
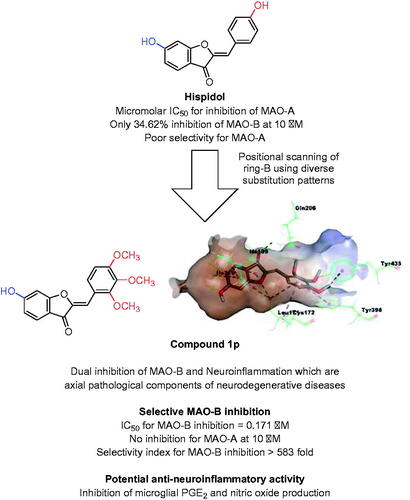
Abstract
Multifunctional molecules might offer better treatment of complex multifactorial neurological diseases. Monoaminergic pathways dysregulation and neuroinflammation are common convergence points in diverse neurodegenerative and neuropsychiatric disorders. Aiming to target these diseases, polypharmacological agents modulating both monoaminergic pathways and neuroinflammatory were addressed. A library of analogues of the natural product hispidol was prepared and evaluated for inhibition of monoamine oxidases (MAOs) isoforms. Several molecules emerged as selective potential MAO B inhibitors. The most promising compounds were further evaluated in vitro for their impact on microglia viability, induced production of proinflammatory mediators and MAO-B inhibition mechanism. Amongst tested compounds, 1p was a safe potent competitive reversible MAO-B inhibitor and inhibitor of microglial production of neuroinflammatory mediators; NO and PGE2. In-silico study provided insights into molecular basis of the observed selective MAO B inhibition. This study presents compound 1p as a promising lead compound for management of neurodegenerative disease.
Graphical Abstract
1. Introduction
Healthy adult brain is estimated to have 100 billion neurons forming 100 trillion neuronal connections (synapses)Citation1. Signals flow through these massive brain’s neuronal circuits require chemical neurotransmitters which affect emotions, thoughts, movements, and memories. Disruption of such intricate brain networking results in divergent neurological disorders including neurodegenerative and neuropsychiatric diseases which afflicts millions of peoplesCitation2. Unfortunately, the currently available therapeutics are still far from realising complete cure of these neurological diseases and, in addition, such diseases impose a high socioeconomic burdenCitation3,Citation4.
Despite the complex etiopathologies of neurodegenerative and neuropsychiatric diseases, common converging mechanisms and manifestations were found shared by these divergent diseases pathwaysCitation5–12. Amongst known aetiological factors, monoaminergic pathway abnormalities were demonstrated as a major contributor and/or manifestation of numerous neurodegenerative and neuropsychiatric diseasesCitation10–12. Synaptic transmission in monoaminergic system involves dopamine (DA), serotonin (5-HT), noradrenaline (NA), and histamine as chemical neurotransmitters which are catabolised by monoamine oxidases (MAOs). MAOs are mitochondrial membrane-bound enzymes that convert monoamines into the corresponding aldehydes producing hydrogen peroxide, ammonia products of the same reaction. Two isoforms of MAO have been identified; MAO-A which is mainly associated with catabolism of catecholaminergic and serotonergic neurotransmitters; and MAO-B which is mainly associated with catabolism of dopaminergic and histaminergic neurotransmitters. Impairment of monoaminergic signalling via upregulation of MAO activity results not only in lowering neurotransmitters’ levels, but also the products of the enzymatic reaction involving reactive oxygen species (ROS) induce oxidative stress; a well-known cause of degenerative neuronal cells’ deathCitation13,Citation14. Consequently, monoamine oxidase inhibitors (MAOIs) would enhance the monoaminergic neurotransmission within the brain’s circuits and also protect neuronal cells against cytotoxicity associated with increased MAO activity/expression. Because of limitations of irreversible inhibitors of MAOs including hypertension crisis arising from irreversible inhibition of MAO-A coupled with dietary amines’ intake, development of selective MAO-B inhibitors was addressedCitation15. Nevertheless, long-term treatment of neurodegenerative diseases with irreversible MAO-B inhibitors fails due to compensatory mechanismsCitation16. In contrast, selective reversible inhibitors have proven to elicit a better therapeutic profile. Successfully, moclobemide; a reversible MAO-A inhibitor, has been approved for treatment of neuropsychiatric disorders, such as depression, whereas safinamide, a reversible MAO-B inhibitor, has been approved for neurodegenerative disorders, such as Parkinson’s disease (PD) and Alzheimer’s disease (AD).
Neuroinflammation is another commonality shared in neurodegenerative and neuropsychiatric diseases. CNS neuroinflammatory response is mainly triggered by microglia releasing inflammatory mediators, such as nitric oxide (NO), PGE2, and cytokines as defence mechanisms. However, abnormal activation results in neurotoxic effects and eventually neuronal cells deathCitation17. Therefore, neuroinflammation is a component in several neurodegenerative diseases including PD, AD, amyotrophic lateral sclerosis (ALS), and frontotemporal dementia (FTD)Citation17. This is further supported by multiple evidences demonstrated targeting the neuroinflammatory component in neurodegenerative diseases as a valid treatment approachCitation18–20. In addition, neuroinflammation was found as a key contributor to the development and progression of several neuropsychiatric diseases including schizophrenia, depression, and bipolar disordersCitation21,Citation22. A microglia hypothesis for neuropsychiatric disorders was shaped in recognition of their central rolesCitation23. Eventually, recent literature reports attempted targeting neuroinflammation towards treatment of neuropsychiatric diseasesCitation24,Citation25.
Inflammation is an important component of the vicious cycle that exists in several complex diseases such as neurological disorders and cancersCitation26,Citation27. For example, neurodegeneration triggers neuroinflammation which in turn results in the formation of neurotoxic substances that trigger more neurodegeneration. Therefore, the efficacy of a single targeting therapy might be limited for treatment. A polypharmacological molecule impacting multiple pathways involved in such complex diseases might be more effective strategy to break this vicious cycle and achieve better therapeutic effectsCitation28–33.
Because of the high cost of new drugs development, repositioning or repurposing of previously developed or failed molecules has emerged as a promising strategy in drug discovery and developmentCitation2,Citation34. In fact, literature reports several successful discoveries after repurposing strategyCitation35–38. Herein, we report our efforts to develop multifunctional molecules inhibiting both of human MAO and neuroinflammation based on rational repurposing approach.
2. Results and discussion
2.1. Design of focussed hispidol analogues library
Considering that drug discovery of naturally inspired compounds might be associated with higher success rates including MAOIsCitation39–42, hispidol (); a 6-hydroxyaurone-based natural product, was chosen as a starting lead compound. It was recently reported to inhibit in vitro MAO-A and MAO-B with more selectivity towards MAO-A inhibition as well as to elicit in vivo antidepressant-like effectsCitation43,Citation44. Previously, aurone was showed as possible scaffold for development of MAOIsCitation45. Considering that aurones, as well as other flavonoids, exist in natural with different oxygenation patterns of rings A and B, literature was searched for MAO inhibition by natural/synthetic oxygenated aurones. In contrast to hispidol, the search outcome showed that reported unsubstituted-ring A, 5-hydroxy/methoxy or 4-hydroxy derivatives of aurones with ring B bearing simple hydroxylation/methoxylation patterns elicit almost low selectivity for inhibition of MAO-B ()Citation46,Citation47. Based on the reported activity of hispidol, the effort reported herein aims to conduct positional analogue scanningCitation48 of hispidol analogues employing hydroxylation/methoxylation patterns of ring B towards identification of molecules with interesting MAO inhibitory profiles and activities. In addition, a recent report has shown some synthetic hispidol analogues to elicit anti-inflammatory effects in in vitro LPS-induced macrophages’ modelCitation49. These points might provide a rationale for hispidol analogues repurposing for development of multifunctional agents modulating monoaminergic signalling and neuroinflammation; both are axial components in the neurodegenerative and neuropsychiatric diseases. Towards this goal, hispidol was selected as a starting point to establish a focussed library of analogues. While the hydroxy moiety is a polar moiety that can act as hydrogen bond donor or acceptor, the methoxy moiety is less polar as well as can act only as hydrogen bond donor. In addition, both hydroxy and methoxy moieties are ubiquitous in natural products. Therefore, and in attempt to gain insights of structure–activity relationship and, as illustrated in , the characteristic C-6 hydroxy substitution pattern of ring A found in hispidol was maintained or converted to the natural products ubiquitous methoxy moiety in the designed library. While hispidol possesses one hydroxy moiety at ring B located at C-4′ of the aurone scaffold, this hydroxy was translocated to C-2′ and C-3′ position to explore activity of regioisomers. In addition, members having diversely dihydroxylated ring B were enumerated in the designed library to pattern explore the impact of different dihydroxylation pattern on the activity. Furthermore, inclusion of trihydroxylated ring B among explored patterns was planned. Moreover, exploration of monomethoxylated ring B regiosomers, diverse dimethoxylation patterns of ring B, and trimethoxylated ring B as well as the protruding 4′-methoxymethoxy moiety were considered in the designed library to investigate the impact of these structural modifications on the activity.
Figure 1. Positional scanning of hispidol’s ring-B towards generation of hispidol analogues’ focussed library based on monoamine oxidases inhibition and selectivity of hispidol and other methoxylated-aurones.

2.2. Synthesis of targeted library members
Economies of the synthesis including step and atom economy are important consideration in achieving a concise practical synthesisCitation50–52. In lieu of this, hispidol analogues 1a–q possessing C-6 hydroxy feature were synthesised in single step via acid-catalysed or base-catalysed cross-aldol condensation of commercially available or 6-hydroxy-3-coumaranone with diversely substituted aldehydes similar to reported procedure to afford the desired compounds (Scheme 1)Citation49. To access hispidol analogues 1r–y possessing C-6 methoxy feature, 6-hydroxy-3-coumaranone was methylated using methyl iodide in presence of potassium carbonate as a base and dimethylformamide as a solvent to yield 6-methoxy-3-coumaranone. Similar to the previously reported procedure, acid-catalysed or base-catalysed cross-aldol condensation of 6-methoxy-3-coumaranone with diversely substituted aldehydes yielded the desired C-6 methoxy members 1r–y of the desired hispidol analogues libraryCitation49. The structures of unreported compounds were elucidated by 1H NMR, 13 C NMR, and HRMS analyses. For known compounds 1a, 1b, 1d, 1e, 1f, 1h–1t, and 1v–1y, spectroscopic data were in agreement with literatureCitation43,Citation49,Citation53–58.
Scheme 1. Reagents and reaction conditions: (a) 12 N HCl, ethanol, 60–70 °C, for the specified time; (b) 50% KOH, methanol, 60 °C, or the specified time; (c) methyl iodide, K2CO3, DMF, and rt, overnight.

2.3. Biological evaluation
2.3.1. Evaluation of inhibition of different monoamine oxidase isoforms
Two different MAO isoforms are known. Each isoform is correlated with different neurological disorders. Therefore, the designed focussed library of hispidol analogues were evaluated for inhibitory activities against both recombinant human MAO-A and MAO-B isoforms employing previously reported spectrophotometric methodCitation59. The screening results are summarised in and discussed in the following sections.
Table 1. Results of hMAO-A and hMAO-B inhibition and selectivity of the synthesised compounds (1a−1y).
2.3.1.1. Structure–activity relationship of hMAO-A inhibitory activity
The structure of hispidol involves a hydroxy substituent on ring A at C-6-position and mono- hydroxy substituent on ring B at para-position. These molecular features enabled hispidol to trigger micromolar MAO-A inhibitory activity in our assay system (IC50 was close to 5.28 µM, ). Structure–activity analysis for C-6-hydroxy compounds with mono-hydroxylation pattern (versus hispidol) suggested an important role for ring-B para-position or a deleterious effect of ring-B ortho- or meta-positions mono-hydroxy substitution pattern. Thus, compounds 1a and 1b were of very low MAO-A inhibitory activity. Combining two-hydroxy substituents at ring-B para- and ortho-positions (compound 1d) decreased MAO-A inhibitory activity relative to hispidol while at both ring-B para- and meta-positions maintained almost same level of MAO-A inhibitory activity (compound 1e). Interestingly, vicinal ortho- and meta-dihydroxylation pattern at 2′,3′-positions should low activity (compound 1c) while ortho- and meta-dihydroxylation pattern at 2′,5′-positions was more capable of inhibiting MAO-A activity but with lower potency relative to hispidol (compound 1f). However, two hydroxy moieties at both meta-positions and ring-B as well as three hydroxy moieties at para- and both meta-positions elicited low MAO-A inhibitory activities (compounds 1g and 1h). Based on activities of ring-B positional scanning for hydroxy moieties in C-6 hydroxy compounds 1a–1h, it might be deduced that ring-B para-position is the most important for eliciting MAO-A inhibition. Replacing the polar hydrogen bond donor and acceptor hydroxy moieties of ring-B with the less polar hydrogen bond donor only methoxy moieties (compounds 1i–1p) reinforced the assumption of the importance of para-position. Thus, only compound 1k having monomethoxy substituent at para-position of ring-B showed potential MAO-A inhibitory activity. As a methoxy substituent is more steric than hydroxy substituent and is also unable to accept hydrogen indicates some steric tolerance of this position as well as negates establishment of a donor-hydrogen bond at this position. However, such steric tolerance might be limited as compound 1k showed relatively lower activity than hispidol. The finding that compound 1q having the protruding methoxymethoxy substituent at para-position of ring-B showed limited MAO-A inhibitory activity provides further supported to this found steric tolerance. It was notable to find out that C-6 methoxy-substituted ring-A compounds 1r–1y show MAO-A inhibitory activity profile distinct from compounds 1a–1q having C-6 hydroxy-substituted ring-A. While the monomethoxy-substituted ring-B derivatives (1x and 1 y) possessed very low to almost no MAO-A inhibitory activity similar to the trend found for C-6 hydroxy-substituted ring-A compounds, the activity trend was changed for monohydroxy-substituted ring-B derivatives (1r–1t) as no MAO-A inhibitory activity was triggered by para-substituted ring-B. In addition, the vicinal ortho- and meta-dihydroxylation pattern at 2′,3′-positions as well as para- and ortho-positions in C-6 hydroxy-substituted ring-A compounds (1c and 1d, respectively) which were of low activity were found to trigger potential MAO-A inhibitory activity in C-6 methoxy-substituted ring-A compounds (1 u and 1v, respectively). This activity trend change could reflect different binding modes for C-6 methoxy-substituted ring-A compounds.
2.3.1.2. Structure–activity relationship of hMAO-B inhibitory activity
As human MAO-B isoform is different from human MAO-A, different structure-activity relationship trend might be identified which might assist in achieving selective inhibitors better than the poorly selective hispidol. Amongst compounds having ring-A C-6-hydroxy substitution pattern, ortho-monohydroxy substituted ring-B (compound 1a) showed some increased MAO-B inhibitory activity relative to hispidol but the activity still under 50% inhibition. However, meta-monohydroxy substituted ring-B (compound 1b) showed very weak MAO-B inhibition which was less than hispidol. In general, combining two-hydroxy substituents at ring-B resulted in slight to significant increase in MAO-B inhibitory activity (compounds 1c–1f) except for combining two meta-hydroxy substituents on ring-B as well as three hydroxy moieties at both two meta-and para-positions (compounds 1g and 1h) which showed marked decreased activity. In fact, vicinal ortho- and meta-dihydroxylation pattern at 2′,3′-positions, as well as ortho- and meta-dihydroxylation pattern at 2′,5′-positions or para- and meta-dihydroxylation of ring-B afforded potential MAO-B inhibitors eliciting low micro molar IC50 values. It might be concluded that steric requirements are tolerated for dihydroxylation of ring-B at these positions. Replacing the polar hydrogen bond donor and acceptor hydroxy moieties of ring-B with the less polar hydrogen bond donor only methoxy moieties (compounds 1i–1p) afforded an interesting structure-activity relationship outcome. There were significant increases in MAO-B inhibitory activities of monomethoxylated patterns of ring-B not only for para-methoxy substituent on ring-B (compound 1k), which exhibited a submicromolar potency, but also ortho- or meta-methoxy substitution patterns at ring-B afforded potential MAO-B inhibitors eliciting low micro molar IC50 values (compounds 1i and 1j). This suggested not only a tolerance of steric requirements of the more demanding methoxy groups, but also negated establishment of hydrogen bond acceptor interactions and, further, might indicate hydrophobic interactions. Except for compound 1 l having vicinal ortho- and meta-dimethoxy substituents at 2′,3′-positions, substitution patterns combining two methoxy substituents at ring-B (compounds 1 m–1o) resulted in lowering MAO-A inhibitory activity under 50% inhibition. Such results suggested that steric demand of vicinal ortho- and meta-dimethoxy substituents at 2′,3′-positions of ring-B, but not other patterns, might be well-tolerated. Combining such vicinal ortho- and meta-dimethoxy substituents with a third methoxy group at para-position of ring-B, which was revealed to afford submicromolar potent compound 1k, resulted in a more potent MAO-B inhibitor 1p eliciting submicromolar activity comparable to safinamide. Such outcome suggested steric tolerance or favourable hydrophobic interactions of such trimethoxylated pattern of compound 1p. The finding that compound 1q having the protruding methoxymethoxy moiety at para-position of ring-B elicited potent submicromolar activity reinforces that conceived conclusion that MAO-B inhibitory activity might tolerate steric demanding substituents at this position. Next, structure–activity relationship for compounds having C-6-methoxy substitution pattern were analysed. For compounds, 1r–1t bearing monohydroxy substitution pattern showed that only compound 1t possessing this moiety at para-position elicited high inhibition and displayed low micromolar potency. Combining the two ortho- and meta-dihydroxy substituents the vicinal 2′,3′-positions as well as the meta- and para-positions in C-6-methoxy derivatives 1u and 1w, respectively, showed more or less similar activity to analogous derivatives in C-6-hydroxy derivatives 1c and 1e, respectively. However, the ortho- and para-dihydroxy groups at ring-B in C-6-methoxy derivative 1v showed enhancement of activity relative to analogous derivative 1d in C-6-hydroxy derivatives. The enhancement of activity was more remarkable for ring-B monomethoxylated derivatives 1x and 1y bearing ortho- or meta-substitution pattern, respectively, which elicited potent submicromolar activities. Remarkably, potency of compound 1y possessing meta-methoxylation patterns of ring-B was comparable to safinamide.
2.3.1.3. Selectivity of MAO-B inhibition
Analysis of activity data of hispidol and the synthesised 25 hispidol analogues against MAO-A and MAO-B isoforms revealed interesting outcome. Similar to recently reported benzo[b]tiophen-3-ol derivative that revealed dependence of the triggered MAO inhibitor activity on position and chemical nature of the substituentCitation60, it became evident from our results that amongst ring-A C-6-hydroxy analogues 1a–1q, there might be steric limitations for substituents at ortho- and meta- positions of ring-B. This was reflected in lowered MAO-A inhibitory activities of compounds having the sterically more demanding methoxy groups more than those having the less bulkier hydroxyl groups (e.g. compounds 1i and 1j versus compounds 1a and 1b). In contrast, MAO-B inhibition activity tolerated substituents at these positions and even derivatives incorporating the bulkier methoxy moieties at these positions elicited potential MAO-B inhibition. It was found that both potential MAO-A and MAO-B inhibition activities tolerated and were associated with substituents at para-position of ring-B (e.g. compounds 1k and hispidol). Accordingly, derivatives having such substitution pattern, although were potential MAO inhibitors, showed low selectivity values. However, it was found that steric tolerance of MAO-A isoform is limited relative to tolerance of MAO-B. Accordingly, incorporation the more steric protruding methoxymethoxy at para-position of ring-B afforded potential MAO-B inhibitor 1q showing high selectivity (, selectivity index > 168). Another strategy was revealed to afford selective MAO-B inhibition based on combining more than one substituent at ring-B to exceed the steric requirements of MAO-A but still tolerated by MAO-B. This strategy might be indicated by the highly selective potent MAO-B inhibition, yet almost no MAO-A inhibition, by compound 1p (). This derivative combines para-methoxy moiety at ring-B; which was found to trigger both MAO-B and MAO-A inhibitory activities, with vicinal ortho- and meta-methoxy moieties at ring-B; which were found to abolish MAO-A inhibitory activity. Switching to ring-A C-6-methoxy analogues 1r–1y, it was found again that MAO-B isoform, in contrast to MAO-A isoform, tolerated substituents at ortho- and meta-positions of ring-B. In addition, sterically more demanding and less polar methoxy moieties were efficient in triggering submicromolar MAO-B IC50 values while showing very low MAO-A inhibition. Consequently, compounds 1x and 1y were identified as highly selective potent MAO-B inhibitors. Collectively, four compounds; 1p, 1q, 1x, and 1y, were found to trigger highly selective potent MAO-B inhibition.
2.3.2. Evaluation of absence of cells’ viability disruption
Promising antineurodegenrative candidate molecules should not impair viability of CNS cells. Microglia are critical cells within CNS involved in microglia–neuron communications and are responsible for maintenance of brain integrity and healthCitation61,Citation62. However, under neuroinflammatory conditions, aberrant activation of microglia contributes to neurodegeneration and disease progressionCitation63. In this regard, suppression of the abnormal activation while maintenance of viability of microglia is important considerations. To assess this later crucial requirement, the identified potent MAO-B inhibitor compounds that showed selective MAO-B inhibition by 168-fold or more were evaluated for their impact on viability of BV2 cells; a valid in vitro model for microgliaCitation64. The viability assay was carried out using the water-soluble tetrazolium reagent WST-1 which eliminates the formazan dissolution step requested in case of MTTCitation65 employing multiple doses of 1, 10, 30, and 50 µM. On the opposite to revealed cytotoxic effects for safinamide and hispidol at high doses (), the results indicated that none of the tested concentrations for all of the employed four compounds 1p, 1q, 1x, and 1y triggered significant impairment of BV2 cells’ viability. In lieu of this, it might be inferred that these compounds showing effective inhibition of MAO-B activity and triggering no impairment of microglia viability are good candidates might be advanced for further evaluation towards the development of multifunctional molecules for neurodegenerative diseases such as PD.
Figure 2. Dose-dependent viabilities % upon treatment with the evaluated candidate hispidol-analogues as well as safinamide and hispidol.

2.3.3. Investigation of anti-neuroinflammatory activity
Neuroinflammation is a crucial component in neurodegenerative diseases. Chronic activation of microglia, which are the brain-resident form of macrophages, results in sustained production of several proinflammatory mediators including glial NO, PGE2, and others molecules that are implicated in multiple neurodegenerative diseasesCitation66–68. Considering the that LPS-induced production of NO and PGE2 in RAW 264.7 macrophages was found to be inhibited by this class of compoundsCitation49, the ability of these compounds to inhibit the production of proinflammatory mediators by CNS microglia was checked. In addition to safinamide and hispidol, which were used as controls, both of compounds 1p and 1y which emerged as potent (IC50 = 0.171 and 0.201 µM, respectively), highly selective MAO-B inhibitors (selectivity indices were >583 and >496 respectively) and, in addition, have no detrimental effects of cells viability were employed for these evaluations. In this regard, LPS-treated BV2 cells were incubated with or without different concentrations of each compound. As shown in , compounds 1p, safinamide, and hispidol but not compound 1y triggering significant inhibition of NO production by BV2 cells. Interestingly, safinamide showed undesirable dose-dependent increase in PGE2 production at tested concentrations. Meanwhile, compound 1p and hispidol effectively suppressed the induced-PGE2 production by BV2 cells at low concentrations (). Based on collective results of antineuroinflammatory activity evaluation, in conjugation with above-conducted MAO inhibitions’ assays, compound 1p might be claimed as a promising lead compound eliciting polypharmacological effects combining inhibition of neuroinflammation and MAO-B activity; both processes are crucial components of neurodegenerative diseases such as PD.
Figure 3. In vitro anti-neuroinflammatory activity in BV2 Cells: A) dose-dependent effects of compounds 1p and 1y as well as safinamide and hispidol against LPS-induced production of nitric oxide; B) Dose-dependent effects of compounds 1p and 1y as well as safinamide and hispidol against LPS-induced production of PGE2.

2.4. In vitro MAO-B inhibition kinetic studies and evaluation of reversibility
To get insights into MAO-B inhibition reaction by compounds 1p and 1y, a kinetic study and reversibility testing were conducted. As illustrated in , Michaelis–Menten graphs were constructed using different concentrations of compounds 1p and 1y employing different concentrations benzylamine as a substrate. Calculation of the reaction kinetics showed that compounds 1p and 1y are potent MAO-B inhibitors showing approximately similar Vmax, Km, and Ki values (6.89e+7 versus 6.92e+7, 144.1 versus 139.3 µM, and 36.5 versus 29.1 nM for Vmax, Km, and Ki of compounds 1p versus 1y, respectively). Establishing Lineweaver–Burk plots indicated that both compounds 1p and 1y are competitive inhibitors. Reversibility testing () showed that washing restored the enzymatic activity in case of both of compounds 1p and 1y but not for selegiline (an irreversible MAO-B inhibitor) proving that both compounds are reversible MAO-B inhibitors.
Figure 4. In vitro kinetic study and reversibility testing for inhibition of MAO-B by compounds 1p and 1y: (A) Kinetics of inhibition of MAO-B by different concentrations of compound 1p employing different concentrations benzylamine as a substrate; (B) Kinetics of inhibition of MAO-B by different concentrations of compound 1y employing different concentrations benzylamine as a substrate; (C) Reversibility testing for inhibition of MAO-B by compounds 1p and 1y.

2.5. In-silico simulation study
To get insights into the molecular interactions of the discovered compounds potentially active selective compounds, in-silico calculation of the possible binding modes of compounds 1p and 1y with MAO-B and MAO-A was conducted. The substrate-binding site of MAO-A is around 550 Å3 while MAO-B has a 420 Å3 substrate-binding cavity connected to a 290 Å3 hydrophobic entrance cavity. Amino acid residues Ile199 and Tyr326 of MAO-B are gating residues that can open to connect the two site into a single elongate cavity as in safinamide-MAO-B complex (, PDB code: 2V5Z). In case of MAO-A, Ile335, and Phe208 are the gating residues which correspond to Tyr326 and Ile199 of MAO-B (, harmine-MAO-A complex, PDB code: 2Z5X). The differences in size, shape and gating amino acid residues of substrate/inhibitor binding pocket might be the basis of selectivity of some inhibitorsCitation69–71.
Figure 5. (A) Interactions network of safinamide within MAO-B pocket as found in the co-crystallised complex (PDB: 2V5Z); (B) Interactions network of harmine within MAO-A pocket as found in the co-crystallised complex (PDB: 2Z5X).

As illustrated in , in-silico calculations predicted that compound 1p well-fits in the substrate pocket of MAO-B showing an orientation that positions the 2,3,4-trimethoxybenzylidene moiety towards the aromatic cage formed of Tyr398 and Tyr435 in front of the flavin, while the coumaranone moiety extends into the remainder elongated hydrophobic pocket. Meanwhile compound 1y well-fits in the substrate pocket of MAO-B in flipped orientation where the coumaranone moiety was directed towards the aromatic cage formed of Tyr398 and Tyr435 in front of the flavin, while the 3-methoxybenzylidene moiety extended into the remainder elongated hydrophobic pocket (). Both compounds established important interactions with aromatic cage amino acids; Tyr398 and Tyr435 forming π–σ interaction through the 4′-methoxy group and π–π interaction through the benzylidene ring of compound 1p, and through π–σ interaction with the 6-methoxy group and π–π interactions through rings A and C of compound 1y. In addition, they formed favourable interaction with the gating amino acid residue Ile199 through π-alkyl interaction with rings A and C of compound 1p and the ring C of compound 1y. However, the docked hispidol did not show the important interactions with aromatic cage amino acids; Tyr398 and Tyr435 but showed interactions with the gating amino acid residue Ile199 (), which might explain its poor MAO-A inhibitory activity. The most potent compound 1p showed also further interactions including hydrogen bonding through the carbonyl moiety with the other gating amino acid residue Tyr326. Such interaction was not noticed for compound 1y. In addition, the calculated scores for docked poses showed slight favourable energy difference for compound 1p over compound 1y (−7.84287 versus − 7.79966, respectively). The predicted interaction network suggests important contribution of the methoxy groups in conjugation of rings A − C to the overall calculated binding mode.
Figure 6. (A) Predicted binding mode of compound 1p within MAO-B pocket (PDB: 2V5Z); (B) Predicted binding mode of compound 1y within MAO-B pocket (PDB: 2V5Z); (C) Predicted binding mode of hispidol within MAO-B pocket (PDB: 2V5Z).

As illustrated in , attempts to dock safinamide; a MAO-B selective drug, into MAO-A revealed that MAO-A pocket size is smaller to accommodate it and produced a docked pose showing bumping of safinamide into the boundary of MAO-A pocket. This might provide a basis for the known selectivity of safinamide for MAO-B over MAO-A. Meanwhile, hispidol upon docking showed a docking pose that is free from bumping interactions and, furthermore, established important interactions with the aromatic cage residues (), which explains it activity as potential MAO-A rather than MAO-B inhibitor. Similar to safinamide, in silico calculations showed that the size of compound 1p is larger than the substrate pocket size of MAO-A (). The calculation showed that the best pose of compound 1p within MAO-A adopted in similar orientation to that observed in case of MAO-B. Nevertheless, the smaller substrate pocket of MAO-A resulted in unfavourable bumping into the pocket’s boundary of MAO-A which was absent in case of MAO-B. This size limitation might explain the high selectivity of compounds 1p. On contrast, the size limit was less influential in case of compound 1y which possessed smaller size and, thus, showed better fitting (). However, only one π–σ interaction with the 3′-methoxy group and one π–π interaction through the benzylidene ring of compound 1y were predicted with the aromatic cage which is much less than the three π–π interactions through rings A and C of compound 1y and through π–σ interaction with the 6-methoxy group. In addition, the larger distance of 5.20 Å for the interaction with gating Phe208 of MAO-A compared with a distance of 4.83 Å for interaction with the corresponding gating Tyr326 of MAO-B might be an additional factor contributing to its decreased propensity to inhibit MAO-A.
Figure 7. (A) Predicted binding mode of safinamide within MAO-A pocket (PDB: 2Z5X): Yellow surface represents the solvent-accessible surface of safinamide; (B) Predicted binding mode of hispidol within MAO-A pocket (PDB: 2Z5X): Yellow surface represents the solvent-accessible surface of hispidol; (C) Predicted binding mode of compound 1p within MAO-A pocket (PDB: 2Z5X): Yellow surface represents the solvent-accessible surface of compounds 1p; (D) Predicted binding mode of compound 1y within MAO-A pocket (PDB: 2Z5X): Yellow surface represents the solvent-accessible surface of compounds 1y.

3. Conclusion
Neurological disorders including neurodegenerative and neuropsychiatric diseases are diverse complex diseases. Nevertheless, they share commonalities including anomalous monoaminergic pathways and neuroinflammation. To break the vicious circle of the disease, it might address multitargeted therapy instead of monotargeted therapy. In this regard, polypharmacological molecules might provide promising performance. Considering these points, this work was conducted in search for multifunctional molecules inhibiting MAOs and alleviating neuroinflammation. Hispidol, which is an aurone natural product reported to inhibit MAO-A with low selectivity was selected as a starting point to prepare a library of 25 analogues bearing different substitution patterns at ring-B. While previously reported aurones bearing only hydroxy/methoxy functions showed low selectivity for MAO inhibition, several members among the evaluated library members were identified as highly selective MAO-B inhibitors. As promising agents for treatment of neurodegenerative diseases should not impair cells viability, the most promising molecules were evaluated for their impact on viability of CNS microglia which presented them as safe compounds. Evaluation of the most promising compounds for anti-neuroinflammatory effects presented compound 1p as a promising compound selectively inhibit MAO-B and downregulating neuroinflammation. Study of kinetics of the enzymatic inhibition reaction coupled with reversibility testing proved that compounds 1p and 1y are competitive reversible MAO-B inhibitors. Collectively, this report reveals compound 1p as a promising multifunctional lead molecule for further development into possible anti-neurodegenerative therapeutics.
4. Experimental
4.1. Chemistry
General: All solvents and reagents have been purchased from commercial suppliers and used without any further purification. NMR spectra were acquired on Bruker Avance 400 spectrometer (400 MHz) or JEOL JNM-ECZ500R spectrometer (500 MHz). 1H NMR spectra were referenced to tetramethylsilane (δ = 0.00 ppm) as an internal standard. High-resolution mass spectra (HRMS) were recorded on Jeol AccuTOF (JMS-T100TD) equipped with a DART (direct analysis in real time) ion source from ionsense, Tokyo, Japan in the positive modes. TLC was carried out using glass sheets pre-coated with silica gel 60F254 purchased by Merk and spots were visualised under UV lamp or using staining solutions, such as p-anisaldehyde solution, ninhydrin solution. 6-Methoxy-3-coumaranone and compounds 1a, 1b, 1d, 1e, 1f, 1h–1t, and v–1y were in agreement with reported literature (Supplementary materials)Citation43,Citation49,Citation53–58.
4.1.1. General method for acid-catalysed cross-aldol condensation to prepare hispidol analogues
HCl was added dropwise to a solution of the appropriate 3-coumaranone derivative in methanol or ethanol. After complete dissolution, the appropriate benzaldehyde derivative was added dropwise. The mixture was heated at 60 °C for the specified time. After completion of the reaction (TLC), the reaction mixture was diluted with water, filtered and the collected precipitate was collected by filtration and dried under reduced vacuum before purification of the obtained crude products using column chromatography.
4.1.2. General method for base-catalysed cross-aldol condensation to prepare hispidol analogues
KOH was added dropwise to a solution of the appropriate 3-coumaranone derivative in methanol or ethanol. After complete dissolution, the appropriate benzaldehyde derivative was added dropwise. The mixture was heated at 60 °C for the specified time. After completion of the reaction (TLC), the reaction was quenched by water, evaporated under reduced pressure and the obtained residue was partitioned between water and ethyl acetate, extracted with ethyl acetate, dried over MgSO4, evaporated under reduced pressure, and purified using column chromatography.
4.1.2.1. (Z)-2–(2,3-Dihydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one (1c)
Compound 1c was obtained according to general procedure 4.1.2 using 6-hydroxy-3-coumaranone (300.6 mg, 2.0 mmol), methanol (30 ml), KOH (50%, 3 ml) and 2,3-dihydroxybenzaldehyde (276.2 mg, 2.0 mmol). Reaction time was 6 h and the crude product was purified by column chromatography (silica gel, EtOAc/n-hexane = 1:3) to afford the above titled compound 1c (80.7 mg, 19% yield).
1H NMR (500 MHz, MeOD-d4): δ 7.67 (d, J = 1.4 Hz, 1H), 7.66 (d, J = 1.4 Hz, 1H), 7.60 (d, J = 1.1 Hz, 1H), 7.59 (d, J = 1.2 Hz, 1H), 7.33 (s, 1H), 6.80 (dd, J = 8.0, 1.7 Hz, 2H'), 6.74 (t, J = 8.0 Hz, 2H), 6.68 (s, 1H), 6.66 (d, J = 1.7 Hz, 1H); 13 C NMR (125 MHz, MeOD-d4): δ 183.4, 168.7, 167.1, 147.5, 146.2, 145.1, 125.6, 122.0, 119.5, 119.2, 116.3, 113.5, 112.7, 106.9, 98.1; HRMS calcd for C15H11O5 [M + H]+ 271.0607, found 271.0601.
4.1.2.2. (Z)-2–(3,5-Dihydroxybenzylidene)-6-hydroxybenzofuran-3(2H)-one (1g)
Compound 1 g was obtained according to general procedure 4.1.1 using 6-hydroxy-3-coumaranone (144.8 mg, 1.0 mmol), ethanol (30 ml), HCl (12 N, 3 ml) and 3,5-dihydroxybenzaldehyde (133.3 mg, 1.0 mmol). Reaction time was 6 h and it was purified by column chromatography (silica gel, EtOAc/n-hexane = 1:2) to afford the above titled compound 1 g (25.3 mg, 7% yield).
1H NMR (500 MHz, DMSO-d6): δ 11.2 (brs, 1H), 9.47 (s, 2H), 7.59 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 2.0 Hz, 2H), 6.68 (m, 2H), 6.52 (s, 1H), 6.27 (t, J = 2.2 Hz, 1H'); 13 C NMR (125 MHz, DMSO-d6): δ 182.0, 168.4, 167.1, 159.1, 147.7, 133.8, 126.6, 113.6, 113.3, 111.6, 109.8, 105.0, 98.9; HRMS calcd for C15H11O5 [M + H]+ 271.0607, found 271.0623.
4.1.2.3. (Z)-2–(2,3-Dihydroxybenzylidene)-6-methoxybenzofuran-3(2H)-one (1u)
Compound 1u was obtained according to general procedure 4.1.1 using 6-methoxy-3-coumaranone (328.9 mg, 2.0 mmol), ethanol (10 ml), HCl (12 N, 3 ml) and 2,3-dihydroxybenzaldehyde (277.7 mg, 2.0 mmol). Reaction time was 8 h and the crude product was purified by column chromatography (silica gel, EtOAc/n-hexane = 1:2) to afford the above titled compound 1u (147.1 mg, 26% yield).
1H NMR (500 MHz, DMSO-d6): δ 7.64 (d, J = 8.6 Hz, 1H), 7.57 (dd, J = 8.0, 1.5 Hz, 1H), 7.14 (s, 1H), 7.11 (d, J = 2.2 Hz, 1H), 6.82–6.79 (m, 2H), 6.71 (t, J = 7.9 Hz, 1H), 3.88 (s, 3H), 3.12 (s, 1H), 2.49 (s, 1H); 13 C NMR (125 MHz, DMSO-d6): δ 182.1, 168.3, 167.7, 147.2, 146.8, 146.1, 125.9, 121.8, 119.9, 119.8, 117.4, 114.6, 113.1, 106.5, 97.7, 40.9; HRMS calcd for C16H13O5 [M + H]+ 285.0763, found 285.0764.
4.2. Biological evaluations
4.2.1. In vitro assay of human monoamine oxidases inhibition
The inhibitory activities of the compounds against hMAO-A and hMAO-B enzymes were evaluated referring to a previously described method (Supporting information)Citation49.
4.2.2. In vitro evaluation of cell viability
The impact of the compounds on the viability of cells was performed by the well-known WST-1 assay method as described in the Supporting information.
4.2.3. In vitro evaluation of inhibition of LPS-induced BV2 microglial cells production of nitric oxide and PGE2
The abilities of compounds to inhibit the induced production of NO and PGE2 were assessed by well-known protocol as indicated in the Supporting information.
4.2.4. In vitro MAO-B inhibition kinetic studies and evaluation of reversibility
Kinetics of the inhibition of MAO-B and testing of reversibility were conducted according to literature-reported protocolsCitation59 as indicated in the Supporting information.
4.3. Molecular modelling study
Molecular docking was conducted using MOE employing MAO-B crystal structure (PDB: 2V5Z) and MAO-A crystal structure (PDB: 2Z5X) similar to literature reported protocolCitation72.
Supplemental Material
Download PDF (835 KB)Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- ASA. Alzheimer’s disease facts and figures. Alzheimer’s Dement 2020;16:391–460.
- Lüscher Dias T, Schuch V, Beltrão-Braga PCB, et al. Drug repositioning for psychiatric and neurological disorders through a network medicine approach. Transl Psychiatry 2020;10:141.
- Yang W, Hamilton JL, Kopil C, et al. Current and projected future economic burden of Parkinson’s disease in the US. NPJ Parkinsons Dis 2020;6:15.
- Farag AK, Hassan AHE, Jeong H, et al. First-in-class DAPK1/CSF1R dual inhibitors: Discovery of 3,5-dimethoxy-N-(4-(4-methoxyphenoxy)-2-((6-morpholinopyridin-3-yl)amino)pyrimidin-5-yl)benzamide as a potential anti-tauopathies agent. Eur J Med Chem 2019;162:161–75.
- Gilhus NE, Deuschl G. Neuroinflammation - a common thread in neurological disorders. Nat Rev Neurol 2019;15:429–30.
- Brambilla R. Neuroinflammation, the thread connecting neurological disease: cluster: “neuroinflammatory mechanisms in neurodegenerative disorders.” Acta Neuropathol 2019;137:689–91.
- Skaper SD, Facci L, Zusso M, Giusti P. An inflammation-centric view of neurological disease: beyond the neuron. Front Cell Neurosci 2018;12:72.
- Thibaut F. Psychiatric disorders: neurodevelopmental disorders, neurodegenerative disorders, or both? Dialogues Clin Neurosci 2018;20:251–2.
- Xie A, Gao J, Xu L, Meng D. Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed Res Int 2014;2014:648740.
- Morgese MG, Trabace L. Monoaminergic System Modulation in Depression and Alzheimer's Disease: A New Standpoint? Front Pharmacol 2019;10:483–483.
- Šimić G, Babić Leko M, Wray S, et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog Neurobiol 2017;151:101–38.
- Fišar Z. Drugs related to monoamine oxidase activity. Prog Neuropsychopharmacol Biol Psychiatry 2016;69:112–24.
- Kumar MJ, Nicholls DG, Andersen JK. Oxidative alpha-ketoglutarate dehydrogenase inhibition via subtle elevations in monoamine oxidase B levels results in loss of spare respiratory capacity: implications for Parkinson's disease. J Biol Chem 2003;278:46432–9.
- Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 2013;3:461–91.
- Ramsay RR, Tipton KF. Assessment of enzyme inhibition: a review with examples from the development of monoamine oxidase and cholinesterase inhibitory drugs. Molecules 2017;22:1192.
- Park JH, Ju YH, Choi JW, et al. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci Adv 2019;5:eaav0316
- Dorothée G. Neuroinflammation in neurodegeneration: role in pathophysiology, therapeutic opportunities and clinical perspectives. J Neural Transm (Vienna) 2018;125:749–50.
- Kim N, Yoo HS, Ju YJ, et al. Synthetic 3',4'-dihydroxyflavone exerts anti-neuroinflammatory effects in BV2 microglia and a mouse model. Biomol Ther (Seoul) 2018;26:210–17.
- Yan A, Liu Z, Song L, et al. Idebenone alleviates neuroinflammation and modulates microglial polarization in LPS-stimulated BV2 cells and MPTP-induced Parkinson’s disease mice. Front Cell Neurosci 2018;12:529–9.
- R, Gordon TM, Woodruff Chapter 3 - Neuroinflammation as a therapeutic target in neurodegenerative diseases. In: Baekelandt V, Lobbestael E, eds. Disease-modifying targets in neurodegenerative disorders. Cambridge (MA): Academic Press; 2017:49–80.
- Dunn GA, Loftis JM, Sullivan EL. Neuroinflammation in psychiatric disorders: an introductory primer. Pharmacol Biochem Behav 2020;196:172981.
- Almeida PGC, Nani JV, Oses JP, et al. Neuroinflammation and glial cell activation in mental disorders. Brain Behav Immun 2020;2:100034.
- Mattei D, Notter T. Basic concept of microglia biology and neuroinflammation in relation to psychiatry. Curr Top Behav Neurosci 2020;44:9–34.
- Radtke FA, Chapman G, Hall J, Syed YA. Modulating neuroinflammation to treat neuropsychiatric disorders. Biomed Res Int 2017;2017:5071786.
- Echeverria V, Grizzell JA, Barreto GE. Neuroinflammation: a therapeutic target of cotinine for the treatment of psychiatric disorders? Curr Pharm Des 2016;22:1324–33.
- Gao HM, Hong JS. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol 2008;29:357–65.
- Wattenberg MM, Beatty GL. Overcoming immunotherapeutic resistance by targeting the cancer inflammation cycle. Semin Cancer Biol 2020;65:38–50.
- Savelieff MG, Nam G, Kang J, et al. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chem Rev 2019;119:1221–322.
- Van der Schyf CJ, Geldenhuys WJ, Youdim MB. Multifunctional drugs with different CNS targets for neuropsychiatric disorders. J Neurochem 2006;99:1033–48.
- Wong EH, Yocca F, Smith MA, Lee CM. Challenges and opportunities for drug discovery in psychiatric disorders: the drug hunters’ perspective. Int J Neuropsychopharmacol 2010;13:1269–84.
- Milelli A, Turrini E, Catanzaro E, et al. Perspectives in designing multifunctional molecules in antipsychotic drug discovery, drug. Drug Dev Res 2016;77:437–43.
- Alam MM, Hassan AHE, Kwon YH, et al. Design, synthesis and evaluation of alkylphosphocholine-gefitinib conjugates as multitarget anticancer agents. Arch Pharm Res 2018;41:35–45.
- Alam MM, Hassan AHE, Lee KW, et al. Design, synthesis and cytotoxicity of chimeric erlotinib-alkylphospholipid hybrids. Bioorg Chem 2019;84:51–62.
- Würth R, Thellung S, Bajetto A, et al. Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds. Drug Discov Today 2016;21:190–9.
- Crisan L, Istrate D, Bora A, Pacureanu L. Virtual screening and drug repurposing experiments to identify potential novel selective MAO-B inhibitors for Parkinson’s disease treatment. Mol Divers 2021;25:1775–94.
- Hassan AHE, Yoo SY, Lee KW, et al. Repurposing mosloflavone/5,6,7-trimethoxyflavone-resveratrol hybrids: discovery of novel p38-α MAPK inhibitors as potent interceptors of macrophage-dependent production of proinflammatory mediators. Eur J Med Chem 2019;180:253–67.
- Farag AK, Hassan AHE, Chung KS, et al. Diarylurea derivatives comprising 2,4-diarylpyrimidines: discovery of novel potential anticancer agents via combined failed-ligands repurposing and molecular hybridization approaches. Bioorg Chem 2020;103:104121.
- Farag AK, Hassan AHE, Ahn BS, et al. Reprofiling of pyrimidine-based DAPK1/CSF1R dual inhibitors: identification of 2,5-diamino-4-pyrimidinol derivatives as novel potential anticancer lead compounds. J Enzyme Inhib Med Chem 2020;35:311–24.
- Carradori S, D’Ascenzio M, Chimenti P, et al. Selective MAO-B inhibitors: a lesson from natural products. Mol Divers 2014;18:219–43.
- Hassan AHE, Choi E, Yoon YM, et al. Natural products hybrids: 3,5,4'-Trimethoxystilbene-5,6,7-trimethoxyflavone chimeric analogs as potential cytotoxic agents against diverse human cancer cells. Eur J Med Chem 2019;161:559–80.
- Shen B. A new golden age of natural products drug discovery. Cell 2015;163:1297–300.
- Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov 2015;14:111–29.
- Baek SC, Lee HW, Ryu HW, et al. Selective inhibition of monoamine oxidase A by hispidol. Bioorg Med Chem Lett 2018;28:584–88.
- Oh JM, Lee HS, Baek SC, et al. Antidepressant-like activities of hispidol and decursin in mice and analysis of neurotransmitter monoamines. Neurochem Res 2020;45:1930–40.
- Geldenhuys WJ, Funk MO, Van der Schyf CJ, Carroll RT. A scaffold hopping approach to identify novel monoamine oxidase B inhibitors. Bioorg Med Chem Lett 2012;22:1380–3.
- Morales-Camilo N, Salas CO, Sanhueza C, et al. Synthesis, biological evaluation, and molecular simulation of chalcones and aurones as selective MAO-B inhibitors. Chem Biol Drug Des 2015;85:685–95.
- Badavath VN, Nath C, Ganta NM, et al. Design, synthesis and MAO inhibitory activity of 2-(arylmethylidene)-2,3-dihydro-1-benzofuran-3-one derivatives. Chin Chem Lett 2017;28:1528–32.
- Pennington LD, Aquila BM, Choi Y, et al. Positional analogue scanning: an effective strategy for multiparameter optimization in drug design. J Med Chem 2020;63:8956–76.
- Shin SY, Shin MC, Shin JS, et al. Synthesis of aurones and their inhibitory effects on nitric oxide and PGE2 productions in LPS-induced RAW 264.7 cells. Bioorg Med Chem Lett 2011;21:4520–3.
- Seo JM, Hassan AHE, Lee YS. An expeditious entry to rare tetrahydroimidazo[1,5-c]pyrrolo[1,2-a]pyrimidin-7(8H)-ones: a single-step gateway synthesis of glochidine congeners. Tetrahedron 2019;75:130760.
- Gaich T, Baran PS. Aiming for the Ideal Synthesis. J Org Chem 2010;75:4657–73.
- Jo H, Hassan AHE, Jung SY, et al. Construction of 8-Azabicyclo[3.2.1]octanes via sequential DDQ-mediated oxidative Mannich reactions of N-Aryl pyrrolidines. Org Lett 2018;20:1175–8.
- Łączkowski KZ, Pakulski MM, Krzemiński MP, et al. Asymmetric synthesis of N-substituted N-hydroxyureas. Tetrahedron Asymmetr 2008;19:788–95.
- Lee YH, Shin MC, Yun YD, et al. Synthesis of aminoalkyl-substituted aurone derivatives as acetylcholinesterase inhibitors. Bioorg Med Chem 2015;23:231–40.
- Siah M, Farzaei MH, Ashrafi-Kooshk MR, et al. Inhibition of guinea pig aldehyde oxidase activity by different flavonoid compounds: an in vitro study. Bioorg Chem 2016;64:74–84.
- Liew KF, Chan KL, Lee CY. Blood-brain barrier permeable anticholinesterase aurones: synthesis, structure-activity relationship, and drug-like properties. Eur J Med Chem 2015;94:195–210.
- Cheng H, Zhang L, Liu Y, et al. Design, synthesis and discovery of 5-hydroxyaurone derivatives as growth inhibitors against HUVEC and some cancer cell lines. Eur J Med Chem 2010;45:5950–7.
- Lee CY, Chew EH, Go ML. Functionalized aurones as inducers of NAD(P)H:quinone oxidoreductase 1 that activate AhR/XRE and Nrf2/ARE signaling pathways: Synthesis, evaluation and SAR. Eur J Med Chem 2010;45:2957–71.
- Choi JW, Jang BK, Cho NC, et al. Synthesis of a series of unsaturated ketone derivatives as selective and reversible monoamine oxidase inhibitors. Bioorg Med Chem 2015;23:6486–96.
- Guglielmi P, Secci D, Petzer A, et al. Benzo[b]tiophen-3-ol derivatives as effective inhibitors of human monoamine oxidase: design, synthesis, and biological activity. J Enzyme Inhib Med Chem 2019;34:1511–25.
- York EM, Bernier L-P, MacVicar BA. Microglial modulation of neuronal activity in the healthy brain. Dev Neurobiol 2018;78:593–603.
- Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 2018;18:225–42.
- Angelova DM, Brown DR. Microglia and the aging brain: are senescent microglia the key to neurodegeneration? J Neurochem 2019;151:676–88.
- Henn A, Lund S, Hedtjärn M, et al. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 2009;26:83–94.
- Adachi K, Kakigawa K, Tazuke Y, Tsukada Y. Development of an automatic assay method for sulfated bile acids in urine using water-soluble tetrazolium WST-1. Japanese J Clin Chem 1997;26:95–100.
- Yuste JE, Tarragon E, Campuzano CM, Ros-Bernal F. Implications of glial nitric oxide in neurodegenerative diseases. Front Cell Neurosci 2015;9:322.
- Ghasemi M, Fatemi A. Pathologic role of glial nitric oxide in adult and pediatric neuroinflammatory diseases. Neurosci Biobehav Rev 2014;45:168–82.
- Anna LB, Klaus LL. Cyclooxygenase and Neuroinflammation in Parkinsons Disease Neurodegeneration. Curr Neuropharmacol 2010;8:62–8.
- Binda C, Newton-Vinson P, Hubalek F, et al. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol 2002;9:22–6.
- Gaweska H, Fitzpatrick PF. Structures and mechanism of the monoamine oxidase family. Biomol Concepts 2011;2:365–77.
- Son SY, Ma J, Kondou Y, et al. Structure of human monoamine oxidase A at 2.2-A resolution: the control of opening the entry for substrates/inhibitors. Proc Natl Acad Sci USA 2008; 105: 5739–44.
- Khattab SN, Haiba NS, Asal AM, et al. Synthesis and evaluation of quinazoline amino acid derivatives as mono amine oxidase (MAO) inhibitors. Bioorg Med Chem 2015;23:3574–85.