
Abstract
In this study, different assortments of 2-arylquinolines and 2,6-diarylquinolines have been developed. Recently, we have developed a new series of 6,7-dimethoxy-4-alkoxy-2-arylquinolines as Topoisomerase I (TOP1) inhibitors with potent anticancer activity. Utilising the SAR outputs from this study, we tried to enhance anticancer and TOP1 inhibitory activities. Though target quinolines demonstrated potent antiproliferative effect, specifically against colorectal cancer DLD-1 and HCT-116, they showed weak TOP1 inhibition which may be attributable to their non-coplanarity. Thereafter, screening against kinase panel revealed their dual inhibitory activity against EGFR and FAK. Quinolines 6f, 6h, 6i, and 20f were the most potent EGFR inhibitors (IC50s = 25.39, 20.15, 22.36, and 24.81 nM, respectively). Meanwhile, quinolines 6f, 6h, 6i, 16d, and 20f exerted the best FAK inhibition (IC50s = 22.68, 14.25, 18.36, 17.36, and 15.36 nM, respectively). Finally, molecular modelling was employed to justify the promising EGFR/FAK inhibition. The study outcomes afforded the first reported quinolines with potent EGFR/FAK dual inhibition.
Graphical Abstract
1. Introduction
Cancer is a major health obstacle in the world threating the life of millions of people annuallyCitation1,Citation2. The universal burden of cancer has been expected to rise to 21.6 million in 2023 compared to 14.1 million in 2012 and was predicted to increase to 28.4 million in 2040 with 47% increment relative to 2020Citation3–5. In 2020, 19.3 million new cancer cases have been diagnosed and 10 million cancer patients passed away. As established by WHO in 2019, cancer was estimated to be the first or second dominant cause of death for the ages <70 years in 112 countries, while it was projected to be the third or fourth death cause in 23 countries. In general, the incidence and mortality of cancer are growing rapidly worldwideCitation4,Citation6. Subsequently, enormous attempts have been implemented to develop potent anticancer drugs through investigations of diverse scaffolds against numerous potential chemotherapeutic targetsCitation7–9.
Epidermal growth factor receptor (EGFR) is a member of tyrosine kinase family in which the endogenous ligand binds to the extracellular domain leading to conformational changes and dimerisation of EGFR resulting in its activation which subsequently stimulates its intrinsic intracellular protein-tyrosine kinase activityCitation10. EGFR is over-expressed in many solid tumours and is related to cancer cell proliferation, angiogenesis and metastasis, so it has a critical role in cancer growth. Therefore, EGFR has been validated as an efficient target for anticancer drug discovery. In the last two decades, different 4-anilinoquinazoline-based EFGR inhibitors, such as Gefitinib, Erlotinib, Afatinib, and Dacomitinib (), have been FDA-approved for clinical use in treatment of non-small cell lung cancerCitation11,Citation12.
Figure 1. Some reported EGFR and FAK inhibitors.

Focal adhesion kinase (FAK) is a cytoplasmic non-receptor tyrosine kinase involved in signal transductions from cell adhesions to regulate different biological cell functions including survival and cell migrationCitation13,Citation14. Also, it is activated and overexpressed in diverse cancer types controlling cancer proliferation, survival and metastasis. Thus, FAK has been identified as a promising druggable target for targeted cancer therapy. Currently, several FAK inhibitors, such as 2,4-diaminopyridine derivative GSK2256098 and 2,4-diaminopyrimidine derivative Defactinib (), are currently being evaluated in clinical trials for cancer treatment, in addition to the 2,4-diaminopyrimidine derivative TAE-226 () which displayed potent antitumor impact in different cancer types in vivo and in vitro and usually used as a reference drugCitation7,Citation15,Citation16. Noteworthy, it was established that the most affected colorectal cancer expressed high levels of EGFR and FAK that particularly correlated with tumour angiogenesis, cancer aggressiveness and poor prognosisCitation17,Citation18.
Thus, dual EGFR/FAK inhibition mechanism is an efficient strategy to fight cancer that could be attributed to a non-overlapping downstream signalling/inhibitionCitation19,Citation20. For example, the kinase inhibitor APG-2449 () was reported to improve the antitumor effect of Ibrutinib via EGFR/FAK inhibition mechanism in oesophageal squamous cell carcinomaCitation19. Also, combined EGFR/FAK inhibition caused higher radiosensitization than either approach aloneCitation21. Interestingly, few studies have succeeded to develop dual EGFR/FAK small molecule inhibitors. In 2020, Ai et al. has exploited a fragment-based drug design approach to identify novel series of 2,4-diaminopyrimidines as potent dual EGFR/FAK inhibitors with good in vitro and in vivo antitumor effectsCitation20.
Quinoline is an outstanding planar heterocyclic motif playing a distinctive role in anticancer drug discovery. So far, assortments of quinoline-based small molecules have been developed and investigated against numerous biological targets for cancer treatment displaying exquisite outcomesCitation22–25. It is worth stressing that plenty of quinoline derivatives provoked their anticancer impact through different mechanisms of action, such as inhibition of DNA repair, tubulin polymerisation, and inhibition of various enzymes implicated in critical cancer cell proliferation prominently kinases enzymes (EGFR, VEGFR, pim-1 kinase, c-Met factor, and PI3K) which stood out as one of the most significant targets implemented in cancer therapy due to their functions in cellular signal transductionCitation26–30. Of special interest, Pelitinib (EKB-569, ) is a 4-anilinoquinoline derivative which is a potent irreversible inhibitor of EGFR in the clinical trials as an anticancer candidateCitation24. In addition, several 4-aminoquinoline derivatives, such as compounds I–III (), were reported as promising EGFR inhibitors endowed with effective anticancer activities. Accordingly, quinoline stands out as a significant privileged scaffold in anticancer drug discovery to develop many efficient kinases inhibitorsCitation24,Citation31–35.
Figure 2. Some reported quinolines with potential anticancer activity.

Recently, we have developed a new series of 6,7-dimethoxy-4-alkoxy-2-arylquinolines as potential Topoisomerase I (TOP1) inhibitorsCitation36. The TOP1-mediated DNA cleavage assay was utilised to assess the ability of the reported compounds to stabilise TOP1-DNA cleavage complexes (TOP1ccs). The assay outcomes revealed a moderate TOP1 inhibitory activity of compounds IVa,b (). Interestingly, the developed quinolines showed outstanding anti-proliferative profile upon evaluation at the Developmental Therapeutics Program (DTP) of the NCI-USA. Noteworthy, the weightiness of incorporation of p-substituted phenyl at C-2, as well as propyl linker at C-4 of the quinoline scaffold, was highlighted by the SAR study.
As a continuation for our previous studyCitation36 novel five sets of 4-propoxy-2-arylquinolines (6a–o, 8a,b, 10a,b, 12a–d, 16a–d), and 4-propoxy-2,6-diarylquinolines (20a–f) are herein designed and synthesised, exploiting the deduced SARs from the previous study, with the aim to afford more potent anticancer TOP1 inhibitors (). Different structural modification strategies were adopted seeking to enhance both anticancer and TOP1 inhibitory activities of the lead compounds IVa,b ().
Figure 3. Different structural modification strategies adopted for design of target quinoline derivatives in this study.

First, diverse secondary alicyclic and aromatic amines, in addition to different primary amines were appended to the propyl linker to illuminate the influence of these moieties on the activity. Also, we introduced the electron donating methyl group as well as electron withdrawing Cl and CF3 for more elucidation of the electronic impact of p-substituent of the 2-phenyl. Likewise, the heterocycles: 2-furyl and 2-thienyl were appended to para position of the C2-phenyl moiety to explore their electronic and size impact on the desired activity. Afterward, the 6,7-dimethoxy groups were removed in some synthesised analogs to confirm their significance. Moreover, the ring system was extended via fusion of the quinoline motif with 1,3-dioxolane in attempt to enhance the planar structure requisite for DNA intercalation which may potentiate both anticancer and TOP1 poisoning effects. Finally, a structural extension approach was utilised via grafting the HBA-bearing 2-furyl and 4-methoxyphenyl moieties at C6 of quinoline scaffold, hoping to enhance the hydrophobic interactions (). The designed 4-propoxy-2-arylquinolines were prepared employing different synthetic procedures, and then investigated for their anticancer and TOP1 inhibitory activities.
Although the target quinolines demonstrated potent antiproliferative effect against different cancer cell lines, they showed no or weak TOP1 poisoning influence. Accordingly, the promising anticancer activity prompted us to search for the plausible molecular mechanism for herein reported quinolines.
The diverse well-reported kinase inhibitory activities of quinoline-based small molecules, as mentioned above, motivated us to explore the potential inhibitory activity of target 4-propoxy-2-arylquinolines (6a–o, 8a,b, 10a,b, 12a–d, and 16a–d) and 4-propoxy-2,6-diarylquinolines (20a–f) against various kinases (EGFR, FAK, FRK, IGF-1R, BTK, c-Src, VEGFR-1, VEGFR-2, HER-2). Strikingly, the investigated quinolines exhibited promising dual inhibitory effect towards EGFR and FAK kinases. Moreover, the apoptotic impact of the most potent anti-proliferative agents in this study was investigated on DLD-1 cells exploiting AV/PI dual staining assay. Finally, in silico molecular modelling techniques, including docking and molecular dynamics studies, were exploited to justify and support results obtained from the biological evaluations.
2. Results and discussion
2.1. Chemistry
The synthetic routes adopted for the synthesis of the target 4-propoxy-2-arylquinolines (6a–o, 8a,b, 10a,b, 12a–d, 16a–d) and 4-propoxy-2,6-diarylquinolines 20a–f are illustrated in Schemes 1–3. Regarding Scheme 1, the benzamides 3a–c were prepared by reacting 4,5-dimethoxy-2′-aminoacetophenone 1 with p-substituted benzoyl derivatives 2a–c in dry THF and Et3N. The latter were cyclized in refluxing dioxane and NaOH to afford the corresponding quinolones 4a–c in excellent yields which then were subjected to O-alkylation with 1-bromo-3-chloropropane using our previously confirmed procedure to yield the respective 4-propoxy key intermediates 5a–cCitation36. Finally, these key intermediates 5a–c were converted to the target 4-prpoxy-2-arylquinolines 6a–o in good to excellent yields (70–80%) through nucleophilic substitution with the appropriate amine in dry DMF and potassium carbonate anhydrous using catalytic amount of potassium iodide at 90 °C.
Scheme 1. Synthesis of target 4-propoxy-2-arylquinolines 6a–o; Reagents and conditions: (i) Et3N, THF, 0 °C then rt, overnight; (ii) dry dioxane, NaOH, reflux under N2, 110 °C, 4 h; (iii) KI, KOH, 1-Bromo-3-chloropropane, dry DMF, rt, 24 h; (iv) KI, K2CO3 anhydrous, appropriate amine, dry DMF, reflux, 90 °C, 12 h.

In Scheme 2(A), the bromo analog of 4-propoxy-N-methylpiperazine-2-arylquinoline 7 was obtained based on the same pathway for 6a–o using p-bromobenzoyl chloride. Then, this bromo analog 7 has been transformed into the target heterocyclic derivatives 8a,b in good yields (70–72%) under Suzuki cross coupling condition through its reaction with the appropriate boronic acid derivative in dioxane using tetrakis catalyst and 2 M sodium carbonate under N2 at 90 °C. In Scheme 2(B,C), syntheses of the target demethoxylated analogs 10a,b and 1,3-dioxolo analogs 12a–d have been accomplished in good to excellent yields (72–87%) adopting the same synthetic routes described for 6a–o using the respective starting compounds, intermediates, and amines.
Scheme 2. Synthesis of target 4-propoxy-2-arylquinolines (A) 8a,b; (B) 10a,b; (C) 12a–d; Reagents and conditions: (i) Pd(PPh3)4, 2 M Na2CO3, Arylboronic acid, dioxane, 90 °C under N2, 16 h. (ii) KI, K2CO3 anhydrous, the respective amine, dry DMF, reflux, 90 °C, 12 h.

Concerning Scheme 3, initiated from 5-bromo-2′-aminoacetophenone 13, the target 6-bromo analogs 16a–d have been afforded in good to excellent yields (78–87%) applying the synthetic routes utilised for 6a–o. Moreover, 5-bromo-2′-aminoacetophenone 13 was transferred to the 6-aryl derivatives 17a,b via Suzuki coupling reaction by its reaction with the appropriate boronic acid derivative in dioxane/water in the presence of tetrakis catalyst and potassium carbonate under N2 at 100 °C. Subsequently, these 6-aryl derivatives 17a,b have been similarly converted to the target 4-propoxy-2,6-diarylquinolnes 20a–f in good to excellent yields (74–89%) exploiting the same experimental pathways adopted for 6a–o.
Scheme 3. Synthesis of the target 6-bromo-4-propoxy-2-arylquinolines 16a–d and 4-propoxy-2,6-diarylquinolines 20a–f; Reagents and conditions: (i) the respective benzoyl chloride, Et3N, THF, 0 °C then rt, overnight; (ii) dry dioxane, NaOH, reflux under N2, 110 °C, 4 h; (iii) KI, KOH, 1-Bromo-3-chloropropane, dry DMF, rt, 24 h; (iv) KI, K2CO3 anhydrous, the respective amine, dry DMF, reflux, 90 °C, 12 h; (v) K2CO3, Pd(PPh3)4, Arylboronic acid, dioxane/H2O (1:1), 100 °C under N2, 4 h.

The structures of all synthesised compounds have been authenticated employing one- dimensional (1D) and two-dimensional (2D) NMR (for 2D NMR, see Supplementary Data), in addition to high resolution mass spectrometry (HRMS).
2.2. 2.2. In vitro anticancer activity
2.2.1. Antiproliferative activity against different cancer cell lines
The in vitro antiproliferative activity of the synthesised 4-propoxy-2-arylquinolines (6a–o, 8a,b, 10a,b, 12a–d, and 16a–d) and 4-propoxy-2,6-diarylquinolines (20a–f) was preliminary investigated by their screening at one dose level (10 µM) against five cancer cell lines representing three cancer types; colorectal cancer (DLD-1 and HCT-116), breast cancer (MDMBA-231 and MCF-7) and cervical cancer (HeLa). After the incubation of the tested compounds for 24 h, the percent growth inhibition (GI%) was calculated. For DLD-1 cell line, twelve compounds exerted good to excellent growth inhibition ranging from 71.94 to 95.36%, from them compounds 6f, 6h, 6i, 16d, 20e, and 20f displayed the highest GI%. Similarly, HCT-116 cell line established good to excellent sensitivity to the tested compounds and thirteen compounds exhibited GI% ranging from 64.26 to 97.48%, from them compounds 6d, 6f, 6h, 6i, 12c, 16b, 16d, 20e, and 20f demonstrated the best sensitivity ().
Table 1. % Growth inhibition (GI%) of all target compounds 6a–o, 8a,b, 10a,b, 12a–d, 16a–d, and 20a–f against different cancer cell lines at 10 µM dose level.
Regarding breast cancer cell lines, GI% ranged from 75.34 to 84.76% and 71.97 to 87.36% for MDMBA-231 and MCF-7 cell lines, respectively. While five compounds showed good inhibitory activity towards MDMBA-231, ten compounds possessed good inhibition to the growth of MCF-7 cancer cell line. For HeLa cell line, seven compounds had good GI% from 65.73 to 90.78%. Based on the preceding screening results, it was revealed that colorectal cancer cell lines (DLD-1 and HCT-116) were the most sensitive to the tested compounds, therefore they were selected for further antiproliferative assay at six doses levels ().
The growth inhibitory activity of the tested 4-propoxy-2-arylquinolines (6a–o, 8a,b, 10a,b, 12a–d, and 16a–d) and 4-propoxy-2,6-diarylquinolines 20a–f exerted on the colorectal cancer cell lines (DLD-1 and HCT-116) was assessed using MTT assay. DLD-1 and HCT-116 cell lines were incubated for 24 h with increasing concentrations (0.5, 1, 10, 30, 50, and 100 µM) of the tested compounds. Gefitinib (EGFR inhibitor) and TAE226 (FAK inhibitor) were used as reference drugs. The results were presented as half maximal growth inhibitory concentration (IC50) which represents the concentration of a drug exhibiting 50% growth inhibition of the cell line compared to the negative control ().
Table 2. The half maximal growth inhibitory concentration (IC50) of all target compounds 6a–o, 8a,b, 10a,b, 12a–d, 16a–d, and 20a–f against two colorectal cancer cell lines (DLD1 and HCT-116) compared to Gefitinib and TAE226.
The antiproliferative investigations against DLD-1 revealed that compound 6h emerged as the most potent counterpart showing IC50 = 1.79 µM surpassing the activity of Gefitinib by 5-folds which possessed IC50 = 10.24 µM. Thereafter, compounds 6f, 6i, 16d, and 20f displayed 4-folds superior activity compared to Gefitinib with IC50 values = 2.25, 2.48, 2.18, and 2.09 µM, respectively. In addition, compounds 6d, 6l, 6n, 8b, 12b, 12c, 16a, 16b, 20b, and 20e exerted better inhibitory activity than Gefitinib demonstrating IC50 values = 8.15, 6.34, 6.11, 7.34, 8.36, 9.12, 8.22, 8.69, 9.65, and 4.46 µM, respectively. The rest of compounds had week to moderate or no activity relative to Gefitinib.
Concerning HCT-116 cell line, it was found that compounds 6f, 6h, 16d, and 20f established twice inhibitory activity compared to Gefitinib (IC50 = 6.94 µM) displaying IC50 values = 3.09, 3.28, 2.43, and 2.96 µM, respectively. Furthermore, compounds 6d, 6i, 12c, 16a, 16b, and 20e exhibited higher growth inhibitory activities than Gefitinib with IC50 values = 4.67, 5.68, 6.37, 6.15, 4.22, and 4.75 µM, respectively. The remaining compounds possessed week to moderate or no activity compared to Gefitinib.
It is noteworthy that appending p-CF3 to the 2-phenyl enhanced the antiproliferative activity compared to p-Cl and p-CH3, except for the cyclohexylamine analogs 6b, 6g, and 6l in which the p-CH3 substituent is preferred for antiproliferative activity. Remarkably, the incorporation of p-(2-furyl) to the 2-phenyl abolished the growth inhibitory activity, while the introduction of p-(2-thienyl) increased the activity compared to p-Cl and p-CH3 analogs. In the context of impact of amine substituents on the antiproliferative activity, it was proved that 4-amino-N-methylpiperidine is preferred for anticancer activity, then piperazine, tetrahydrofurfurylamine and cyclohexylamine. Notably, grafting imidazole along with 6,7-dimethoxy substituents abolished the antiproliferative activity for all derivatives.
Besides, the removal of 6,7-dimethoxy groups from the quinoline scaffold decreased or abolished activity. While the fusion of 1,3-dioxolo to the quinoline scaffold along with p-CF3 substitution on phenyl dramatically potentiated the anticancer activity of the imidazole derivatives, the replacement of imidazole with morpholine decreased the activity of p-CF3 analogs and markedly increased the activity of p-Cl counterparts. On the other hand, appending of electron withdrawing (Br) to position 6 of quinoline parallel with p-Cl substitution on 2-phenyl greatly enhanced the activity of the imidazole derivatives while, p-F substitution on the 2-phenyl extremely decreased the anticancer activity of imidazole derivatives. Also, the replacement of imidazole with morpholine almost had no impact on the p-Cl analogs, but tremendously elevated the anticancer activity of p-F derivatives.
Furthermore, the incorporation of 4-methoxyphenyl or 2-furyl to position 6 of quinoline along with p-Cl or F on the 2-phenyl enhanced the activity of the imidazole derivatives compared to the dimethoxy analogs, but the 2-furyl derivatives exhibited better activity. Moreover, the replacement of imidazole with morpholine elevated the antiproliferative activity of p-Cl analogs while, diminished the activity of p-F derivatives.
Finally, the deduced structure activity relationships indicated that the substitution pattern on positions 6 and 7 of quinoline and position 4 of the 2-phenyl, in addition to the amine substitution on the 4-propoxy linker are crucial elements for the anticancer activity. In general, incorporation of dimethoxy groups at positions 6 and 7 of quinoline along with p-CF3 at 2-phenyl and 4-amino-N-methylpiperidine, piperazine or tetrahydrofurfurylamine on the 4-propoxy linker, in addition to grafting of electron withdrawing (Br) or 2-furyl at position 6 of quinoline along with p-F or Cl on the 2-phenyl and morpholine on the propoxy linker resulted in the most potent antiproliferative agents in this study.
2.2.2. Annexin V-FITC/propidium iodide apoptosis assay (AV/PI)
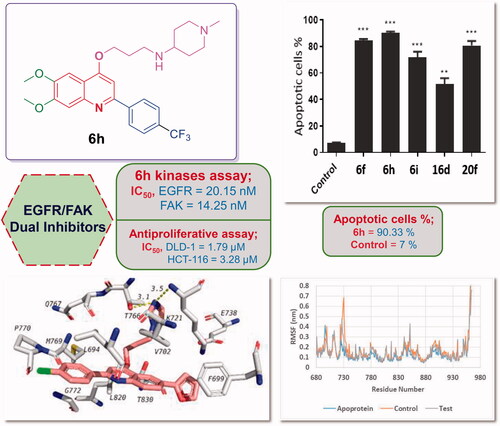
The apoptotic impact of the most potent antiproliferative agents 6f, 6h, 6i, 16d, and 20f on DLD-1 colorectal cancer cell line was investigated exploiting AV/PI dual staining assay. The assay outcomes proved that the tested compounds elicited apoptosis of such cell line as indicated by significant rise in the total percentage of AV positive apoptotic DLD-1 cells compared to the control (). Compound 6h increased the total percentage of apoptotic cells from 7% for the control to 90.33%. Also, compounds 6f, 6i, 16d, and 20f exerted potential apoptotic effect elevating the total percentage of apoptotic cells to 84.33, 71.66, 51.66, and 80.66%, respectively.
Figure 4. Influence of the promising compounds on the total percentage of AV-FITC positive staining in DLD1 cancer cell line.

2.3. Topoisomerase I-mediated DNA cleavage assay
TOP1 poisoning activity of all target compounds has been estimated utilising TOP1-mediated DNA cleavage assay that determines the TOP1 poisoning activity relative to 1 µM Camptothecin (CPT)Citation37. The tested compounds were incubated at 0.1, 1, 10, and 100 µM with recombinant human TOP1 enzyme and a 3′-[32P]-labeled 117-bp DNA oligonucleotideCitation38. TOP1 poisoning agents specifically trap TOP1ccs leading to their stabilisation and DNA cleavage. The drug-induced stabilised TOP1ccs are visualised by gel electrophoresis demonstrating specific DNA cleavage patterns. Then a semiquantitative scoring system by visual comparison between lanes induced by the target compounds and 1 µM CPT was used to score the compounds (, see Supplementary Data for the results of gel electrophoresis)Citation39–41.
Table 3. The TOP1 inhibitory activity of all target compounds 6a–o, 8a,b, 10a,b, 12a–d, 16a–d, and 20a–f compared to Camptothecin (CPT).
Compounds 6a–e, 6h, 6l, 6m, 12a, and 12b exhibited weak TOP1 poisoning effect (-/+) displaying DNA cleavage activity equal to 0–25% of the activity of 1 µM CPT, while compound 16c demonstrating activity (+) equals to 25–50% of the activity of 1 µM CPT. The rest of compounds possessed no cleavage activity. Despite the target compounds showing promising antiproliferative activity against cancer cell lines, their TOP1 poisoning activities were not encouraging for further development as potent TOP1 inhibitors. Accordingly, the synthesised compounds have been evaluated for the plausible mechanism by which they provoked the antiproliferative activity.
2.4. Kinase inhibitory activities of target quinolines
2.4.1. Kinase profiling
The non-significant TOP1 poisoning activities of target compounds obtained from Topoisomerase I-mediated DNA cleavage assay motivated us to search for the plausible molecular mechanism for herein reported 4-propoxy-2-arylquinolines.
The potential inhibitory activity of the target 4-propoxy-2-arylquinolines (6a–o, 8a,b, 10a,b, 12a–d, 16a–d) and 4-propoxy-2,6-diarylquinolines (20a–f) was explored against a panel of nine kinases representing different signalling pathways; EGFR, FAK, FRK, IGF-1R, BTK, c-Src, VEGFR-1, VEGFR-2 and HER-2 (see Supplementary Data, Table S1). The half maximal inhibitory concentration (IC50) values were calculated for each kinase and presented in Table S1 and . Strikingly, the screening outcomes revealed that the investigated quinolines exhibited promising dual inhibitory effect towards EGFR and FAK kinases.
Table 4. The half maximal inhibitory concentration (IC50) of all target compounds 6a–o, 8a,b, 10a,b, 12a–d, 16a–d, and 20a–f against EGFR and FAK kinase activity compared to Gefitinib and TAE226.
2.4.2. EGFR and FAK kinase inhibitory activity
All the newly prepared 4-propoxy-2-arylquinolines (6a–o, 8a,b, 10a,b, 12a–d, and 16a–d) and 4-propoxy-2,6-diarylquinolines (20a–f) were examined for their potential EGFR and FAK inhibitory activities. Gefitinib and TAE-226 were used as reference EGFR and FAK inhibitors, respectively. The results are reported as half maximal inhibitory concentration values (IC50), as determined from triplicate measurements and are presented in .
Results in revealed that the examined quinolines displayed moderate to potent inhibitory activity towards EGFR (IC50 values ranging between 20.15 ± 1.07 and 485.46 ± 11.37 nM, ). In particular, trifluoromethyl phenyl-bearing 6,7-dimethoxy-2-arylquinolines 6f, 6h, and 6i, as well as 6-furanyl-2-arylquinoline 20f emerged as the most efficient EGFR inhibitors with two-digits nanomolar IC50s (IC50 = 25.39 ± 3.49, 20.15 ± 1.07, 22.36 ± 2.05, and 24.81 ± 2.71 nM, respectively). Notably, these four derivatives displayed 2-fold higher activity than the reference EGFR inhibitor Gefitinib (IC50 = 48.52 ± 3.64 nM). In addition, compounds 6b, 6d, 6l, 6n, 8b, 16d, and 20e exhibited potent EGFR inhibitory activity, as the measured IC50 values ranged between 33.65 ± 1.02 and 46.37 ± 4.09 nM, which are slightly improved or comparable to that of the reference drug Gefitinib (). Moreover, compounds 6c and 12b showed 2-fold decreased activity (IC50 = 85.67 ± 6.46 and 95.36 ± 2.05 nM, respectively) than Gefitinib against EGFR. The remaining examined quinolines possessed moderate EGFR inhibitory activity (IC50 range: 121.74 ± 9.40–485.46 ± 11.37 nM) compared to Gefitinib (). Strikingly, the inclusion of 4-amino-N-methylpiperidine, tetrahydrofurfurylamine and N-methylpiperazine along with 2-(p-CF3 phenyl) and 6,7-dimethoxy substituents (6f, 6h, and 6i), in addition to the grafting of morpholine together with 2-(p-Cl phenyl) and 6–(2-furyl) 20f afforded the most potent EGFR inhibitors in this study displaying IC50 range from 20.15 ± 1.07 to 25.39 ± 3.49 nM.
On the other hand, as depicted in , FAK kinase was efficiently inhibited by all 4-propoxy-2-arylquinolines (6a–o, 8a,b, 10a,b, 12a–d, and 16a–d) and 4-propoxy-2,6-diarylquinolines (20a–f) herein reported in the nanomolar range (IC50 range: 14.25 ± 2.72–298.74 ± 1.94 nM). Superiorly, p-CF3-phenyl-bearing 6,7-dimethoxy-2-arylquinolines 6f, 6h, and 6i, as well as, morpholine-bearing 6-bromo-2-arylquinoline 16d and 6-furanyl-2-arylquinoline 20f were the most potent FAK inhibitors in this study with IC50 values equal 22.68 ± 2.38, 14.25 ± 2.72, 18.36 ± 3.17, 17.36 ± 2.15, and 15.36 ± 0.98 nM, respectively (). Moreover, compounds 6c, 6d, 6l, 6n, 8b, 16a, 16b, and 20e exerted potent FAK inhibitory activity with IC50 spanning in the range 25.36 ± 3.48–50.36 ± 4.81 nM.
Further analysis of the obtained results in revealed that compounds 6b, 12b, and 20a–c exhibited two-digit nanomolar IC50s; 98.16 ± 4.67, 70.85 ± 3.16, 77.25 ± 4.37, 63.25 ± 3.25, and 91.03 ± 5.85 nM, respectively, whereas the remaining derivatives displayed moderate inhibitory activity against FAK kinase (IC50 range: 111.06 ± 8.94–298.74 ± 1.94 nM) (). Interestingly, the incorporation of 4-amino-N-methylpiperidine, tetrahydrofurfurylamine and N-methylpiperazine along with 2-(p-CF3 phenyl) and 6,7-dimethoxy substituents (6f, 6h, and 6i), besides the appending of morpholine with 2-(p-F phenyl) and 6-Br 16d, as well as the addition of morpholine in conjunction with 2-(p-Cl phenyl) and 6–(2-furyl) 20f provided the most potent FAK inhibitors in this study demonstrating IC50 values ranging from 14.25 ± 2.72 to 22.68 ± 2.38 nM.
It is worth stressing that 4-propoxy-2-arylquinolines 6f, 6h, 6i, and 20f emerged not only as the most potent dual EGFR/FAK inhibitors in this study, but also as the most efficient anti-proliferative agents towards the examined colorectal cancer (DLD-1 and HCT-116) cell lines.
2.5. In silico molecular docking
2.5.1. Docking into EGFR binding site
The molecular docking approach was utilised to investigate the potential binding of herein reported 4-propoxy-2-arylquinolines to EGFR binding site (PDB: 1M17). The docking procedure was validated through the redocking of the co-crystalised ligand. The correct pose was predicted accurately with RMSD of 1.498 between the docked and co-crystalised ligand using DockRMSD server ()Citation42. In addition, docking was able to maintain hydrogen bonding seen in the co-crystalised ligand with NH of M769 (2.7 Å) and with the NH of G772 (3.2 Å). Furthermore, hydrophobic interactions with residues in the active site were also maintained. These include interactions between K721 and ethyne benzene moiety, and L694, L768, and L820 with hydrophobic part of the quinazoline ring ().
Figure 5. Docking of target quinolines in the active site EGFR. (a) validation of docking procedure showing overlapping of crystalised (blue) and docked (pink) poses; (b) interactions of Erlotinib with EGFR; (c) docking pose of 20c; (d) docking pose of 20e.

Docking scores of the tested compounds with EGFR are shown in . The docking score of the co-crystalised ligand was −7.2 kcal/mol, whereas all the tested quinolines have shown better docking scores than that of the co-crystalised ligand (−7.9 to −9.7 kcal/mol). Best docking scores were seen with quinolines form the series 20. Compound 20c showed the best binding energy to EGFR with docking score of −9.7 kcal/mol and its binding pose is shown in . The compound has formed 2 hydrogen bonds between imidazole ring and both K721 (3.5 Å) and T766 (3.1 Å). Also, several hydrophobic interactions have been seen, which included interactions between quinoline ring and side chains of V702 and T830. The phenyl ring at position 2 formed hydrophobic interactions with L694 and L820 which are also common with the co-crystalised ligand. In addition, π- π stacking between the ring at quinoline position 6 and F699.
Table 5. Docking results of target compounds with EGFR and FAK.
Another compound from this series is compound 20e which was selected as a representative example because it has shown good biological results with both EGFR and FAK. The docking pose of this compound is shown in showing a similar docking pose to 20c. The same hydrogen bonds with K721 (3.5 Å) and T766 (3.1 Å) were maintained as well as hydrophobic interactions with V702 and T830 as well as with L694 and L820. The π–π stacking was also seen between F699 and the furan ring of 20e. This compound, in complex with EGFR, was subjected to further investigation using molecular dynamics to study the stability of its complex with EGFR as will be discussed later.
2.5.2. Docking into FAK binding site
Potential binding of target quinolines to FAK was also investigated using docking studies (PDB: 2JKM). Initially, the docking procedure was validated through the redocking of the co-crystalised ligand (AZW592). The docking searching algorithm was able to correctly predict the binding pose with acceptable accuracy with RMSD of 1.318 between the docked and co-crystalised ligands as predicted by DockRMSD server ()Citation42. The docked structure was able to maintain same hydrogen bonds that are in the crystal structure including those between the sulphamoyl moiety oxygen and the terminal amino group of K454 (Å) and the hydrogen bond with the α-carbonyl group of C502. In addition, several hydrophobic interactions have been also seen with residues in the active site including I428, V436, V484, L501, and L553 ().
Figure 6. Docking of target compounds in the active site FAK. (a) validation of docking procedure showing overlapping of crystalised (blue) and docked (pink) poses; (b) interactions of AZW592 with FAK; (c) general binding of 16 and 20 compound series; (d) docking pose of 20e.

Next, target synthesised quinolines were docked in the active site of the FAK after their preparation. The docking scores of tested compounds are shown in . The docking score of co-crystalised ligand (AZW592) was found to be −8.5 kcal/mol. Some of tested compounds have shown scores that are comparable to the co-crystalised ligand. Best results were seen with 12 and 20 and some of the 16 series. Docking poses of these compounds were similar with most of the docked compounds as can be seen in which shows docking pose of some compounds from these series.
Docking pose of compound 20e which was chosen as representative example is shown in . The compound was able to form 3 hydrogen bonds with N551 (3.1 Å), D564 (3.1 Å), and E506 (3.3 Å). In addition, several hydrophobic interactions were also seen, such as the hydrophobic interaction between quinoline ring and L553 and between phenyl ring at position 2 and I428 which are common with the co-crystallized ligand. This pose was selected for further investigation of the complex stability using molecular dynamics study.
2.6. Molecular dynamics (MD) simulation
The stability of compound 20e complexes with both EGFR and FAK was investigated using 100 ns molecular dynamics studies. With each complex, the results were compared with the co-crystalised ligand complex as a control and with the apoprotein (the protein alone with no ligands). The missing loops in both targets were built using Swiss-Model serverCitation43 before starting the dynamics to ensure correct results. All complexes were equilibrated under NVT then NPT conditions for 1 ns each and the analysis was done on the production run.
Analysis of the production runs trajectories for 20e in the active site of EGFR demonstrated stability comparable to the co-crystalised ligand. Radius of gyration (Rg) is a measure of the compactness of the complexes. Stable Rg suggested the stability of the protein or complex under investigation. shows a plot of Rg of 20e, co-crystalised ligand and apoprotein. The average Rg was found to be 2.01 ± 0.02, 2.03 ± 0.02, and 2.02 ± 0.01 nm for apoprotein, control, and 20e, respectively. In addition, Root mean square fluctuation (RMSF) of protein residue () for all the three complexes showing similar patterns. The average RMSF for co-crystalised ligand and 20e was found to be 0.19 and 0.18 nm, respectively which is slightly higher than that of the apoprotein (0.15 nm). Although root mean square deviation (RMSD) of ligand heavy atoms for 20e is slightly higher than that of the co-crystalised ligand (), the value is <1 nm for most of the trajectory. This value cannot be calculated for the apoprotein as it has no ligand in the system. Finally, the number of hydrogen bonds between ligands and protein are shown in , which showed that 20e formed extra hydrogen bonds during at least 50% of the production run time. These results suggested that 20e complex with EGFR is at least of comparable stability when compared to the complex with the co-crystalised ligand; Erlotinib.
Figure 7. Molecular dynamics analysis of the production run trajectory of 20e in the active site of EGFR compared to control and apoprotein. (a) Radius of gyration; (b) Root mean square fluctuation of residues; (c) Root mean square deviation of ligand heavy atoms; (d) Number of hydrogen bonds between ligand and protein.

Complexes of FAK with 20e, its co-crystalised ligand and the apoprotein showed similar pattern to that of the EGFR (). This includes the radius of gyration (Rg), which showed an average of 2.00 ± 0.01, 1.99 ± 0.01, and 1.96 ± 0.01 nm for apoprotein, co-crystalised ligand, and 20e, respectively (). Also, RMSF of protein residues was found to follow similar patterns for all the three studied systems (). The average RMSF for the three systems was found to be 0.12 ± 0.07, 0.13 ± 0.09, and 0.12 ± 0.07 nm for apoprotein, co-crystalised ligand, and 20e, respectively. In addition, plotting of RMSD of ligand heavy atoms () showed minimal fluctuation for both with and average RMSD of 0.19 ± 0.05 and 0.57 ± 0.16 nm for co-crystalised ligand and 20e, respectively. Although, the value for 20e is higher but it is within acceptable range (<1 nm). Finally, plotting of hydrogen bonds between ligands and target protein () showed that the number of hydrogen bonds is higher in case of the co-crystalised ligand compared to 20e. Being said, 20e was still able to maintain an average of 1.93 ± 0.45 hydrogen bonds during the 100 ns production run.
Figure 8. Molecular dynamics analysis of the production run trajectory of 20e in the active site of FAK compared to control and apoprotein. (a) Radius of gyration; (b) Root mean square fluctuation of residues; (c) Root mean square deviation of ligand heavy atoms; (d) Number of hydrogen bonds between ligand and protein.

These results collectively suggested the stability of compound 20e complexes with both EGFR and FAK compared to the corresponding co-crystalised ligands in each target. This in general supported the dual mechanism similar to the enzymatic inhibition data.
3. Conclusion
Different series of 4-propoxy-2-arylquinolines (6a–o, 8a,b, 10a,b, 12a–d, and 16a–d) and 4-propoxy-2,6-diarylquinolines (20a–f) have been designed and synthesised as potential anticancer agents. The quinolines 6f, 6h, 6i, 16d, and 20f demonstrated the most potent antiproliferative effect against DLD-1 colorectal cancer with respective IC50 values = 2.25, 1.79, 2.48, 2.18, and 2.09 µM with 4- to 5-folds potency compared to Gefitinib (IC50 = 10.24 µM). Additionally, compounds 6f, 6h, 16d, and 20f possessed twice growth inhibitory impact as Gefitinib (IC50 = 6.94 µM) displaying IC50 values = 3.09, 3.28, 2.43, and 2.96 µM against HCT-116 cell line, respectively. Moreover, compounds 6f, 6h, 6i, 16d, and 20f significantly elevated the total percentage of DLD-1 apoptotic cells. Furthermore, the quinolines 6f, 6h, 6i, and 20f exerted potent EGFR inhibitory effects with IC50 values = 25.39 ± 3.49, 20.15 ± 1.07, 22.36 ± 2.05, and 24.81 ± 2.71 nM, respectively compared to Gefitinib (IC50 = 48.52 ± 3.64 nM). In a similar fashion, the quinolines 6f, 6h, 6i, 16d, and 20f displayed the best FAK inhibitory actions with IC50 values = 22.68 ± 2.38, 14.25 ± 2.72, 18.36 ± 3.17, 17.36 ± 2.15, and 15.36 ± 0.98 nM, respectively. Molecular docking and molecular dynamics simulation rationalised EGFR/FAK dual inhibition providing different quinolines being as the first reported quinolines possessing potential EGFR/FAK dual inhibition. The latter compounds can be used as lead compounds for the development of more potent EGFR/FAK dual inhibitors as potential anticancer agents.
4. Experimental
4.1. Chemistry
4.1.1. General
Melting points have been measured by Yanaco melting point device and were uncorrected. NMR spectra were measured using Bruker Advance III HD at 400 MHz for 1H NMR, 100 MHz for 13C NMR and 376 MHz for 19F NMR in deuterated CDCl3 or DMSO-d6 using tetramethyl silane (TMS) as an internal standard. Coupling constant values (J) were determined in Hertz (Hz) and chemical shifts (δ) were expressed in ppm. High resolution mass spectrometry (HRMS) have been measured by Thermo Fisher Scientific LTQ Orbitrap XL spectrophotometer using electrospray ionisation (ESI) and the results were expressed as [M + H]+ or [M + Na]+ at Natural Science Research and Development Centre, Hiroshima University, Japan. The purities of all biologically tested compounds were determined by HPLC and were found to be ≥95%. HPLC analysis was performed utilising JASCO 880-PU HPLC system (Japan spectroscopic Co. Ltd) connected to a diode array detector with detection at a wavelength of 254 nm. The column exploited in the HPLC analysis was Inertsil ODS-3 column with dimensions of 250 × 4.6 mm and 5 µm particle size (GL SCIENCES INC., Japan). The mobile phase employed for HPLC analysis was acetonitrile/water/TFA (29.9/70/0.1, v/v) at a flow rate of 0.5 ml/min. The reactions have been monitored by thin layer chromatography (TLC) using Merck silica gel 60F254 aluminium sheets. Column chromatography has been performed utilising silica gel 60 N, 63–210 µm that was purchased from Kanto Chemical Co. Inc., Japan using dichloromethane/methanol (100/0 to 90/10, v/v) and hexane/ethylacetate (100/0 to 90/10, v/v). Unless otherwise stated, all chemicals and solvents were available commercially and have been used without further purification.
4.1.2. Synthesis of 1-(2-amino-4,5-dimethoxyphenyl)ethan-1-one (1)
2′-Aminoacetophenone derivative 1 was prepared using the reported methodCitation44,Citation45.
Yellow solid, yield 70%, m.p. 100–102 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.53 (s, 3H, CH3), 3.85 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 6.12 (s, 1H, phenyl CH), 6.27 (br s, 2H, NH2), 7.12 (s, 1H, phenyl CH).
4.1.3. General procedure for the synthesis of benzamides (3a–c)
In dry THF (8 ml) and Et3N (2 ml), 1–(2-amino-4,5-dimethoxyphenyl)ethan-1-one 1 (0.976 g, 5 mmol) was dissolved and cooled in ice bath. Then, a solution of the respective p-substituted benzoyl chloride 2a–c (5.1 mmol) in dry THF (2 ml) was added dropwise while cooling in ice bath. The reaction mixture was stirred in ice bath for 30 min and then overnight at room temperature. After that, reaction mixture was poured into ice/water and the resulting solid was filtered off and washed excessively with water and methanol to afford the corresponding 3a–c.
The spectral characterisation of the benzamides 3a,b were reported in our previous studyCitation36.
4.1.3.1. N-(2-acetyl-4,5-dimethoxyphenyl)-4-methylbenzamide (3c)
White solid, yield 70%, m.p. 165–167 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.42 (s, 3H, CH3), 2.65 (s, 3H, CH3C=O), 3.91 (s, 3H, OCH3), 4.02 (s, 3H, OCH3), 7.29 (d, 2H, benzoyl 2CH, J = 8.1 Hz), 7.30 (s, 1H, phenyl CH), 7.96 (d, 2H, benzoyl 2CH, J = 8.1 Hz), 8.77 (s, 1H, phenyl CH), 12.93 (s, 1H, NH); 13 C NMR (100 MHz, CDCl3) δ (ppm): 21.48, 28.37, 56.23, 56.43, 103.52, 113.90, 114.52, 127.45, 129.48, 132.01, 138.49, 142.49, 143.57, 154.89, 166.19 (C=O), 201 (C=O); HRESIMS (m/z): [M + H]+ Calcd for C18H20NO4, 314.13868; found, 314.13901.
4.1.4. General procedures for synthesis of the quinolones (4a–c)
Under N2 atmosphere, the benzamides 3a–c and three equivalents NaOH were refluxed in dry dioxane at 110 °C for 4 h and then cooled to room temperature. Small amount of water and excess amount of hexane were added to the reaction mixture. The resulting mixture was subjected to sonication for 2 min and then neutralised using 1 M HCl. The separated solid was filtered off and washed excessively with water to give the corresponding quinolones 4a–c.
The spectral data of the quinolones 4a,b have been reported in our previous studyCitation36.
4.1.4.1. 6,7-dimethoxy-2-(p-tolyl)quinolin-4(1H)-one (4c)
Yellow solid, Yield 92%, m.p. > 250 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.43 (s, 3H, CH3), 3.93 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 7.03 (s, 1H, vinyl CH), 7.47 (d, 2H, p-toluoyl 2CH, J = 8.1 Hz), 7.51 (s, 1H, phenyl CH), 7.62 (s, 1H, phenyl CH), 7.84 (d, 2H, p-toluoyl 2CH, J = 8.1 Hz); 13 C NMR (100 MHz, DMSO-d4) δ (ppm): 21.43, 56.39, 56.62, 100.57, 102.37, 104.13, 115.90, 128.22, 130.19, 130.34, 137.47, 142.04, 149.30, 151.20, 155.22, 170. 10 (C=O); HRESIMS (m/z): [M + H]+ Calcd for C18H18NO3, 296.12812; found, 296.12827.
4.1.5. General procedure for synthesis of the key intermediates (5a–c)
The quinolones 4a–c (3 mmol), KI (0.498 g, 3 mmol) and KOH (1.009 g, 18 mmol) were stirred for 2 h in dry DMF (30 ml) and then 1-bromo-3-chloropropane (2.361 g, 15 mmol) was added to the mixture and stirred at room temperature for 24 h. Thereafter, the mixture was poured into ice/water and the separated solid was filtered off and washed with water then hexane to furnish the key intermediates 5a–c which were used without further purification.
The spectral data of 5a,b have been reported in our previous studyCitation36.
4.1.5.1. 4-(3-chloropropoxy)-6,7-dimethoxy-2-(p-tolyl)quinoline (5c)
White solid, Yield 90%, m.p. 140–142 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.42 (s, 3H, CH3), 2.43 (p, 2H, CH2, J = 6.1 Hz), 3.83 (t, 2H, CH2Cl, J = 6.2 Hz), 4.02 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 4.42 (t, 2H, CH2-O, J = 6 Hz), 7.10 (s, 1H, vinyl CH), 7.30 (d, 2H, p-toluoyl 2CH, J = 8.1 Hz), 7.36 (s, 1H, phenyl CH), 7.44 (s, 1H, phenyl CH), 7.96 (d, 2H, p-toluoyl 2CH, J = 8.1 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 21.30, 32.01, 41.40, 56.05, 56.11, 64.81, 97.57, 99.58, 108.37, 114.64, 127.14, 129.43, 137.66, 138.87, 146.28, 148.88, 152.61, 156.97, 160.56; HRESIMS (m/z): [M + H]+ Calcd for C21H23ClNO3, 372.13610; found, 372.13623.
4.1.6. General procedure for synthesis of the target 4-propoxy-2-arylquinolines (6a–o)
To a stirred mixture of 5a–c (1 mmol), anhydrous K2CO3 (1.38 g, 10 mmol) and KI (0.83 g, 5 mmol) in dry DMF (20 ml), the respective amine (10 mmol) was added. Then, the mixture was refluxed at 90 °C for 12 h and poured into ice/water (50 ml). The separated solid was filtered off then washed with water and hexane. The products were purified by silica gel column chromatography using DCM/MeOH to furnish the pure target compounds 6a–o.
4.1.6.1. 2-(4-chlorophenyl)-6,7-dimethoxy-4–(3-(4-methylpiperazin-1-yl)propoxy)quinoline (6a)
White solid, Yield 71%, m.p. 140–142 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.15 (p, 2H, CH2, J = 6.8 Hz), 2.29 (s, 3H, CH3-N), 2.36–2.66 (br s, 8H, piperazinyl 4CH2), 2.62 (t, 2H, CH2-N, J = 7.3 Hz), 4.01 (s, 3H, OCH3), 4.02 (s, 3H, OCH3), 4.31 (t, 2H, CH2-O, J = 6.3 Hz), 7.03 (s, 1H, vinyl CH), 7.37 (s, 1H, phenyl CH), 7.40 (s, 1H, phenyl CH), 7.44 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.99 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.59, 46.02, 53.31, 55.11, 56.04, 56.11, 66.65, 97.38, 99.68, 108.23, 114.93, 128.55, 128.82, 134.90, 139.00, 146.18, 149.07, 152.72, 155.59, 161.08; HRESIMS (m/z): [M + H]+ Calcd for C25H31ClN3O3, 456.20485; found, 456.20474; HPLC purity: 97.65%.
4.1.6.2. N-(3-((2–(4-chlorophenyl)-6,7-dimethoxyquinolin-4-yl)oxy)propyl)cyclohexanamine (6b)
Grey solid, Yield 80%, m.p. 118–120 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.02–1.13 (m, 2H, cyclohexyl 2CHH′), 1.14–1.19 (m, 1H, cyclohexyl CHH′), 1.20–1.30 (m, 2H, cyclohexyl 2CHH′), 1.44 (br s, 1H, NH), 1.59–1.63 (m, 1H, cyclohexyl CHH′), 1.70–1.74 (m, 2H, cyclohexyl 2CHH′), 1.89–1.92 (m, 2H, cyclohexyl 2CHH′), 2.14 (p, 2H, CH2, J = 6.5 Hz), 2.43–2.50 (m, 1H, cyclohexyl CH-NH), 2.93 (t, 2H, CH2-NH, J = 6.9 Hz), 4.02 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 4.35 (t, 2H, CH2-O, J = 6.1 Hz), 7.06 (s, 1H, vinyl CH), 7.39 (s, 1H, phenyl CH), 7.41 (s, 1H, phenyl CH), 7.45 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 8 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 25.06, 26.14, 30.00, 33.68, 43.90, 56.05, 56.12, 56.91, 66.94, 97.42, .99.67, 108.23, 114.94, 128.55, 128.82, 134.90, 138.98, 146.18, 149.07, 152.71, 155.61, 161.07; HRESIMS (m/z): [M + H]+ Calcd for C26H32ClN2O3, 455.20960; found, 455.20932; HPLC purity: 98.81%.
4.1.6.3. N-(3-((2–(4-chlorophenyl)-6,7-dimethoxyquinolin-4-yl)oxy)propyl)-1-methylpiperidin-4-amine (6c)
White solid, Yield 70%, m.p. 124–126 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.35–1.45 (m, 3H, piperidinyl 2CHH′, NH), 1.89 (d, 2H, piperidinyl 2CHH′, J = 12.6 Hz), 1.97 (t, 2H, piperidinyl 2CHH′, J = 11.7 Hz), 2.14 (p, 2H, CH2, J = 6.5 Hz), 2.25 (s, 3H, N-CH3), 2.43–2.51 (m, 1H, piperidinyl CH-NH), 2.80 (d, 2H, piperidinyl 2CHH′, J = 11.7 Hz), 2.92 (t, 2H, CH2-NH, J = 6.9 Hz), 4.01 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 4.35 (t, 2H, CH2-O, J = 6.1 Hz), 7.05 (s, 1H, vinyl CH), 7.37 (s, 1H, phenyl CH), 7.41 (s, 1H, phenyl CH), 7.44 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.99 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 29.96, 32.91, 43.73, 46.23, 54.50, 54.64, 56.06, 56.12, 66.83, 97.39, 99.64, 108.25, 114.92, 128.54, 128.83, 134.91, 138.97, 146.19, 149.08, 152.72, 155.60, 161.05; HRESIMS (m/z): [M + H]+ Calcd for C26H333ClN3O3, 470.22050; found, 470.21988; HPLC purity: 96.18%.
4.1.6.4. 3-((2–(4-chlorophenyl)-6,7-dimethoxyquinolin-4-yl)oxy)-N-((tetrahydrofuran-2-yl)methy- l)propan-1-amine (6d)
White solid, Yield 74%, m.p. 108–110 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.48–1.57 (m, 1H, furyl CHH′), 1.66 (s, 1H, NH), 1.83–1.92 (m, 2H, furyl CH2), 1.93–2 (m, 1H, furyl CHH′), 2.16 (p, 2H, CH2, J = 6.6 Hz), 2.67 (dd, 1H, furfuryl CHH′-NH, J = 8, 12 Hz) , 2.75 (dd, 1H, furfuryl CHH′-NH, J = 3.6, 12 Hz) , 2.92 (t, 2H, CH2-NH, J = 6.9 Hz), 3.70–3.75 (m, 1H, furyl CHH′-O), 3.80–3.85 (m, 1H, furyl CHH′-O), 4 (p, 1H, furyl CH-O, J = 3.6 Hz), 4.02 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 4.36 (t, 2H, CH2-O, J = 6.2 Hz), 7.06 (s, 1H, vinyl CH), 7.39 (s, 1H, phenyl CH), 7.41 (s, 1H, phenyl CH), 7.44 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 8 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 25.78, 29.32, 29.67, 46.99, 54.64, 56.08, 66.80, 67.95, 78.31, 97.41, 99.80, 108.25, 114.97, 128.55, 128.80, 134.88, 139.02, 146.20, 149.09, 152.73, 155.58, 161.12; HRESIMS (m/z): [M + H]+ Calcd for C25H30ClN2O4, 457.18886; found, 457.18936; HPLC purity: 97.34%.
4.1.6.5. 4-(3-(1H-imidazol-1-yl)propoxy)-2–(4-chlorophenyl)-6,7-dimethoxyquinoline (6e)
White solid, Yield 75%, m.p. 209–211 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.43 (p, 2H, CH2, J = 6.2 Hz), 4.03 (s, 3H, OCH3), 4.04 (s, 3H, OCH3), 4.22 (t, 2H, CH2-N, J = 5.8 Hz), 4.28 (t, 2H, CH2-O, J = 6.6 Hz), 6.94 (s, 1H, imidazole CH), 6.96 (s, 1H, vinyl CH), 7.07 (s, 1H, imidazole CH), 7.33 (s, 1H, phenyl CH), 7.43 (s, 1H, phenyl CH), 7.44 (d, 2H, chlorophenyl 2CH, J = 8.5 Hz), 7.50 (s, 1H, imidazole CH), 7.96 (d, 2H, chlorophenyl 2CH, J = 8.5 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 30.51, 43.59, 56.12, 64.48, 97.30, 99.33, 108.44, 114.67, 118.87, 128.51, 128.85, 130.03, 135.05, 137.23, 138.73, 146.34, 149.36, 152.94, 155.55, 160.43; HRESIMS (m/z): [M + H]+ Calcd for C23H23ClN3O3, 424.14225; found, 424.14264; HPLC purity: 99.50%.
4.1.6.6. 6,7-dimethoxy-4–(3-(4-methylpiperazin-1-yl)propoxy)-2–(4-(trifluoromethyl)phenyl)quin-oline (6f)
White solid, Yield 71%, m.p. 84–86 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.17 (p, 2H, CH2, J = 6.8 Hz), 2.29 (s, 3H, CH3-N), 2.36–2.71 (br s, 8H, piperazinyl 4CH2), 2.63 (t, 2H, CH2-N, J = 7.3 Hz), 4.02 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 4.33 (t, 2H, CH2-O, J = 6.3 Hz), 7.09 (s, 1H, vinyl CH), 7.39 (s, 1H, phenyl CH), 7.43 (s, 1H, phenyl CH), 7.73 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz), 8.16 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.59, 46.03, 53.34, 55.12, 56.07, 56.13, 66.72, 97.71, 99.64, 108.28, 115.18, 124.26 (CF3, q, J = 271.7 Hz), 125.59 (CH-C-CF3, q, J = 3.9 Hz), 127.59, 130.59 (CH-C-CF3, q, J = 32.4 Hz), 143.94, 146.24, 149.32, 152.84, 155.25, 161.18; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.47 (s); HRESIMS (m/z): [M + H]+ Calcd for C26H31F3N3O3, 490.23120; found, 490.23087; HPLC purity: 98.93%.
4.1.6.7. N-(3-((6,7-dimethoxy-2–(4-(trifluoromethyl)phenyl)quinolin-4-yl)oxy)propyl)cyclohexan- amine (6g)
Grey solid, Yield 75%, m.p. 131–133 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.02–1.12 (m, 2H, cyclohexyl 2CHH′), 1.13–1.19 (m, 1H, cyclohexyl CHH′), 1.20–1.30 (m, 2H, cyclohexyl 2CHH′), 1.37 (br s, 1H, NH), 1.59–1.63 (m, 1H, cyclohexyl CHH′), 1.70–1.74 (m, 2H, cyclohexyl 2CHH′), 1.89–1.92 (m, 2H, cyclohexyl 2CHH′), 2.15 (p, 2H, CH2, J = 6.5 Hz), 2.43–2.50 (m, 1H, cyclohexyl CH-NH), 2.93 (t, 2H, CH2-NH, J = 6.9 Hz), 4.03 (s, 3H, OCH3), 4.04 (s, 3H, OCH3), 4.37 (t, 2H, CH2-O, J = 6.1 Hz), 7.11 (s, 1H, vinyl CH), 7.40 (s, 1H, phenyl CH), 7.43 (s, 1H, phenyl CH), 7.73 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz), 8.16 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 25.05, 26.14, 30.03, 33.71, 43.87, 56.07, 56.13, 56.90, 67.01, 97.73, 99.64, 108.30, 115.19, 124.26 (CF3, q, J = 272 Hz), 125.59 (CH-C-CF3, q, J = 3.7 Hz), 127.57, 130.62 (CH-C-CF3, q, J = 33.9 Hz), 143.92, 146.24, 149.33, 152.84, 155.25, 161.17; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.47 (s); HRESIMS (m/z): [M + H]+ Calcd for C27H32F3N2O3, 489.23595; found489.23578; HPLC purity: 99.37%.
4.1.6.8. N-(3-((6,7-dimethoxy-2–(4-(trifluoromethyl)phenyl)quinolin-4-yl)oxy)propyl)-1-methyl p- iperidin-4-amine (6h)
White solid, Yield 78%, m.p. 137–139 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.36–1.45 (m, 3H, piperidinyl 2CHH′, NH), 1.89 (d, 2H, piperidinyl 2CHH′, J = 12.5 Hz), 1.97 (t, 2H, piperidinyl 2CHH′, J = 11.7 Hz), 2.14 (p, 2H, CH2, J = 6.5 Hz), 2.25 (s, 3H, N-CH3), 2.43–2.51 (m, 1H, piperidinyl CH-NH), 2.80 (d, 2H, piperidinyl 2CHH′, J = 11.7 Hz), 2.92 (t, 2H, CH2-NH, J = 6.9 Hz), 4.02 (s, 3H, OCH3), 4.04 (s, 3H, OCH3), 4.37 (t, 2H, CH2-O, J = 6.1 Hz), 7.10 (s, 1H, vinyl CH), 7.39 (s, 1H, phenyl CH), 7.43 (s, 1H, phenyl CH), 7.73 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz), 8.16 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 29.95, 32.89, 43.68, 46.21, 54.50, 54.63, 56.08, 56.14, 66.89, 97.71, 99.61, 108.31, 115.17, 124.27 (CF3, q, J = 272.6 Hz), 125.60 (CH-C-CF3, q, J = 3.9 Hz), 127.57, 130.64 (CH-C-CF3, q, J = 32.7 Hz), 143.90, 146.25, 149.34, 152.85, 155.26, 161.15; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.47 (s); HRESIMS (m/z): [M + H]+ Calcd for C27H33F3N3O3, 504.24685; found, 504.24692; HPLC purity: 99.75%.
4.1.6.9. 3-((6,7-dimethoxy-2–(4-(trifluoromethyl)phenyl)quinolin-4-yl)oxy)-N-((tetrahydrofuran-2-yl)methyl)propan-1-amine (6i)
White solid, Yield 76%, m.p. 105–107 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.49–1.57 (m, 1H, furyl CHH′), 1.69 (s, 1H, NH), 1.83–1.92 (m, 2H, furyl CH2), 1.93–2 (m, 1H, furyl CHH′), 2.18 (p, 2H, CH2, J = 6.6 Hz), 2.68 (dd, 1H, furfuryl CHH′-NH, J = 8, 12 Hz) , 2.75 (dd, 1H, furfuryl CHH′-NH, J = 3.5, 12 Hz), 2.93 (t, 2H, CH2-NH, J = 6.9 Hz), 3.70–3.75 (m, 1H, furyl CHH′-O), 3.80–3.86 (m, 1H, furyl CHH′-O), 4 (p, 1H, furyl CH-O, J = 3.5 Hz), 4.03 (s, 3H, OCH3), 4.04 (s, 3H, OCH3), 4.38 (t, 2H, CH2-O, J = 6.2 Hz), 7.11 (s, 1H, vinyl CH), 7.41 (s, 1H, phenyl CH), 7.43 (s, 1H, phenyl CH), 7.43 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz), 8.16 (d, 2H, chlorophenyl 2CH, J = 8.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 25.78, 29.32, 29.64, 47.00, 54.67, 56.11, 56.13, 66.87, 67.97, 78.28, 97.74, 99.72, 108.26, 115.20, 124.28 (CF3, q, J = 272 Hz), 125.58 (CH-C-CF3, q, J = 3.8 Hz), 127.58, 130.59 (CH-C-CF3, q, J = 32.3 Hz), 143.92, 146.23, 149.31, 152.82, 155.24, 161.20; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.46 (s); HRESIMS (m/z): [M + H]+ Calcd for C26H30F3N2O4, 491.21522; found, 491.21533; HPLC purity: 99.63%.
4.1.6.10. 4-(3-(1H-imidazol-1-yl)propoxy)-6,7-dimethoxy-2–(4-(trifluoromethyl)phenyl)quinoline (6j)
White solid, Yield 88%, m.p. 217–219 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.45 (p, 2H, CH2, J = 6.2 Hz), 4.05 (s, 3H, OCH3), 4.06 (s, 3H, OCH3), 4.24 (t, 2H, CH2-N, J = 5.8 Hz), 4.29 (t, 2H, CH2-O, J = 6.6 Hz), 6.94 (s, 1H, imidazole CH), 7.01 (s, 1H, vinyl CH), 7.08 (s, 1H, imidazole CH), 7.35 (s, 1H, phenyl CH), 7.46 (s, 1H, phenyl CH), 7.51 (s, 1H, imidazole CH), 7.73 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz), 8.13 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 30.49, 43.58, 56.15, 56.18, 64.54, 97.63, 99.24, 108.47, 114.90, 118.88, 124.23 (CF3, q, J = 272 Hz), 125.63 (CH-C-CF3, q, J = 3.7 Hz), 127.56, 130.05, 130.72 (CH-C-CF3, q, J = 32.5 Hz), 137.25, 143.64, 146.38, 149.59, 153.03, 155.22, 160.51; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.48 (s); HRESIMS (m/z): [M + H]+ Calcd for C24H23F3N3O3, 458.16860; found, 458.16879; HPLC purity: 99.78%.
4.1.6.11. 6,7-dimethoxy-4–(3-(4-methylpiperazin-1-yl)propoxy)-2-(p-tolyl)quinoline (6k)
White solid, Yield 70%, m.p. 128–130 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.15 (p, 2H, CH2, J = 6.8 Hz), 2.29 (s, 3H, CH3-N), 2.41 (s, 3H, tolyl CH3), 2.41–2.64 (br s, 8H, piperazinyl 4CH2), 2.62 (t, 2H, CH2-N, J = 7.3 Hz), 4.01 (s, 3H, OCH3), 4.02 (s, 3H, OCH3), 4.31 (t, 2H, CH2-O, J = 6.3 Hz), 7.07 (s, 1H, vinyl CH), 7.44 (d, 2H, tolyl 2CH, J = 8 Hz), 7.38 (s, 1H, phenyl CH), 7.43 (s, 1H, phenyl CH), 7.94 (d, 2H, tolyl 2CH, J = 8 Hz); 13C NMR (100 MHz, CDCl3) δ (ppm): 21.29, 26.62, 46.03, 53.33, 55.12, 55.17, 56.02, 56.09, 66.56, 97.58, 99.72, 108.30, 114.76, 127.16, 129.39, 137.79, 138.77, 146.20, 148.77, 152.52, 157.01, 160.91; HRESIMS (m/z): [M + H]+ Calcd for C26H34N3O3, 436.25947; found, 436.25943; HPLC purity: 96.05%.
4.1.6.12. N-(3-((6,7-dimethoxy-2-(p-tolyl)quinolin-4-yl)oxy)propyl)cyclohexanamine (6l)
Buff solid, Yield 73%, m.p. 126–128 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.03–1.14 (m, 2H, cyclohexyl 2CHH′), 1.15–1.21 (m, 1H, cyclohexyl CHH′), 1.23–1.30 (m, 2H, cyclohexyl 2CHH′), 1.59–1.63 (m, 2H, cyclohexyl CHH′, NH), 1.70–1.75 (m, 2H, cyclohexyl 2CHH′), 1.89–1.93 (m, 2H, cyclohexyl 2CHH′), 2.15 (p, 2H, CH2, J = 6.6 Hz), 2.41 (s, 3H, tolyl CH3), 2.44–2.51 (m, 1H, cyclohexyl CH-NH), 2.93 (t, 2H, CH2-NH, J = 7 Hz), 4.01 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 4.34 (t, 2H, CH2-O, J = 6.1 Hz), 7.08 (s, 1H, vinyl CH), 7.29 (d, 2H, tolyl 2CH, J = 8 Hz), 7.39 (s, 1H, phenyl CH), 7.43 (s, 1H, phenyl CH), 7.95 (d, 2H, tolyl 2CH, J = 8 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 21.29, 25.05, 26.13, 29.96, 33.62, 43.93, 56.03, 56.09, 56.91, 66.84, 97.60, 99.71, 108.32, 114.77, 127.15, 129.40, 137.77, 138.78, 146.21, 148.79, 152.52, 157.01, 160.88; HRESIMS (m/z): [M + H]+ Calcd for C27H35N2O3, 435.26422; found, 435.26422; HPLC purity: 97.90%.
4.1.6.13. N-(3-((6,7-dimethoxy-2-(p-tolyl)quinolin-4-yl)oxy)propyl)-1-methylpiperidin-4-amine (6m)
White solid, Yield 76%, m.p. 66–68 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.35–1.45 (m, 3H, piperidinyl 2CHH′, NH), 1.89 (d, 2H, piperidinyl 2CHH′, J = 12.5 Hz), 1.96 (t, 2H, piperidinyl 2CHH′, J = 11.8 Hz), 2.13 (p, 2H, CH2, J = 6.5 Hz), 2.24 (s, 3H, N-CH3), 2.41 (s, 3H, tolyl CH3), 2.43–2.50 (m, 1H, piperidinyl CH-NH), 2.80 (d, 2H, piperidinyl 2CHH′, J = 11.8 Hz), 2.91 (t, 2H, CH2-NH, J = 6.9 Hz), 4.01 (s, 3H, OCH3), 4.02 (s, 3H, OCH3), 4.34 (t, 2H, CH2-O, J = 6.1 Hz), 7.08 (s, 1H, vinyl CH), 7.28 (d, 2H, tolyl 2CH, J = 8 Hz), 7.37 (s, 1H, phenyl CH), 7.43 (s, 1H, phenyl CH), 7.94 (d, 2H, tolyl 2CH, J = 8 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 21.29, 29.97, 32.90, 43.79, 46.22, 54.49, 54.64, 56.03, 56.09, 66.74, 97.58, 99.68, 108.33, 114.75, 127.14, 129.40, 137.76, 138.79, 146.21, 148.79, 152.53, 157.00, 160.87; HRESIMS (m/z): [M + H]+ Calcd for C27H36N3O3, 450.27512; found, 450.27481; HPLC purity: 95.52%.
4.1.6.14. 3-((6,7-dimethoxy-2-(p-tolyl)quinolin-4-yl)oxy)-N-((tetrahydrofuran-2-yl)methyl)propa- n-1-amine (6n)
Yellow solid, Yield 85%, m.p. 60–62 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.48–1.57 (m, 1H, furyl CHH′), 1.83–2.00 (m, 4H, furyl CH2, furyl CHH′, NH), 2.16 (p, 2H, CH2, J = 6.5 Hz), 2.41 (s, 3H, tolyl CH3), 2.67 (dd, 1H, furfuryl CHH′-NH, J = 8, 12 Hz), 2.74 (dd, 1H, furfuryl CHH′-NH, J = 3.6, 12 Hz), 2.92 (t, 2H, CH2-NH, J = 6.8 Hz), 3.69–3.75 (m, 1H, furyl CHH′-O), 3.79–3.85 (m, 1H, furyl CHH′-O), 4 (p, 1H, furyl CH-O, J = 3.6 Hz), 4.02 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 4.35 (t, 2H, CH2-O, J = 6.2 Hz), 7.08 (s, 1H, vinyl CH), 7.29 (d, 2H, tolyl 2CH, J = 8 Hz), 7.39 (s, 1H, phenyl CH), 7.43 (s, 1H, phenyl CH), 7.94 (d, 2H, tolyl 2CH, J = 8 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 21.28, 25.77, 29.33, 29.61, 47.06, 54.61, 56.06, 56.08, 66.70, 67.95, 78.23, 97.64, 99.80, 108.22, 114.78, 127.18, 129.39, 137.76, 138.78, 146.16, 148.78, 152.53, 157.03, 160.93; HRESIMS (m/z): [M + H]+ Calcd for C26H33N2O4, 437.24348; found, 437.24353; HPLC purity: 97.50%.
4.1.6.15. 4-(3-(1H-imidazol-1-yl)propoxy)-6,7-dimethoxy-2-(p-tolyl)quinoline (6o)
White solid, Yield 83%, m.p. 203–205 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.41 (s, 3H, tolyl CH3), 2.42 (p, 2H, CH2, J = 6.3 Hz), 4.03 (s, 6H, 2CH3O), 4.22 (t, 2H, CH2-N, J = 5.8 Hz), 4.27 (t, 2H, CH2-O, J = 6.7 Hz), 6.93 (s, 1H, imidazole CH), 6.99 (s, 1H, vinyl CH), 7.07 (s, 1H, imidazole CH), 7.28 (d, 2H, tolyl 2CH, J = 8.1 Hz), 7.34 (s, 1H, phenyl CH), 7.45 (s, 1H, phenyl CH), 7.51 (s, 1H, imidazole CH), 7.92 (d, 2H, tolyl 2CH, J = 8.1 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 21.29, 30.55, 43.62, 56.10, 56.13, 64.37, 97.50, 99.35, 108.48, 114.48, 118.92, 127.12, 129.44, 129.97, 137.22, 137.51, 138.96, 146.34, 149.03, 152.71, 156.97, 160.25; HRESIMS (m/z): [M + H]+ Calcd for C24H26N3O3, 404.19687; found, 404.19724; HPLC purity: 99.42%.
4.1.7. Synthesis of 2-(4-bromophenyl)-6,7-dimethoxy-4-(3-(4-methylpiperazin-1-yl)propoxy)qu-inoline (7)
The bromo analog of 4-propoxy-N-methylpiperazine-2-arylquinoline 7 has been prepared using the same synthetic procedure as 6a–o utilising p-bromobenzoyl chloride.
White solid, Yield 77%, m.p. 148–150 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.15 (p, 2H, CH2, J = 6.8 Hz), 2.29 (s, 3H, CH3-N), 2.36–2.66 (br s, 8H, piperazinyl 4CH2), 2.62 (t, 2H, CH2-N, J = 7.3 Hz), 4.01 (s, 3H, OCH3), 4.02 (s, 3H, OCH3), 4.31 (t, 2H, CH2-O, J = 6.3 Hz), 7.03 (s, 1H, vinyl CH), 7.37 (s, 1H, phenyl CH), 7.41 (s, 1H, phenyl CH), 7.60 (d, 2H, bromophenyl 2CH, J = 8.6 Hz), 7.92 (d, 2H, bromophenyl 2CH, J = 8.6 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.57, 45.96, 53.24, 55.07, 56.04, 56.10, 66.65, 97.34, 99.71, 108.23, 114.97, 123.23, 128.85, 131.77, 139.45, 146.20, 149.11, 152.76, 155.62, 161.10; HRESIMS (m/z): [M + H]+ Calcd for C25H31BrN3O3, 500.15433; found, 500.15466.
4.1.8. General procedure for synthesis of heterocyclic analogs of the bromo derivative (8a,b)
The bromo derivative 7 (125 mg, 0.25 mmol) was taken with 2-furylboronic acid or 2-thienylboronic acid (0.5 mmol) and Pd(PPh3)4 (0.05 equivalent, 15 mg), then dioxane (5 ml) and 2 M Na2CO3 (0.3 ml) were added to the mixture under N2 atmosphere. The reaction mixture was refluxed under N2 at 90 °C for 16 h. Then, the reaction mixture was poured into ice/water (50 ml), the aqueous layer was extracted with ethyl acetate (50 × 3) and the organic layers were washed with water and brine. After evaporation of the organic solvent under vacuum, the residue was purified by silica gel column chromatography using DCM/MeOH to furnish 8a,b in pure form.
4.1.8.1. 2-(4-(furan-2-yl)phenyl)-6,7-dimethoxy-4-(3-(4-methylpiperazin-1-yl)propoxy)quinoline (8a)
Yellow solid, Yield 70%, m.p. 135–137 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.17 (p, 2H, CH2, J = 6.8 Hz), 2.34 (s, 3H, CH3-N), 2.43–2.78 (br s, 8H, piperazinyl 4CH2), 2.66 (t, 2H, CH2-N, J = 7.3 Hz), 4.02 (s, 3H, OCH3), 4.04 (s, 3H, OCH3), 4.33 (t, 2H, CH2-O, J = 6.3 Hz), 6.50 (dd, 1H, furyl CH, J = 1.8, 3.4 Hz), 6.73 (dd, 1H, furyl CH, J = 0.5, 3.4 Hz), 7.11 (s, 1H, vinyl CH), 7.38 (s, 1H, phenyl CH), 7.44 (s, 1H, phenyl CH), 7.50 (dd, 1H, furyl CH-O, J = 0.5, 1.8 Hz), 7.79 (d, 2H, 2-phenyl 2CH, J = 8.5 Hz), 8.09 (d, 2H, 2-phenyl 2CH, J = 8.5 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.53, 45.73, 52.86, 54.91, 55.02, 56.07, 56.12, 66.51, 97.49, 99.69, 105.63, 108.28, 111.83, 114.89, 124.02, 127.58, 131.21, 139.24, 142.35, 146.23, 148.95, 152.65, 153.71, 156.27, 160.95; HRESIMS (m/z): [M + H]+ Calcd for C29H34N3O4, 488.25438; found, 488.25446; HPLC purity: 96.06%.
4.1.8.2. 6,7-dimethoxy-4–(3-(4-methylpiperazin-1-yl)propoxy)-2–(4-(thiophen-2-yl)phenyl)quinol-ine (8b)
Buff solid, Yield 72%, m.p. 155–157 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.17 (p, 2H, CH2, J = 6.8 Hz), 2.29 (s, 3H, CH3-N), 2.34–2.70 (br s, 8H, piperazinyl 4CH2), 2.63 (t, 2H, CH2-N, J = 7.3 Hz), 4.02 (s, 3H, OCH3), 4.03 (s, 3H, OCH3), 4.33 (t, 2H, CH2-O, J = 6.3 Hz), 7.09 (dd, 1H, thienyl CH, J = 3.7, 5.1 Hz), 7.10 (s, 1H, vinyl CH), 7.30 (dd, 1H, thienyl CH, J = 1.1, 5.1 Hz), 7.38 (dd, 1H, thienyl CH-S, J = 1.1, 3.7 Hz), 7.39 (s, 1H, phenyl CH), 7.44 (s, 1H, phenyl CH), 7.73 (d, 2H, 2-phenyl 2CH, J = 8.4 Hz), 8.08 (d, 2H, 2-phenyl 2CH, J = 8.4 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.62, 46.05, 53.35, 55.13, 55.17, 56.04, 56.12, 66.62, 97.49, 99.72, 108.30, 114.94, 123.39, 125.12, 126.09, 127.76, 128.13, 134.80, 139.52, 144.01, 146.25, 148.95, 152.64, 156.20, 160.99; HRESIMS (m/z): [M + H]+ Calcd for C29H34N3O3S, 504.23154; found, 504.23141; HPLC purity: 98.96%.
4.1.9. Synthesis of 4-(3-chloropropoxy)-2-(4-(trifluoromethyl)phenyl)quinoline (9)
The demethoxylated key intermediate 9 has been synthesised using the same synthetic procedure for 5a–c starting from 2-aminoacetophenone.
White solid, Yield 76%, m.p. 98–100 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.45 (p, 2H, CH2, J = 6 Hz), 2.87 (t, 2H, CH2-N, J = 6.2 Hz), 4.46 (t, 2H, CH2-O, J = 5.8 Hz), 7.20 (s, 1H, vinyl CH), 7.52 (ddd, 1H, phenyl CH, J = 1.2, 6.9, 8.3 Hz), 7.74 (ddd, 1H, phenyl CH, J = 1.3, 6.9, 8.4 Hz), 7.77 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz), 8.11 (d, 2H, phenyl CH, J = 8.4 Hz), 8.19 (dd, 1H, phenyl CH, J = 1.3, 8.3 Hz), 8.22 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 31.94, 41.23, 64.86, 98.50, 121.56, 124.20 (CF3, q, J = 272 Hz), 125.67 (CH-C-CF3, q, J = 3.7 Hz), 125.96, 127.87, 129.42, 130.31, 131.08 (CH-C-CF3, q, J = 32.4 Hz), 143.56, 149.23, 157.12, 162.07; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.53 (s); HRESIMS (m/z): [M + H]+ Calcd for C19H16ClF3NO, 366.08670; found, 366.08725.
4.1.10. Synthesis of the target demethoxylated 4-propoxy-2-arylquinolines (10a,b)
The demethoxylated 4-propoxy-2-arylquinolines 10a,b were prepared according to the general synthetic procedure for 6a–o using the respective key intermediate 9.
4.1.10.1. 4-(3-(4-methylpiperazin-1-yl)propoxy)-2-(4-(trifluoromethyl)phenyl)quinoline (10a)
White solid, Yield 87%, m.p. 110–112 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.16 (p, 2H, CH2, J = 6.7 Hz), 2.29 (s, 3H, CH3-N), 2.37–2.71 (br s, 8H, piperazinyl 4CH2), 2.64 (t, 2H, CH2-N, J = 7.2 Hz), 4.34 (t, 2H, CH2-O, J = 6.2 Hz), 7.16 (s, 1H, vinyl CH), 7.50 (ddd, 1H, phenyl CH, J = 1.1, 6.9, 8.2 Hz), 7.72 (ddd, 1H, phenyl CH, J = 1.5 6.9, 8.3 Hz), 7.75 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz), 8.09 (d, 2H, phenyl CH, J = 8.3 Hz), 8.2 (m, 3H, phenyl CH, CF3-phenyl 2CH); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.54, 46.04, 53.30, 55.00, 55.13, 66.79, 98.49, 120.63, 121.74, 124.21 (CF3, q, J = 272 Hz), 125.63 (CH-C-CF3, q, J = 3.8 Hz), 125.82 127.89, 129.32, 130.21, 130.99 (CH-C-CF3, q, J = 32.6 Hz), 143.72, 149.19, 157.14, 162.43; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.53 (s); HRESIMS (m/z): [M + Na]+ Calcd for C21H33ClF3NONa, 430.20950; found, 430.20993; HPLC purity: 98.63%.
4.1.10.2. N-(3-((2-(4-(trifluoromethyl)phenyl)quinolin-4-yl)oxy)propyl)cyclohexanamine (10b)
Grey solid, Yield 78%, m.p. 68–70 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 1.03–1.13 (m, 2H, cyclohexyl 2CHH′), 1.14–1.19 (m, 1H, cyclohexyl CHH′), 1.20–1.31 (m, 3H, cyclohexyl 2CHH′, NH), 1.60–1.63 (m, 1H, cyclohexyl CHH′), 1.71–1.75 (m, 2H, cyclohexyl 2CHH′), 1.91 (d, 2H, cyclohexyl 2CHH′, J = 10.2 Hz), 2.15 (p, 2H, CH2, J = 6.5 Hz), 2.43–2.50 (m, 1H, cyclohexyl CH-NH), 2.94 (t, 2H, CH2-NH, J = 6.9 Hz), 4.38 (t, 2H, CH2-O, J = 6.1 Hz), 7.18 (s, 1H, vinyl CH), 7.51 (ddd, 1H, phenyl CH, J = 1.1, 7, 8.1 Hz), 7.72 (ddd, 1H, phenyl CH, J = 1.4 7, 8.3 Hz), 7.75 (d, 2H, CF3-phenyl 2CH, J = 8.7 Hz), 8.09 (d, 2H, phenyl CH, J = 8.3 Hz), 8.21 (d, 3H, phenyl CH, CF3-phenyl 2CH, J = 8.1 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 25.08, 26.16, 30.05, 33.71, 43.77, 56.94, 67.04, 98.51, 120.64, 121.74, 124.25 (CF3, q, J = 271.7 Hz), 125.64 (CH-C-CF3, q, J = 3.7 Hz), 125.82, 127.88, 129.33, 130.20, 130.99 (CH-C-CF3, q, J = 32.4 Hz), 143.70, 149.20, 157.15, 162.42; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.53 (s); HRESIMS (m/z): [M + H]+ Calcd for C25H28F3N2O, 429.21482; found, 429.21420; HPLC purity: 99.22%.
4.1.11. Synthesis of 1,3-dioxoloarylquinolines key intermediates (11a,b)
Starting from 6′-amino-3′,4′-(methylenedioxy)acetophenone and the respective p-substituted benzoyl chloride, the key intermediates 11a,b were prepared according to the synthetic route for 5a–c.
4.1.11.1. 6-(4-chlorophenyl)-8–(3-chloropropoxy)-[1,3]dioxolo[4,5-g]quinoline (11a)
White solid, Yield 90%, m.p. 186–188 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.40 (p, 2H, CH2, J = 6 Hz), 3.83 (t, 2H, CH2Cl, J = 6.2 Hz), 4.39 (t, 2H, CH2-O, J = 5.8 Hz), 6.09 (s, 2H, dioxolo CH2), 7.05 (s, 1H, vinyl CH), 7.37 (s, 1H, phenyl CH), 7.39 (s, 1H, phenyl CH), 7.45 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 8 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 31.99, 41.27, 64.68, 97.50, 101.68, 106.01, 116.23, 128.53, 128.84, 135.06, 138.63, 147.30, 147.48, 151.04, 155.48, 161.15; HRESIMS (m/z): [M + H]+ Calcd for C19H16Cl2NO3, 376.05018; found, 376.05045.
4.1.11.2. 8-(3-chloropropoxy)-6-(4-(trifluoromethyl)phenyl)-[1,3] dioxolo[4,5-g]quinoline (11b)
White solid, Yield 93%, m.p. 145–147 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.41 (p, 2H, CH2, J = 6 Hz), 3.84 (t, 2H, CH2Cl, J = 6.2 Hz), 4.41 (t, 2H, CH2-O, J = 5.8 Hz), 6.10 (s, 2H, dioxolo CH2), 7.10 (s, 1H, vinyl CH), 7.39 (s, 1H, phenyl CH), 7.40 (s, 1H, phenyl CH), 7.73 (d, 2H, CF3-phenyl 2CH, J = 8.1 Hz), 8.16 (d, 2H, CF3-phenyl 2CH, J = 8.1 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 31.96, 41.24, 64.74, 97.50, 97.83, 101.75, 106.09, 116.50, 124.25 (CF3, q, J = 271.8 Hz), 125.61 (CH-C-CF3, q, J = 3.7 Hz), 127.56, 130.72 (CH-C-CF3, q, J = 32.4 Hz), 143.55, 147.54, 147.57, 151.16, 155.13, 161.25; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.50 (s); HRESIMS (m/z): [M + H]+ Calcd for C20H16ClF3NO3, 410.07653; found, 410.07736.
4.1.12. Synthesis of the target propoxy derivatives of 1,3-dioxoloarylquinolines (12a–d)
The target dioxolo derivatives 12a–d have been synthesised utilising the synthetic procedures used for 6a–o using imidazole or morpholine with the appropriate key intermediate 11a,b.
4.1.12.1. 8-(3-(1H-imidazol-1-yl)propoxy)-6-(4-chlorophenyl)-[1,3] dioxolo[4,5-g]quinoline (12a)
White solid, Yield 77%, m.p. 146–148 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.39 (p, 2H, CH2, J = 6.2 Hz), 4.16 (t, 2H, CH2-N, J = 5.7 Hz), 4.28 (t, 2H, CH2-O, J = 6.7 Hz), 6.10 (s, 2H, dioxolo CH2), 6.93 (s, 1H, imidazole CH), 6.94 (s, 1H, vinyl CH), 7.07 (s, 1H, imidazole CH), 7.38 (s, 1H, phenyl CH), 7.39 (s, 1H, phenyl CH), 7.43 (d, 2H, chlorophenyl 2CH, J = 8.7 Hz), 7.49 (s, 1H, imidazole CH), 7.95 (d, 2H, chlorophenyl 2CH, J = 8.7 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 30.54, 43.43, 64.18, 97.23, 97.46, 101.76, 106.13, 116.07, 118.90, 128.51, 128.85, 129.98, 135.12, 137.27, 138.49, 147.46, 147.53, 151.13, 155.48, 160.83; HRESIMS (m/z): [M + H]+ Calcd for C22H19ClN3O3, 408.11095; found, 408.11179; HPLC purity: 99.60%.
4.1.12.2. 6-(4-chlorophenyl)-8-(3-morpholinopropoxy)-[1,3]dioxolo [4,5-g]quinoline (12b)
White solid, Yield 72%, m.p. 153–155 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.12 (p, 2H, CH2, J = 6.7 Hz), 2.49 (t, 4H, morpholinyl 2CH2, J = 4.6 Hz), 2.61 (t, 2H, CH2-N, J = 7.2 Hz), 3.73 (t, 4H, morpholinyl 2CH2, J = 4.6 Hz), 4.29 (t, 2H, CH2-O, J = 6.2 Hz), 6.08 (s, 2H, dioxolo CH2), 7.03 (s, 1H, vinyl CH), 7.36 (s, 1H, phenyl CH), 7.42 (s, 1H, phenyl CH), 7.44 (d, 2H, chlorophenyl 2CH, J = 8.7 Hz), 7.99 (d, 2H, chlorophenyl 2CH, J = 8.7 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.27, 53.82, 55.47, 66.43, 66.98, 97.50, 97.66, 101.63, 105.97, 116.35, 128.53, 128.82, 135.00, 138.76, 147.21, 147.44, 150.97, 155.51, 161.49; HRESIMS (m/z): [M + H]+ Calcd for C23H24ClN2O4, 427.14191; found, 427.14221; HPLC purity: 98.94%.
4.1.12.3. 8-(3-(1H-imidazol-1-yl)propoxy)-6-(4-(trifluoromethyl)phenyl)-[1,3]dioxolo[4,5-g]qui-noline (12c)
White solid, Yield 81%, m.p. 125–127 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.41 (p, 2H, CH2, J = 6.2 Hz), 4.19 (t, 2H, CH2-N, J = 5.7 Hz), 4.29 (t, 2H, CH2-O, J = 6.7 Hz), 6.12 (s, 2H, dioxolo CH2), 6.93 (s, 1H, imidazole CH), 7 (s, 1H, vinyl CH), 7.08 (s, 1H, imidazole CH), 7.41 (s, 2H, phenyl 2CH), 7.49 (s, 1H, imidazole CH), 7.72 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz), 8.12 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 30.52, 43.42, 64.26, 97.22, 97.79, 101.83, 106.22, 16.34, 118.88, 124.22 (CF3, q, J = 272.7 Hz), 125.61 (CH-C-CF3, q, J = 3.7 Hz), 127.55, 130.01, 130.79 (CH-C-CF3, q, J = 33.2 Hz), 137.28, 143.41, 147.60, 147.73, 151.26, 155.15, 160.92; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.52 (s); HRESIMS (m/z): [M + H]+ Calcd for C23H19F3N3O3, 442.13730; found, 442.13754; HPLC purity: 99.35%.
4.1.12.4. 8-(3-morpholinopropoxy)-6-(4-(trifluoromethyl)phenyl)-[1,3]dioxolo[4,5-g]quinoline (12d)
White solid, Yield 76%, m.p. 138–140 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.14 (p, 2H, CH2, J = 6.7 Hz), 2.50 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 2.62 (t, 2H, CH2-N, J = 7.2 Hz), 3.73 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 4.32 (t, 2H, CH2-O, J = 6.2 Hz), 6.10 (s, 2H, dioxolo CH2), 7.08 (s, 1H, vinyl CH), 7.39 (s, 1H, phenyl CH), 7.44 (s, 1H, phenyl CH), 7.73 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz), 8.16 (d, 2H, CF3-phenyl 2CH, J = 8.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.27, 53.82, 55.45, 66.51, 66.98, 97.65, 97.83, 101.70, 106.05, 116.62, 124.23 (CF3, q, J = 272.3 Hz), 125.59 (CH-C-CF3, q, J = 3.9 Hz), 127.57, 130.70 (CH-C-CF3, q, J = 32.2 Hz), 143.70, 147.48, 147.51, 151.10, 155.18, 161.59; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −62.50 (s); HRESIMS (m/z): [M + H]+ Calcd for C24H24F3N2O4, 461.16827; found, 461.16837; HPLC purity: 99.62%.
4.1.13. Synthesis of 1-(2-amino-5-bromophenyl)ethan-1-one (13)
The 5-bromo derivative of 2-aminoacetophenone 13 has been synthesised based on the reported procedureCitation46,Citation47.
Yellow solid, Yield 96%, m.p. 77–79 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.55 (s, 3H, CH3), 6.29 (s, 2H, NH2), 6.54 (d, 1H, phenyl CH, J = 8.8 Hz), 7.31 (dd, 1H, phenyl CH, J = 2.3, 8.8 Hz), 7.78 (d, 1H, phenyl CH, J = 2.3 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 27.83, 106.63, 118.99, 119.40, 134.11, 136.99, 149.08, 199.64; HRESIMS (m/z): [M + H]+ Calcd for C8H9CBrNO, 213.98620; found, 213.98599.
4.1.14. Synthesis of 6-bromo-2-arylquinolones (14a,b)
1-(2-amino-5-bromophenyl)ethan-1-one 13 was benzoylated with p-chloro or fluorobenzoyl chloride using the same procedure for 3a–c. Then, the resulted benzoyl derivatives have been subjected to ring closure reaction according to the synthetic route for 4a–c to afford the corresponding 6-bromo-2-arylquinolones 14a,b.
4.1.14.1. 6-bromo-2-(4-chlorophenyl)quinolin-4(1H)-one (14a)
Yellow solid, Yield 95%, m.p. > 250 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 6.41 (s, 1H, vinyl CH), 7.67 (d, 2H, chlorophenyl 2CH, J = 8.5 Hz), 7.72 (d, 1H, bromophenyl CH, J = 8.9 Hz), 7.84 (dd, 1H, bromophenyl CH, J = 1.7, 8.9 Hz), 7.87 (d, 2H, chlorophenyl 2CH, J = 8.5 Hz), 8.17 (d, 1H, bromophenyl CH, J = 1.7 Hz), 11.91 (s, 1H, NH); 13 C NMR (100 MHz, CDCl3) δ (ppm): 108.25, 116.52, 121.86, 126.76, 127.35, 129.53, 129.81, 133.14, 135.15, 135.96, 139.85, 149.64, 176.06; HRESIMS (m/z): [M + H]+ Calcd for C15H10BrClNO, 333.96288; found, 333.96295.
4.1.14.2. 6-bromo-2-(4-fluorophenyl)quinolin-4(1H)-one (14b)
Yellow solid, Yield 93%, m.p. > 250 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 6.39 (s, 1H, vinyl CH), 7.44 (t, 2H, fluorophenyl 2CH, J = 8.7 Hz), 7.72 (d, 1H, bromophenyl CH, J = 8.8 Hz), 7.84 (dd, 1H, bromophenyl CH, J = 2.3, 8.8 Hz), 7.91 (dd, 2H, fluorophenyl 2CH, J = 5.4, 8.7 Hz), 8.18 (d, 1H, bromophenyl CH, J = 2.3 Hz), 11.88 (s, 1H, NH); 13 C NMR (100 MHz, CDCl3) δ (ppm): 108.14, 116.44, 116.50 (CH-C-F, d, J = 21.8 Hz), 121.83, 126.72, 127.35, 130.43 (CH-CH-C-F, d, J = 8.7 Hz), 130.85, 135.07, 139.85, 149.88, 163.95 (C-F, d, J = 248.3 Hz), 176.02; 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −110.21 (s); HRESIMS (m/z): [M + H]+ Calcd for C15H10BrFNO, 317.99243; found, 317.99265.
4.1.15. Synthesis of 6-bromo-2-arylquinolines key intermediates (15a,b)
The key intermediates 15a,b have been prepared from 14a,b according to the synthetic route for 5a–c.
4.1.15.1. 6-bromo-2-(4-chlorophenyl)-4-(3-chloropropoxy)quinoline (15a)
White solid, Yield 96%, m.p. 134–136 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.44 (p, 2H, CH2, J = 6 Hz), 3.86 (t, 2H, CH2Cl, J = 6.1 Hz), 4.43 (t, 2H, CH2-O, J = 5.9 Hz), 7.15 (s, 1H, vinyl CH), 7.47 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.76 (dd, 1H, bromophenyl CH, J = 2.2, 9 Hz), 7.92 (d, 1H, bromophenyl CH, J = 9 Hz), 8.04 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 8.26 (d, 1H, bromophenyl CH, J = 2.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 31.84, 41.23, 65.02, 98.77, 119.50, 121.47, 124.04, 128.76, 128.99, 131.03, 133.57, 135.78, 138.13, 147.77, 157.72, 160.98; HRESIMS (m/z): [M + H]+ Calcd for C18H15BrCl2NO, 409.97088; found, 409.97205.
4.1.15.2. 6-bromo-4-(3-chloropropoxy)-2-(4-fluorophenyl)quinoline (15b)
White solid, Yield 92%, m.p. 163–165 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.44 (p, 2H, CH2, J = 6 Hz), 3.86 (t, 2H, CH2Cl, J = 6.1 Hz), 4.43 (t, 2H, CH2-O, J = 5.9 Hz), 7.15 (s, 1H, vinyl CH), 7.19 (t, 2H, fluorophenyl 2CH, J = 8.8 Hz), 7.75 (dd, 1H, bromophenyl CH, J = 2.3, 9 Hz), 7.93 (d, 1H, bromophenyl CH, J = 9 Hz), 8.09 (dd, 2H, fluorophenyl 2CH, J = 5.4, 8.8 Hz), 8.27 (d, 1H, bromophenyl CH, J = 2.3 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 31.85, 41.22, 65.00, 98.84, 115.77 (CH-C-F, d, J = 21.7 Hz), 119.33, 121.37, 124.03, 129.38 (CH-CH-C-F, d, J = 8.6 Hz), 130.98, 133.52, 135.91 (C-CH-CH-C-F, d, J = 3 Hz), 147.78, 157.97, 160.94, 163.89 (C-F, d, J = 249.4 Hz); 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −111.96 (s); HRESIMS (m/z): [M + H]+ Calcd for C18H15BrClFNO, 394.00041; found, 394.00122.
4.1.16. Synthesis of the target 6-bromo-4-propoxy-2-arylquinolines (16a–d)
The synthesis of the target 6-bromo-4-propoxy-2-arylquinolines 16a–d has been accomplished based on the general synthetic route for 6a–o utilising imidazole or morpholine with the respective key intermediate 15a,b.
4.1.16.1. 4-(3-(1H-imidazol-1-yl)propoxy)-6-bromo-2-(4-chlorophenyl)quinoline (16a)
White solid, Yield 84%, m.p. 149–151 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.44 (p, 2H, CH2, J = 6.2 Hz), 4.19 (t, 2H, CH2-N, J = 5.7 Hz), 4.31 (t, 2H, CH2-O, J = 6.6 Hz), 6.95 (s, 1H, imidazole CH), 7.04 (s, 1H, vinyl CH), 7.09 (s, 1H, imidazole CH), 7.46 (d, 2H, chlorophenyl 2CH, J = 8.7 Hz), 7.50 (s, 1H, imidazole CH), 7.78 (dd, 1H, bromophenyl CH, J = 2.2, 9 Hz), 7.94 (d, 1H, bromophenyl CH, J = 9 Hz), 8 (d, 2H, chlorophenyl 2CH, J = 8.7 Hz), 8.27 (d, 1H, bromophenyl CH, J = 2.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 30.42, 43.41, 64.59, 98.77, 118.87, 119.69, 121.33, 123.79, 128.75, 129.00, 130.09, 131.15, 133.71, 135.86, 137.28, 137.99, 147.81, 157.74, 160.67; HRESIMS (m/z): [M + H]+ Calcd for C21H18BrClN3O, 442.03163; found, 442.03214; HPLC purity: 98.60%.
4.1.16.2. 4-(3-((6-bromo-2-(4-chlorophenyl)quinolin-4-yl)oxy)propyl)morpholine (16b)
White solid, Yield 78%, m.p. 137–139 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.15 (p, 2H, CH2, J = 6.7 Hz), 2.50 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 2.62 (t, 2H, CH2-N, J = 7.1 Hz), 3.73 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 4.33 (t, 2H, CH2-O, J = 6.3 Hz), 7.13 (s, 1H, vinyl CH), 7.47 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.75 (dd, 1H, bromophenyl CH, J = 2.2, 9 Hz), 7.92 (d, 1H, bromophenyl CH, J = 9 Hz), 8.03 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 8.30 (d, 1H, bromophenyl CH, J = 2.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.14, 53.81, 55.37, 66.84, 66.98, 98.76, 119.38, 121.63, 124.21, 128.77, 128.97, 130.96, 133.49, 135.71, 138.28, 147.76, 157.75, 161.34; HRESIMS (m/z): [M + H]+ Calcd for C22H23BrClN2O2, 461.06259; found, 461.06314; HPLC purity: 99.92%.
4.1.16.3. 4-(3-(1H-imidazol-1-yl)propoxy)-6-bromo-2-(4-fluorophenyl)quinoline (16c)
White solid, Yield 80%, m.p. 144–146 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.43 (p, 2H, CH2, J = 6.2 Hz), 4.19 (t, 2H, CH2-N, J = 5.7 Hz), 4.31 (t, 2H, CH2-O, J = 6.6 Hz), 6.94 (s, 1H, imidazole CH), 7.03 (s, 1H, vinyl CH), 7.09 (s, 1H, imidazole CH), 7.17 (t, 2H, fluorophenyl 2CH, J = 8.7 Hz), 7.50 (s, 1H, imidazole CH), 7.77 (dd, 1H, bromophenyl CH, J = 2.3, 9 Hz), 7.94 (d, 1H, bromophenyl CH, J = 9 Hz), 8.04 (dd, 2H, fluorophenyl 2CH, J = 5.4, 8.7 Hz), 8.27 (d, 1H, bromophenyl CH, J = 2.3 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 30.42, 43.41, 64.55, 98.83, 115.78 (CH-C-F, d, J = 21.6 Hz), 118.88, 119.51, 121.22, 123.78, 129.37 (CH-CH-C-F, d, J = 8.5 Hz), 130.07, 131.10, 133.65, 135.75 (C-CH-CH-C-F, d, J = 3.1 Hz), 137.28, 147.80, 157.97, 160.61, 163.91 (C-F, d, J = 249.5 Hz); 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −111.81 (s); HRESIMS (m/z): [M + H]+ Calcd for C21H18BrFN3O, 426.06118; found, 426.06125; HPLC purity: 99.65%.
4.1.16.4. 4-(3-((6-bromo-2-(4-fluorophenyl)quinolin-4-yl)oxy)propyl)morpholine (16d)
White solid, Yield 87%, m.p. 132–134 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.16 (p, 2H, CH2, J = 6.7 Hz), 2.51 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 2.62 (t, 2H, CH2-N, J = 7.1 Hz), 3.73 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 4.33 (t, 2H, CH2-O, J = 6.2 Hz), 7.12 (s, 1H, vinyl CH), 7.19 (t, 2H, fluorophenyl 2CH, J = 8.7 Hz), 7.75 (dd, 1H, bromophenyl CH, J = 2.3, 9 Hz), 7.92 (d, 1H, bromophenyl CH, J = 9 Hz), 8.08 (dd, 2H, fluorophenyl 2CH, J = 5.4, 8.7 Hz), 8.17 (d, 1H, bromophenyl CH, J = 2.3 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.14, 53.81, 55.37, 66.81, 66.97, 98.83, 115.74 (CH-C-F, d, J = 21.7 Hz), 119.21, 121.52, 124.19, 129.37 (CH-CH-C-F, d, J = 8.5 Hz), 130.91, 133.44, 136.05 (C-CH-CH-C-F, d, J = 2.6 Hz), 147.76, 158.01, 161.28, 163.85 (C-F, d, J = 249.4 Hz); 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −112.05 (s); HRESIMS (m/z): [M + H]+ Calcd for C22H23BrFN2O2, 445.09215; found, 445.09241; HPLC purity: 99.15%.
4.1.17. Synthesis of 5-aryl-2-aminoacetophenones (17a,b)
To a mixture of 1–(2-amino-5-bromophenyl)ethan-1-one 13 (1.07 g, 5 mmol), 4-methoxyphenylboronic acid or 2-furylboronic acid (5.5 mmol), K2CO3 (2.28 g, 16.5 mmol) and Pd(PPh3)4 (0.02 equivalent, 116 mg), dioxane (14 ml) and H2O (14 ml) were added under N2 atmosphere. The reaction mixture was refluxed under N2 at 100 °C for 4 h. Then, the reaction mixture was poured into ethylacetate (50 ml) and the organic layer was separated. The aqueous layer was extracted with ethylacetate (30 × 3) and the organic layers were collected and washed with 1 M HCl (100 × 3) then brine ((100 × 3). After evaporation of the organic solvent under vacuum, the residue was purified by silica gel column chromatography using hexane/ethylacetate to afford the corresponding compounds 17a,b.
4.1.17.1. 1-(4-amino-4′-methoxy-[1,1′-biphenyl]-3-yl)ethan-1-one (17a)
Yellow solid, Yield 86%, m.p. 101–103 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.63 (s, 3H, CH3-C=O), 3.85 (s, 3H, CH3-O), 6.29 (s, 2H, NH2), 6.71 (d, 1H, phenyl CH, J = 8.6 Hz), 6.97 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.45 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.48 (dd, 1H, phenyl CH, J = 2.2, 8.6 Hz), 7.76 (d, 1H, phenyl CH, J = 2.2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 27.93, 55.39, 114.29, 117.73, 118.44, 127.36, 128.80, 129.83, 133.08, 133.24, 149.15, 158.66, 200.79; HRESIMS (m/z): [M + H]+ Calcd for C15H16NO2, 242.11756; found, 242.11717.
4.1.17.2. 1-(2-amino-5-(furan-2-yl)phenyl)ethan-1-one (17b)
Yellow solid, Yield 70%, m.p. 87–89 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.63 (s, 3H, CH3), 6.36 (s, 2H, NH2), 6.44–6.46 (m, 2H, furyl 2CH), 6.66 (d, 1H, phenyl CH, J = 8.6 Hz), 7.41 (dd, 1H, furyl CH, J = 0.9, 1.6 Hz), 7.55 (dd, 1H, phenyl CH, J = 2, 8.6 Hz), 8.02 (d, 1H, phenyl CH, J = 2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 27.91, 102.61, 111.55, 117.61, 118.02, 119.47, 127.24, 130.41, 141.09, 149.57, 153.73, 200.74; HRESIMS (m/z): [M + H]+ Calcd for C12H12NO2, 202.08626; found, 202.08594.
4.1.18. Synthesis of 2,6-diarylquinolones (18a–c)
The synthesis of 2,6-diarylquinolones 18a–c has been accomplished using 5-aryl-2-aminoacetophenones 17a,b and the respective p-chloro or fluorobenzoyl chloride according to the synthetic route used for 6-bromo-2-arylquinolones 14a,b. The 2,6-diarylquinolones 18a,b have poor solubility for NMR spectral analysis, so the NMR spectral analysis of the soluble 6-furyl analog 18c was used for their structural authentication in addition to HRMS for 18a,b. Moreover, the corresponding 4-propoxy analogs 19a–c exhibited good solubility and their spectral characterisation was enough for structural confirmation of 2,6-diarylquinolones 18a–c.
4.1.18.1. 2-(4-chlorophenyl)-6-(4-methoxyphenyl)quinolin-4(1H)-one (18a)
Yellow solid, Yield 97%, m.p. > 250 °C; HRESIMS (m/z): [M + Na]+ Calcd for C22H16ClNO2Na, 384.07618; found, 384.07623.
4.1.18.2. 2-(4-fluorophenyl)-6-(4-methoxyphenyl)quinolin-4(1H)-one (18b)
Yellow solid, Yield 90%, m.p. > 250 °C; HRESIMS (m/z): [M + H]+ Calcd for C22H16FNO2Na, 368.10573; found, 368.10574.
4.1.18.3. 2-(4-chlorophenyl)-6-(furan-2-yl)quinolin-4(1H)-one (18c)
Yellow solid, Yield 93%, m.p. > 250 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 6.68 (dd, 1H, furyl CH, J = 1.6, 3.4 Hz), 7.04 (s, 1H, vinyl CH), 7.19 (d, 1H, furyl CH, J = 3.4 Hz), 7.73 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.86 (d, 1H, furyl CH, J = 1.6 Hz), 7.99 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 8.18 (d, 1H, phenyl CH, J = 8.9 Hz), 8.26 (dd, 1H, phenyl CH, J = 2. 8.9 Hz), 8.43 (d, 1H, phenyl CH, J = 2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 106.42, 108.09, 112.95, 117.53, 121.27, 122.67, 128.11, 129.59, 129.71, 130.49, 132.12, 136.85, 140.05, 144.39, 151.67, 152.32, 172.86; HRESIMS (m/z): [M + H]+ Calcd for C19H13ClNO2, 322.06293; found, 322.06299.
4.1.19. Synthesis of 4-propoxy-2,6-diarylquinolines key intermediates (19a–c)
The key intermediates 4-propoxy-2,6-diarylquinolines 19a–c were synthesised from 2,6-diarylquinolones 18a–c utilising the synthetic route used for 5a–c.
4.1.19.1. 2-(4-chlorophenyl)-4-(3-chloropropoxy)-6-(4-methoxyphenyl)quinoline (19a)
Off-white solid, Yield 93%, m.p. 183–185 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.45 (p, 2H, CH2, J = 6 Hz), 3.86 (t, 2H, CH2Cl, J = 6.2 Hz), 3.88 (s, 3H, CH3-O), 4.45 (t, 2H, CH2-O, J = 5.8 Hz), 7.04 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.15 (s, 1H, vinyl CH), 7.48 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.66 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.94 (dd, 1H, phenyl CH, J = 2, 8.8 Hz), 8.07 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 8.11 (d, 1H, phenyl CH, J = 8.8 Hz), 8.26 (d, 1H, phenyl CH, J = 2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 31.95, 41.34, 55.41, 64.90, 98.43, 114.41, 118.46, 120.52, 128.48, 128.76, 128.93, 129.59, 129.68, 133.16, 135.43, 138.06, 138.60, 148.31, 156.99, 159.47, 161.94; HRESIMS (m/z): [M + H]+ Calcd for C25H22Cl2NO2, 438.10221; found, 438.10242.
4.1.19.2. 4-(3-chloropropoxy)-2-(4-fluorophenyl)-6-(4-methoxyphenyl)quinoline (19b)
Off-white solid, Yield 86%, m.p. 174–176 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.46 (p, 2H, CH2, J = 6 Hz), 3.86 (t, 2H, CH2Cl, J = 6.2 Hz), 3.88 (s, 3H, CH3-O), 4.46 (t, 2H, CH2-O, J = 5.8 Hz), 7.04 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.16 (s, 1H, vinyl CH), 7.20 (t, 2H, fluorophenyl 2CH, J = 8.7 Hz), 7.66 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.93 (dd, 1H, phenyl CH, J = 2, 8.8 Hz), 8.09–8.13 (m, 3H, fluorophenyl 2CH, phenyl CH), 8.27 (d, 1H, phenyl CH, J = 2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 31.96, 41.35, 55.41, 64.88, 98.51, 114.41, 115.68 (CH-C-F, d, J = 21.7 Hz), 118.47, 120.40, 128.48, 129.34 (CH-CH-C-F, d, J = 8.5 Hz), 129.55, 129.63, 133.22, 136.38 (C-CH-CH-C-F, d, J = 3 Hz), 137.94, 148.32, 157.26, 159.44, 161.90, 163.75 (C-F, d, J = 248.8 Hz); 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −112.55 (s); HRESIMS (m/z): [M + H]+ Calcd for C25H22ClFNO2, 422.13176; found, 422.13123.
4.1.19.3. 2-(4-chlorophenyl)-4-(3-chloropropoxy)-6-(furan-2-yl)quinoline (19c)
Yellow solid, Yield 74%, m.p. 144–146 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.47 (p, 2H, CH2, J = 6 Hz), 3.88 (t, 2H, CH2Cl, J = 6.2 Hz), 4.44 (t, 2H, CH2-O, J = 5.8 Hz), 6.53 (dd, 1H, furyl CH, J = 1.7, 3.4 Hz), 6.79 (d, 1H, furyl CH, J = 3.4 Hz), 7.14 (s, 1H, vinyl CH), 7.47 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.55 (d, 1H, furyl CH, J = 1.7 Hz), 7.98 (dd, 1H, phenyl CH, J = 1.9, 8.8 Hz), 8.06 (d, 3H, chlorophenyl 2CH, phenyl CH, J = 8.6 Hz), 8.38 (d, 1H, phenyl CH, J = 1.9 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 31.94, 41.36, 64.95, 98.60, 106.15, 111.95, 115.65, 120.52, 126.56, 128.02, 128.72, 128.93, 129.71, 135.50, 138.43, 142.63, 148.55, 153.66, 157.06, 161.94; HRESIMS (m/z): [M + H]+ Calcd for C22H18Cl2NO2, 398.07091; found, 398.07150.
4.1.20. Synthesis of the target 4-propoxy-2,6-diarylquinolines (20a–f)
The synthesis of the target 4-popoxy-2,6-diarylquinolines 20a–f has been conducted by reaction of imidazole or morpholine with the corresponding key intermediate 19a–c under the same conditions used for synthesis of 6a–o.
4.1.20.1. 4-(3-(1H-imidazol-1-yl)propoxy)-2-(4-chlorophenyl)-6-(4-methoxyphenyl)quinoline (20a)
White solid, Yield 80%, m.p. 210–212 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.45 (p, 2H, CH2, J = 6.2 Hz), 3.88 (s, 3H, CH3-O), 4.22 (t, 2H, CH2-N, J = 5.7 Hz), 4.31 (t, 2H, CH2-O, J = 6.6 Hz), 6.94 (s, 1H, imidazole CH), 7.04 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.05 (s, 1H, vinyl CH), 7.08 (s, 1H, imidazole CH), 7.47 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.50 (s, 1H, imidazole CH), 7.67 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.95 (dd, 1H, phenyl CH, J = 2, 8.8 Hz), 8.03 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 8.13 (d, 1H, phenyl CH, J = 8.8 Hz), 8.27 (d, 1H, phenyl CH, J = 2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 30.50, 43.45, 55.43, 64.38, 98.41, 114.47, 118.18, 118.90, 120.40, 128.50, 128.74, 128.94, 129.74, 129.83, 130.05, 133.11, 135.51, 137.30, 138.30, 138.45, 148.35, 156.97, 159.54, 161.61; HRESIMS (m/z): [M + H]+ Calcd for C28H25ClN3O2, 470.16298; found, 470.16299; HPLC purity: 99.88%.
4.1.20.2. 4-(3-((2-(4-chlorophenyl)-6-(4-methoxyphenyl)quinolin-4-yl)oxy)propyl)morpholine (20b)
White solid, Yield 89%, m.p. 173–175 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.18 (p, 2H, CH2, J = 6.7 Hz), 2.50 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 2.63 (t, 2H, CH2-N, J = 7.1 Hz), 3.73 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 3.88 (s, 3H, CH3-O), 4.35 (t, 2H, CH2-O, J = 6.3 Hz), 7.03 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.13 (s, 1H, vinyl CH), 7.48 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.67 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.93 (dd, 1H, phenyl CH, J = 2.1, 8.8 Hz), 8.06 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 8.10 (d, 1H, phenyl CH, J = 8.8 Hz), 8.30 (d, 1H, phenyl CH, J = 2.1 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.22, 53.83, 55.41, 55.48, 66.64, 66.98, 98.40, 114.40, 118.59, 120.66, 128.44, 128.77, 128.90, 129.48, 129.63, 133.20, 135.37, 137.91, 138.74, 148.30, 157.02, 159.44, 162.28; HRESIMS (m/z): [M + H]+ Calcd for C29H30ClN2O3, 489.19395; found, 489.19415; HPLC purity: 96.78%.
4.1.20.3. 4-(3-(1H-imidazol-1-yl)propoxy)-2-(4-fluorophenyl)-6-(4-methoxyphenyl)quinoline (20c)
White solid, Yield 79%, m.p. 176–178 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.44 (p, 2H, CH2, J = 6.2 Hz), 3.88 (s, 3H, CH3-O), 4.22 (t, 2H, CH2-N, J = 5.7 Hz), 4.30 (t, 2H, CH2-O, J = 6.6 Hz), 6.94 (s, 1H, imidazole CH), 7.04 (s, 1H, vinyl CH), 7.06 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.07 (s, 1H, imidazole CH), 7.18 (t, 2H, fluorophenyl 2CH, J = 8.8 Hz), 7.50 (s, 1H, imidazole CH), 7.67 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.95 (dd, 1H, phenyl CH, J = 2, 8.8 Hz), 8.07 (dd, 2H, fluorophenyl 2CH, J = 5.4, 8.8 Hz), 8.13 (d, 1H, phenyl CH, J = 8.8 Hz), 8.27 (d, 1H, phenyl CH, J = 2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 30.51, 43.46, 55.42, 64.36, 98.48, 114.46, 115.69 (CH-C-F, d, J = 21.7 Hz), 118.19, 118.91, 120.27, 128.49, 129.33 (CH-CH-C-F, d, J = 8.3 Hz), 129.70, 129.77, 130.02, 133.15, 136.22, (C-CH-CH-C-F, d, J = 3.3 Hz), 137.29, 138.16, 148.34, 157.23, 159.52, 161.57, 163.77 (C-F, d, J = 249.2 Hz); 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −112.39 (s); HRESIMS (m/z): [M + H]+ Calcd for C28H25FN3O2, 454.19326; found, 454.19244; HPLC purity: 99.21%.
4.1.20.4. 4-(3-((2-(4-fluorophenyl)-6-(4-methoxyphenyl)quinolin-4-yl)oxy)propyl)morpholine (20d)
White solid, Yield 82%, m.p. 168–170 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.18 (p, 2H, CH2, J = 6.7 Hz), 2.50 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 2.63 (t, 2H, CH2-N, J = 7.1 Hz), 3.73 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 3.88 (s, 3H, CH3-O), 4.36 (t, 2H, CH2-O, J = 6.3 Hz), 7.03 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.13 (s, 1H, vinyl CH), 7.20 (t, 2H, fluorophenyl 2CH, J = 8.7 Hz), 7.67 (d, 2H, methoxyphenyl 2CH, J = 8.8 Hz), 7.93 (dd, 1H, phenyl CH, J = 2, 8.8 Hz), 8.09–8.12 (m, 3H, fluorophenyl 2CH, phenyl CH), 8.30 (d, 1H, phenyl CH, J = 2 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.23, 53.83, 55.40, 55.48, 66.62, 66.98, 98.48, 114.40, 115.65 (CH-C-F, d, J = 21.5 Hz), 118.59, 120.54, 128.43, 129.34, (CH-CH-C-F, d, J = 8.5 Hz), 129.44, 129.57, 133.25, 136.51 (C-CH-CH-C-F, d, J = 3 Hz), 137.79, 148.30, 157.29, 159.42, 162.24, 163.71 (C-F, d, J = 248.8 Hz); 19 F NMR (376.46 MHz, CDCl3) δ (ppm): −112.63 (s); HRESIMS (m/z): [M + H]+ Calcd for C29H30FN2O3, 473.22422; found, 473.22357; HPLC purity: 99.75%.
4.1.20.5. 4-(3-(1H-imidazol-1-yl)propoxy)-2-(4-chlorophenyl)-6-(furan-2-yl)quinoline (20e)
Yellow solid, Yield 79%, m.p. 130–132 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.47 (p, 2H, CH2, J = 6.2 Hz), 4.22 (t, 2H, CH2-N, J = 5.7 Hz), 4.34 (t, 2H, CH2-O, J = 6.6 Hz), 6.55 (dd, 1H, furyl CH, J = 1.6, 3.3 Hz), 6.82 (d, 1H, furyl CH, J = 3.3 Hz), 6.96 (s, 1H, imidazole CH), 7.04 (s, 1H, vinyl CH), 7.09 (s, 1H, imidazole CH), 7.46 (d, 2H, chlorophenyl 2CH, J = 8.5 Hz), 7.53 (s, 1H, imidazole CH), 7.56 (d, 1H, furyl CH, J = 1.6 Hz), 7.99 (dd, 1H, phenyl CH, J = 1.8, 8.7 Hz), 8.02 (d, 2H, chlorophenyl 2CH, J = 8.5 Hz), 8.07 (d, 1H, phenyl CH, J = 8.7 Hz), 8.41 (d, 1H, phenyl CH, J = 1.8 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 30.48, 43.47, 64.40, 98.63, 106.32, 112.03, 115.33, 118.94, 120.41, 126.70, 128.19, 128.71, 128.95, 129.87, 129.96, 135.58, 137.29, 138.30, 142.72, 148.59, 153.57, 157.09, 161.62; HRESIMS (m/z): [M + H]+ Calcd for C25H21ClN3O2, 430.13168; found, 430.13174; HPLC purity: 95.27%.
4.1.20.6. 4-(3-((2-(4-chlorophenyl)-6-(furan-2-yl)quinolin-4-yl)oxy)propyl)morpholine (20f)
Yellow solid, Yield 74%, m.p. 136–138 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 2.19 (p, 2H, CH2, J = 6.7 Hz), 2.52 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 2.65 (t, 2H, CH2-N, J = 7.1 Hz), 3.74 (t, 4H, morpholinyl 2CH2, J = 4.4 Hz), 4.35 (t, 2H, CH2-O, J = 6.3 Hz), 6.53 (dd, 1H, furyl CH, J = 1.8, 3.4 Hz), 6.79 (dd, 1H, furyl CH, J = 0.5, 3.4 Hz), 7.12 (s, 1H, vinyl CH), 7.47 (d, 2H, chlorophenyl 2CH, J = 8.6 Hz), 7.54 (dd, 1H, furyl CH, J = 0.5, 1.8 Hz), 7.98 (dd, 1H, phenyl CH, J = 1.9, 8.8 Hz), 8.05 (d, 3H, chlorophenyl 2CH, phenyl CH, J = 8.6 Hz), 8.42 (d, 1H, phenyl CH, J = 1.9 Hz); 13 C NMR (100 MHz, CDCl3) δ (ppm): 26.19, 53.84, 55.49, 66.71, 67.00, 98.60, 106.04, 111.94, 115.86, 120.67, 126.49, 127.93, 128.73, 128.91, 129.67, 135.44, 138.60, 142.58, 148.56, 153.75, 157.12, 162.29; HRESIMS (m/z): [M + H]+ Calcd for C26H26ClN2O3, 449.16265; found, 449.16275; HPLC purity: 98.79%.
4.2. In vitro anticancer activity
4.2.1. In vitro antiproliferative assay
The antiproliferative assay against five cancer cell lines representing three different tumour subpanels, including colorectal (DLD-1, HCT-116), breast (MDMBA-231, MCF-7), and cervical (HeLa) cell lines was conducted using MTT assay. Cells were seeded at 1 × 104 cells/well and cultured overnight in a 96-well plate. The cells were treated with either 10 µМ of tested quinolines 6a–o, 8a,b, 10a,b, 12a–d, 16a–d, and 20a–f, or DMSO as a negative control. After 24 h, the cells washed with phosphate buffered saline (PBS, Invitrogen Gibco) and incubated with 20 µl of MTT solution (2 mg/ml) for 4 h at 37 °C. Then, 150 µl DMSO was used to solubilise MTT formazan crystals. Finally, the plates were shaken, and the optical density was determined at 570 nm using ELISA plate reader. At least, three independent experiments were performed. Percentage of growth inhibition was determined as (1-[OD of treated cells/OD of control cells]). On the other hand, using the MTT assay, we tested the effect of different concentrations (0.5, 1, 10, 30, 50, and 100 µM)) of the synthesised compounds on colorectal cancer cell lines (DLD-1 and HCT-116), using DMSO as a negative control, whereas Gefitinib and TAE226 were used as positive controls. The IC50 values were calculated using Prism v0.8 software (GraphPad Software Inc., La Jolla, CA).
4.2.2. Apoptosis assay
The apoptotic effect of the most potent antiproliferative agents 6f, 6h, 6i, 16d, and 20f on DLD-1 colorectal cancer cell line was investigated using the annexin V/propidium iodide (AV/PI) staining kit (BioLegend, San Diego, CA, USA) according to the manufacturer's instructions. DLD-1 cells were treated with 3 µM of the most potent antiproliferative compounds 6f, 6h, 6i, 16d, and 20f or DMSO as a negative control then incubated for 24 h. Flow cytometric analysis was conducted using FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA).
4.3. Topoisomerase I-mediated DNA cleavage assay
A 3′-[32P]-labeled 117-bp DNA substrate oligonucleotide was prepared as described previouslyCitation39. Radiolabeled DNA was incubated with recombinant human TOP1 in 20 µL reaction buffer (10 mmol/L Tris-HCl, pH 7.5, 50 mmol/L KCl, 5 mmol/L MgCl2, 0.1 mmol/L EDTA, and 15 µg/mL BSA) at 30 °C for 20 min in the presence of the indicated drug concentrations. Reactions were terminated by adding SDS (0.5% final concentration) followed by the addition of two volumes of loading dye (80% formamide, 10 mmol/L sodium hydroxide, 1 mmol/L sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of reaction mixtures were subjected to 20% denaturing PAGE. Gels were dried and visualised by using PhosphorImager and Image Quant software (Molecular Dynamics).
4.4. Kinases inhibitory assay
The IC50 values of the tested compounds, Gefitinib and TAE226 on different nine kinases (EGFR, FAK, FRK, IGF-1R, BTK, c-Src, VEGFR-1, VEGFR-2, HER-2) were estimated utilising Z`-LYTE® technology, which is based on FRET (Invitrogen/Life Technologies).
4.5. In silico molecular docking
Ligands were converted to 3D structures and minimised using AvogadroCitation48. Ligands were prepared and converted to pdbqt files using PyRxCitation49. Protein targets were downloaded from the protein data bank under the codes 1M17 for EGFR and 2JKM for FAKCitation50. Co-crystalised ligands were extracted form pdb files and prepared similar to the tested ligands. Docking was done using Autodock VinaCitation51 in a grid box of 253 Å3 centred on the co-crystalised ligand with exhaustiveness of 16. Visualisation and 3D images were prepared using PyMolCitation52.
4.6. Molecular dynamics (MD) simulation
Missing loops in the 3D structures of the protein were constructed using Swiss-ModelCitation43 before starting molecular dynamics steps. All atom molecular dynamics simulations were performed using GROMACS 2020.3Citation53 for the selected protein-ligand complexes as reported earlierCitation54. In brief, SwissParam serverCitation55 was used for ligands parameterisation while Charmm36 all-atom force fieldCitation56 was used to generate topology files for the protein. Ligand coordinates obtained from docking studies were used to build complexes which were boxed in a dodecahedron box and then solvated with TIP3PCitation57 explicit water. Systems were neutralised by the addition of required number of Na+ or Cl− ions. Systems energy was minimised with a maximum force of 1000 kJ mol−1 nm−1 using steepest descent algorithm. Equilibration using NVT and NPT ensembles for 1 ns each was done afterward then production run was done for 50 ns. Temperature was kept at 300 K using the V-rescale algorithmCitation58 while pressure was controlled using the Parrinello-Rahman barostatCitation59 as required. The LINear Constraint Solver (LINCS) algorithmCitation60 and Particle mesh Ewald (PME) methodCitation61 were used for bond’s length constraints and long-range electrostatics calculations, respectively. Two femtosecond timestep was used for all simulations. Van der Waals distance cut-off (rvdw) was set to 1.2 nm. Trajectories from the production run were used for analysis using trajconv after correction of periodic boundary condition (PBC).
Supplemental Material
Download PDF (17 MB)Disclosure statement
The authors have declared no conflict of interest.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (10.1080/14756366.2022.2024999).
Additional information
Funding
References
- Peng CK, Zeng T, Xu XJ, et al. Novel 4-(4-substituted amidobenzyl)furan-2(5H)-one derivatives as topoisomerase I inhibitors. Eur J Med Chem 2017;127:187–99.
- Mohassab AM, Hassan HA, Abdelhamid D, et al. Design and synthesis of novel quinoline/chalcone/1,2,4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAFV600E kinases. Bioorg Chem 2021;106:104510.
- Kovvuri J, Nagaraju B, Nayak VL, et al. Design, synthesis and biological evaluation of new β-carboline-bisindole compounds as DNA binding, photocleavage agents and topoisomerase I inhibitors. Eur J Med Chem 2018;143:1563–77.
- Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49.
- Khodair AI, Elbadawi MM, Elsaady MT, Abdellatif KRA. Design, synthesis, molecular docking and cytotoxicity evaluation of some novel 5-arylidene-3-(substituted phenyl)-2-(p-tolylamino)-4-imidazolones. J Appl Pharm Sci 2017;7:58–68.
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424.
- Xie H, Lin X, Zhang Y, et al. Design, synthesis and biological evaluation of ring-fused pyrazoloamino pyridine/pyrimidine derivatives as potential FAK inhibitors. Bioorganic Med Chem Lett 2020;30:127459.
- Elbadawi MM, Eldehna WM, Nocentini A, et al. Identification of N-phenyl-2-(phenylsulfonyl)acetamides/propanamides as new SLC-0111 analogues: synthesis and evaluation of the carbonic anhydrase inhibitory activities. Eur J Med Chem 2021;218:113360.
- Abdellatif KRA, Elbadawi MM, Elsaady MT, et al. Design, synthesis and cytotoxicity evaluation of new 3, 5-disubstituted-2-thioxoimidazolidinones. Anticancer Agents Med Chem 2018;18:573–82.
- Ismail RSM, Abou-Seri SM, Eldehna WM, et al. Novel series of 6-(2-substitutedacetamido)-4-anilinoquinazolines as EGFR-ERK signal transduction inhibitors in MCF-7 breast cancer cells. Eur J Med Chem 2018;155:782–96.
- Wang Z, Wu X, Wang L, et al. Facile and efficient synthesis and biological evaluation of 4-anilinoquinazoline derivatives as EGFR inhibitors. Bioorg Med Chem Lett 2016;26:2589–93.
- Hu S, Xie G, Zhang DX, et al. Synthesis and biological evaluation of crown ether fused quinazoline analogues as potent EGFR inhibitors. Bioorg Med Chem Lett 2012;22:6301–5.
- Dawson JC, Serrels A, Stupack DG, et al. Targeting FAK in anticancer combination therapies. Nat Rev Cancer 2021;21:313–24.
- Dao P, Jarray R, Coq JL, et al. Synthesis of novel diarylamino-1,3,5-triazine derivatives as FAK inhibitors with anti-angiogenic activity. Bioorg Med Chem Lett 2013;23:4552–6.
- Wang R, Zhao X, Yu S, et al. Discovery of 7H-pyrrolo[2,3-d]pyridine derivatives as potent FAK inhibitors: design, synthesis, biological evaluation and molecular docking study. Bioorg Chem 2020;102:104092.
- Li B, Li Y, Tomkiewicz-Raulet C, et al. Design, synthesis, and biological evaluation of covalent inhibitors of focal adhesion kinase (FAK) against human malignant glioblastoma. J Med Chem 2020;63:12707–24.
- Gavriil ES, Doukatas A, Karampelas T, et al. Design, synthesis and biological evaluation of novel substituted purine isosters as EGFR kinase inhibitors, with promising pharmacokinetic profile and in vivo efficacy. Eur J Med Chem 2019;176:393–409.
- McLean GW, Carragher NO, Avizienyte E, et al. The role of focal-adhesion kinase in cancer – a new therapeutic opportunity. Nat Rev Cancer 2005;5:505–15.
- Luo QY, Zhou SN, Pan WT, et al. A multi-kinase inhibitor APG-2449 enhances the antitumor effect of ibrutinib in esophageal squamous cell carcinoma via EGFR/FAK pathway inhibition. Biochem Pharmacol 2021;183:114318.
- Ai M, Wang C, Tang Z, et al. Design and synthesis of diphenylpyrimidine derivatives (DPPYs) as potential dual EGFR T790M and FAK inhibitors against a diverse range of cancer cell lines. Bioorg Chem 2020;94:103408.
- Eke I, Cordes N. Dual targeting of EGFR and focal adhesion kinase in 3D grown HNSCC cell cultures. Radiother Oncol 2011;99:279–86.
- Yadav P, Shah K. Quinolines, a perpetual, multipurpose scaffold in medicinal chemistry. Bioorg Chem 2021;109:104639.
- Karnik KS, Sarkate AP, Tiwari SV, et al. Computational and synthetic approach with biological evaluation of substituted quinoline derivatives as small molecule L858R/T790M/C797S triple mutant EGFR inhibitors targeting resistance in non-small cell lung cancer (NSCLC). Bioorg Chem 2021;107:104612.
- Matada BS, Pattanashettar R, Yernale NG. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg Med Chem 2021;32:115973.
- Weyesa A, Mulugeta E. Recent advances in the synthesis of biologically and pharmaceutically active quinoline and its analogues: a review. RSC Adv 2020;10:20784–93.
- Afzal O, Kumar S, Haider MR, et al. A review on anticancer potential of bioactive heterocycle quinoline. Eur J Med Chem 2015;97:871–910.
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci 2015;36:422–39.
- He R, Xu B, Ping L, Lv X. Structural optimization towards promising β-methyl-4-acrylamido quinoline derivatives as PI3K/mTOR dual inhibitors for anti-cancer therapy: The in vitro and in vivo biological evaluation. Eur J Med Chem 2021;214:113249.
- Nan X, Li HJ, Fang SB, et al. Structure-based discovery of novel 4-(2-fluorophenoxy)quinoline derivatives as c-Met inhibitors using isocyanide-involved multicomponent reactions. Eur J Med Chem 2020;193:112241.
- Li K, Li Y, Zhou D, et al. Synthesis and biological evaluation of quinoline derivatives as potential anti-prostate cancer agents and Pim-1 kinase inhibitors. Bioorg Med Chem 2016;24:1889–97.
- Pannala M, Kher S, Wilson N, et al. Synthesis and structure-activity relationship of 4-(2-aryl-cyclopropylamino)-quinoline-3-carbonitriles as EGFR tyrosine kinase inhibitors. Bioorg Med Chem Lett 2007;17:5978–82.
- Pawar VG, Sos ML, Rode HB, et al. Synthesis and biological evaluation of 4-anilinoquinolines as potent inhibitors of epidermal growth factor receptor. J Med Chem 2010;53:2892–901.
- Li S, Hu L, Li J, et al. Design, synthesis, structure-activity relationships and mechanism of action of new quinoline derivatives as potential antitumor agents. Eur J Med Chem 2019;162:666–78.
- Abouzid K, Shouman S. Design, synthesis and in vitro antitumor activity of 4-aminoquinoline and 4-aminoquinazoline derivatives targeting EGFR tyrosine kinase. Bioorg Med Chem 2008;16:7543–51.
- Tsou HR, Overbeek-Klumpers EG, Hallett WA, et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J Med Chem 2005;48:1107–31.
- Elbadawi MM, Eldehna WM, Wang W, et al. Discovery of 4-alkoxy-2-aryl-6,7-dimethoxyquinolines as a new class of topoisomerase I inhibitors endowed with potent in vitro anticancer activity. Eur J Med Chem 2021;215:113261.
- Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol 2013;8:82–95.
- Marzi L, Agama K, Murai J, et al. Novel fluoroindenoisoquinoline non-camptothecin topoisomerase I inhibitors. Mol Cancer Ther 2018;17:1694–704.
- Dexheimer TS, Pommier Y. DNA cleavage assay for the identification of topoisomerase I inhibitors. Nat Protoc 2008;3:1736–50.
- Antony S, Jayaraman M, Laco G, et al. Differential induction of topoisomerase I-DNA cleavage complexes by the indenoisoquinoline MJ-III-65 (NSC 706744) and camptothecin: base sequence analysis and activity against camptothecin-resistant topoisomerases I. Cancer Res 2003;63:7428–35.
- Elsayed MSA, Su Y, Wang P, et al. Design and synthesis of chlorinated and fluorinated 7-azaindenoisoquinolines as potent cytotoxic anticancer agents that inhibit topoisomerase I. J Med Chem 2017;60:5364–76.
- Bell EW, Zhang Y. DockRMSD: an open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J Cheminform 2019;11:1–9.
- Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018;46:W296–303.
- Zhao S, hong He Y, Wu D, Guan Z. A new general approach to 4-substituted-3-halo-2-quinolones. J Fluor Chem 2010;131:597–605.
- Butin AV, Smirnov SK, Stroganova TA, et al. Simple route to 3-(2-indolyl)-1-propanones via a furan recyclization reaction. Tetrahedron 2007;63:474–91.
- Huang Z, Yang Y, Xiao Q, et al. Auto-tandem catalysis: synthesis of acridines by Pd-catalyzed C=C bond formation and C(sp2)-N cross-coupling. Eur J Org Chem 2012;2012:6586–93.
- Mamidala R, Subramani MS, Samser S, et al. Chemoselective alkylation of aminoacetophenones with alcohols by using a palladacycle-phosphine catalyst. Eur J Org Chem 2018;2018:6286–96.
- Hanwell MD, Curtis DE, Lonie DC, et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 2012;4:17.
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol 2015;1263:243–50.
- Protein data bank, n.d. Available from: https://www.rcsb.org/
- Trott O, Olson AJ. Software news and update AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61.
- The PyMOL molecular graphics system, n.d.
- Abraham MJ, Murtola T, Schulz R, et al. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015;1–2:19–25.
- Said MA, Albohy A, Abdelrahman MA, Ibrahim HS. Importance of glutamine 189 flexibility in SARS-CoV-2 main protease: lesson learned from in silico virtual screening of ChEMBL database and molecular dynamics. Eur J Pharm Sci 2021;160:105744.
- Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 2011;32:2359–68.
- Huang J, Mackerell AD. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 2013;34:2135–45.
- Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A 2001;105:9954–60.
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys 2007;126:014101.
- Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 1981;52:7182–90.
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem 1997;18:1463–72.
- Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys 1993;98:10089–92.