
Abstract
The advent of proteolysis-targeting chimaeras (PROTACs) mandates that new ligands for the recruitment of E3 ligases are discovered. The traditional immunomodulatory drugs (IMiDs) such as thalidomide and its analogues (all based on the phthalimide glutarimide core) bind to Cereblon, the substrate receptor of the CRL4ACRBN E3 ligase. We designed a thalidomide analogue in which the phthalimide moiety was replaced with benzotriazole, using an innovative synthesis strategy. Compared to thalidomide, the resulting “benzotriazolo thalidomide” has a similar binding mode, but improved properties, as revealed in crystallographic analyses, affinity assays and cell culture.
Graphical Abstract
Introduction
The approach to eliminating dysregulated proteins via targeted protein degradation is rapidly gaining momentum as an alternative to small-molecule inhibitorsCitation1. One of the most promising and powerful molecular tools to achieve that are the so-called proteolysis-targeting chimaeras (PROTACs) in which two recruiter moieties are joined with a linkerCitation2. One is a ligand of the protein of interest (POI, i.e. the one to be degraded) and the other a ligand of an E3 ligase. Once the two proteins (POI and E3 ligase) are brought in proximity by the PROTAC molecule, this triggers poly-ubiquitination of the POI by the E3 ligase, which makes the former a client for proteasomal degradationCitation3. Cereblon (CRBN) is one of the most important E3 ligases that has been employed for PROTAC developmentCitation4 so far. The ligand space of CRBN mostly includes phthalimide-based thalidomide (1) and its analogsCitation5. In addition to these, structural requirements for CRBN ligand binding have recently been elucidatedCitation6.
The binding mode of thalidomide (1) includes a hydrogen bond between one of the phthalimide carbonyl groups and a conserved asparagine residue of the protein, which contributes to the affinity between the two moleculesCitation7. However, the other phthalimide carbonyl is not involved in any specific interactions, not even with water molecules, and thus represents an unsatisfied polar group that potentially lowers the binding energy. We hypothesised that replacing the phthalimide core with benzotriazole, thereby removing both carbonyl groups, would possibly eliminate the possibility of hydrogen bonding with the conserved asparagine, but also rid the molecule of the “dissatisfied” carbonyl group. As the net result, the affinity to CRBN may be retained, assuming that the introduced triazole nitrogen is less unfavoured than the carbonyl group due to its decreased polarity with only a single free electron pair. Herein, we report on the verification of this hypothesis.
Results and discussion
The synthesis of the benzotriazole analogue 2 of thalidomide was achieved as detailed below. Commercially available glutarimide 3 was (dimethylamino)methylenated at the α-position using the Brederick’s reagent (4)Citation8. The resulting derivative 5 readily entered the Regitz diazo transfer reactionCitation9 with 4-nitrophenylsulfonyl azide (NsN3) to give hitherto undescribed 3-diazopiperidine-2,6-dione (6) in excellent yield. α-Diazocarbonyl compounds were recently established to regioselectively alkylate benzotriazoles at N2 when activated as Rh(II) carbenesCitation10. Indeed, when α-diazoglutarimide (6) was activated by Rh(II) espionate (bis[rhodium(α,α,α′,α′-tetramethyl-1,3-benzenedipropionic acid)]) (1 mol%) and reacted with benzotriazole, desired ‘benzotriazolo thalidomide’ 2 was obtained in excellent yield and complete regioselectivity (Scheme 1).
Scheme 1. Synthesis of ‘benzotriazolo thalidomide’ 2.

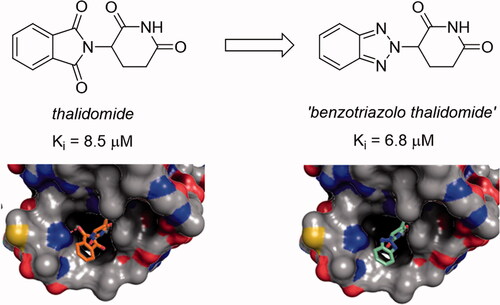
To our delight, when evaluated for affinity to the thalidomide-binding domain of human CRBN in comparison to thalidomide (1, Ki 8.5 ± 0.8 µM), using the recently reported thermophoresis-based assayCitation11, the benzotriazolo analogue 2 displayed an improvement in affinity with a Ki value of 6.8 ± 1.6 µM.
To gain insight into the binding mode of 2, we employed our previously established crystal soaking system based on the bacterial CRBN homologue Magnetospirillum gryphiswaldense Cereblon Isoform 4 (MsCI4)Citation6. The obtained crystal structure revealed that 2 binds in the same overall orientation as thalidomide, but lacks any specific hydrogen-bonding interactions with the target other than those mediated by the glutarimide moiety ()Citation12. None of the benzotriazole nitrogen is involved in hydrogen bonds, not even to water, suggesting that the retained affinity could indeed be due to their lower polarity as compared to the ‘unsatisfied’ carbonyl groups in thalidomide.
Figure 1. Binding mode of ‘benzotriazolo thalidomide’ (2) compared to thalidomide. Left: two views of 2 bound to MsCI4 with an FO-FC omit map contoured at 4σ. Three tryptophan residues and a conserved asparagine residue of the binding site are indicated. Of note, the asparagine does not form interactions with 2. Right: The binding of 2 compared to thalidomide in surface representation coloured by atom type. The hydrogen bond of thalidomide to the conserved asparagine is indicated. Residue numbering according to the MsCI4 sequence.

Usage of 2 in PROTAC design mandates that a functionalised version of it (akin to lenalidomideCitation13 or pomalidomideCitation14) is developed. Before taking steps into that direction, we were keen to determine the cytotoxicity and apoptosis-inducing profile of “benzotriazolo thalidomide” 2, in comparison to thalidomide (1) itself. shows the cytotoxicity of the two compounds towards MOLP-8Citation15 and KMS-12-PECitation16 multiple myeloma cell lines, which clearly demonstrates the absence of any appreciable cytotoxicity at concentrations as high as 250 µM.
Figure 2. Cytotoxicity profile of compounds 1 and 2 against two multiple myeloma cell lines.

As to the apoptosis-inducing ability (evaluated by flow cytometry in MOLP-8 cells), compound 2 showed a clear advantage compared to thalidomide (1): at 300 µM, it preserved a substantially higher population of live cells (). This clearly shows the promise of the ‘benzotriazolo thalidomide’ scaffold reported herein for the future use in the design of PROTACs.
Table 1. Apoptosis induction by compounds 1 and 2 (300 µM, MOLP-8 cells, 48 h incubation time).
Conclusion
In summary, we have described a promising novel benzotriazolo analogue of thalidomide. Despite the absence of the carbonyl group involved in hydrogen bonding with CRBN, it retained affinity, likely due to the relief from the other, ‘dissatisfied’ carbonyl group. These assumptions are corroborated by the crystal structure of the complex between 2 and CRBN. Compound 2 is distinctly non-cytotoxic towards multiple myeloma cell lines (at concentrations as high as 250 µM) and preserves more live cell population in apoptosis-induction experiments, compared to thalidomide (1). Development of a functionalised version of 2 for the use in the PROTAC design is highly desirable and is currently underway in our laboratories.
Supplemental Material
Download PDF (1 MB)Acknowledgements
The authors thank the staff of beamline X10SA of the Swiss Light Source (PSI, Villigen, Switzerland) for the excellent technical support. The authors are grateful to the Russian Foundation for Basic Research for financial support (project grant 19–03-00775). The authors thank the Research Center for Magnetic Resonance, the Center for Chemical Analysis and Materials Research, and Research Resource Center for Molecular and Cell Technologies of Saint Petersburg State University Research Park for obtaining the analytical data.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Konstantinidou M, Li J, Zhang B, et al. PROTACs- a game-changing technology. Expert Opin Drug Discov 2019;14:1255–68.
- Donoghue C, Cubillos-Rojas M, Guitierrez-Prat N, et al. Optimal linker length for small molecule PROTACs that selectively target p38α and p38β for degradation. Eur J Med Chem 2020;201:112451.
- Zhang Z, Chang X, Zhang C, et al. Identification of probe-quality degraders for Poly(ADP-ribose) polymerase-1 (PARP-1). J Enzyme Inhib Med Chem 2020;35:1606–15.
- (a) Girardini M, Maniaci C, Hughes SJ, et al. Cereblon versus VHL: hijacking E3 ligases against each other using PROTACs. Bioorg Med Chem 2019;27:2466–79. (b) Steinebach C, Sosič I, Lindner S, et al. A MedChem toolbox for cereblon-directed PROTACs. Med Chem Comm 2019;10:1037–41.
- Xiao D, Wang YJ, Wang HL, et al. Design and synthesis of new lenalidomide analogs via Suzuki cross‐coupling reaction. Arch Pharm 2020;353:1900376.
- (a) Boichenko I, Bär K, Deiss S, et al. Chemical ligand space of cereblon. ACS Omega 2018;3:11163–71. (b) Heim C, Pliatsika D, Mousavizadeh F, et al. De-novo design of cereblon (CRBN) effectors guided by natural hydrolysis products of thalidomide derivatives. J Med Chem 2019;62:6615–29. (c) Heim C, Maiwald S, Steinebach C, et al. On the correlation of cereblon binding, fluorination and antiangiogenic properties of immunomodulatory drugs. Biochem Biophys Res Commun 2021;534:67–72.
- Hartmann MD, Boichenko I, Coles M, et al. Structural dynamics of the cereblon ligand binding domain. PLoS One 2015;10:e0128342.
- Rosso GB. tert-Butoxy bis(dimethyl-amino)methane (Bredereck's reagent). Synlett 2006;809–810.
- Cui X, Zhang X, Wang W, et al. Regitz diazo transfer reaction for the synthesis of 1,4,5-trisubstituted 1,2,3-triazoles and subsequent regiospecific construction of 1,4-disubstituted 1,2,3-triazoles via C-C bond cleavage. J Org Chem 2021;86:4071–80.
- Wang K, Chen P, Ji D, et al. Rhodium-catalyzed regioselective N2 -alkylation of benzotriazoles with diazo compounds/enynones via a nonclassical pathway. Angew Chem Int Ed Engl 2018;57:12489–93.
- Maiwald S, Heim C, Alvarez BH, Hartmann MD. Sweet and blind spots in E3 ligase ligand space revealed by a thermophoresis-based assay. ACS Med Chem Lett 2021;12:74–81.
- (a) Fischer ES, Böhm K, Lydeard JR, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014;512:49–53. (b) Hartmann MD, Boichenko I, Coles M, et al. Thalidomide mimics uridine binding to an aromatic cage in cereblon. J Struct Biol 2014;188:225–32.
- Armoiry X, Aulagner G, Facon T. Lenalidomide in the treatment of multiple myeloma: a review. J Clin Pharm Ther 2008;33:219–26.
- Lentzsch S, Rogers MS, LeBlanc R, et al. S-3-Amino-phthalimido-glutarimide inhibits angiogenesis and growth of B-cell neoplasias in mice. Cancer Res 2002;62:2300–5.
- Matsuo Y, Drexler HG, Harashima A, et al. Induction of CD28 on the new myeloma cell line MOLP-8 with t(11;14)(q13;q32) expressing delta/lambda type immunoglobulin. Leuk Res 2004;28:869–77.
- Ohtsuki T, Yawata Y, Wada H, et al. Two human myeloma cell lines, amylase-producing KMS-12-PE and amylase-non-producing KMS-12-BM, were established from a patient, having the same chromosome marker, t(11;14)(q13;q32). Br J Haematol 1989;73:199–204.