. J Med Chem 2016;59:2083–93.
- Maren TH. Direct measurements of the rate constants of sulfonamides with carbonic anhydrase. Mol Pharmacol 1992;41:419–26.
- (a) Maren TH, Clare BW, Supuran CT. Structure-activity studies of sulfonamide carbonic anhydrase inhibitors. Roum Chem Quart Rev 1994;2:259–82. (b) Maren TH, Conroy CW. A new class of carbonic anhydrase inhibitor. J Biol Chem 1993;268:26233–9.
- Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;46:7697–9.
- McDonald PC, Chia S, Bedard PL, et al. A phase 1 study of SLC-0111, a novel inhibitor of carbonic anhydrase IX, in patients with advanced solid tumors. Am J Clin Oncol 2020;43:484–90.
- Lock FE, McDonald PC, Lou Y, et al. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 2013;32:5210–9.
- Kreuzer M, Banerjee A, Birts CN, et al. Glycolysis, via NADH-dependent dimerisation of CtBPs, regulates hypoxia-induced expression of CAIX and stem-like breast cancer cell survival. FEBS Lett 2020;594:2988–3001.
- Gibadulinova A, Bullova P, Strnad H, et al. CAIX-mediated control of LIN28/let-7 axis contributes to metabolic adaptation of breast cancer cells to hypoxia. Int J Mol Sci 2020;21:4299.
- Swayampakula M, McDonald PC, Vallejo M, et al. The interactome of metabolic enzyme carbonic anhydrase IX reveals novel roles in tumor cell migration and invadopodia/MMP14-mediated invasion. Oncogene 2017;36:6244–61.
- Lee SH, Griffiths JR. How and why are cancers acidic? Carbonic anhydrase IX and the homeostatic control of tumour extracellular pH. Cancers (Basel) 2020;12:1616.
- Ciccone V, Filippelli A, Angeli A, et al. Pharmacological inhibition of CA-IX impairs tumor cell proliferation, migration and invasiveness. Int J Mol Sci 2020;21:2983.
- Pastorekova S, Gillies RJ. The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev 2019;38:65–77.
- Lee SH, McIntyre D, Honess D, et al. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. Br J Cancer 2018;119:622–30.
- McDonald PC, Chafe SC, Brown WS, et al. Regulation of pH by carbonic anhydrase 9 mediates survival of pancreatic cancer cells with activated KRAS in response to hypoxia. Gastroenterology 2019;157:823–37.
- Persi E, Duran-Frigola M, Damaghi M, et al. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat Commun 2018;9:2997.
- Boyd NH, Walker K, Fried J, et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017;2:92928.
- Iessi E, Logozzi M, Mizzoni D, et al. Rethinking the combination of proton exchanger inhibitors in cancer therapy. Metabolites 2017;8:2.
- McDonald PC, Swayampakula M, Dedhar S. Coordinated regulation of metabolic transporters and migration/invasion by carbonic anhydrase IX. Metabolites 2018;8:20.
- Chafe SC, McDonald PC, Saberi S, et al. Targeting hypoxia-induced carbonic anhydrase IX enhances immune-checkpoint blockade locally and systemically. Cancer Immunol Res 2019;7:1064–78.
- Damgaci S, Ibrahim-Hashim A, Enriquez-Navas PM, et al. Hypoxia and acidosis: immune suppressors and therapeutic targets. Immunology 2018;154:354–62.
- Kuchuk O, Tuccitto A, Citterio D, et al. pH regulators to target the tumor immune microenvironment in human hepatocellular carcinoma. Oncoimmunology 2018;7:e1445452.
- Ward C, Meehan J, Gray M, et al. Carbonic anhydrase IX (CAIX), cancer, and radiation responsiveness. Metabolites 2018;8:13.
- Doyen J, Parks SK, Marcié S, et al. Knock-down of hypoxia-induced carbonic anhydrases IX and XII radiosensitizes tumor cells by increasing intracellular acidosis. Front Oncol 2012;2:199.
- Serbian I, Schwarzenberger P, Loesche A, et al. Ureidobenzenesulfonamides as efficient inhibitors of carbonic anhydrase II. Bioorg Chem 2019;91:103123.
- Vanchanagiri K, Emmerich D, Bruschke M, et al. Synthesis and biological investigation of new carbonic anhydrase IX (CAIX) inhibitors. Chem Biol Interact 2018;284:12–23.
- Federici C, Lugini L, Marino ML, et al. Lansoprazole and carbonic anhydrase IX inhibitors sinergize against human melanoma cells. J Enzyme Inhib Med Chem 2016;31:119–25.
- Andreucci E, Peppicelli S, Carta F, et al. Carbonic anhydrase IX inhibition affects viability of cancer cells adapted to extracellular acidosis. J Mol Med (Berl) 2017;95:1341–53.
- Bryant JL, Gieling RG, Meredith SL, et al. Novel carbonic anhydrase IX-targeted therapy enhances the anti-tumour effects of cisplatin in small cell lung cancer. Int J Cancer 2018;142:191–201.
- Logsdon DP, Grimard M, Luo M, et al. Regulation of HIF1α under hypoxia by APE1/Ref-1 impacts CA9 expression: dual targeting in patient-derived 3D pancreatic cancer models. Mol Cancer Ther 2016;15:2722–32.
- Peppicelli S, Andreucci E, Ruzzolini J, et al. The Carbonic Anhydrase IX inhibitor SLC-0111 as emerging agent against the mesenchymal stem cell-derived pro-survival effects on melanoma cells. J Enzyme Inhib Med Chem 2020;35:1185–93.
- Genah S, Angeli A, Supuran CT, Morbidelli L. Effect of carbonic anhydrase IX inhibitors on human endothelial cell survival. Pharmacol Res 2020;159:104964.
- Riemann A, Güttler A, Haupt V, et al. Inhibition of carbonic anhydrase IX by ureidosulfonamide inhibitor U104 reduces prostate cancer cell growth, but does not modulate daunorubicin or cisplatin cytotoxicity. Oncol Res 2018;26:191–200.
- Güttler A, Theuerkorn K, Riemann A, et al. Cellular and radiobiological effects of carbonic anhydrase IX in human breast cancer cells. Oncol Rep 2019;41:2585–94.
- Lee JY, Alexeyev M, Kozhukhar N, et al. Carbonic anhydrase IX is a critical determinant of pulmonary microvascular endothelial cell pH regulation and angiogenesis during acidosis. Am J Physiol Lung Cell Mol Physiol 2018;315:L41–L51.
- Lee JY, Onanyan M, Garrison I, et al. Extrinsic acidosis suppresses glycolysis and migration while increasing network formation in pulmonary microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol 2019;317:L188–L201.
- Bernardino RL, Dias TR, Moreira BP, et al. Carbonic anhydrases are involved in mitochondrial biogenesis and control the production of lactate by human Sertoli cells. Febs J 2019;286:1393–406.
- Petrenko M, Güttler A, Funtan A, et al. Combined 3-O-acetylbetulin treatment and carbonic anhydrase IX inhibition results in additive effects on human breast cancer cells. Chem Biol Interact 2021;333:109326.
- Venkateswaran G, Dedhar S. Interplay of carbonic anhydrase IX with amino acid and acid/base transporters in the hypoxic tumor microenvironment. Front Cell Dev Biol 2020;8:602668.
- Brown WS, McDonald PC, Nemirovsky O, et al. Overcoming adaptive resistance to KRAS and MEK inhibitors by co-targeting mTORC1/2 complexes in pancreatic cancer. Cell Rep Med 2020;1:100131.