
Abstract
Considering the allosteric regulation of mGlu receptors for potential therapeutic applications, we developed a group of 1,2,4-oxadiazole derivatives that displayed mGlu4 receptor positive allosteric modulatory activity (EC50 = 282–656 nM). Selectivity screening revealed that they were devoid of activity at mGlu1, mGlu2 and mGlu5 receptors, but modulated mGlu7 and mGlu8 receptors, thus were classified as group III-preferring mGlu receptor agents. None of the compounds was active towards hERG channels or in the mini-AMES test. The most potent in vitro mGlu4 PAM derivative 52 (N-(3-chloro-4-(5-(2-chlorophenyl)-1,2,4-oxadiazol-3-yl)phenyl)picolinamide) was readily absorbed after i.p. administration (male Albino Swiss mice) and reached a maximum brain concentration of 949.76 ng/mL. Five modulators (34, 37, 52, 60 and 62) demonstrated significant anxiolytic- and antipsychotic-like properties in the SIH and DOI-induced head twitch test, respectively. Promising data were obtained, especially for N-(4-(5-(2-chlorophenyl)-1,2,4-oxadiazol-3-yl)-3-methylphenyl)picolinamide (62), whose effects in the DOI-induced head twitch test were comparable to those of clozapine and better than those reported for the selective mGlu4 PAM ADX88178.
Graphical Abstract
Introduction
Targeting the allosteric regulation of G protein-coupled receptors (GPCRs) has introduced a new paradigm for drug discovery. Among the metabotropic glutamate receptors (mGlu receptors), the first developed mGlu receptor ligands influenced the receptor response by competing with endogenous glutamate at the orthosteric binding siteCitation1,Citation2. However, the high level of conservation at the glutamate binding pocket hampered the discovery of truly selective ligands for receptors belonging to groups I–III of the mGlu receptors family. As improvement of receptor selectivity was pointed out among the main advantages of the allosteric mode of action, screening campaigns into the identification of new selective mGlu receptors ligands were oriented towards molecules that bind to sites other than the orthosteric region of the receptorCitation3–6. These compounds, identified as allosteric modulators, affected mGlu receptor activity either positively by potentiating the response of the receptor (i.e. positive allosteric modulators – PAMs) or negatively by antagonising the activity of orthosteric agonists (negative allosteric modulators – NAMs)Citation6,Citation7.
From a historical point of view, interest in mGlu receptor ligand discovery has concentrated mostly on group I (mGlu1 and mGlu5) and II (mGlu2 and mGlu3) receptors, leading to several potential drug candidates in clinical trials for various central nervous system (CNS) diseasesCitation6,Citation8–14. However, in recent years, the therapeutic potential of group III (mGlu4, mGlu6–8) receptors, especially type 4 receptors, has become a focus of research at a number of pharmaceutical companies, such as AddexCitation15–18, MerckCitation17,Citation19,Citation20, LundbeckCitation21–24, Prexton TherapeuticsCitation7,Citation25, Domain TherapeuticsCitation26, Hoffmann-La RocheCitation27, Boehringer Ingelheim/EvotecCitation28,Citation29 as well as Vanderbilt UniversityCitation23,Citation25,Citation30–36, which holds leading position in the development of mGlu receptor allosteric modulators.
The first ligands identified as PAMs for mGlu4 receptor, MPEP (1) and SIB-1893 (2)Citation34,Citation37 (), were weak and not selective, as they showed cross-reactivity with mGlu5 receptor and mGlu1 receptor. Similarly, the ligand (–)-PHCCC (3, mGlu4 EC50 = 1.4 µM), originally thought to be a breakthrough, proved to be non-selectiveCitation20, showing partial antagonist activity towards mGlu1 receptor as well as agonist activity towards mGlu6Citation34. However, further pharmacological studies with the use of (–)–PHCCC (3) revealed its efficacy in animal models of Parkinson’s diseaseCitation7,Citation20,Citation33,Citation35,Citation38–44, depressionCitation45,Citation46, anxietyCitation47,Citation48, epilepsyCitation49,Citation50, neuroprotectionCitation51 and oncologyCitation52 but also showed a poor pharmacokinetic profile, limited brain exposure and low aqueous solubilityCitation2,Citation53,Citation54.
Figure 1. Chemical structures of various classes of mGlu4 PAMs. Compounds 1 and 2 were the first ligands identified as PAMs for mGlu4 receptor, 11 is currently in phase II clinical trials, and 12 has advanced as a preclinical development candidate.

Modification of the (–)-PHCCC (3) structure by replacing the phenyl amide with a 2-pyridyl amide led to VU0359516 (4, EC50 = 0.38 µM) (), a more potent, efficacious and highly selective mGlu4 receptor PAM devoid of mGlu1 receptor activityCitation55. This discovery was a starting point for extensive structure-activity relationship (SAR) studies initiated at Vanderbilt University and continued by others for the identification of a number of new PAMs of mGlu4 receptor of various chemotypesCitation53,Citation56,Citation57.
The reported PAMs of mGlu4 receptor represent various chemical classes of compounds and different scaffoldsCitation33, such as picolinamides (e.g. VU0361737 (5))Citation9,Citation15,Citation31,Citation58, sulphonamides (e.g. VU0364439 (6))Citation19,Citation59, phthalimides (e.g. VU0400195 (7))Citation60, cyclohexyl amides (e.g. (1S,2R)-Lu AF21934 (8))Citation21, triaryl amines (ADX88178 (9))Citation61–63, pyrazolo[4,3-b] pyridines (e.g. VU0418506 (10))Citation31, benzisoxazolesCitation64,Citation65, and other polyheterocyclesCitation16,Citation22,Citation66,Citation67.
Although most of the developed mGlu4 PAMs suffered from poor physicochemical and pharmacokinetic properties, limited CNS exposure and/or CYP inhibition/induction issues, their pharmacological investigation did provide in vivo target validation and further increased interest in the mechanism of mGlu4 modulation.
The first mGlu4 PAM clinical candidate, PXT002331 (11), a chemical analogue of (–)-PHCCC (3) developed by Prexton/Domain TherapeuticsCitation7 had very good mGlu4 PAM potency (EC50 = 46 nM), improved pharmacokinetics, high CNS penetration with preferential exposure in the brain, and significant anti-Parkinson’s activity in vivo in rodent models of motor symptoms of the disease, however, Lundbeck has announced in April 2020 that the phase IIa study (AMBLED) of its novel selective positive allosteric modulator of the glutamate 4 receptor, Foliglurax, for the treatment of Parkinson’s disease did not meet the primary study endpoint and the study was stopped. The second potential clinical candidate has been recently reported by Vanderbilt researchesCitation35. Valiglurax (12) represents an isoquinoline-based series and has shown mGlu4 receptor PAM potency (EC50 = 64.6 nM) comparable to PXT002331. Moreover, 12 exhibits excellent pharmacokinetic properties, an acceptable CYP profile and affords robust oral efficacy in a pharmacodynamic model.
Considering the importance of mGlu4 receptor modulation for potential therapeutic applications, we focussed on searching for mGlu4 receptor PAM activity among the group of 1,2,4-oxadiazole derivatives. In addition to identifying new PAMs with high activity for mGlu4 receptor, we also evaluated their mGlu receptor selectivity profile and therapeutic potential with respect to anxiolytic, antipsychotic and antidepressant properties in preliminary in vivo tests. The presented results also include a determination of the cardiotoxic risk and potential mutagenic properties as well as a preliminary metabolic and pharmacokinetic profile for the most active mGlu4 receptor PAMs (34, 37, 49, 52, 60 and 62).
Results and discussion
Chemistry and in vitro activity
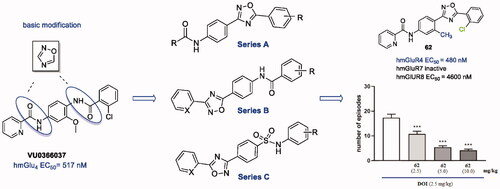
The bioisosteric approach was used to design a pilot series of compounds, and VU0366037 (EC50 = 517 nM, ), the PAM of mGlu4 receptor discovered at Vanderbilt University, was selected as the parent derivative for structural modifications. The basic bioisosteric replacement focussed on exchanging one of the amide groups for the 1,2,4-oxadiazole systemCitation68 and coupling it with various substituted aryl or heteroaryl moieties, in such a way that a distinct series of compounds (A–C) were developed ().
Figure 2. Design of three new series of compounds generated via bioisosteric replacement.

The designed compounds representing all three chemotypes were synthesised according to the procedures outlined in Schemes 1–3.
Series A ligands were synthesised by a four-step pathway (Scheme 1) starting from the nucleophilic addition of hydroxylamine hydrochloride to 4-nitrobenzonitrile (13). The obtained benzamide oxime (14) was coupled with the corresponding acyl chloride to give 1,2,4-oxadiazoles (15). Further nitro group reduction led to amines (16), which in the last step were reacted with the appropriate acyl chlorides, resulting in final amides formation (17a–g).
Scheme 1. Reagents and conditions: (a) NH2OH·HCl, NaOHaq, EtOH, reflux, 1–5 h; (b) R1COCl, toluene, K2CO3, MW 170 °C, 10 min; (c) Fe, CH3COOH, EtOH, water, 60 °C, 1–2 h or SnCl2, 5 N HCl, EtOH, reflux, 2 h; (d) R2COCl, py, rt, overnight.

The second type of ligands (series B) were prepared in a similar way starting from benzonitrile or pyridine-2-carbonitrile (18, Scheme 2). The reaction of N'-hydroxybenzimidamide (19) with 4-nitrobenzoyl chloride led to the corresponding 1,2,4-oxadiazoles (20), which were reduced to amines (21) and finally converted into amides (22a–e).
Scheme 2. Reagents and conditions: (a) NH2OH·HCl, NaOHaq, EtOH, reflux, 1–5 h; (b) 4-nitrobenzoyl chloride, toluene, K2CO3, MW 170 °C, 10 min; (c) Fe, CH3COOH, EtOH, water, 60 °C, 1–2 h or Raney Ni, NH2-NH2 aq, MeOH/THF, 60 °C, 30 min; (d) R2COCl, py, rt, overnight.

Synthesis of the series C ligands started from sulfonylation of the appropriate amine (23a or 23 b) with 4-cyanobenzenesulfonyl chloride (24) to give the corresponding 4-cyanosulfonamides (25a and 25 b). In the next step, the sulphonamides reacted with hydroxylamine hydrochloride to give benzamide oximes (26a and 26 b), which were coupled with various acyl chlorides, leading to the final 1,2,4-oxadiazoles (27a–c) (Scheme 3).
Scheme 3. Reagents and conditions: (a) py, rt, overnight; (b) NH2OH·HCl, NaOHaq, EtOH, reflux, 1–5 h; (c) R2COCl, toluene, K2CO3, MW 170 °C, 10 min or rt, 30 min than reflux, 6 h.

The activity of the compounds was assessed in vitro in a forskolin-stimulated cAMP assay in T-REx 293 cells expressing human mGlu4 receptors in the presence of an EC20 concentration of L-Glu; a PAM was identified if it potentiated cAMP inhibition by the Gi/o-dependent pathway of mGlu4 receptor activation; i.e. if it was able to produce a leftward shift in the potency of the endogenous agonist.
Among all tested compounds, only 17d belonging to chemotype A exhibited PAM activity at mGlu4 receptor (EC50 = 3700 nM). The introduction of a moiety other than the picolinamide moiety on the left side (R2) of the 1,2,4-oxadiazole core in series A was not tolerated (17c, 17e–g) as well as all derivatives from series B and C were found to be inactive.
Taking into account that substituents R1 and R2 of compound 17d, i.e. 2-chloro and 2-pyridyl, respectively, were identical to the parent molecule VU0366037, 17d became the subject of subsequent modifications (), and a new series of analogues (28–62) was synthesised.
Figure 3. Chemical optimisation strategy for compound 17d.

Two synthetic paths were employed to allow modifications of the aromatic ring on the left, centre and right sides of molecule 17d.
In the first path, the method already described for the synthesis of preliminary series A was applied to obtain compounds 28–32. Additionally, commercially available 4-nitrobenzonitriles substituted at the 2 position were used to obtain derivatives 33–51 and 59–62. For the second method, the synthetic approach described for the type C series was used. The starting 4-cyanosulfonyl chloride (24) was replaced by pinacolyl chloride, which was reacted with substituted 4-cyanoanilines to provide compounds 52–58.
The results of the in vitro cAMP assay of derivatives 28–32 () showed that the change in the position of the nitrogen atom (or its removal) on the left side of the pyridyl fragment as well as the additional substitution of fluorine or chlorine atoms to the 2-pyridyl ring led to a complete loss of activity. Thus, the picolinamide system was again identified as an important pharmacophore element for the allosteric modulation of mGlu4 receptor in the 1,2,4-oxadiazole-based derivatives.
Table 1. In vitro mGlu4 receptor PAM activity of 1,2,4-oxadiazole derivatives.
Therefore, using the 2-pyridyl group as the basic fragment on the left side of the molecule, we modified the central ring by introducing various substituents at R3, such as OMe, Cl, F, CF3 and Me, at position 3 relative to the amide moiety, according to the SAR data of known mGlu4 receptor PAMsCitation9. Modification to the 2-position (relative to the amide group) was not tolerated as previously indicatedCitation57. Additionally, modifications on the right side of the molecule (R1) were surveyed in parallel by the attachment of fluorine (instead of chlorine) at the 2 position of the phenyl ring or the introduction of an additional fluorine at the 4 positionCitation23,Citation57,Citation69.
Among compounds 33–48 containing a methoxy group attached to the central ring, only three compounds did not potentiate cAMP inhibition in response to mGlu4 receptor activation (40, 42, and 48), while the rest of the derivatives presented various levels of PAM activity for mGlu4 receptor (EC50 = 393–5300 nM). In comparison to compound 17d without the methoxy substituent (EC50 = 3700 nM, ), the most active derivative, 37, with R1 = 2-F significantly enhanced positive modulation of the mGlu4 receptor (EC50 = 393 nM) and was also more active than VU0366037 (EC50 = 517 nM). The change from the fluorine atom to the chlorine atom (R1) weakened the potency of 34 (EC50 = 656 nM) and halogen removal (33), position change (35, 36) or the introduction of an additional 4-fluorine substituent (38) caused an even more pronounced decrease in activity. Modifications within the 2-pyridyl fragment by the introduction of an additional fluorine and/or chlorine atoms to the 3, 5 and/or 6 positions generally weakened the activity of compounds 39–44. In this group, only two derivatives, 6-F-2-pyridyl 44 (EC50 = 780 nM) and 3-F-2-pyridyl 39 (EC50 = 920 nM), showed a similar level of PAM potency as the corresponding 2-pyridyl analogue 34 (EC50 = 656 nM).
Subsequent modifications were made by replacing the OMe group at the R3 position with the following substituents: Cl, F, CF3 or CH3 (derivatives: 49–62; ); most of the compounds contained a picolinamide moiety on the left side, and two derivatives (50, 51) had an additional fluorine or chlorine atom at the 3 position of the 2-pyridyl group (these last two compounds were, however, unable to modulate mGlu4 receptor activity).
Among the group of chlorinated derivatives, 49 (EC50 = 352 nM) and 52 (EC50 = 282 nM) were found to be the most active PAMs of mGlu4 receptor while analogues with R1 substituents other than 2-Cl (53, 54) presented significantly lower levels of PAM potency (EC50 = 1350 and 750 nM, respectively).
Introduction of a fluorine atom to the central ring led to inactive compounds (55–58), and only derivative 55 (EC50 = 2560 nM) displayed some degree of activity at mGlu4 receptor. On the other hand, it seems that the presence of a trifluoromethyl (59, 60) in this position could restore PAM activity since compound 60 with an EC50 = 390 nM was among the most active derivatives. Of the two compounds with a methyl substituent at the R3 position, 62 showed a significant level of PAM mGlu4 activity, whereas analogue 61 was less active (EC50 = 308 vs 1650 nM, respectively).
In summary, as a result of the in vitro evaluation of the mGlu4 receptor activity of the 1,2,4-oxadiazole derivatives, five compounds, 37, 49, 52, 60 and 62, showed an increase in mGlu4 receptor PAM potency compared with VU0366037, and one derivative (34) was found slightly less active. All derivatives contained the preferred picolinamide fragment on the left side of the 1,2,4-oxadiazole moiety, different small R3 substituents at the central aryl ring (OMe, Cl, CF3, Me), and small R1 groups on the right side (Cl, F, OMe).
mGlu receptor selectivity
Further in vitro characterisation included an assessment of potential direct agonist activation of mGlu4 receptor by the six most potent mGlu4 receptor PAMs (34, 37, 49, 52, 60, 62) and determination of their selectivity for group III mGlu receptor members, i.e. mGlu7 and mGlu8 (), as well as other types of mGlu receptors from group I (mGlu1, mGlu5) and group II (mGlu2) (Supplemental material Figures S3 and S4). The compounds were tested in the agonist, PAM (in the presence of EC20 concentration of L-Glu) or NAM (with EC80 of L-Glu) modes of forskolin-induced cAMP using T-REx 293 cells expressing the protein of a given receptor.
Figure 4. The activity of compounds 34, 37, 49, 52, 60, 62 and reference drugs MMPIP (a selective NAM of mGlu7 receptor) and AZ12216052 (a PAM of mGlu8 receptor) tested at a concentration of 10 μM in the cAMP accumulation assay in cells expressing mGlu7 and mGlu8 receptors. The percent activity refers to two extreme FRET signals: 0% corresponds to forskolin treatment alone and 100% to maximal FRET signal for the control cells (without any treatment). A/D – agonistic activity; B/E – PAM activity measured in the presence of L-Glu at the EC20 concentration (0.1 mM for mGlu7 receptor, and 1 μM for mGlu8 receptor); C/F – NAM activity measured in the presence of L-Glu at the EC80 concentration (1 mM for mGlu7 receptor, 10 μM for Glu8 receptor).

Only two derivatives, 34 and 37, showed some agonist activity for mGlu4 receptor (EC50 = 1.11 µM and 3.94 µM, respectively), although their potencies were weaker than those observed for PAM activity and comparable to the agonist activity of the reference ago-PAM of mGlu4 receptor VU0155041 (EC50 = 2.5 µM)Citation54.
With respect to mGlu7 and mGlu8 receptors, in addition to the pronounced PAM properties of all compounds, direct agonistic activity of 34, 37, 52, and 60 at both receptors and direct agonistic activity of 49 at mGlu7 receptor were detected (). At the same time, there was no significant cross-reactivity with mGlu1,2 and mGlu5 receptors (Supplemental material Figures S3 and S4).
Generally, the evaluated 1,2,4-oxadiazole derivatives positively modulated the group III mGlu receptors in a non-specific way, and their activity profile can be described as group III mGlu receptor selective. Of note is that derivative 62 lacked agonistic action in relation to any of the investigated members of the group III mGlu receptors and presented the properties of pure positive allosteric modulation. Additionally, none of the compounds examined caused a decrease in activity in the presence of the EC80 concentration of L-Glu at any of the investigated glutamate receptors, which indicates the lack of antagonistic properties of the discovered group III mGlu receptors allosteric modulators (Supplemental material Figures S2, S3, and S4).
Preliminary safety and pharmacokinetic screening
The primary assessment of the cardiotoxic risk and mutagenic potential of the six selected 1,2,4-oxadiazole derivatives was performed in the hERG channel assay and the mini-AMES test, respectively. The mutagenicity potential was determined in strains TA98 (frameshift mutation) and TA100 (base-pair substitution) of Salmonella typhimurium in the presence and absence of an exogenous metabolic activation system (rat liver S9 fraction) containing mammalian microsomal enzymes.
All tested 1,2,4-oxadiazole derivatives (at 10 µM concentration) were inactive against hERG channels (Supplemental material Figures S8) and did not show mutagenic activity towards S. typhimurium TA98 and TA100 with and without S9 (at a concentration of ≤ 30 µM) (Supplemental material Figures S9).
Additionally, the most potent mGlu4 receptor PAM in vitro, derivative 52, was tested for its ability to inhibit six key cytochrome P450 family isoenzymes involved in drug metabolism, showing only mild inhibition of the CYP2C9 and CYP2C19 isoenzymes (3.3 < IC50<10 µM).
The pharmacokinetic properties and brain uptake of 52 were further determined in mice after i.p. administration. The compound was administered at a dose of 10 mg/kg in 3% DMSO + 20% Captisol in water. The concentration of 52 was monitored in the plasma and the brain at six time points over 6 h ().
Figure 5. The time course of 52 after i.p. administration in mice: (A) plasma and (B) brain concentration-time profiles.

The pharmacokinetic analysis () showed that 52 was readily absorbed after i.p. administration (Tmax = 30 min in the brain) (). The maximum brain concentration (949.76 ng/mL, 2.12 µM/L) was almost four times higher than the maximum plasma concentration (243.22 ng/mL, 0.54 µM/L). The analysed compound penetrated the blood-brain barrier after i.p. administration and peaked in the brain for longer than in plasma ().
Table 2. Pharmacokinetic parameters in the mouse plasma and brain after a 10 mg/kg i.p. dose of 52.
In vivo profiling
Although efforts to determine the therapeutic potential of group III mGlu receptor have focussed mainly on the role of the mGlu4 receptor subtype in Parkinson’s disease, it has been suggested that non-selective activation of group III mGlu receptors can be a promising therapeutic approach for the treatment of various neuropsychiatric disordersCitation8,Citation70. For example, the non-selective group III mGlu receptor agonist ACTP-I shows antianxiety-like properties in the mouse elevated plus maze and stress-induced hyperthermia testsCitation71, antipsychotic-like activity in amphetamine-induced, MK-801-induced hyperactivity in rats and in DOI-induced head twitches in miceCitation72, as well as antidepressant-like potency in the forced swimming test in ratsCitation73,Citation74.
Taking into account the rather non-selective group III mGlu receptors character of the discovered PAMs, we investigated the anxiolytic, antipsychotic, and antidepressant-like potential of the selected 1,2,4-oxadiazole derivatives (34, 37, 49, 52, 60, 62) in the relevant in vivo behavioural models, i.e. stress-induced hyperthermia (SIH) test, DOI-induced head twitch response (DOI-induced HTR), and tail suspension test (TST), respectively, according to previously described methodsCitation48,Citation75.
Modified stress-induced hyperthermia in singly housed mice
Reflecting autonomic aspects involved in anxiety and fear processes, SIH is an extremely useful procedure to measure potential anxiolytic-like effects of new drug candidatesCitation76. In the performed experiments, an injection of diazepam (5 mg/kg), used as a positive control, significantly reduced SIH (). One-way ANOVA followed by Dunnett's post hoc analysis revealed different magnitudes of SIH response observed after administration of the selected 1,2,4-oxadiazole derivatives ( and Supplemental material Figure S10). None of the administered doses of compound 42 statistically affected the SIH, and weak activity was detected for 37 at a dose of 10 mg/kg. In the case of 34, dose-dependent efficacy was observed; however, a statistically significant reduction in SIH was observed only at higher doses (10 and 20 mg/kg) (Supplemental material Figure S10). Derivatives 52 and 62 significantly reduced SIH responses at all three doses (), whereas for 60, statistically significant changes in the SIH test were caused by the administration of two extreme doses, 5 and 20 mg/kg (Supplemental material Figure S10). At the same time, the investigated compounds had no effect on the basal core temperature.
Figure 6. The effects of compounds 52 and 62 on (A, B) stress-induced hyperthermia and (D, E) DOI-induced HTR. Doses are indicated as mg/kg. Bars represent the mean ± SEM ***p < 0.001 vs. vehicle. Compounds 52 and 62 at all doses significantly reduced the SIH response: 52 [F(3.34) = 9.35; p < 0.001], 62 [F(3.36)=17.12; p < 0.001]; and DOI-induced HTR: 52 [F(3.36) = 28.14; p < 0.001]), 62 [F (3.16) = 38.24; p < 0.001]. The control drugs are presented in panels C (diazepam) and F (clozapine). ***p < 0.001 and **p < 0.01.
![Figure 6. The effects of compounds 52 and 62 on (A, B) stress-induced hyperthermia and (D, E) DOI-induced HTR. Doses are indicated as mg/kg. Bars represent the mean ± SEM ***p < 0.001 vs. vehicle. Compounds 52 and 62 at all doses significantly reduced the SIH response: 52 [F(3.34) = 9.35; p < 0.001], 62 [F(3.36)=17.12; p < 0.001]; and DOI-induced HTR: 52 [F(3.36) = 28.14; p < 0.001]), 62 [F (3.16) = 38.24; p < 0.001]. The control drugs are presented in panels C (diazepam) and F (clozapine). ***p < 0.001 and **p < 0.01.](/cms/asset/152fc2db-26eb-4466-9308-93fef34c5ca8/ienz_a_1998022_f0006_b.jpg)
DOI-induced HTR
The head-twitch behavioural response in rodents induced by the administration of the selective 5-HT2A/2C agonist 1–(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI) has been proposed as an animal model of symptoms associated with a variety of behavioural and psychiatric conditions, including hallucinations. Due to the readily identifiable behaviour, it can be used to assess potential antipsychotic properties in the pre-clinical evaluation of CNS-active new agents.
All newly synthesised compounds, except 49 for which no influence on DOI-induced HTR was observed (Supplemental material Figure S11), significantly attenuated the number of head shakes in mice compared with the vehicle-treated control group. Clear dose-response effects were observed for derivatives 52 and 62 (), and the latter was the strongest blocking agent of the head twitch frequency, presenting activity comparable to that of clozapine, which was used in our study as a reference drug at a dose of 5 mg/kg (). For 37, dose-dependent reverse effects were noticed, while for 34 and 60, significant reductions in the HTR score were seen at two of the administered doses (2.5 and 10 mg/kg, respectively) (Supplemental material Figure S11).
Tail suspension test (TST)
The tail suspension is one of the most commonly used behavioural models for assessing antidepressant-like activity in mice. In contrast to imipramine (20 mg/kg) used as a reference drug, none of the tested compounds at doses of 5, 15 or 20 mg/kg significantly affected the immobilisation time of the mice (Supplemental material Figure S12).
In general, it has been observed that the 1,2,4-oxadiazole derivatives exhibit variable efficacy when tested in vivo. This is most likely due to different pharmacokinetic properties of examined compounds, but firm conclusions can only be drawn after more careful profiling of their drug-like parameters. Nevertheless, the obtained results of in vivo studies clearly indicate the anxiolytic and antipsychotic properties of the tested PAMs of mGlu group III receptors. Indeed, as shown in previous literature reports, group III mGlu receptors are thought to be implicated in such pathological conditionsCitation8,Citation70. In particular, the anxiolytic effects of the abovementioned non-selective group III agonist ACTP-ICitation71, as well as a number of mGlu4 receptor PAMs such as Lu AF21934, have been observed in SIH and other acute models used to study anxiety-related behaviour in rodentsCitation48. Moreover, both agents have been shown to have an antipsychotic-like profile in various rodent models, including DOI-induced HTR in miceCitation72,Citation77. Thus, except for compound 42 which failed to alter SIH and block DOI-induced HTR in mice, the pharmacological in vivo profiles of 34, 37, 52, 60 and 62 resemble that of the non-selective group III agonist ACTP-I and various mGlu4 receptor PAMs investigated to dateCitation78. On the other hand, the lack of antidepressant-like effects of the 1,2,4-oxadiazole derivatives in the TST is similar to our earlier studies, in which we did not find such activity for either the group III agonist ACTP-ICitation71 or the selective mGlu4 receptor PAM Lu AF21934Citation48.
Considering the in vivo properties, the effects of compound 62 deserves special mention in the DOI-induced HTR compared with the efficacy of ADX88178 reported by Addex Therapeutics as a selective and brain-penetrable mGlu4 receptor PAM. In contrast to 62, which presented a clear dose-dependent response and high potency, ADX88178, when tested in doses 3, 10 and 30 mg/kg, lacked dose dependency and showed a flat profile in the reduction of DOI-induced HTR that did not exceed 30% for the medium doseCitation62 only.
Despite the strongly postulated role of glutamate hyperactivity in the pathophysiology of schizophreniaCitation8,Citation79, animal data linking mGlu receptor group III function to this disease are limited. The majority of the currently known results come from our studiesCitation72,Citation77,Citation80 and describe the efficacy of group III mGlu receptor allosteric modulators, indicating that mGlu4 receptor is the primary target for their antipsychotic-like activityCitation78,Citation81. However, the strong in vivo potency of 62, revealed from the DOI-induced HTR, shows the antipsychotic potential of less selective group III mGlu receptor agents. This seems to be confirmed by recently published results describing the important role of group III mGlu receptor activation in the clozapine mechanism of actionCitation82. Therapeutically relevant concentrations of clozapine inhibited thalamocortical hyperglutamatergic transmission by activating presynaptic inhibitory group III mGlu receptors in the medial prefrontal cortexCitation82. Thus, the stimulation of group III mGlu receptors on the glutamatergic terminals of thalamocortical neurons counteracting glutamatergic hyperactivity in schizophrenia might contribute to the efficacy of specific agents, which can be observed in behavioural models predicting antipsychotic activity.
Conclusions
Although the present study was aimed at obtaining selective PAMs of mGlu4 receptor active compounds from the series of 1,2,4-oxadiazole derivatives should be classified as group III mGlu receptor-preferring PAMs. To some extent, this may be due to the highest amino acid sequence homology observed between members of the same mGlu receptor group. The group III mGlu receptor subtypes share more than 94% sequence identity in the transmembrane binding pocket, while a comparison of the corresponding amino acid sequence between the group III mGlu receptors and group I and II mGlu receptor subtypes shows similarity in the range of 65–81%Citation83. Nevertheless, in the identified group III mGlu receptor PAMs, we discovered potent in vivo compounds evoking a decrease in anxiety-related behaviour assessed using the SIH test, and antipsychotic-like properties from the DOI-induced HTR. Moreover, our finding that the antipsychotic-like effect of 62 was comparable to that of clozapine in the DOI-induced head twitch test and better than that reported for the selective mGlu4 receptor PAM ADX88178 might be clinically relevant and requires further pharmacological evaluation.
Materials and methods
Chemistry
Chemicals
All organic reagents were purchased from commercial suppliers and were used without purification. Solvents and inorganic reagents were acquired from Chempur or POCh (Poland). Reaction progress was monitored by TLC on Merck Silica Gel 60 F254 on aluminium plates or Merck Aluminium oxide 60 F254, neutral on aluminium plates and visualised with UV light (254 nm). Column chromatography was performed on Merck Silica Gel 60 (0.063–0.200 mm; 70–230 mesh ASTM) or on Merck Aluminium oxide 90 active neutral (0.063–0.200 mm; 70–230 mesh ASTM).
Software
MarvinSketch software was used for drawing, displaying and characterising chemical structures, substructures and reactions, Marvin 17.24.0, 2017, ChemAxon. JChem Base was used for structure searching and chemical database access and management, JChem 18.3.0, 2018, ChemAxon (www.chemaxon.com).
General procedures
General procedure 1 for carboximidamide formation
Method ACitation84: In a round-bottom flask, the starting benzonitrile (10.0 mmol, 1.0 eq) was dissolved in EtOH (40 ml), and hydroxylamine hydrochloride (20.0 mmol, 2.0 eq) was added followed by a solution of NaOH (20.0 mmol, 2.0 eq) in water (10 ml) (Schemes 1(a) and Citation2(a)). The reaction mixture was refluxed for 6 h (TLC control), and then evaporated to dryness. Water (50 ml) was added, and the reaction mixture was acidified with 1 N HCl. At this stage, for a few compounds, a yellow solid was precipitated, filtered and identified as a corresponding benzamide byproduct. The filtrate was extracted with AcOEt, and the water layer was alkalised with NH3aq. The crude product was extracted with CHCl3 or filtered directly as a precipitated solid and further purified by maceration from iPrOH/hexane (1:3).
Method BCitation85: In a round-bottom flask, the starting benzonitrile (16.0 mmol, 1.0 eq) was dissolved in EtOH (80 ml) and hydroxylamine hydrochloride (80.0 mmol, 5.0 eq) was added followed by TEA (96.0 mmol, 6.0 eq). The reaction mixture was refluxed for 3 h (TLC control), and the EtOH was evaporated in vacuo. Water (100 ml) was added, and the reaction mixture was extracted with AcOEt. After evaporation of the solvent, the crude product was purified by column chromatography over silica gel using a gradient of CHCl3 to CHCl3/MeOH (49:1) followed by trituration with iPrOH/hexane (1:2).
General procedure 2 for 1,2,4-oxadiazole formation
To a round-bottom flask containing toluene (15 ml), a carboximidamide (2.0 mmol, 1.0 eq) and the required acid chloride (2.6 mmol, 1.3 eq) was added followed by K2CO3 (2.6 mmol, 1.3 eq)Citation86,Citation87 (Schemes 1(b) and Citation2(b)). After 30 min of stirring at rt, the reaction mixture was refluxed until the TLC showed the end of the reaction (5–25 h). The reaction mixture was poured into water (40 ml) and extracted with CHCl3. The crude product was purified by column chromatography or by maceration in iPrOH/hexane (1:2). The reaction was also carried out in a MW oven by suspending all reagents in toluene (4 ml) in a reactor flask and heating to 170 °C for 10 min to complete the reaction.
General procedure 3 for reduction of the NO2 group
Method A: Reduction was carried out with Fe powder in the presence of 90% CH3COOH according to the method described in the literatureCitation88(Schemes 1(c) and Citation2(c)).
Method B: To a solution of nitrooxadiazole (1.0 mmol, 1eq) in EtOH (15 ml), SnCl2 (4.0 mmol, 4.0 eq) dissolved in 5 N HCl (1.5 ml) was added dropwise. The reaction mixture was refluxed for 5 h (TLC control), and the solvent was removed in vacuo. Water (30 ml) was added, and the reaction mixture was alkalised with 2 M NaOH to pH = 9. The resulting suspension was extracted with AcOEt. Evaporation of the solvent afforded the crude amino derivative, which was further purified by column chromatography followed by maceration from iPrOH/hexane (1:2).
Method C: Reduction was performed in MeOH in the presence of Raney Ni and hydrazine hydrate according to known proceduresCitation89.
General procedure 4 for amide formation from acyl chlorides
The starting substituted aniline (0.75 mmol, 1.0 eq) was dissolved in pyridine (3 ml). The corresponding acyl chloride (0.98 mmol, 1.3 eq) was added in one portion. After stirring overnight at room temperature, the reaction mixture was poured into water (50 ml). The precipitated solid was filtered and dried. The crude product was purified by column chromatography or maceration.
General procedure 5 for amide formation from carboxylic acids
BOP (0.88 mmol, 1.6 eq) and the corresponding carboxylic acid (0.83 mmol, 1.5 eq) were dissolved under argon in anhydrous MeCN (10 ml). TEA (1.10 mmol, 2.0 eq) was added dropwise, and after 30 min of stirring at rt, a solution of the required aniline (0.55 mmol, 1.0 eq) in MeCN (5 ml) was added to the reaction mixture. Stirring continued until TLC showed disappearance of the starting aniline. The precipitated solid was filtered, washed with MeCN and dried. In cases where the reaction mixture was homogenous, water (50 ml) was added, and the product was extracted with CHCl3. The organic layer was dried, and the solvent was evaporated in vacuo. The crude product was purified by column chromatography or maceration.
In vitro pharmacology
Drugs
Reference compounds: AZ 12216052, CBiPES, VU0155041, VU0469650, VU1545, MPIPP, and Ro 67–4853 were purchased from Tocris Bioscience, and the mGlu receptor agonist L-Glu was purchased from Sigma Aldrich. 3–(2-Pyridyl)-5–(2-chlorophenyl)-1,2,4-oxadiazole was synthesised in-house according to the method previously describedCitation90.
Generation of cDNA constructs and cell lines
Generation the of cell lines with tetracycline-inducible expression of human metabotropic receptors 4, 7 and 8 was described in detail by Chruścicka et al.Citation91 (mGlu4 receptor NM_000841, mGlu7 receptor NM_000844.2, and mGlu8 receptor NM_000845) (Figure S5 and Figure S6, Table S1). Additionally, a new cell line with the human mGlu2 receptor was obtained. The sequence of GRM2 (NM_000839) was subcloned from pcDNA3.1+ (supplied from the University of Missouri–Rolla U.S.) into the multicloning side of the pcDNA5/FTR/TO vector (Invitrogen, Carlsbad, CA) using BamHI and XhoI restriction enzymes. After restriction analysis, T-REx 293 cells were transfected with the received plasmid and expression of mGlu2 receptor in the cells was analysed by RT-PCR and Western blot (Figure S5 and Figure S6). In a similar way, cells with inducible expression of GRM1 (NM-000838.2) and GRM5 (NM_000842.1) were prepared. GRM1 was subcloned into the pcDNA5/FTR/TO vector by the XhoI enzyme (blunt ends) and GRM5 by XhoI and XbaI (blunt ends). Both were inserted initially in the pCMV6-XL vector (Origene). Cells were cotransfected with pcDNA5/FRT/TO-GRM and pOG44 plasmid coding Flp recombinase. Next, stably transfected clones were established by antibiotic selection (hygromycin). The presence of the hmGlu1 receptor or hmGlu5 receptor protein was detected in cell lysates by Western blotting with mouse anti-human mGlu1 or mGlu5 receptors monoclonal antibodies (both from R&D Systems) (Figure S7). Cells were grown under standard cell culture conditions (37 °C, 5% CO2) in DMEM supplemented with 10% tetracycline-free FBS. The expression was induced with tetracycline added to culture medium at 0.75 μg/mL.
Forskolin-induced cAMP production assay
Determination of the intracellular cAMP through a homogeneous time-resolved fluorescence (HTRF) cAMP dynamic 2 kit from Cisbio (Codolet, France) was described previously by Chruścicka et al.Citation91. Briefly, cells were grown in DMEM supplemented with 10% foetal bovine serum (FBS) (tetracycline free). Forty-eight hours before the experiments, mGlu receptor expression was induced by the addition of 0.75 µg/mL tetracycline. Twenty hours before the experiment, FBS and L-Glu were removed from the medium. Thereafter, the cells were scraped and centrifuged. A cell pellet was suspended in Hanks-HEPES (130 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, 0.9 mM NaH2PO4, 20 mM HEPES, and 3.25 mM glucose; pH 7.4). Then, the cell suspension was incubated in the presence of 10 µM forskolin (or 3 µM forskolin for mGlu7 receptor), the agonist L-glutamate and a compound for 5 min. Next, 10 µl of the cell suspension was mixed with 5 µl of cAMP-d2 conjugate and 5 µL of anti-cAMP cryptate conjugate. After 1 h of incubation at rt, the fluorescence at 620 nm and 665 nm was read (Tecan Infinite M1000). The results were calculated as the 665 nm/620 nm ratio multiplied by 104. Each sample was prepared in triplicate.
Calcium flux assay
Cells, 7000 per well, were plated two days before the assay in 384-well clear bottom black wall plate (BD PureCoat Amine, 354719) in FluoroBrite DMEM with 2 mM L-Glu, sodium pyruvate, 10% dialysed FBS, blasticidin and hygromycin. Tetracycline (0.75 mg/mL) was added to cells for 3 h, deprived from FBS and induced for 1 h in the presence of tetracycline (0.75 mg/mL), 2 mM Fluo-8 AM, 0.1% Pluronic F-127, 2 mM probenecid, and 0.04% trypan blue in Hanks-HEPES buffer. Cells were incubated with Fluo-8 loading for 20 min at 37 °C, followed by 15 min at RT. The fluorescence signal was measured on a uCell (Hammamatsu; Japan). Modulators were added after 40 s of the measurement procedure, and L-glutamate was added at 4 min 30 s for a Vfinal = 60 μL. The total time of measurement was 7 min 30 s (0.5 s intervals). For analysis, the data used were from 4 min 30 s to 7 min 30 s.
Statistical calculations were performed using GraphPad Prism 5.04 software (GraphPad Software, La Jolla, CA, USA). The curves were fitted to a 3-parametric logistic equation, allowing for the determination of EC50 values. Each data point was analysed in triplicate.
Safety screening
In vitro binding to hERG assay
The propensity of the tested compounds to block human hERG potassium channels was investigated in a whole cell patch clamp assay in CHO-K1 cells expressing hERG channels by BLIRT, Gdańsk, Poland. Experiments were performed using CHO-K1 cells stably transfected with the hERG potassium channel. hERG potassium current was recorded with whole-cell patch-clamp technique at room temperature using an Axopatch 200B amplifier (Axon Instruments, CA, USA) and a CV203BU head‐stage. Data were acquired through a DigiData 1200 Series (Axon Instruments, CA, USA). Solutions were perfused using a rapid solution changer RSC-200 (Bio-Logic - Science Instruments, Claix, France) connected to an EVH-9 system. Patch‐pipettes were pulled using a PP-830 pipette puller (Narishige, Tokyo, Japan) and polished in a microforge MF-830 (Narishige, Tokyo, Japan). The extracellular recording solution was: 140 mM NaCl, 2,8 mM KCl, 2 mM, CaCl2, 2 mM MgCl2, 10 mM glucose, 10 mM HEPES; titrated to pH 7.3 with NaOH. During the experiments, the tested cell was washed with a control solution (extracellular solution with the addition of 0.1% DMSO) or with a tested compound solution (extracellular solution with the addition of tested compound in concentration: 10 µM dissolved in DMSO, the total amount of DMSO was 0.1% and did not influence on hERG channels).Citation92 In the protocol of the experiments, all recordings were performed in duplicate. In the first recording, the cell was washed with a control solution, in the second recording with a solution containing a test compound (the cell was washed with the test compound at least 1 min before starting recording). During the analysis, the intensity of the current flowing through the membrane at a given potential was measured and then normalised to the maximum intensity observed for a given control recording. Then the change of the normalised current was calculated. The experiments and controls were repeated several times using different cells each time. In the end, the average value of normalised current at the given potential was calculated. In all the experiments, the characteristics of potassium currents flowing through the cell membrane changed with time. Therefore, a single experiment consisted of two records comparing the change in the intensity of the current.
Inhibition of the cytochrome P450 isoform activity (screening)
The assays for cytochrome P450 inhibition facilitate the identification of drug candidates with a lower potential for drug-drug interactions. In vitro experiments conducted to determine whether a drug inhibits a specific CYP enzyme involve an incubation of the drug with probe substrates for the CYP enzymes.
Recombinant cytochrome P450 isoforms available for the assay: CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 isoform with various probe substrates enabling fluorescence detection.
The examined compound was prepared as a 10 mM stock solution in DMSO. The following three concentrations of the test compound were used for screening determination: 1.1, 3.3 and 10 μM. Reference compounds were prepared as a stock solution in DMSO at the following concentration: KTZ 0.025 mM (3A4), SFZ 0.5 mM (2C9), TCP 5 mM (2C19), TCP 25 mM (2B6), Furafylline 2.5 mM (1A2), Quinidine 25 μM (2D6). The assay was performed on 96-well microtiter plates (Microplate 96 well, PS, F-botton, black, non-binding, Greiner BIO-ONE). Plates filled with solution of cofactors (NADP, G6P, MgCl2, G6PDH) as well as examined compound were scanned with a fluorescence plate reader in order to eliminate false results originating from autofluorescence of the test compounds. Isoform-specific substrates: 0.2 mM BFC (3A4), 0.15 mM MFC (2C9), 0.05 mM CEC (2C19), 0.01 mM CEC (1A2), 25 mM EFC (2B6), 7.7 × 10−5 mM AMMC (2D6) were prepared in 0.5 M PBS buffer pH = 7.4 and incubated at 37 °C individually with CYP enzymes (Supersomes Human, Corning). After 10 min of preincubation, isoform/substrate solutions were transferred to black plates which were incubated 30 min for 3A4, 2C19, 2B6, 1A2, 2D6 and 45 min for 2C9. At the end of the incubation, the reaction was stopped and the fluorescence of the product was determined continuously with the specific for every product excitation as well as emission values with the use of multiwell plate scanner equipped with fluorescence measurement (Fluorescence EnSpire Multimode Plate Reader).
Mini AMES test
The Ames microplate fluctuation protocol (MPF) assay was performed with Salmonella typhimurium strains TA98 and TA100, enabling the detection of base-pair substitution. The bacterial strain as well as the exposure and indicator medium were obtained from Xenometrix AG (Allschwil, Switzerland). The mutagenic potential of the tested structures was evaluated by incubation of the bacteria with the test compound at concentrations of 1 mM and 10 mM for 90 min (37 °C) in exposure medium containing a limited amount of histidine. After the addition of indicator medium, each well of the 24-well plate was aliquoted into 48 wells of a 384-well plate.
The occurrence of reversion events to histidine prototrophy was observed as the growth of bacteria in the indicator medium without histidine after 72 h of incubation at 37 °C. Bacterial growth in 384-well plates was visualised by a colour change of medium from violet to yellow due to the addition of pH indicator dye. The absorbance was measured with a microplate reader (EnSpire) at 420 nm. The reference mutagen NQNO (0.5 mM) was used as a positive control in the experiments. The medium control baseline (MCB) was calculated, as derived from the mean number of revertants in the medium control plus one standard deviation.
Pharmacokinetic studies
The method described below was successfully applied to a pharmacokinetic study of 52 in mice (male Albino Swiss) after i.p. injection. Compound 52 was administered to mice at a dose of 10 mg/kg i.p. At 0.25, 0.50, 1.0, 2.0, 4.0, and 6.0 h (three animals were used for one time point) the mice were anaesthetised by the use of Morbital (Biowet, Puławy, Poland), and the blood was collected from the portal vein to tubes containing 5% EDTA. The mice were then perfused with 0.1 M PBS to remove remaining blood from the body, and the brains were taken out for the analysis. Blood was centrifuged at 2000 rpm for 10 min at 4 °C, and the plasma was collected and frozen at −80 °C for further analysis. Plasma and tissue samples from all drug-treated animals were thawed at room temperature prior to use. Standard protocol of sample preparation: 200 µL of acetonitrile was added to the Eppendorf tubes with 50 µL of studied plasma samples or tissue homogenate. Samples were mixed for 5 min on a mixer at 25 °C and 1400 rpm. Tubes were then centrifuged at 2000 × g for 15 min at 4 °C. A total of 180 µL of each supernatant was transferred to a plate well. Finally, each sample was injected onto the LC-MS column. Calibration curve serial dilution method: Plasma was spiked with a standard at different concentrations. Acetonitrile was added. After mixing and centrifugation, the supernatant was collected.
LC-MS analysis
Chromatographic conditions
Plasma and tissue samples from all drug-treated animals at selected time points were analysed using a previously developed non-validated LC-MS/MS method. A sensitive and highly selective liquid chromatography-tandem mass spectrometry (LC-MS) method was used to determine the drug concentration in mouse plasma samples or tissue homogenates. LC-MS analysis was carried out on a Bruker amaZon SL mass spectrometer using positive/negative ion ESI mode. Chromatographic separation was achieved on an Ascentis Express C18 column (5 cm × 2.1 mm, 2.7 µM, Supelco Technologies) at room temperature with a thermostatted column oven. A gradient elution of eluents A (acetonitrile (LiChrosolv, Reag. Ph Eur) + 0.1% formic acid (Sigma Aldrich, 98–100%)) and B (water +0.1% formic acid) was used for separation. The flow rate was set at 1 ml/min. The injection volume was 20 µL, and the time of injection was 4 min.
Mass spectrometric conditions
An ion trap mass spectrometer (Bruker amaZon SL) was equipped with an electrospray source operating in positive/negative ion mode. Data were collected and processed using Bruker Quant Analysis software. Quantification of the analytes was performed in SIM mode.
In vivo pharmacology
Animals and housing
Male Albino Swiss mice (20–25 g) and male C57BL/6J mice (20–22 g) were used in DOI-induced HTR and SIH tests and in the TST, respectively. All mice were 5–6 weeks old and were purchased from Charles-River Company (Germany). The animals were housed under standard laboratory conditions of lighting (light phase 6:00–18:00 h), temperature (19–21 °C) and humidity of 50% with food and water freely available. The experimental groups consisted of five to ten animals, depending on the experimental protocol. The experiments were carried out between 10:00–14:00 h by an observer blind to the treatment. All procedures were conducted according to the guidelines of the National Institutes of Health Animal Care and Use Committee and were approved by the Ethics Committee of the Institute of Pharmacology, Polish Academy of Sciences in Krakow.
Drugs and treatment
The compounds were dissolved in a small amount of 100% EtOH and then adjusted with 20% cyclodextrin, and the pH was adjusted to 6.0. The investigated compounds were administered intraperitoneally (i.p.) 60 min before the behavioural test. Control drugs i.e. diazepam, clozapine and imipramine (Sigma–Aldrich, St. Louis, USA), were administered i.p. 60 min before the behavioural test. Cyclodextrin (20%) was used as a vehicle. All solutions were prepared immediately prior to the experiments and were administered at a constant volume of 10 ml/kg.
Modified stress-induced hyperthermia in singly housed mice
The procedure for modified stress-induced hyperthermia was adapted from Van der HeydenCitation93 and based on the procedure introduced by BorsiniCitation94. Each experimental group consisted of eight to ten animals. The animals were housed individually in a 26 × 21 × 14 cm Macolon cage 24 h before testing. For this assay, the body temperature was measured for each mouse at t = 0 min (T1) and t = +15 min (T2). Albino Swiss mice were placed into a new cage immediately following T1, with the difference in temperature (T2−T1) used as the measure of stress-induced hyperthermia. Pilot studies by Spooren demonstrated that a T2−T1 interval of 15 min is optimal for SIH assaysCitation95. A comparison between T1 in vehicle-treated mice and those administered the test compound was used to determine whether the agent affects body temperature alone. Diazepam (5 mg/kg) was used as the positive control. The rectal temperature was measured to the nearest 0.1 °C with a Physitemp Theralert thermometer, TH-5, Clifton NJ, USA, with the temperature sensor for mice, Type T, Copper-Constantan Thermocouple, Braintree Scientific Inc. The lubricated thermistor probe (2 mm diameter) was inserted 20 mm into the rectum. The mouse was held at the base of the tail during this determination, and the thermistor probe was left in place until a constant reading was obtained for 15 s. The effects of the investigated compounds on the SIH response were investigated after administration of the compounds at doses of 5, 10 and 20 mg/kg. The mean basal temperature (T1) of the mice did not differ between the groups. The vehicle used (20% (2-hydroxypropyl)-β-cyclodextrin) did not have any influence on the basal body temperature, which was typically between 36 and 37 °C for the Albino Swiss mice used in our laboratory.
DOI-induced head twitch test
The experiments were performed according to the procedure described in our previous studiesCitation75,Citation80. Briefly, to habituate mice to the experimental environment, each animal was transferred to a 12 cm (diameter) × 20 cm (height) glass cage lined with sawdust 30 min before treatment. Test compounds were administered intraperitoneally (i.p.) at doses of 2.5, 5, and 10 mg/kg body weight 60 min before the test was performed. The head twitch response (HTR) in mice was induced by DOI (2.5 mg/kg, i.p.). Immediately after treatment, the number of head twitches was counted during a 20 min session. Haloperidol and clozapine were used as the reference compounds at active doses of 0.2 and 5 mg/kg, respectively.
Tail suspension test
The tail suspension test was performed according to the procedure of SteruCitation96. Imipramine (20 mg/kg, i.p.) was used as a reference drug. C57BL/6J mice were individually suspended by their tails by a plastic string that was positioned horizontally 75 cm above the tabletop using adhesive tape placed approximately 1 cm from the tip of the tail. The immobility duration was recorded for 6 min. The mice were considered immobile only when they hung down passively and were completely motionless.
Supplemental Material
Download PDF (7.2 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Flor PJ, Acher FC. Orthosteric versus allosteric GPCR activation: the great challenge of group-III mGluRs. Biochem Pharmacol 2012;84:414–24.
- Lindsley CW, Emmitte KA, Hopkins CR, et al. Practical strategies and concepts in GPCR allosteric modulator discovery: recent advances with metabotropic glutamate receptors. Chem Rev 2016;116:6707–41.
- Feng Z, Ma S, Hu G, et al. Allosteric binding site and activation mechanism of class C G-protein coupled receptors: metabotropic glutamate receptor family. Aaps J 2015;17:737–53.
- Gregory KJ, Noetzel MJ, Niswender CM. Pharmacology of metabotropic glutamate receptor allosteric modulators: structural basis and therapeutic potential for CNS disorders. Prog Mol Biol Transl Sci 2013;115:61–121.
- Gregory KJ, Dong EN, Meiler J, et al. Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacology 2011;60:66–81.
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 2009;8:41–54.
- Charvin D, Pomel V, Ortiz M, et al. Discovery, structure-activity relationship, and antiparkinsonian effect of a potent and brain-penetrant chemical series of positive allosteric modulators of metabotropic glutamate receptor 4. J Med Chem 2017;60:8515–37.
- Golubeva AV, Moloney RD, O'Connor RM, et al. Metabotropic glutamate receptors in central nervous system diseases. Curr Drug Targets 2016;17:538–616.
- Jones CK, Engers DW, Thompson AD, et al. Discovery, synthesis, and structure-activity relationship development of a series of N-4-(2,5-dioxopyrrolidin-1-yl)phenylpicolinamides (VU0400195, ML182): characterization of a novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu(4)) with oral efficacy in an antiparkinsonian animal model. J Med Chem 2011;54:7639–47.
- Bahi A, Fizia K, Dietz M, et al. Pharmacological modulation of mGluR7 with AMN082 and MMPIP exerts specific influences on alcohol consumption and preference in rats. Addict Biol 2012;17:235–47.
- Gasparini F, Kuhn R, Pin JP. Allosteric modulators of group I metabotropic glutamate receptors: novel subtype-selective ligands and therapeutic perspectives. Curr Opin Pharmacol 2002;2:43–9.
- Knoflach F, Mutel V, Jolidon S, et al. Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci USA 2001;98:13402–7.
- Schlumberger C, Pietraszek M, Gravius A, et al. Comparison of the mGlu(5) receptor positive allosteric modulator ADX47273 and the mGlu(2/3) receptor agonist LY354740 in tests for antipsychotic-like activity. Eur J Pharmacol 2009;623:73–83.
- Rodriguez AL, Grier MD, Jones CK, et al. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol 2010;78:1105–23.
- Bolea C, Addex Pharma SA, Amido derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors, WO 2009/010454 A2; 2009.
- Bolea C, Celanire S, Addex Pharma SA, Heterotricyclic compounds as positive allosteric modulators of metabotropic glutamate receptors, WO 2010/079238 A1; 2010.
- Liverton N, Bolea C, Celanire S, et al., Merck Sharp & Dohme Corp, Addex Pharma SA, Tricyclic compounds as allosteric modulators of metabotropic glutamate receptors, WO 2012/006760 A1; 2012.
- Celanire S, Campo B. Recent advances in the drug discovery of metabotropic glutamate receptor 4 (mGluR4) activators for the treatment of CNS and non-CNS disorders. Expert Opin Drug Discov 2012;7:261–80.
- Mccauley JA, Butcher JW, Hess JW, et al. Merck & Co., Inc., Sulfonamide derivative metabotropic glutamate R4 ligands, WO 2010/033350 A1; 2010.
- Marino MJ, Williams DL Jr., O'Brien JA, et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson's disease treatment. Proc Natl Acad Sci USA 2003;100:13668–73.
- Bennouar KE, Uberti MA, Melon C, et al. Synergy between L-DOPA and a novel positive allosteric modulator of metabotropic glutamate receptor 4: implications for Parkinson's disease treatment and dyskinesia. Neuropharmacology 2013;66:158–69.
- Hong SP, Liu KG, Ma G, et al. Tricyclic thiazolopyrazole derivatives as metabotropic glutamate receptor 4 positive allosteric modulators. J Med Chem 2011;54:5070–81.
- Robichaud AJ, Engers DW, Lindsley CW, et al. Recent progress on the identification of metabotropic glutamate 4 receptor ligands and their potential utility as CNS therapeutics. ACS Chem Neurosci 2011;2:433–49.
- Jimenez HN, Liu KG, Hong SP, et al. 4-(1-Phenyl-1H-pyrazol-4-yl)quinolines as novel, selective and brain penetrant metabotropic glutamate receptor 4 positive allosteric modulators. Bioorg Med Chem Lett 2012;22:3235–9.
- Charvin D, Di Paolo T, Bezard E, et al. An mGlu4-positive allosteric modulator alleviates Parkinsonism in primates. Mov Disord 2018;33:1619–31.
- Schann S, Mayer S, Morice C, et al., Domain Therapeutics, Prestwick Chemical, Inc., Novel oxime derivatives and their use as allosteric modulators of metabotropic glutamate receptors, WO 2011/051478 A1; 2011.
- Biemans B, Jaeschke G, Ricci A, et al., Hoffmann-La Roche Inc., Ethynyl derivatives, WO 2018/015235 A1; 2018.
- East SP, Bamford S, Dietz MG, et al. An orally bioavailable positive allosteric modulator of the mGlu4 receptor with efficacy in an animal model of motor dysfunction. Bioorg Med Chem Lett 2010;20:4901–5.
- East SP, Gerlach K. mGluR4 positive allosteric modulators with potential for the treatment of Parkinson's disease: WO09010455. Expert Opin Ther Pat 2010;20:441–5.
- Reed CW, McGowan KM, Spearing PK, et al. VU6010608, a Novel mGlu7 NAM from a Series of N-(2-(1H-1,2,4-Triazol-1-yl)-5-(trifluoromethoxy)phenyl)benzamides. ACS Med Chem Lett 2017;8:1326–30.
- Engers DW, Blobaum AL, Gogliotti RD, et al. Discovery, synthesis, and preclinical characterization of N-(3-chloro-4-fluorophenyl)-1H-pyrazolo[4,3-b]pyridin-3-amine (VU0418506), a novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu4). ACS Chem Neurosci 2016;7:1192–200.
- Jalan-Sakrikar N, Field JR, Klar R, et al. Identification of positive allosteric modulators VU0155094 (ML397) and VU0422288 (ML396) reveals new insights into the biology of metabotropic glutamate receptor 7. ACS Chem Neurosci 2014;5:1221–37.
- Bollinger SR, Engers DW, Panarese JD, et al. Discovery, structure-activity relationship, and biological characterization of a novel series of 6-((1 H-pyrazolo[4,3- b]pyridin-3-yl)amino)-benzo[d]isothiazole-3-carboxamides as positive allosteric modulators of the metabotropic glutamate receptor 4 (mGlu4). J Med Chem 2019;62:342–58.
- Lindsley CW, Hopkins CR. Metabotropic glutamate receptor 4 (mGlu4)-positive allosteric modulators for the treatment of Parkinson's disease: historical perspective and review of the patent literature. Expert Opin Ther Pat 2012;22:461–81.
- Panarese JD, Engers DW, Wu YJ, et al. The discovery of VU0652957 (VU2957, Valiglurax): SAR and DMPK challenges en route to an mGlu4 PAM development candidate. Bioorg Med Chem Lett 2019;29:342–6.
- Conn PJ, Lindsley CW, Felts AS, et al., Vanderbilt University, Indazole compounds as mGluR4 allosteric potentiators, compositions, and methods of treating neurological dysfunction, WO 2019/036534 A1; 2019.
- Mathiesen JM, Svendsen N, Brauner-Osborne H, et al. Positive allosteric modulation of the human metabotropic glutamate receptor 4 (hmGluR4) by SIB-1893 and MPEP. Br J Pharmacol 2003;138:1026–30.
- Rovira X, Malhaire F, Scholler P, et al. Overlapping binding sites drive allosteric agonism and positive cooperativity in type 4 metabotropic glutamate receptors. Faseb J 2015;29:116–30.
- Williams R, Johnson KA, Gentry PR, et al. Synthesis and SAR of a novel positive allosteric modulator (PAM) of the metabotropic glutamate receptor 4 (mGluR4). Bioorg Med Chem Lett 2009;19:4967–70.
- Battaglia G, Busceti CL, Molinaro G, et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurosci 2006;26:7222–9.
- Litim N, Morissette M, Di Paolo T. Metabotropic glutamate receptors as therapeutic targets in Parkinson's disease: An update from the last 5 years of research. Neuropharmacology 2017;115:166–79.
- Maj M, Bruno V, Dragic Z, et al. (-)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology 2003;45:895–906.
- Dickerson JW, Conn PJ. Therapeutic potential of targeting metabotropic glutamate receptors for Parkinson's disease. Neurodegener Dis Manag 2012;2:221–32.
- Annoura H, Fukunaga A, Uesugi M, et al. A novel class of antagonists for metabotropic glutamate receptors, 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylates. Bioorg Med Chem Lett 1996;6:763–6.
- Hovelso N, Sotty F, Montezinho LP, et al. Therapeutic potential of metabotropic glutamate receptor modulators. Curr Neuropharmacol 2012;10:12–48.
- Klak K, Palucha A, Branski P, et al. Combined administration of PHCCC, a positive allosteric modulator of mGlu4 receptors and ACPT-I, mGlu III receptor agonist evokes antidepressant-like effects in rats. Amino Acids 2007;32:169–72.
- Stachowicz K, Chojnacka-Wojcik E, Klak K, et al. Anxiolytic-like effects of group III mGlu receptor ligands in the hippocampus involve GABAA signaling. Pharmacol Rep 2006;58:820–6.
- Sławińska A, Wierońska JM, Stachowicz K, et al. Anxiolytic- but not antidepressant-like activity of Lu AF21934, a novel, selective positive allosteric modulator of the mGlu4 receptor. Neuropharmacology 2013;66:225–35.
- Teall M, White K, Mack S, et al., mGluR7 agonist compounds for treating mGluR7- regulated diseases, disorders, or conditions, WO 2018/092921 A1; 2018.
- Ngomba RT, Ferraguti F, Badura A, et al. Positive allosteric modulation of metabotropic glutamate 4 (mGlu4) receptors enhances spontaneous and evoked absence seizures. Neuropharmacology 2008;54:344–54.
- Canudas AM, Di Giorgi-Gerevini V, Iacovelli L, et al. PHCCC, a specific enhancer of type 4 metabotropic glutamate receptors, reduces proliferation and promotes differentiation of cerebellar granule cell neuroprecursors. J Neurosci 2004;24:10343–52.
- Shin SS, Martino JJ, Chen S. Metabotropic glutamate receptors (mGlus) and cellular transformation. Neuropharmacology 2008;55:396–402.
- Williams R, Niswender CM, Luo Q, et al. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4). Part II: challenges in hit-to-lead. Bioorg Med Chem Lett 2009;19:962–6.
- Niswender CM, Johnson KA, Weaver CD, et al. Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol 2008;74:1345–58.
- Williams R, Zhou Y, Niswender CM, et al. Re-exploration of the PHCCC scaffold: discovery of improved positive allosteric modulators of mGluR4. ACS Chem Neurosci 2010;1:411–9.
- Niswender CM, Lebois EP, Luo Q, et al. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4): Part I. Discovery of pyrazolo[3,4-d]pyrimidines as novel mGluR4 positive allosteric modulators. Bioorg Med Chem Lett 2008;18:5626–30.
- Engers DW, Gentry PR, Williams R, et al. Synthesis and SAR of novel, 4-(phenylsulfamoyl)phenylacetamide mGlu4 positive allosteric modulators (PAMs) identified by functional high-throughput screening (HTS). Bioorg Med Chem Lett 2010;20:5175–8.
- Engers DW, Niswender CM, Weaver CD, et al. Synthesis and evaluation of a series of heterobiarylamides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs). J Med Chem 2009;52:4115–8.
- Conn PJ, Lindsley CW, Hopkins CR, et al., Vanderbilt University, Substituted benzoimidazolesulfonamides and substituted indolesulfonamides as mGluR4 potentiators, WO 2011/011722 A1; 2011.
- McCauley JA, Hess JW, Liverton NJ, et al., Merck & Co. Inc., Phthalimide derivative metabotropic glutamate R4 ligands, WO 2010/033349 A1; 2010.
- Kalinichev M, Rouillier M, Girard F, et al. ADX71743, a potent and selective negative allosteric modulator of metabotropic glutamate receptor 7: in vitro and in vivo characterization. J Pharmacol Exp Ther 2013;344:624–36.
- Kalinichev M, Le Poul E, Bolea C, et al. Characterization of the novel positive allosteric modulator of the metabotropic glutamate receptor 4 ADX88178 in rodent models of neuropsychiatric disorders. J Pharmacol Exp Ther 2014;350:495–505.
- Cheung YY, Zamorano R, Blobaum AL, et al. Solution-phase parallel synthesis and SAR of homopiperazinyl analogs as positive allosteric modulators of mGlu4). ACS Comb Sci 2011;13:159–65.
- Conn PJ, Lindsley CW, Hopkins CR, et al., Vanderbilt University, Benzisoxazoles and azabenzisoxazoles as mGluR4 allosteric potentiators, compositions, and methods of treating neurological dysfunction, WO 2011/100614 A1; 2011.
- Conn PJ, lindsley CW, Hopkins CR, et al., Vanderbilt University, mGluR4 allosteric potentiators, compositions, and methods of treating neurological dysfunction, WO 2011/029104 A1; 2011.
- Bolea C, Celanire S, Addex Pharma SA, Novel heteroaromatic derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors, WO 2009/010455 A2; 2009.
- Biemans B, Guba W, Jaeschke G, et al., F. Hoffmann-La Roche AG, 3-(4-Ethynylphenyl)hexahydropyrimidin-2,4-dione derivatives as modulators of mGluR4. WO 2016/146600 A1. 2016.
- Biernacki K, Daśko M, Ciupak O, et al. Novel 1,2,4-oxadiazole derivatives in drug discovery. Pharmaceuticals 2020;13:111.
- Engers DW, Field JR, Le U, et al. Discovery, synthesis, and structure-activity relationship development of a series of N-(4-acetamido)phenylpicolinamides as positive allosteric modulators of metabotropic glutamate receptor 4 (mGlu(4)) with CNS exposure in rats. J Med Chem 2011;54:1106–10.
- Palucha A, Pilc A. Metabotropic glutamate receptor ligands as possible anxiolytic and antidepressant drugs. Pharmacol Ther 2007;115:116–47.
- Stachowicz K, Kłodzińska A, Palucha-Poniewiera A, et al. The group III mGlu receptor agonist ACPT-I exerts anxiolytic-like but not antidepressant-like effects, mediated by the serotonergic and GABA-ergic systems. Neuropharmacology 2009;57:227–34.
- Pałucha-Poniewiera A, Kłodzińska A, Stachowicz K, et al. Peripheral administration of group III mGlu receptor agonist ACPT-I exerts potential antipsychotic effects in rodents. Neuropharmacology 2008;55:517–24.
- Palucha A, Tatarczynska E, Branski P, et al. Group III mGlu receptor agonists produce anxiolytic- and antidepressant-like effects after central administration in rats. Neuropharmacology 2004;46:151–9.
- Palucha A, Klak K, Branski P, et al. Activation of the mGlu7 receptor elicits antidepressant-like effects in mice. Psychopharmacology (Berl) 2007;194:555–62.
- Cieślik P, Woźniak M, Kaczorowska K, et al. Negative allosteric modulators of mGlu7 receptor as putative antipsychotic drugs. Front Mol Neurosci 2018;11:316.
- Olivier B, Zethof T, Pattij T, et al. Stress-induced hyperthermia and anxiety: pharmacological validation. Eur J Pharmacol 2003;463:117–32.
- Sławińska A, Wierońska JM, Stachowicz K, et al. The antipsychotic-like effects of positive allosteric modulators of metabotropic glutamate mGlu4 receptors in rodents. Br J Pharmacol 2013;169:1824–39.
- Wierońska JM, Zorn SH, Doller D, et al. Metabotropic glutamate receptors as targets for new antipsychotic drugs: Historical perspective and critical comparative assessment. Pharmacol Ther 2016;157:10–27.
- Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012;37:4–15.
- Wieronska JM, Slawinska A, Lason-Tyburkiewicz M, et al. The antipsychotic-like effects in rodents of the positive allosteric modulator Lu AF21934 involve 5-HT1A receptor signaling: mechanistic studies. Psychopharmacology (Berl) 2015;232:259–73.
- Wierońska JM, Acher FC, Sławińska A, et al. The antipsychotic-like effects of the mGlu group III orthosteric agonist, LSP1-2111, involves 5-HTıA signalling. Psychopharmacology 2013;227:711–25.
- Fukuyama K, Kato R, Murata M, et al. Clozapine normalizes a glutamatergic transmission abnormality induced by an impaired NMDA Receptor in the thalamocortical pathway via the activation of a group III metabotropic glutamate receptor. Biomolecules 2019;9:234.
- Harpsøe K, Isberg V, Tehan BG, et al. Selective negative allosteric modulation of metabotropic glutamate receptors – a structural perspective of ligands and mutants. Sci Rep 2015;5:13869.
- Bedford CD, Howd RA, Dailey OD, et al. Nonquaternary cholinesterase reactivators. 3. 3(5)-Substituted 1,2,4-oxadiazol-5(3)-aldoximes and 1,2,4-oxadiazole-5(3)-thiocarbohydroximates as reactivators of organophosphonate-inhibited eel and human acetylcholinesterase in vitro. J Med Chem 1986;29:2174–83.
- Xia G, You X, Liu L, et al. Design, synthesis and SAR of piperidyl-oxadiazoles as 11β-hydroxysteroid dehydrogenase 1 inhibitors . Eur J Med Chem 2013;62:1–10.
- Nowrouzi N, Khalili D, Irajzadeh M. One-pot synthesis of 1,2,4-oxadiazoles from carboxylic acids using 4-(dimethylamino)pyridinium acetate as efficient, regenerable, and green catalyst with ionic liquid character. Journal of the Iranian Chemical Society 2015;12:801–6.
- Rostamizadeh S, Ghaieni HR, Aryan R, et al. Clean one-pot synthesis of 1,2,4-oxadiazoles under solvent-free conditions using microwave irradiation and potassium fluoride as catalyst and solid support. Tetrahedron 2010;66:494–7.
- Selva A, Zerilli LF, Cavalleri B, et al. Mass spectrometry of heterocyclic compounds. V—Substituent effects on the fragmentation pathways of 3,5-diphenyl-1,2,4-oxadiazole derivatives. Org Mass Spectrom 1974;9:558–66.
- Balcom D, Furst A. Reductions with Hydrazine Hydrate Catalyzed by Raney Nickel. I. Aromatic Nitro Compounds to Amines1,2. J Am Chem Soc 1953;75:4334.
- Van Wagenen B, Stormann TM, Moe ST, et al., NPS Pharmaceuticals Inc., Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists, US 2003/0055085 A1; 2003.
- Chruścicka B, Burnat G, Brański P, et al. Tetracycline-based system for controlled inducible expression of group III metabotropic glutamate receptors. J Biomol Screen 2015;20:350–8.
- Walker BD, Singleton CB, Bursill JA, et al. Inhibition of the human ether-a-go-go-related gene (HERG) potassium channel by cisapride: affinity for open and inactivated states. Br J Pharmacol 1999;128:444–450.
- Van der Heyden JA, Zethof TJ, Olivier B. Stress-induced hyperthermia in singly housed mice. Physiol Behav 1997;62:463–70.
- Borsini F, Lecci A, Volterra G, et al. A model to measure anticipatory anxiety in mice? Psychopharmacology 1989;98:207–11.
- Spooren WP, Schoeffter P, Gasparini F, et al. Pharmacological and endocrinological characterisation of stress-induced hyperthermia in singly housed mice using classical and candidate anxiolytics (LY314582, MPEP and NKP608). Eur J Pharmacol 2002;435:161–70.
- Steru L, Chermat R, Thierry B, et al. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 1985;85:367–70.