
Abstract
We have carried out the design, synthesis, and evaluation of a small library of 2-aminobenzoxazole-appended coumarins as novel inhibitors of tumour-related CAs IX and XII. Substituents on C-3 and/or C-4 positions of the coumarin scaffold, and on the benzoxazole moiety, together with the length of the linker connecting both units were modified to obtain useful structure-activity relationships. CA inhibition studies revealed a good selectivity towards tumour-associated CAs IX and XII (Ki within the mid-nanomolar range in most of the cases) in comparison with CAs I, II, IV, and VII (Ki > 10 µM); CA IX was found to be slightly more sensitive towards structural changes. Docking calculations suggested that the coumarin scaffold might act as a prodrug, binding to the CAs in its hydrolysed form, which is in turn obtained due to the esterase activity of CAs. An increase of the tether length and of the substituents steric hindrance was found to be detrimental to in vitro antiproliferative activities. Incorporation of a chlorine atom on C-3 of the coumarin moiety achieved the strongest antiproliferative agent, with activities within the low micromolar range for the panel of tumour cell lines tested.
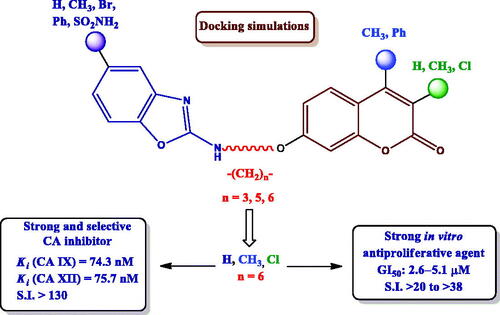
Graphical Abstract
1. Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous (Metallo)enzymes distributed across all life kingdoms and encoded into eight genetic familiesCitation1: α-(primarily invertebrates, but also in protozoa, algae, the cytoplasm of green plants, and numerous Gram-negative bacteria), β- (bacteria, fungi, algae, some archea, and chloroplasts of mono-and di-cotyledons), γ-(most types of bacteria), δ-, ζ-(marine diatoms), η-(protozoa), θ-(marine diatoms), ι-(marine phytoplankton and bacteria), the last one just recently discoveredCitation2. In turn, mammalian α-CAs are categorised into 16 isoforms, classified according to their tissue distribution and kinetic propertiesCitation3: cytosolic (CA I, II, III, VII, XIII), mitochondrial (CA VA, VB), membrane-bound (CA IV, IX, XII, XIV, XV), secreted from saliva and colostrum (CA VI) and CA-related proteins (CARP)Citation4, which are catalytically inactive (CA VIII, X, XI).
The biological role of these enzymes is to catalyse the reversible hydration of CO2 to furnish bicarbonate and a proton; this simple process, which is too slow under physiological conditions to meet metabolic requirementsCitation1, was found to be essential for many biological events, like respiration (by dissolving CO2 in blood as HCO3-)Citation5, maintenance of pH homeostasisCitation6, ureagenesis or gluconeogenesisCitation7. From a pharmacological point of view, a plethora of therapeutic involvements of CAs have been reported, in connection with glaucomaCitation8, epilepsyCitation9, neuropathic painCitation10, ischaemiaCitation11, obesityCitation12, cancerCitation13, and more recently, neurodegenerative disorders, like Alzheimer’s diseaseCitation14. Therefore, the development of CA inhibitorsCitation15 and activatorsCitation16 is a research area with an increasing interest in the medicinal chemistry area.
Although some metals, e.g. Cd(II), Co(II), Fe(II), Mn(II), have been identified as prosthetic groups in the active site of CAs, the most frequent one is Zn(II)Citation17; it has been demonstrated that metal coordination geometries, together with their capacity to orchestrate the dynamics of the surrounding water network through long-range electrostatic effects, can modulate the catalytic efficiencyCitation18.
The most common family of CA inhibitors is comprised of sulphonamides and their isosteres (sulfamates, sulfamides), which behave as strong inhibitors by chelating the Zn2+ ion in the active siteCitation17; nevertheless, they are frequently endowed with moderate selectivity, which leads to a series of side-effects. Consequently, the search for alternative chemotypes of CA inhibitors is a hot topic nowadays. In this context, coumarins (2H-chromen-2-ones), which are abundant phytochemicalsCitation19, but also present in bacteria and fungi (more than 1300 natural structures have been identified so far) emerged as an interesting new family of CA inhibitorsCitation20.
Coumarins are considered as a privileged structure in medicinal chemistryCitation21, exhibiting a plethora of bioactivitiesCitation22, such as antioxidantCitation23, anti-inflammatoryCitation24, antimicrobialCitation25, anti-Alzheimer’sCitation26,Citation27, or antiproliferativeCitation28,Citation29 properties. Conjugation of coumarins with a second pharmacophore is currently gaining attention to access multitarget drugsCitation30. Many of such activities are the result of the inhibition of key enzymes by coumarin-containing derivativesCitation20,Citation31–38, either natural or synthetic; this is due to their peculiar planar structure and to the possibility of establishing strong non-covalent interactions involving the lactone moiety (hydrogen bonding, dipole-dipole) and the aromatic scaffold (π-π and cation-π interactions)Citation21. Regarding CAs, the slow inhibition mode observed for coumarins compared to sulfonamido-derivatives suggested that they might behave as suicide inhibitorsCitation39. Kinetic, crystallographic and computational data revealed that coumarins act in fact as prodrugsCitation40,Citation41: they undergo hydrolysis on their lactone functionality by the esterase activity of the CAs, and the corresponding 2-hydroxycinnamic acids occlude the entrance to the enzyme active site. In particular, coumarin derivatives usually behave as selective inhibitors of CAs IX and XII, which are upregulated in several hypoxic tumoursCitation3,Citation42, and are responsible for the acidic microenvironment in tumour cells. hCA IX expression is limited in normal tissues and is considered to be a marker of aggressive and resistant tumoursCitation43. Regarding hCA XII, its inhibition has been associated with the inactivation of the P-gp machinery, one of the mechanisms for eliminating xenobiotics, and therefore, correlated with the development of resistance towards chemotherapeutic drugsCitation44.
We envisioned the preparation of a small library of the hitherto unknown coumarin-benzoxazole hybrids depicted in to develop novel inhibitors of hCAs IX and XII. The numerous pharmacological properties associated with the benzoxazole skeleton, together with the reduced toxicities of its derivativesCitation45, stimulated us to incorporate such scaffold and analyse the possibility of interactions with CAs.
Figure 1. The general structure of the coumarin-benzoxazole hybrids is prepared herein.

The structure of these novel hybrids is comprised of three key structural motifs: the coumarin skeleton, acting as a prodrug against CAs, and decorated with different substituents on C-3 and C-4 positions; the 2-aminobenzoxazole scaffold, that might establish non-covalent interactions with both, the hydrophobic and the hydrophilic regions of the enzyme (π-π interactions, hydrogen bonding); and the linker, a hydrocarbon chain with different lengths, providing conformational flexibility to the heterocyclic residues.
2. Materials and methods
2.1. General procedures
TLCs (Merck 60 F254, gel thickness 0.25 mm) were performed using aluminium-coated sheets, using the eluant indicated in the experimental section. Spots were visualised by UV light (λ = 254 nm), and by charring with 10% ethanolic vainillin containing 1% H2SO4, or with 5% ethanolic phosphomolybdic acid.
Column chromatography purifications were performed using silica gel stationary phase (Merck 60, particle size 40‒63 µm), eluting by gravity, or with mild pressure, using the eluant indicated in the experimental section.
NMR spectra were registered in the Centro de Investigación, Tecnología e Innovación de la Universidad de Sevilla (CITIUS), using Bruker Avance III 300 and 500 spectrometers (300 and 500 MHz for 1H, 75.5 and 125.7 MHz for 13 C), and the deuterated solvents indicated in each case. Chemical shifts (δ) are expressed in ppm, and coupling constants (J), in Hz. Residual signals from the solvent are used as internal referencesCitation46. Mass spectra were registered using a Qexactive spectrometer, using Electrospray Ionisation (ESI).
2.2. Chemistry
2.2.1. General procedure for the preparation of azides 5a–f
To a solution of the corresponding ω-bromoderivative 4a–f (1.0 equiv.) in DMF (5 ml) was added NaN3 (3.0 equiv.), and the corresponding mixture was heated at 70 °C for 3–4 h. After that, it was extracted with EtOAc (3 × 30 ml); the organic layer was washed with brine (3 × 20 ml), H2O (3 × 20 ml), dried over Na2SO4, and filtered. The filtrate was concentrated to dryness under reduced pressure to give 5a–f in quantitative yields, which were used for the next step without any further purification.
2.2.2. General procedure for the preparation of amines 6a–f
To a solution of 5a–f (106–240 mg) in MeOH (5 ml) was added Pd(OH)2/C (10–20 mg). The resulting suspension was subjected to standard hydrogenolysis at rt and 1 atm H2 for 4 h. The catalyst was removed by filtration through a Celite® pad, and the filtrate was concentrated to dryness to give 6a–f, which were obtained in quantitative yields and used directly for the next step without any further purification.
2.2.3. General procedure for the preparation of isothiocyanates 7b,c,e,f
To a vigorously stirred solution of the amines 6b,c,e,f (1.0 equiv.) in a 1:1 CH2Cl2/H2O mixture (20 ml) were added CaCO3 (3.0 equiv.) and thiophosgene (1.5 equiv.); the corresponding mixture was kept stirring for 30 min. Then, it was filtered through a Celite® pad and the filtrate was extracted with CH2Cl2 (3 × 30 ml) and washed with brine (3 × 20 ml) and H2O (3 × 20 ml). The organic layer was dried over Na2SO4, filtered and the filtrate was concentrated to dryness to give 7b,c,e,f which were used for the next step without any further purification.
2.2.4. General procedure for the preparation of benzoxazoles 8a–j
Method A. To a solution of amines 6a,d in CH2Cl2 (5 ml), thiocarbonyldiimidazole (TCDI) (1.5 equiv.) and DMAP (1.0 equiv.) were added; the corresponding mixture was stirred at rt and under Ar for 2 h. After that, it was concentrated to dryness under reduced pressure and redissolved in THF (5 ml); to this solution, the corresponding 2-aminophenol (1 equiv.) was added and it was refluxed for 17–40 h. Then, my mixture was allowed to cool down to rt, and TBAI (cat.) and 30% H2O2 (2.0 equiv.) were added; the mixture was stirred at rt for 1–2 h. After that, it was concentrated to dryness and the residue was purified by column chromatography (7:3 Cyclohexane–EtOAc) to give derivatives 8a,d (See Supplementary Material).
Method B. To a solution of isothiocyanates 7b,c,e,f in THF (5 ml) the corresponding 2-aminophenol (1.0 equiv.) was added and the mixture was refluxed for 17–40 h. After cooling down to rt, 30% H2O2 (2.0 equiv.) and TBAI (cat.) were added. This mixture was stirred at rt for 1–2 h; then, it was concentrated to dryness and the residue was purified by column chromatography (7:3 Cyclohexane–EtOAc) to give derivatives 8b,c,e–j (See Supplementary Material).
2.3. CA inhibition assays
A stopped-flow CO2 hydrase assay has been employed as reported earlierCitation10–12. All enzymes were recombinant proteins obtained in-house as reportedCitation10–12 and their concentrations in the assay system were in the range of 5–12 nM.
2.4. Antiproliferative assays
The antiproliferative assays were conducted following the protocol of the US National Cancer Institute (NCI), with minor modificationsCitation47.
2.5. Docking simulations
Structures for all proteins (CA IX: PDBid 5FL4; CA XII: PDBid 4HT2) were retrieved from the Protein DataBankCitation48. Crystal structures were optimised using the QuickPrep protocol from MOE (Chemical Computing Group). All ligands were drawn, hydrogens added, and geometry optimised with MOE. For the docking calculations, performed with MOE, in the placement stage, we used the Triangle Matcher algorithm with the London dG scoring scheme. In the refinement stage, we kept the receptor rigid and used the GBVI/WSA dG scoring scheme.
Citation3. Results and discussion
3.1. Chemistry
The retrosynthetic analysis for accessing the coumarin-benzoxazole hybrids proposed herein is depicted in Scheme 1; the key starting materials for accessing such compounds are 4-substituted 2-aminophenols, resorcinol, β-ketoesters and α,ω-dibromoalkanes.
Scheme 1. Retrosynthetic analysis for the preparation of coumarin-benzoxazole hybrids

The synthetic pathway started with the acid-catalysed Pechmann condensationCitation49 involving resorcinol 1 and substituted β-ketoesters 2 to furnish umbelliferone derivatives 3a–d, bearing different substituents on C-3 and/or C-4 positions of the coumarin moiety (Scheme 2). It has been reported that alkyl substitution on those positions decreases the potential hepatotoxicity of such derivatives, by decreasing the rate of the formation of a transient 3,4-epoxide moiety upon metabolisationCitation50.
Scheme 2. Preparation of ω-azidoalkyl derivatives 5a–f. Reagents and conditions: (a) H2SO4, 0 °C→rt; (b) α,ω-Dibromoalkane, anyh. K2CO3, CH3CN, reflux; (c) NaN3, DMF, 70 °C.

Next, the hydroxyl group on C-7 position was subjected to a Williamson synthesis, using an excess of an α,ω-dibromoalkane to favour the monosubstitution process, under mild basic conditions (Scheme 2). Subsequent nucleophilic substitution with NaN3, followed by Pd-catalysed hydrogenolysis of the corresponding azido derivative 5a–f afforded amino-alkyl counterparts 6a–f (Scheme 3). Transformation of the amino moiety into the corresponding isothiocyanate (alternatively with TCDI or CSCl2), coupling with an o-aminophenol to furnish a transient and not isolated thiourea, and final H2O2/TBAI-promoted cyclodesulfurizationCitation51 furnished target benzoxazoles 8a–j (Scheme 3).
Scheme 3. Preparation of coumarin-benzoxazole hybrids 8a–j. Reagents and conditions: (a) H2, Pd(OH)2, MeOH; (b) CSCl2, CaCO3, 1:1 CH2Cl2−H2O, rt; (c) TCDI, DMAP, CH2Cl2, rt; (d) Corresponding o-aminophenol, TFH, reflux; (e) H2O2, TBAI, THF, rt.

TCDI was tentatively used as a green substitute for thiophosgene in the preparation of isothiocyanates. Attempts to isolate the corresponding isothiocyanate upon isothiocyanation reaction of 6a,d gave rather modest yields (roughly 17%, Scheme 3); increase of the reaction times led to decomposition of the isothiocyanate. It was then assumed that reaction of TCDI with coumarin-amino derivatives 6a,d was not complete, and presumably, a thioureido derivative involving the amino-coumarin and one of the imidazole units from TCDI was obtained as the major compound instead of the heterocumulenes 7. Interestingly, the addition of 2-aminophenol to the crude reaction mixture gave the corresponding transient thiourea, as evidenced by TLC. Final in situ iodide-catalyzed oxidative cyclodesulfurization reaction by treatment of thioureas with H2O2 and a catalytic amount of TBAI (Scheme 3) allowed the isolation of benzoxazoles 8a and 8d in a 45% and 34% yield for the one-pot three-step procedure.
Access to benzoxazoles turned out to be more practical, with fewer side-products, when CSCl2 was used as the thionating agent, as amino derivatives 6b,c,e,f could be transformed quantitatively into the expected isothiocyanates 7b,c,e,f (Scheme 3), in a three-phase medium (H2O-CH2Cl2-CaCO3) and under mild conditions. Isothiocyanates were isolated from the crude reaction mixture just by liquid-liquid extraction, without the need for chromatographic purification. Transformation of the heterocumulenes into the transient and non-isolated thioureas upon coupling with substituted o-aminophenols, followed by in situ cyclodesulfurization reaction afforded target benzoxazoles 8b,c,e–j in moderate to good yields (25–67%, two steps, Scheme 3). For derivatives 8h and 8j, a Fisher-Porter tube was required for the formation of the thioureas; reduced reactivity of the corresponding o-aminophenols might be due to the electron-withdrawing effects of the bromine and sulfonamido substituents.
Cyclodesulfurization was confirmed by 1H- and 13 C-NMR; thus, resonances at 4.97‒5.95 (assigned to the NH proton), and at 157.6‒161.7 ppm (C = N) demonstrated the proposed structures. Moreover, spectra of compounds 8i and 8j, registered in CDCl3/CD3OD mixtures, evidenced the absence of the NH proton, due to chemical exchange with the solvent. The absence of a C = S moiety in 13 C at roughly 180 ppm, confirmed the disappearance of the thioureido motif.
3.2. Biological assessments
3.2.1. Carbonic anhydrase inhibition
The 10 new benzoxazole-coumarin hybrids prepared herein have been evaluated in vitro as potential inhibitors of therapeutically relevant hCAs using the stopped-flow CO2 hydration assay () using the drug acetazolamide (AAZ) as control. Two different groups of such metalloenzymes have been used: cytosolic isoforms I (off-target), II (related to glaucomaCitation52) and VII (involved in epilepsy and neuropathic painCitation53) and membrane-bound isoforms IV (involved in rheumatoid arthritisCitation54), IX, XII (both of them overexpressed in hypoxic tumoursCitation40).
Table 1. Inhibition data (Ki, nM) of compounds 8a–j against human CAs I, II, IV, VII, IX, and XIIa,b.
The following structure-activity relationships could be established:
Compounds turned out to be inactive (Ki > 10,000 nM) against the cytosolic isoforms (I, II, VII), except for sulfonamido-containing 8j, which exhibited submicromolar activities (Ki= 806, 516 and 381 nM, respectively).
Compounds 8a–h, bearing no substituents on the phenyl residue of the benzoxazole moiety (R3= H), or small ones (R3= CH3, Br) exhibited moderate inhibition of hCA IV (Ki= 762‒984 nM). On the contrary, bulky substituents and/or endowed with strong electron-withdrawing effects (R3= Ph, SO2NH2) led to weak inhibitors of this enzyme (8i, 8j), with inhibition constants within the low micromolar range (6265 and 3468 nM, respectively).
The linker length (n = 3, 5, 6) did not have a profound influence on the inhibition activities. Thus, a comparison of derivatives 8a–c revealed a mild impairment on CA IV and IX inhibition and a moderate improvement on CA XII inhibition for the longest linker.
Coumarin-benzoxazole hybrids behaved as selective inhibitors of tumour-associated hCAs IX and XII, with strong inhibitions (mid-nanomolar range) in most of the cases. In general, CA IX was more sensitive to the substitution pattern of the coumarin and the benzoxazole moieties.
Disubstitution on the coumarin moiety (R2 position) with either a Me (8d) or a chlorine atom (8e) did not have a very appreciable influence. Nevertheless, incorporation of a Ph motif on R1 (monosubstituted derivative 8f) led to a 3- and 5-fold impairment for the inhibition of CA IX and XII, respectively, compared to its Me-counterpart 8c.
Substitution of the benzoxazole moiety (R3) with CH3 and Br (derivatives 8g and 8h, respectively) was found to be detrimental for the activity against CA IX (6- and 11-fold decreased activity, submicromolar activities) when compared to their non-substituted counterpart 8b.
Substitution of the benzoxazole moiety (R3) with a Ph (8i) furnished strong inhibition of the tumour-associated CAs (Ki = 33.2 and 57.1 nM). The use of a sulfonamido motif in R3, despite providing the strongest CA XII inhibitor of the series, led to an outstanding loss of selectivity, due to the inhibition also of the off-target enzyme in the submicromolar range.
Comparison with native 4-methylumbelliferone 3aCitation55 (hCAI, hCAII > 100 µM; hCA IX 560 nM; hCAXII 8100 nM) revealed the outstanding increase in activity achieved with the hybrids reported herein (up to 16.9-fold for CA IX and up to 160.1-fold for CA XII).
3.2.2. Antiproliferative activity
The coumarin-benzoxazole hybrids were also tested as antiproliferative agents against a panel of six human solid tumour cell lines (): A549 (non-small cell lung), HBL-100 (breast), HeLa (cervix), SW1573 (non-small cell lung) as examples of drug-sensitive lines, and T-47D (breast) and WiDr (colon) as multidrug-resistant lines. A non-tumour cell line (BJ-hTert, human fibroblasts) was also used for analysing the selectivity. Chemotherapeutic agents 5-fluorouracyl (5-FU) and cisplatin (CDDP) were included in the study as drug references.
Table 2. GI50 values (µM) for the antiproliferative activity of derivatives 8a–j.
The following structure-activity relationships could be established:
Substitution patterns had a deeper impact than in the inhibition assay.
The order in potency as antiproliferative agents, considering substitution on the benzoxazole moiety (R3) was: 8h > 8j > 8b > 8i > 8g (Br > SO2NH2 > H > Ph > CH3). Unexpectedly incorporation of a Me moiety (8g) completely abolished activity against all cell lines.
Regarding substitution on the coumarin moiety, the use of a bulky substituent (R1=Ph, 8f) completely abolished activity against all cell lines.
Disubstitution with a second Me group (R1=R2=Me, 8d) led to a clear impairment of activity against the multidrug-resistant cell lines in comparison with the monosubstituted counterpart (8c).
Disubstitution with a chorine atom (R1= Me, R2=Cl) led to the strongest compound in the series (8e), with activities in the low micromolar range for all cell lines (GI50 = 2.6 − 5.1 µM); a remarkably increased activity was found for the multidrug-resistant cell lines compared to the chemotherapeutic agents included in the assay (up to 11.7-fold compared to 5-FU and up to 6.2-fold compared to CDDP).
An increase in the tether length (compounds 8a–c) provoked a strong impairment of activity; for line A549 this situation was completely reversed, leading to a strong antiproliferative activity for compound 8c (GI50= 8.3 µM), the one with the longest linker.
Regarding selectivity, most of the tested compounds lacked significant activity (GI50 > 100 µM, derivatives 8b,c,e–h,j) against BJ-hTert cell line. Derivatives 8a and 8d exhibited weak activity against the non-tumour cell line; moreover, benzoxazole 8i (R1=Me, R3=Ph) was a moderate antiproliferative agent against it, and thus the one with the poorest selectivity.
Selectivity ranges of the lead compound of the series (8e, R1=Me, R2=Cl, > 19.6 – >38.5) clearly exceeded those found for the chemotherapeutic agents 5-FU (0.1‒2.5) and CDDP (0.5‒7.8).
3.3.3. Docking studies
In order to get a deeper insight into the inhibition mechanism exerted by coumarin-benzoxazoles hybrids, compounds reported herein were subjected to docking studies with CAs IX and XII.
As aforementioned, CAs can also exert an esterase activity; the water molecule coordinated to the Zn2+ ion is activated by the metal, thus allowing it to act as a strong nucleophileCitation56. Upon hydrolysis of the lactone functionalityCitation39, the corresponding 2-hydroxycinnamic acid might be isomerised to the most stable E-configuration, depending on the sterical hindrance of the substituents on the coumarin core (Scheme 4).
Scheme 4. The mechanism for the inhibition of CAs by coumarins.

Compound 8i, the strongest CA inhibitor within the series was taken as a model compound for the computational study. Firstly, energy minimisation between 8i-CA complex was accomplished, considering both, the coumarin moiety (closed form) and its E-configured hydrolysed product (open form); data are depicted in . Such data are in agreement with previous reportsCitation39 that suggest the strongest interaction (lower docking interaction energies) of the hydrolysed structure with CAs IX and XII.
Table 3. Docking interaction energies of coumarin-benzoxazole hybrids 8i (kcal/mol)
Docking calculations of compound 8i (closed form) complexed with CA IX showed H-arene interactions between Thr200 and the phenyl ring of the benzoxazole scaffold. Furthermore, van der Waals interactions with residues Asn65, Gly67, Gln92, His94, His96, Val121, Val130, Leu134, Val142, Thr201, and Pro203 were also observed. Interestingly, the open form of 8i-CA IX complex revealed coordination of the carboxylate moiety with the Zn2+ ion of the catalytic site. This was also recently observed in molecular modelling of the interaction of psoralen derivatives and CAsCitation57. Additionally, van der Waals interactions between 8i open form and the residues Gln71, Gln92, His94, His96, Val121, Leu199, Th5200, Thr201, and Trp210 were also found.
shows the poses of both forms binding to CA IX, indicating a completely different orientation in both cases; while in the closed-form the benzoxazole moiety is directed towards the enzyme cleft (), the situation is reversed in the open form (), presumably due to the establishment of the strong ionic interaction between the deprotonated form of the cinnamic acid residue at physiological pH, and the Zn2+ cation.
Figure 2. Predicted binding modes of the closed-form (a) and open form (b) of 8i and CA IX.

A similar situation was found for the predicted interaction between 8i and CA XII (). In this case, π-π interactions between the closed-form of 8i and His91 were detected. Furthermore, 8i establishes van der Waals interactions with residues Asn64, Gln89, His93, His117, Val119, Ala129, Ser133, Leu139, Leu197, and Thr198. Docking calculations for the complexation of the open form of 8i with CA XII also revealed coordination of the carboxylate moiety and the Zn2+ ion. Additionally, it established van der Waals interactions with residues Gln89, His91, His93, Glu104, His117, Val119, Ala129, Ser133, Leu139, Val141, Thr198, and Thr199. In this case, unlike CA IX in both structures, the coumarin scaffold is predicted to be directed to the enzyme cleft.
Figure 3. Predicted binding modes of the closed-form (a) and open form (b) of 8i and CA XII.

4. Conclusions
In conclusion, we herein report an unprecedented family of coumarin-2-aminobenzoxazole hybrids as selective inhibitors of tumour-associated hCAs IX and XII. Substituents on the coumarin and benzoxazole scaffolds, as well as the length of the tether connecting both of them, have been modified to obtain valuable structure-activity relationships. These compounds were accessed starting from substituted umbelliferones in a 6-step synthetic approach: monoalkylation on C-7 position with α,ω-dibromoalkanes, nucleophilic displacement with NaN3, hydrogenolysis, conversion of the terminal amino moiety into an isothiocyanate, coupling with o-aminophenols and intramolecular H2O2/tetrabutylammonium iodide (TBAI)-promoted cyclodesulfurization of the transient thioureas. CA inhibition studies revealed that most of title compounds behaved as strong and selective inhibitors of CAs IX and XII, with inhibition constants within the mid-nanomolar range. Coumarin-benzoxazole hybrids exhibited variable in vitro antiproliferative properties against a panel of human tumour cell lines, strongly dependent on the structural pattern. The lead compound (8e) exhibited GI50 values within the low micromolar range, with remarkable selectivities that exceeded the ones found for the control drugs. Therefore, the family of compounds described herein constitutes a promising start point for the future development of CA inhibitors as antiproliferative agents.
Supplemental Material
Download PDF (1.5 MB)Acknowledgements
We thank the Spanish MICINN (PID2020-116460RB-I00, PGC2018-094503-B-C22), the Junta de Andalucía (FQM134), the Gobierno de Canarias (ProID2020010101, ACIISI/FEDER, UE), the Mexican CONACYT (CB-2015/257465) and the Italian Ministry for Education and Science (MIUR), grant PRIN rot. 2017XYBP2R for financial support. M.X.F. thanks Cabildo de Tenerife for a “Agustín de Betancourt” contract and Program FDCAN from Gobierno de Canarias. A.F.-A. thanks the CONACYT (Mexico) for the award of a fellowship. A.P. thanks the EU Social Fund (FSE) and the Canary Islands ACIISI for a predoctoral grant TESIS202001005. We would also like to thank the Servicio de Resonancia Magnética Nuclear, CITIUS (University of Seville) for the performance of NMR experiments.
Disclosure statement
CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry and he was not involved in the assessment, peer review or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- Angeli A, Carta F, Supuran CT. Carbonic anhydrases: versatile and useful biocatalysts in chemistry and biochemistry. Catalyst 2020;10:1008.
- a) Jensen EL, Clement R, Kosta A, et al. A new widespread subclass of carbonic anhydrase in marine phytoplankton. Isme J 2019;13:2094–106.b) Nocentini A, Supuran CT, Capasso C. An overview on the recently discovered iota-carbonic anhydrases. J Enzyme Inhib Med Chem 2021;36:1988–95.
- Kurt BZ, Dag A, Doğan B, et al. Synthesis, biological activity and multiscale molecular modeling studies of bis-coumarins as selective carbonic anhydrase IX and XII inhibitors with effective cytotoxicity against hepatocellular carcinoma. Bioorg Chem 2019;87:838–50.
- Aspatwar A, Tolvanen MEE, Parkkila S. An update on carbonic anhydrase-related proteins VIII, X and XI. J Enzyme Inhib Med Chem 2013;28:1129–42.
- Geers C, Gros G. Carbon dioxide transport and carbonic anhydrase in blood and muscle. Physiol Rev 2000;80:681–715.
- Lee D, Hong JH. The fundamental role of bicarbonate transporters and associated carbonic anhydrase enzymes in maintaining ion and pH homeostasis in non-secretory organs. Int J Mol Sci 2020;21:339.
- a) Henry RP. Multiple roles of carbonic anhydrase in cellular transport and metabolism. Annu Rev Physiol 1996;58:523–38.b) Supuran Novel carbonic anhydrase inhibitors. Future Med Chem 2021;13:1935–7.
- Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47.
- Aggarwal M, Kondeti B, McKenna R. Anticonvulsant/antiepileptic carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:717–24.
- Nocentini A, Alterio V, Bua S, et al. Phenyl(thio)phosphon(amid)ate benzenesulfonamides as potent and selective inhibitors of human carbonic anhydrases II and VII counteract allodynia in a mouse model of oxaliplatin-induced neuropathy. J Med Chem 2020;63:5185–200.
- Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;31:894–9.
- Costa G, Carta F, Ambrosio FA, et al. A computer-assisted discovery of novel potential anti-obesity compounds as selective carbonic anhydrase VA inhibitors. Eur J Med Chem 2019;181:111565.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77.
- Provensi G, Carta F, Nocentini A, et al. A new kid on the block? Carbonic anhydrases as possible new targets in Alzheimer's disease. Int J Mol Sci 2019;20:4724.
- a) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12.b) Supuran CT. Emerging role of carbonic anhydrase inhibitors. Clin Sci (Lond) 2021;135:1233–49.
- Supuran CT. Carbonic anhydrase activators. Future Med Chem 2018;10:561–73.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32.
- Kim JK, Lee C, Lim SW, et al. Elucidating the role of metal ions in carbonic anhydrase catalysis. Nat Commun 2020;11:4557.
- Annunziata F, Pinna C, Dallavalle S, et al. An overview of coumarin as a versatile and readily accessible scaffold with broad-ranging biological activities. Int J Mol Sci 2020;21:4618.
- Supuran CT. Coumarin carbonic anhydrase inhibitors from natural sources. J Enzyme Inhib Med Chem 2020;35:1462–70.
- Stefanachi A, Leonetti F, Pisani L, et al. Coumarin: a natural, privileged and versatile scaffold for bioactive compounds. Molecules 2018;23:250.
- Medina FG, Marrero JG, Macías-Alonso M, et al. Coumarin heterocyclic derivatives: chemical synthesis and biological activity. Nat Prod Rep 2015;32:1472–507.
- Kostova I, Bhatia S, Grigorov P, et al. Coumarins as antioxidants. Curr Med Chem 2011;18:3929–51.
- Miao Y, Yang J, Yun Y, et al. Synthesis and anti-rheumatoid arthritis activities of 3-(4-aminophenyl)-coumarin derivatives. J Enzyme Inhib Med Chem 2021; 36:450–61.
- Hu Y, Shen Y, Wu X, et al. Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents. Eur J Med Chem 2018;143:958–69.
- Mzezewa SC, Omoruyi SI, Zondagh LS, et al. Design, synthesis, and evaluation of 3,7-substituted coumarin derivatives as multifunctional Alzheimer's disease agents. J Enzyme Inhib Med Chem 2021; 36:1607–21.
- Jiang N, Huang Q, Liu J, et al. Design, synthesis and biological evaluation of new coumarin-dithiocarbamate hybrids as multifunctional agents for the treatment of Alzheimer's disease. Eur J Med Chem 2018;146:287–98.
- Wu Y, Xu J, Liu Y, et al. A review on anti-tumor mechanisms of coumarins. Front Oncol 2020;10:592853.
- Al-Warhi T, Sabt A, Elkaeed EB, Eldehna WM. Recent advancements of coumarin-based anticancer agents: an up-to-date review. Bioorg Chem 2020;103:104163.
- Supuran CT. Multitargeting approaches involving carbonic anhydrase inhibitors: hybrid drugs against a variety of disorders. J Enzyme Inhib Med Chem 2021;36:1702–14.
- Koyiparambath VP, Rajappan KP, Rangarajan TM, et al. Deciphering the detailed structure–activity relationship of coumarins as Monoamine oxidase enzyme inhibitors—an updated review. Chem Biol Drug Des 2021;98:655–73.
- Liu H, Xia DG, Chu ZW, et al. Novel coumarin-thiazolyl ester derivatives as potential DNA gyrase inhibitors: design, synthesis, and antibacterial activity. Bioorg Chem 2020;100:103907.
- Era B, Delogu GL, Pintus F, et al. Looking for new xanthine oxidase inhibitors: 3-phenylcoumarins versus 2-phenylbenzofurans. Int J Biol Macromol 2020;162:774–80.
- Hng Y, Lin MH, Lin TS, et al. Design and synthesis of 3-benzylaminocoumarin-7-O-sulfamate derivatives as steroid sulfatase inhibitors. Bioorg Chem 2020;96:103618.
- Xu X-T, Deng X-Y, Chen J, et al. Synthesis and biological evaluation of coumarin derivatives as α-glucosidase inhibitors. Eur J Med Chem 2020;189:112013.
- Lagunes I, Begines P, Silva A, et al. Selenocoumarins as new multitarget antiproliferative agents: synthesis, biological evaluation and in silico calculations. Eur J Med Chem 2019;179:493–501.
- Abdizadeh T, Kalani MR, Abnous K, et al. Design, synthesis and biological evaluation of novel coumarin-based benzamides as potent histone deacetylase inhibitors and anticancer agents. Eur J Med Chem 2017;132:42–62.
- de Souza LG, Rennã MN, Figueroa-Villar JD. Coumarins as cholinesterase inhibitors: a review. Chem Biol Interact 2016;254:11–23.
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62.
- De Luca L, Mancuso F, Ferro S, et al. Inhibitory effects and structural insights for a novel series of coumarin-based compounds that selectively target human CA IX and CA XII carbonic anhydrases. Eur J Med Chem 2018;143:276–82.
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44.
- Krasavin M, Kalinin S, Sharonova T, Supuran CT. Inhibitory activity against carbonic anhydrase IX and XII as a candidate selection criterion in the development of new anticancer agents. J Enzyme Inhib Med Chem 2020;35:1555–61.
- a) Betof AS, Rabbani ZN, Hardee ME, et al. Carbonic anhydrase IX is a predictive marker of doxorubicin resistance in early-stage breast cancer independent of HER2 and TOP2A amplification. Br J Cancer 2012;106:916–22. ‒b) Chafe SC, Vizeacoumar FS, Venkateswaran G, et al. Genome-wide synthetic lethal screen unveils novel CAIX-NFS1/xCT axis as a targetable vulnerability in hypoxic solid tumors. Sci Adv 2021;7:eabj0364.
- Salaroglio IC, Mujumdar P, Annovazzi L, et al. Carbonic anhydrase XII inhibitors overcome P-glycoprotein-mediated resistance to temozolomide in glioblastoma. Mol Cancer Ther 2018;17:2598–609.
- Reddy GM, Kumari AK, Reddy VH, Garcia JR. Novel pyranopyrazole derivatives comprising a benzoxazole core as antimicrobial inhibitors: design, synthesis, microbial resistance and machine aided results. Bioorg Chem 2020;100:103908.
- Fulmer GR, Miller AJM, Sherden NH, et al. NMR chemical shifts of trace impurities: common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010;29:2176–9.
- Elshaflu H, Todorović TR, Nikolić M, et al. Selenazolyl-hydrazones as novel selective MAO inhibitors with antiproliferative and antioxidant activities: experimental and In-silico studies. Front Chem 2018;6:247.
- Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res 2000;28:235–42.
- Zambare AS, Khan FAK, Zambare SP, et al. Recent advances in the synthesis of coumarin derivatives via Pechmann condensation. Curr Org Chem 2016;20:798–828.
- Lake BG, Evans JG, Lewis DFV, Price RJ. Studies on the acute effects of coumarin and some coumarin derivatives in the rat. Food Chem Toxicol 1994;32:357–63.
- Yadav VK, Srivastava VP, Yadav LDS. Iodide catalyzed synthesis of 2-aminobenzoxazoles via oxidative cyclodesulfurization of phenolic thioureas with hydrogen peroxide. Tetrahedron Lett 2018;59:252–5.
- Swain B, Angeli A, Angapelly S, et al. Synthesis of a new series of 3-functionalised-1-phenyl-1,2,3-triazole sulfamoylbenzamides as carbonic anhydrase I, II, IV and IX inhibitors. J Enzyme Inhib Med Chem 2019;34:1199–209.
- a) Briganti F, Pierattelli R, Scozzafava A, Supuran CT, Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996; 31:3. 1001–10.b) Innocenti A, Gülçin I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Antioxidant polyphenols effectively inhibit mammalian isoforms I-XV. Bioorg Med Chem Lett 2010; 20:5050–3.
- Bua S, Lucarini L, Micheli L, et al. Bioisosteric development of multitarget nonsteroidal anti-inflammatory drug-carbonic anhydrases inhibitor hybrids for the management of rheumatoid arthritis. J Med Chem 2020;63:2325–42.
- Touisni N, Maresca A, McDonald PC, et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7.
- Mancuso F, De Luca L, Angeli A, et al. Synthesis, computational studies and assessment of in vitro inhibitory activity of umbelliferon-based compounds against tumour-associated carbonic anhydrase isoforms IX and XII. J Enzyme Inhib Med Chem 2020;35:1442–9.
- Meleddu R, Deplano S, Maccioni E, et al. Selective inhibition of carbonic anhydrase IX and XII by coumarin and psoralen derivatives. J Enzyme Inhib Med Chem 2021;36:685–92.