
Abstract
Inhibiting Cyclin-dependent kinase 2 (CDK2) has been established as a therapeutic strategy for the treatment of many cancers. Accordingly, this study aimed at developing a new set of quinazolinone-based derivatives as CDK2 inhibitors. The new compounds were evaluated for their anticancer activity against sixty tumour cell lines. Compounds 5c and 8a showed excellent growth inhibition against the melanoma cell line MDA-MB-435 with GI% of 94.53 and 94.15, respectively. Cell cycle analysis showed that compound 5c led to cell cycle cessation at S phase and G2/M phase revealing that CDK2 could be the plausible biological target. Thus, the most cytotoxic candidates 5c and 8a were evaluated in vitro for their CDK2 inhibitory activity and were able to display significant inhibitory action. The molecular docking study confirmed the obtained results. ADME study predicted that 5c had appropriate drug-likeness properties. These findings highlight a rationale for further development and optimisation of novel CDK2 inhibitors.
Graphical Abstract
1. Introduction
Cancer prevalence and mortality are promptly increasing worldwide. The growing prominence of cancer; particularly with fast population growth and ageing in many parts of the world partially reveals a marked decline in mortality rates caused by coronary heart disease and stroke, with respect to cancer. Accordingly, cancer rapidly emerges to be the principal cause of death worldwideCitation1.
Cyclin-dependent kinases (Cdks) are serine/threonine kinases. The mammalian cells contain 13 CDKs, among them CDK1, CDK2, CDK3, CDK4, and CDK6 which are involved in regulating the cell cycle checkpoints via initiation of proteinaceous substrates phosphorylation, using adenosine triphosphate (ATP)Citation2,Citation3. Abnormalities in the cell cycle regulation induce unrestricted cell growth, which is usually considered a hallmark of cancerCitation4. Disruption of a deregulated cell cycle, as a result of overactivation of a CDK, has been accepted as a strategy for the treatment of proliferative diseases, especially cancerCitation5.
Cyclin-dependent kinase 2 (CDK2) is a key regulator of the cell cycle transition and progression. CDK2 performs a critical role in controlling various events of the cell division cycle, such as DNA repair, gene transcription, G1-S transition, and modulation of G2 progressionCitation6–8. Additionally, the altered expression of cyclins whose association with CDKs is essential for their catalytic activity can drive aberrant proliferation in cancer. In the same vein, the overexpression of CDK2 regulatory subunits cyclin A and/or E is a key oncogenic process in many types of cancersCitation9,Citation10. Owing to the crucial role of CDK2 in cell cycle regulation, transcription activity, and tumours epigenetic modifications, CDK2 inhibition may confer a therapeutic benefit against certain cancers viz breast cancerCitation11, colo-rectal cancerCitation12,Citation13, glioblastomaCitation14–16, and melanomaCitation17,Citation18.
Several potent CDK2 inhibitors are under clinical evaluation, such as dinaciclib (I)Citation19 (an inhibitor of several CDKs), milciclib (II)Citation20, and roniciclib (III)Citation21 (). Nevertheless, some adverse effects have been observed during clinical trials, revealing that the discovery of new CDK2 inhibitors is still an active field of researchCitation22.
Figure 1. Reported CDK2 inhibitors and structures of the proposed compounds.

Surveying literature revealed that different scaffolds can possess CDK2 inhibitory activity, such as pyrazolopyrimidines (e.g. dinaciclib I), purines [e.g. fadraciclib IV and roscovitine (seliciclib) V]Citation23,Citation24 and quinazolines (e.g. compounds VI and VII)Citation25,Citation26 (). Almost all of these are ATP- competitive inhibitors, interacting with Leu83 residue at the catalytic ATP binding site of the enzyme. Moreover, these CDK2 inhibitors share similar properties, such as hydrophobicity, low molecular weight, and heterocyclic flat structure. Interestingly, quinazolin-4(3H)-one is a privileged basic structure having promising anticancer activity with high human toleranceCitation27–29.
In the course of finding several chemical substances which may serve as leads for developing new anticancer agents, we are particularly interested in the present study with quinazolinone derivatives in an attempt to achieve novel CDK2 inhibitors serving as chemotherapeutic agents. Continuing our studies on quinazolinone analogues as attractive candidates as antitumor agentsCitation30,Citation31, we have designed some new quinazolinone derivatives containing phenoxy methyl group at position 2 and bearing different moieties which are known to enhance the antitumor activity attached to the 3-amino group viz un/substituted phenyl acetamidesCitation32, un/substituted benzene sulfonamidesCitation33, benzylidenesCitation34, and acetyl piperazinesCitation35 (). In this work, the substitution pattern at the 2 and 3 positions of the quinazolinone scaffold was selected to provide a different electronic environment that would affect the lipophilicity, and hence the activity of the target molecules. The aim of constructing these congeners is an attempt to attain active CDK2 inhibitors with potentiated activity against cancerous cells in which CDK2 is overexpressed.
All the newly synthesised quinazolinones were subjected to in vitro anticancer screening by the National Cancer Institute (USA) against 60 different human cell lines. The most potent compounds 5c and 8a were selected to be further studied through the determination of their half-maximal inhibitory concentration (IC50) values against melanoma MDA-MB-435 (5c and 8a) and glioblastoma SNB-75 cell lines (5c). To investigate the mechanistic pathways of the anticancer activity of the most active representatives of this class, they were subjected to in vitro CDK2 inhibition assay. Furthermore, the effect of 5c on the induction of apoptosis in MDA-MB-435 and SNB-75 cell lines was also investigated using flow cytometry and molecular docking study was carried out for the most active compound 5c to identify the binding features required for its antitumor activity.
2. Experimental section
2.1. Chemistry
2.1.1. General
Starting materials and solvents were purchased from commercial sources and were used without further purification. Melting points were determined by open capillary tube method using Electrothermal 9100 melting point apparatus (Staffordshire, UK) and were uncorrected. Elemental analyses and Mass spectra were carried out at the Regional Centre for Mycology and Biotechnology, Al-Azhar University, Egypt. Mass spectral data were given as m/z (relative abundance %). NMR and Infra-red spectra were performed at the Microanalytical Unit-Faculty of Pharmacy, Cairo University. NMR spectra were recorded on a Bruker Ascend 400/R (1H: 400, 13C: 100 MHz) spectrometer and were run in deuterated chloroform (CDCl3) or deuterated dimethylsulphoxide (DMSO‑d6). Chemical shift values (δ) were given in parts per million (ppm). Coupling constants were given in hertz (Hz) and spin multiplicities were given as s (singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet), or m (multiplet). Infra-red spectra (IR) were recorded as Potassium bromide discs on Schimadzu FT-IR 8400S spectrophotometer (Kyoto, Japan) and expressed in wavenumber (υmax, cm−1). TLC was performed on TLC plates UV fluorescent silica gel (60 FZSU), using chloroform: methanol 9:1 as the eluting system, and the spots were visualised by the ultraviolet lamp. The starting compounds 1Citation36, 2Citation37,Citation38, and the key intermediates 3Citation37,Citation38 and 7Citation37,Citation38 were prepared as reported.
2.1.2. General procedure for the synthesis of compounds 4a–c and 5a–c
A mixture of equimolar amounts of the amino derivative 3 and the appropriate 2-chloro-N-phenylacetamide or p-substituted benzenesulfonyl chloride in dry DMF (10 mL) containing anhydrous potassium carbonate (1.5 mmol, 0.2 g) were refluxed for 20 h. The mixture was poured over crushed ice and the precipitated solid was filtered off, washed with water, dried, and recrystallized from the suitable solvent.
2.1.2.1. 2-((4-oxo-2-(phenoxymethyl)quinazolin-3(4H)-yl)amino)-N-phenylacetamide 4a
Yield: 80%, m.p.: 135–137 °C, I.R (KBr, cm−1): υmax 3271 (2NH), 3136 (CH aromatic), 2947 (CH aliphatic), 1678 (2C = O), 1600 (C = N), 1550 (C = C). 1H-NMR (400 MHz, CDCl3): δ 5.30 (s, 2H, CH2), 5.35 (s, 2H, OCH2), 7.03–7.10 (m, 5H, phenyl-H), 7.29–7.36 (m, 5H, phenyl-H), 7.48–7.55 (m, 3H, 1H phenyl-H + 2H NH exchanged by D2O), 7.80 (d, 2H, J = 4.00 Hz, phenyl-H), 8.32 (d, 1H, J = 8.00 Hz, phenyl-H). 13C-NMR: δ 47.7 (CH2 carbon), 53.4 (OCH2 carbon), 114.9, 119.8, 120.6, 121.9, 124.6, 126.5, 127.9, 128.9, 129.1, 129.7, 134.4, 146.5, 151.2 (Ar–Cs), 157.5 (C = N), 158.0 (C–O), 160.9 (C = O), 165.3 (C = O). MS m/z (%): 402.59 (M+ +2, 17.61%), 400.27 (M+, 20.49%), 370.95 (100%). Anal. Calcd. for C23H20N4O3 (400.44): C, 68.99; H, 5.03; N, 13.99. Found: C, 69.16; H, 5.21; N, 14,23.
2.1.2.2. N-(4-methoxyphenyl)-2-((4-oxo-2-(phenoxymethyl)quinazolin-3(4H)-yl)amino)acetamide 4b
Yield: 90%, m.p.: 146–148 °C, I.R (KBr, cm−1): υmax 3448, 3309 (2NH), 3062 (CH aromatic), 2931 (CH aliphatic), 1681 (2C = O), 1604 (C = N), 1500 (C = C). 1H-NMR (400 MHz, DMSO-d6): δ 3.35 (s, 3H, OCH3), 5.35 (s, 2H, CH2), 5.74 (s, 2H, OCH2), 6.91 (d, 1H, J = 8.80 Hz, phenyl-H), 6.96 (t, 1H, J = 7.32 Hz, phenyl-H), 7.05 (d, 2H, J = 8.00 Hz, phenyl-H), 7.31 (t, 2H, J = 7.32 Hz, phenyl-H), 7.53–7.58 (m, 3H, phenyl-H), 7.63 (d, 2H, J = 8.16 Hz, phenyl-H), 7.81 (t, 1H, J = 7.40 Hz, phenyl-H), 8.17 (d, 1H, J = 7.76 Hz, phenyl-H), 9.96 (s, 2H, NH exchanged by D2O). MS m/z (%): 432.08 (M+ +2, 31.71%), 430.82 (M+, 44.07%), 415.76 (100%). Anal. Calcd. for C24H22N4O4 (430.46): C, 66.97; H, 5.15; N, 13.02. Found: C, 67.13; H, 5.41; N, 13.30.
2.1.2.3. N-(4-chlorophenyl)-2-((4-oxo-2-(phenoxymethyl)quinazolin-3(4H)-yl)amino)acetamide 4c
Yield: 80%, m.p.: 165–167 °C, I.R (KBr, cm−1): υmax 3305 (2NH), 3062 (CH aromatic), 2974 (CH aliphatic), 1681 (2C = O), 1600 (C = N), 1492 (C = C), 752 (C–Cl). 1H-NMR (400 MHz, CDCl3): δ 5.30 (s, 2H, CH2), 5.34 (s, 2H, OCH2), 7.02–7.08 (m, 4H, phenyl-H), 7.18 (d, 1H, J = 8.56 Hz, phenyl-H), 7.30–7.40 (m, 3H, phenyl-H), 7.39 (d, 1H, J = 8.64 Hz, phenyl-H), 7.51–7.55 (m, 2H, 1H phenyl-H + 1H NH exchanged by D2O) , 7.78 (d, 2H, J = 8.20 Hz, phenyl-H), 8.29 (d, 1H, J = 7.96 Hz, phenyl-H), 8.88 (s, 1H, NH exchanged by D2O). 13C-NMR: δ 47.7 (CH2 carbon), 68.4 (OCH2 carbon), 114.9, 120.6, 120.9, 121.9, 122.1, 126.5, 127.4, 127.9, 128.9, 129.7, 134.3, 146.5, 151.5 (Ar–Cs), 157.5 (C = N), 158.0 (C–O), 160.9 (C = O), 165.3 (C = O). MS m/z (%): 435.53 (M+ +1, 27.64%), 434.59 (M+, 48.64%), 177.17 (100%). Anal. Calcd. for C23H19ClN4O3 (434.88): C, 63.52; H, 4.40; N, 12.88. Found: C, 63.29; H, 4.62; N, 13.14.
2.1.2.4. N-(4-oxo-2-(phenoxymethyl)quinazolin-3(4H)-yl)benzenesulfonamide 5a
Yield: 70%, m.p.: 114–116 °C, I.R (KBr, cm−1): υmax 3309 (NH), 3062 (CH aromatic), 2978 (CH aliphatic), 1681 (C = O), 1600 (C = N), 1492 (C = C), 1296, 1153 (SO2). 1H-NMR (400 MHz, CDCl3): δ 5.37 (s, 2H, OCH2), 7.01 (t, 1H, J = 6.00 Hz, phenyl-H), 7.08 (d, 1H, J = 8 Hz, phenyl-H), 7.30 (t, 1H, J = 8.00 Hz, phenyl-H), 7.48–7.58 (m, 4H, 3H phenyl-H + 1H NH exchanged by D2O), 7.77 (t, 1H, J = 8.00 Hz, phenyl-H), 7.86 (d, 1H, J = 8.00 Hz, phenyl-H), 7.96 (d, 5H, J = 8.00 Hz, phenyl-H), 8.29 (d, 1H, J = 8.00 Hz, phenyl-H). 13C-NMR: δ 67.9 (OCH2 carbon), 114.9, 120.2, 121.9, 126.7, 127.1, 127.7, 129.3, 129.7, 133.2, 134.6, 141.6 (Ar–Cs), 153.5 (C = N), 157.8 (C–O), 160.5 (C = O). MS m/z (%): 407.75 (M+, 54.00%), 250.15 (100%). Anal. Calcd. for C21H17N3O4S (407.44): C, 61.91; H, 4.21; N, 10.31. Found: C, 61.74; H, 4.34; N, 10.52.
2.1.2.5. 4-methyl-N-(4-oxo-2-(phenoxymethyl)quinazolin-3(4H)-yl)benzenesulfonamide 5b
Yield: 75%, m.p.: 140–142 °C, I.R (KBr, cm−1): υmax 3309 (NH), 3059 (CH aromatic), 2978 (CH aliphatic), 1681 (C = O), 1604 (C = N), 1492 (C = C), 1303, 1153 (SO2). 1H-NMR (400 MHz, CDCl3): δ 2.38 (s, 3H, CH3), 5.28, 5.32 (2 s, 2H, OCH2), 7.01 (t, 1H, J = 6.00 Hz, phenyl-H), 7.08 (d, 2H, J = 8.00 Hz, phenyl-H), 7.28–7.33 (m, 5H, 4H phenyl-H + 1H NH exchanged by D2O) , 7.51 (d, 1H, J = 4.00 Hz, phenyl-H), 7.76–7.81 (m, 4H, phenyl-H), 8.29 (d, 1H, J = 8.00 Hz, phenyl-H). 13C-NMR: δ 21.5 (CH3 carbon), 68.5 (OCH2 carbon), 114.9, 121.9, 126.6, 126.7, 127.2, 127.8, 134.6, 139.1, 143.9, 146.5, 151.5 (Ar–Cs), 157.2 (C = N), 158.1 (C–O), 160.9 (C = O). MS m/z (%): 421.84 (M+, 24.26%), 203.28 (100%). Anal. Calcd. for C22H19N3O4S (421.47): C, 62.70; H, 4.54; N, 9.97. Found: C, 62.59; H, 4.62; N, 10.15.
2.1.2.6. 4-chloro-N-(4-oxo-2-(phenoxymethyl)quinazolin-3(4H)-yl)benzenesulfonamide 5c
Yield: 72%, m.p.: 208–210 °C, I.R (KBr, cm−1): υmax 3329 (NH), 3086 (CH aromatic), 2931 (CH aliphatic), 1681 (C = O), 1602 (C = N), 1496 (C = C), 1373, 1153 (SO2), 752 (C–Cl). 1H-NMR (400 MHz, DMSO-d6): δ 5.22 (s, 2H, OCH2), 6.94–7.09 (m, 4H, phenyl-H), 7.35 (t, 3H, J = 8.00 Hz, phenyl-H), 7.52–7.57 (m, 3H, 2H-phenyl-H + 1H NH exchanged by D2O), 7.82 (br. s, 3H, phenyl-H), 8.31 (d, 1H, J = 8.00 Hz, phenyl-H). 13C-NMR: δ 68.3 (OCH2 carbon), 115.1, 115.3, 121.8, 121.9, 126.4, 127.4, 127.6, 129.8, 130.0, 135.0, 148.6 (Ar–Cs), 153.1 (C = N), 158.3 (C–O), 162.0 (C = O). MS m/z (%): 441.09 (M+, 50.22%), 239.67 (100%). Anal. Calcd. for C21H16ClN3O4S (441.06): C, 57.08; H, 3.65; N, 9.51. Found: C, 57.24; H, 3.71; N, 9.72.
2.1.3. General procedure for the synthesis of compounds 6a and 6b
A mixture of equimolar amounts of compound 3 and the corresponding aromatic aldehyde was refluxed in glacial acetic acid (15 ml) for 8 h. The reaction mixture was poured over crushed ice and the precipitated solid was filtered off, washed with water, dried, and recrystallized from ethanol affording 6a and 6b, respectively.
2.1.3.1. 3-((4-hydroxy-3-methoxybenzylidene)amino)-2-(phenoxymethyl)quinazolin-4(3H)-one 6a
Yield: 83%, m.p.: 191–193 °C, I.R (KBr, cm−1): υmax 3300–3228 (OH), 3070 (CH aromatic), 2939 (CH aliphatic), 2839 (aldehydic CH), 1681 (C = O), 1600 (C = N), 1516 (C = C). 1H-NMR (400 MHz, CDCl3): δ 3.83 (s, 3H, OCH3), 5.35 (s, 2H, OCH2), 6.98–7.10 (m, 3H, phenyl-H), 7.31–7.44 (m, 4H, phenyl-H), 7.49–7.55 (m, 2H, phenyl-H), 7.62–7.64 (m, 1H, phenyl-H), 7.81 (s, 1H, phenyl-H), 8.87 (d, 1H, J = 4.80 Hz, phenyl-H), 9.85 (s, 1H, azomethine-H), 10.80 (s, 1H, OH exchanged by D2O). 13C-NMR: δ 56.1 (CH3O carbon), 68.4 (OCH2 carbon), 108.8, 114.4, 114.9, 121.3, 121.9, 126.8, 127.2, 127.5, 128.2, 129.5, 129.7, 134.4, 134.9, 147.2 (Ar–Cs), 150.1 (C = N), 151.7 (C = N), 163.4 (C–O), 190.9 (C = O). MS m/z (%): 401.27 (M+, 20.76%), 328.41 (100%). Anal. Calcd. for C23H19N3O4 (401.14): C, 68.82; H, 4.77; N, 10.47. Found: C, 68.65; H, 4.89; N, 10.64.
2.1.3.2. 2-(phenoxymethyl)-3-((3-phenylallylidene)amino)quinazolin-4(3H)-one 6b
Yield: 85%, m.p.: 211–213 °C, I.R (KBr, cm−1): υmax 3059 (CH aromatic), 2974 (CH aliphatic), 2850 (aldehydic CH), 1681 (C = O), 1600 (C = N), 1492 (C = C). 1H-NMR (400 MHz, CDCl3): δ 5.32 (s, 2H, OCH2), 7.00 (d, 2H, J = 8.64 Hz, phenyl-H), 7.04 (d, 2H, J = 7.76 Hz, phenyl-H), 7.28–7.32 (m, 3H, phenyl-H), 7.41–7.48 (m, 4H, phenyl-H), 7.53–7.56 (m, 2H, phenyl-H), 7.82 (m, 1H, =C–H), 8.33 (d, 1H, J = 7.84 Hz, =C–H), 8.87 (d, 1H, J = 9.44 Hz, phenyl-H), 9.71 (s, 1H, azomethine-H). 13C-NMR: δ 67.5 (OCH2 carbon), 115.3, 121.6, 123.9, 127.9, 129.5, 130.3, 131.3, 134.5, 135.0, 145.9, 146.7, 151.1, 152.9, 158.2, 158.7, 168.9, 176.1 (Ar–Cs), 193.9 (C = O). MS m/z (%): 381.93 (M+, 17.15%), 302.67 (100%). Anal. Calcd. for C24H19N3O2 (381.44): C, 75.57; H, 5.02; N, 11.02. Found: C, 75.38; H, 5.14; N, 11.25.
2.1.4. General procedure for the synthesis of compounds 8a–c
Equimolar amounts of the chloro derivative 8 and the appropriate piperazine in dry DMF (10 ml) containing anhydrous potassium carbonate (1.5 mmol, 0.2 g), were refluxed for 20 h. The reaction mixture was cooled, poured over crushed ice and the yielded solid was crystallised from methanol to give the corresponding 8a–c.
2.1.4.1. 2–(4-methylpiperazin-1-yl)-N-(4-oxo-2-(phenoxymethyl)quinazolin-3(4H)-yl)acetamide 8a
Yield: 65%, m.p.: 158–160 °C, I.R (KBr, cm−1): υmax 3425 (NH), 3082 (CH aromatic), 2931 (CH aliphatic), 1681 (2C = O), 1620 (C = N), 1496 (C = C). 1H-NMR (400 MHz, DMSO-d6): 3.47–3.65 (m, 13H, 8H piperazinyl-H + 3H CH3 + 2H CH2), 5.03 (s, 2H, OCH2), 6.93–6.97 (m, 2H, phenyl-H), 7.23 (s, 2H, phenyl-H), 7.44–7.70 (m, 5H, 4H phenyl-H + 1H NH exchanged by D2O), 8.14 (s, 1H, phenyl-H). 13C-NMR: δ 44.2 (CH3 carbon), 45.0, 45.2 (piperazine-Cs), 45.5 (CH2 carbon), 72.4 (OCH2 carbon), 119.7, 126.5, 126.6, 131.0, 131.7, 134.4, 139.3 (Ar–Cs), 152.8 (C = N), 157.4 (C–O), 162.6 (C = O), 166.9 (C = O). MS m/z (%): 407.97 (M+, 67.35%), 386.95 (100%). Anal. Calcd. for C22H25N5O3 (407.47): C, 64.85; H, 6.18; N, 17.19. Found: C, 64.71; H, 6.27; N, 17.43.
2.1.4.2. N-(4-oxo-2-(phenoxymethyl)quinazolin-3(4H)-yl)-2–(4-phenylpiperazin-1-yl)acetamid 8b
Yield: 70%, m.p.: 115–117 °C, I.R (KBr, cm−1): υmax 3545 (NH), 3059 (CH aromatic), 2939 (CH aliphatic), 1720, 1693 (2C = O), 1600 (C = N), 1496 (C = C). 1H-NMR (400 MHz, CDCl3): δ 2.90 (s, 2H, CH2), 3.15–3.27 (m, 4H, piperazinyl-H), 3.29–3.38 (m, 4H, piperazinyl-H), 5.02 (d, 1H, J = 11.88 Hz, upfield H of OCH2), 5.15 (d, 1H, J = 11.88 Hz, downfield H of OCH2), 6.89–6.97 (m, 4H, phenyl-H), 7.03–7.07 (m, 2H, phenyl-H), 7.28–7.36 (m, 5H, 4H phenyl-H + 1H NH exchanged by D2O), 7.55 (t, 1H, J = 7.32 Hz, phenyl-H), 7.78–7.85 (m, 2H, phenyl-H), 8.29 (d, 1H, J = 7.64 Hz, phenyl-H). 13C-NMR: δ 48.1, 53.7 (piperazine-Cs), 60.8 (CH2 carbon), 68.2 (OCH2 carbon), 114.6, 116.2, 120.1, 121.9, 127.1, 127.9, 128.1, 129.2, 129.8, 135.1, 146.5, 150.9 (Ar–Cs), 152.1 (C = N), 158.0 (C–O), 159.7 (C = O), 170.2 (C = O). MS m/z (%): 469.48 (M+, 47.44%), 331.43 (100%). Anal. Calcd. for C27H27N5O3 (469.55): C, 69.07; H, 5.80; N, 14.92. Found: C, 68.95; H, 5.97; N, 15.11.
2.1.4.3. 2–(4-(4-methoxyphenyl)piperazin-1-yl)-N-(4-oxo-2-(phenoxymethyl)quinazolin-3(4H)-yl)acetamide 8c
Yield: 70%, m.p.: 100–102 °C, I.R (KBr, cm−1): υmax 3541 (NH), 3143 (CH aromatic), 2951 (CH aliphatic), 1720, 1693 (2C = O), 1598 (C = N), 1496 (C = C). 1H-NMR (400 MHz, CDCl3): δ 2.64 (s, 2H, piperazinyl-H), 2.90 (s, 2H, piperazinyl-H), 3.04 (s, 2H, piperazinyl-H), 3.33 (s, 2H, piperazinyl-H), 3.80 (s, 5H, 3H OCH3 + 2H CH2), 5.01 (d, 1H, J = 12.00 Hz, upfield H of OCH2), 5.14 (d, 1H, J = 12.00 Hz, downfield H of OCH2), 6.85–6.91 (m, 4H, phenyl-H), 7.02 (t, 3H, J = 8.00 Hz phenyl-H), 7.31 (t, 3H, J = 8.00 Hz, phenyl-H), 7.54 (t, 1H, J = 8.00 Hz, phenyl-H), 7.77–7.82 (m, 2H, 1H phenyl-H + 1H NH exchanged by D2O), 8.28 (d, 1H, J = 8.00 Hz, phenyl-H). 13C-NMR: δ 50.7, 53.8 (piperazine-Cs), 55.6 (OCH3 carbon), 60.8 (CH2 carbon), 68.2 (OCH2 carbon), 114.5, 114.6, 118.3, 121.9, 127.1, 127.9, 128.1, 129.8, 135.1, 145.2, 146.5 (Ar–Cs), 152.1 (C–O), 154.0 (C = N), 158.0 (C-O), 159.7 (C = O), 170.2 (C = O). MS m/z (%): 499.66 (M+, 38.98%), 324.50 (100%). Anal. Calcd. for C28H29N5O4 (499.57): C, 67.45; H, 6.09; N, 14.28. Found: C, 67.32; H, 5.85; N, 14.02.
2.2. Biological evaluation
2.2.1. Biological assays
All experimental procedures applied in the mentioned biological assays were conducted as reported earlier; NCI-60 human tumour cell lines screeningCitation39–41, determination of IC50 in MDA-MB-435 and SNB-75 cell linesCitation42,Citation43, Cell cycle analysisCitation44, Apoptosis determinationCitation45, CDK2 inhibition assayCitation46 and have been provided in the Supplementary Materials.
2.2.2. Statistical analysis
The presented data and results were confirmed by at least three independent experiments. The data have been expressed as means ± SD and analysed with GraphPad Prism 8.0 software. Statistical comparisons were made by Student’s t-test. p-Values <0.05 (p < 0.05) was considered statistically significant.
2.3. In silico studies
2.3.1. Molecular docking study
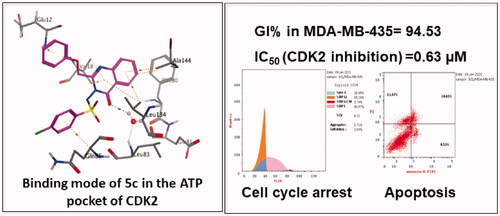
The molecular docking study of the most active compound was achieved using Molecular Operating Environment (MOE, 2010.10) software. The X-ray crystal structure of CDK2 enzyme co-crystallised with the quinazoline derivative DIN-234325 as an inhibitor (PDB code: 2B53) was downloaded from the protein data bankCitation25,Citation47. The enzyme was prepared for docking by removal of all ligands and water molecules that are not involved in the binding. One conserved water molecule involved in the co-crystalized ligand binding to the CDK2 binding site was kept (). The protein structure was prepared for docking study using Protonate 3 D protocol in MOE with default options. The Triangle Matcher placement method and London dG scoring function were used for the evaluation of the binding pattern and binding affinity of the ligands. Docking protocol was first validated by redocking of the co-crystallised ligand (DIN-234325) in the active site of the enzyme with an energy score (S) = −18.24 kcal/mol and root mean square deviation (RMSD) of 0.485 Å. The validated docking protocol was then used to study the ligand-target interactions in the active site for the most potent newly synthesised compounds 5c to predict its binding mode and binding affinity to justify its promising potency.
2.3.2. Drug likeness properties
The free Swiss ADME web tool available from the Swiss Institute of Bioinformatics (SIB) was used for the calculation of the physicochemical descriptors as well as to predict the ADME parameters, pharmacokinetic properties, and drug-like nature of the most active newly synthesised candidate 5cCitation48–50. The SMILES of the tested compound were inserted directly on the webpage followed by running the prediction process.
3. Results and discussion
3.1. Chemistry
The target quinazolin-4(3H)-one derivatives were synthesised as outlined in Schemes 1, 2. The starting compounds 1Citation36, 2Citation37,Citation38, and the key intermediates 3Citation37,Citation38 and 7Citation37,Citation38 were prepared according to the reported procedures. As depicted in Scheme 1, stirring of 3-amino-2-(phenoxymethyl)quinazolin-4(3H)-one 3 with the appropriate 2-chloro-N-phenylacetamide or various 4-substituted benzene sulphonyl chloride in dry Dimethyl formamide at 70 °C in the presence of anhydrous Potassium carbonate afforded compounds 4a–c and 5a–c, respectively. The chemical structures of the new phenyl acetamide derivatives 4a–c were confirmed by the appearance of an additional singlet signal in 1H-NMR spectra around 5.30 ppm assigned to the new methylene group. Furthermore, the 1H-NMR showed characteristic signals in the aromatic region attributed to the additional aromatic protons in compounds 4a–c and 5a–c. 13C NMR also illustrated the carbon skeleton of the target derivatives. Moreover, condensation of the key intermediate 3 with aromatic aldehydes, namely vanillin, and cinnamaldehyde in refluxing glacial acetic acid yielded the corresponding Schiff’s bases 6a,b. 1H NMR spectra of 6a,b illustrated the azomethine proton as a sharp singlet signal at 9.71–9.85 ppm. Additionally, acylation of 3 with chloroacetyl chloride in dry Dimethylformamide produced the chloroacetylamino derivative 7, the key intermediate in Scheme 2. The latter upon reaction with different substituted piperazines in dry Dimethylformamide in the presence of anhydrous Potassium carbonate experienced aromatic nucleophilic substitution reaction and yielded 8a–c. The postulated structures of the products were proved by the appearance of aliphatic signals at δ 3.04–3.48 ppm region ascribed to the piperazine protons. The 13C NMR spectra showed two signals at the range of 159.7–170.2 ppm corresponding to the two carbonyl carbons.
Scheme 1. Preparation of the key starting compound 3 and the target compounds 4a–c, 5a–c, and 6a,b. Reagents and solvents: (a) Methyl anthranilate, dry benzene, (b) Hydrazine hydrate, n-butanol, (c) appropriate 2-chloro-N-phenylacetamide, dry DMF, anhydrous K2CO3, (d) p-substituted benzenesulfonyl chloride, dry DMF, anhydrous K2CO3, (e) cinnamaldehyde or vanillin, glacial acetic acid.

Scheme 2. Synthesis of the target derivatives 8a–c. Reagents and solvents: (a) Chloroacetyl chloride, dry DMF, (b) appropriate piperazine, dry DMF, anhydrous K2CO3.

3.2. Biological evaluation
3.2.1. NCI-60 human tumour cell lines screening
In this research, all of the newly synthesised quinazolinones were subjected to in vitro anticancer screening by the National Cancer Institute (USA) against 60 different human cell lines including leukaemia, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, and breast cancerCitation51. The tested compounds were evaluated at a single dose (10−5 M). The growth inhibition percentages obtained from the single-dose test for the tested compounds are shown in . Screening the anticancer activity of the tested compounds revealed that compounds 5c and 8a were the most active compounds displaying superior activity against the melanoma cell line MDA-MB-435 with growth inhibition percentages 94.53 and 94.15, respectively. Concerning the phenyl acetamide derivatives 4a–c, Compound 4a possessing the unsubstituted phenyl ring showed good potency against glioblastoma cell line SNB-75 with growth inhibition percentages 62.34. Regarding the benzenesulfonamide counterparts, the introduction of an electron-withdrawing group on the benzenesulfonamide moiety increased the inhibitory activity on the glioblastoma cell line SNB-75 as depicted by the chloro derivative 5c (GI% = 63.09) which was higher than that of the unsubstituted derivative 5a (GI% = 40.06) and the methyl derivative 5b (GI% = 33.03). The chloro derivative 5c also displayed high to moderate cytotoxicity against various carcinoma cell lines viz breast cancer cell lines MDA-MB-468 and MCF-7, leukaemia cell lines K-562 and SR, and colon cancer cell lines HCT-15 and KM12 with growth inhibition percentages 59.61, 58.11, 52.75 and 42.42, 45.60 and 44.77, sequentially. It is worth mentioning that the quinazolinone 5c is not only the most active member among this series but also displayed the highest activity among all the target compounds. Interestingly, the piperazine derivative 8a showed potent to moderate cytotoxicity against leukaemia cell lines SR and K-562, breast cancer cell lines MCF-7 and MDA-MB-468 and colon cancer cell lines HCT-15, KM12, and SW-620 with growth inhibition percentages 69.55, 67.80, 57.64, and 49.06, 44.04, 55.03, and 42.80, respectively. Other tested compounds exhibited weak activity against most investigated cell lines. Thus, compounds 5c and 8a were subjected to further testing to determine their IC50 against the most sensitive cell lines.
Table 1. Growth inhibition percent obtained from the single dose (10−5 M) test for the target quinazolinones 4a–c and 5a–c.
Table 2. Growth inhibition percent obtained from the single dose (10−5 M) test for the target quinazolinones 6a,b and 8a–c.
3.2.1.1. Detection of IC50 in melanoma MDA-MB-435, glioblastoma SNB-75 cells, and normal cell line WI-38
Compounds 5c and 8a which displayed superior inhibitory activity against the melanoma cell line were selected to be further studied through the determination of its half-maximal inhibitory concentration (IC50) values against the most sensitive cancer cell lines compared to roscovitine as a reference anticancer drug. The results of the mean values of experiments performed in triplicate were summarised in . The in vitro results confirmed that the most sensitive cell line for compound 5c was melanoma cell line MDA-MB-435 with IC50 = 3.03 µM which is 1.7-folds more potent than roscovitine (IC50 = 5.26 µM). Compound 8a also showed good potency against MDA-MB-435 cell line with IC50 = 6.22 µM approaching the activity of roscovitine. Additionally, the quinazolinone 5c exhibited promising anticancer activity against glioblastoma (SNB-75) cell line with IC50 value of 8.62 µM which is nearly equipotent as roscovitine (IC50 = 7.76 µM).
Table 3. Concentrations required for 50% inhibition of cell viability (IC50) of compound 5c compared to roscovitine.
Furthermore, 5c and 8a were evaluated for their cytotoxic activity in normal fibroblast cells WI-38 to explore the compounds’ selectivity towards tumour cells. As shown in , the tested compounds were found to be more selective to the cancer cells. Interestingly, the quinazolinone 5c showed superior selectivity towards melanoma MDA-MB-435 cell line with the highest selectivity index (SI = 20.63).
Table 4. Cytotoxic activity of compounds 5c, 8a and roscovitine against WI-38 normal cells and their selectivity index (SI).
3.2.2. Flow cytometric analysis and apoptotic studies
Compound 5c showed the highest antiproliferative activity against MDA-MB-435 and SNB-75 cancer cell lines; consequently, flow cytometric analysis was carried out to study its effect on the cell cycle phases in the same cancer cell lines and thus identify its plausible biological target. Besides, apoptotic activity was measured by Annexin V-based flow cytometric analysis.
3.2.2.1. Cell cycle analysis
The role of CDK2 inhibitors in cell cycle arrest and apoptosis induction is well-documentedCitation5,Citation52,Citation53. The chloro quinazolinone 5c that showed promising anticancer activity against MDA-MB-435 and SNB-75 cell lines was subjected to cell cycle analysis and apoptotic assay to define the phases at which cell cycle arrest takes place in both MDA-MB-435 and SNB-75 cell lines and to investigate its effect on induction of apoptosis. The results of the cell cycle analysis of compound 5c are shown in and .
Figure 2. Effect of compound 5c on the cell cycle of melanoma MDA-MB-435 and glioblastoma SNB-75 cell lines.

Table 5. Cell cycle analysis results of compound 5c after 24 h in MDA-MB-435 and SNB-75 cell lines.
The obtained results revealed that compound 5c led to pre G1 apoptosis with a cell growth arrest at S phase in melanoma MDA-MB-435 cells after 24 h. which was confirmed by an increase in the percentage of DNA content (40.97%) after addition of the compound compared to control cells (34.28%). For glioblastoma SNB-75 cells, the results showed that the quinazolinone 5c induced pre G1 apoptosis with a cell growth arrest at G2/M phase which was confirmed by an increase in the percentage of DNA content (31.78%) after the addition of the compound compared to the to that of the untreated control cells (11.59%). Statistical analysis of the different stages of the cell cycle demonstrated the ability of 5c to induce significant levels of pre G1 apoptosis and cell cycle arrest in S phase and G2/M compared to the control. Thus, cell cycle arrest induced by the quinazolinone 5c at S phase and G2/M phase suggests that CDK2 could be the biological target for the newly synthesised compounds.
3.2.2.2. Apoptosis determination
The apoptotic activity of quinazolinone 5c in melanoma MDA-MB-435 and glioblastoma SNB-75 cell lines was further studied using Annexin V-FITC assay. As shown in and , total apoptotic cells percentage was increased in both cell lines (28.65, 44.22%, respectively), compared to the positive control, annexin V (% apoptosis 1.91, 2.53) after 24 h of treatment of MDA-MB-435 and SNB-75 cell lines with 5c at its IC50 concentration. Statistical analysis of the different stages of apoptosis revealed the ability of 5c to induce significant levels of apoptosis in MDA-MB-435 and SNB-75 treated cells (in all stages) compared to the control.
Figure 3. Effect of compound 5c on induction of apoptosis in MDA-MB-435 and SNB-75 cancer cells using PI/Annexin double staining.

Table 6. Apoptosis induction analysis for compound 5c.
3.2.3. In vitro CDK2 inhibitory activity
It was reported that CDK2 is overexpressed in melanomaCitation44,Citation45. Accordingly, the most potent congeners on MDA-MB-435 the melanoma cancer cell line; 5c and 8a were selected as representatives to study the CDK2 inhibitory activity of the newly synthesised compounds. The quinazolinones 5c and 8a were further investigated in vitro for their kinase inhibitory activity on CDK2 and were compared to the CDK2 kinase inhibitor roscovitine as a reference standard. The results were presented in . The anti-proliferative activity of the quinazolinone 5c appeared to correlate well with its ability to inhibit CDK2 at sub-micromolar range with IC50 value 0.63 µM which is twice as potent as roscovitine (IC50 = 1.28 µM). Additionally, Compound 8a exhibited IC50 value equal to 1.74 µM which is slightly higher than that of the reference drug.
Table 7. In vitro inhibitory activity of compound 5c against CDK2.
3.3. In silico studies
3.3.1. Molecular docking study
A molecular docking technique was performed to interpret the observed promising activity of compound 5c on CDK2. Therefore, docking of 5c into the three-dimensional X-ray crystallographic structure of the cyclin-dependent kinase2 (PDB code: 2B53)Citation25 in complex with the quinazoline DIN-234325 (IC50 = 0.6 ± 0.1 µM) was carried out using Molecular Operating Environment (MOE) 2010.10. The inhibitor binds into the ATP binding pocket located in the deep cleft between the upper and lower domains of CDK2. Through two hydrogen bond interactions, the N1 of the quinazoline ring interacts with the key amino acids Glu81 and Leu83 via a water molecule. It is worth mentioning that the two previously mentioned interactions are consistently observed in the hinge region for ATP-competitive CDK2 inhibitorsCitation54,Citation55. Stacking interactions were shaped through the quinazoline ring of the ligand and Val18 and Leu134 residues. Additionally, the aniline ring attached at position 6 of the quinazoline ring forms pi staking interaction with Gln 85.
To confirm the accuracy of the docking procedure, validation was performed by re-docking of the co-crystallised ligand into the catalytic domain of CDK2. The validation results revealed that the used docking protocol is suitable for the proposed docking study. This is demonstrated by the perfect alignment between the coordinates of DIN-234325 and its self-docked pose with root mean square deviation (RMSD) of 0.485 Å, docking score (S) of −18.24 Kcal mol−1. Additionally, the self-docked pose was able to achieve the same binding interactions depicted by the co-crystallised ligand with the main amino acids in the CDK2 binding site ().
Figure 4. 2D interaction diagram showing DIN-234325 interactions with the key amino acids in the CDK2 active site.

Figure 5. 3D representation of the superimposition of the docked pose (red) and the co-crystallised quinazoline ligand (green) in the CDK2 binding site (RMSD = 0.485 Å).

The outcomes of the molecular docking study showed the binding mode of compound 5c within the binding pocket of CDK2 kinase (; binding score (S) of −13.8 Kcal mol−1) indicating that two hydrogen bonds are formed between the quinazolinone moiety and the hinge region. Specifically, the quinazolinone carbonyl oxygen forms two water-mediated hydrogen bonds with the Leu83-NH and Glu81-CO. Moreover, the planar aromatic core makes extensive hydrophobic contacts with Ala31, Phe80, Ile10, Leu134, Val18, and Ala144 in a similar mode to that of the co-crystallised ligand. Additionally, other hydrogen-pi interactions were observed between the phenyl rings attached to the C2 and N2 of the quinazolinone ring and Glu12 and Gln85, respectively.
Figure 6. 2D diagram (A) and 3D representation (B) of quinazolinone 5c showing its interactions with the CDK2 binding site.

3.3.2. Drug likeness properties
The drug-likeness profile for the most potent candidate 5c was predicted using SwissADME serverCitation48. The results of the Swiss ADME prediction of drug-likeness of this compound revealed no alerts for Pan Assay Interfering Substances (PAINS) and no violation to Lipinski and other pharmacokinetics filters (for more details see supporting materials, ). Moreover, the selected quinazolinone 5c showed a predicted log Po/w value in the range of 3.25–4.06 with moderate water solubility, high GIT absorption, and no BBB permeability and thus minimal predicted CNS adverse effects. Furthermore, 5c is not P-glycoprotein substrates (PGP−) so it is not liable to the efflux by this transporter and thus avoiding the drug-resistance mechanism used by some tumour cell linesCitation56 (for boiled egg chart of compound 5c see supporting materials, ). The oral bioavailability radar chart of compound 5c demonstrated its good predicted oral bioavailability (). Collectively the tested compound possesses promising physicochemical and pharmacokinetic properties.
Figure 7. Bioavailability radar chart of compound 5c. The pink area represents the range of the optimal property values for oral bioavailability and the red line is compound 5c predicted properties; saturation (INSATU), size (SIZE), polarity (POLAR), solubility (INSOLU), lipophilicity (LIPO), and flexibility (FLEX).

4. Conclusion
In this study, three series of quinazolin-4(3H)-one derivatives were designed and synthesised as CDK2 inhibitors. All the newly synthesised compounds were screened for their antiproliferative activity against a panel of sixty cancer cell lines. The quinazolinones 5c and 8a displayed excellent growth inhibition against the melanoma cell line MDA-MB-435 (GI% = 94.53 and 94.15, respectively). The quinazolinone 5c also exhibited promising activity against the glioblastoma cell line SNB-75 (GI% = 63.09), while 8a showed potent to moderate activity against leukaemia cell lines SR (GI% = 69.55) and K-562 (GI% = 67.80). Consequently, the IC50 of 5c and 8a against the most sensitive cell line (MDA-MB-435) in this research work was detected. Compound 5c has emerged as the most active member against MDA-MB-435 with IC50 value of 3.03 µM which is 1.7-folds more potent than roscovitine, the reference standard (IC50 = 5.26 µM). Compound 8a also showed good potency on MDA-MB-435 cell line with IC50 value of 6.22 µM which is comparable to that of the reference drug. The quinazolinone 5c triggered cell cycle arrest in the S phase in melanoma MDA-MB-435 cells and in the G2/M phase in glioblastoma SNB-75 cells suggesting that CDK2 could be the potential biological target for the newly synthesised congeners. Furthermore, compound 5c induced apoptosis in MDA-MB-435 and SNB-75 cell lines as indicated by the percent of the total apoptosis of 28.65 and 44.22%, respectively in comparison to the positive control, annexin V (% apoptosis 1.91, 2.53). Accordingly, the most active compounds 5c and 8a were tested for their CDK2 inhibitory activities. Interestingly, the outcomes of CDK2 inhibitory assay were consistent with that of the cytotoxicity data. The quinazolinones 5c and 8a displayed CDK2 IC50 values of 0.63 µM and 1.74 µM, sequentially in comparison with the used reference roscovitine (IC50 = 1.28 µM). The molecular docking study confirmed the biological results via prediction of the possible binding mode of the most active compound 5c with CDK2 active site indicating its ability to form a pair of crucial hydrogen bonds with the CDK2 hinge region (Leu83 and Glu81). Moreover, the most active compound 5c possessed promising pharmacokinetic properties as depicted by the computational ADME study. These outcomes endorsed that the most active candidates 5c and 8a may serve as useful lead compounds in the search for promising anti-melanoma agents acting through CDK2 inhibition.
Supplemental Material
Download PDF (3.6 MB)Acknowledgements
The authors are thankful for the members of the Developmental Therapeutics Program (DTP) at the National Cancer Institute (NCI), Maryland, USA for performing the NCI-60 Human Tumor Cell Lines Screen.
Disclosure statement
The authors report no conflict of interest.
References
- Nagai H, Kim YH. Cancer prevention from the perspective of global cancer burden patterns. J Thorac Dis 2017;9:448–51.
- Fischer PM, Endicott J, Meijer L. Cyclin-dependent kinase inhibitors. Prog Cell Cycle Res 2003;5:235–48.
- Idowu MA. Cyclin-dependent kinases as drug targets for cell growth and proliferation disorders. a role for systems biology approach in drug development. Part II—CDKs as drug targets in hypertrophic cell growth. Modelling of drugs targeting CDKs. Biotechnol Biotechnol Equip 2012;26:2712–5.
- Dictor M, Ehinger M, Mertens F, et al. Abnormal cell cycle regulation in malignancy. Am J Clin Pathol 1999;112:S40–S52.
- Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 2003;36:131–49.
- Ma T, Van Tine BA, Wei Y, et al. Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev 2000;14:2298–313.
- Flores O, Wang Z, Knudsen KE, et al. Nuclear targeting of cyclin-dependent kinase 2 reveals essential roles of cyclin-dependent kinase 2 localization and cyclin E in vitamin D-mediated growth inhibition. Endocrinology 2010;151:896–908.
- De Boer L, Oakes V, Beamish H, et al. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene 2008;27:4261–8.
- Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005;24:2776–86.
- Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature 1999;401:297–300.
- Akli S, Van Pelt CS, Bui T, et al. Cdk2 is required for breast cancer mediated by the low-molecular-weight isoform of cyclin E. Cancer Res 2011;71:3377–86.
- Cam WR, Masaki T, Shiratori Y, et al. Activation of cyclin E-dependent kinase activity in colorectal cancer. Dig Dis Sci 2001;46:2187–98.
- Yamamoto H, Monden T, Miyoshi H, et al. Cdk2/cdc2 expression in colon carcinogenesis and effects of cdk2/cdc2 inhibitor in colon cancer cells. Int J Oncol 1998;13:233–9.
- Juric V, Murphy B. Cyclin-dependent kinase inhibitors in brain cancer: current state and future directions. Cancer Drug Resist 2020;3:48–62.
- Wang J, Yang T, Xu G, et al. Cyclin-dependent kinase 2 promotes tumor proliferation and induces radio resistance in glioblastoma. Transl Oncol 2016;9:548–56.
- Riess C, Irmscher N, Salewski I, et al. Cyclin-dependent kinase inhibitors in head and neck cancer and glioblastoma-backbone or add-on in immune-oncology? Cancer Metastasis Rev 2021;40:153–71.
- Desai BM, Villanueva J, Nguyen T-TK, et al. The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PLOS One 2013;8:e59588.
- Du J, Widlund HR, Horstmann MA, et al. Critical role of CDK2 for melanoma growth linked to its melanocyte-specific transcriptional regulation by MITF. Cancer Cell 2004;6:565–76.
- Kumar SK, LaPlant B, Chng WJ, et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015;125:443–8.
- Villa E, Piscaglia F, Geva R, et al. Phase IIa safety and efficacy of milciclib, a pan-cyclin dependent kinase inhibitor, in unresectable, sorafenib-refractory or -intolerant hepatocellular carcinoma patients. J Clin Oncol 2020;38:e16711.
- Lin S-F, Lin J-D, Hsueh C, et al. Potent effects of roniciclib alone and with sorafenib against well-differentiated thyroid cancer. Endocr Relat Cancer 2018;25:853–64.
- Tadesse S, Caldon EC, Tilley W, et al. Cyclin-dependent kinase 2 inhibitors in cancer therapy: an update. J Med Chem 2019;62:4233–51.
- Frame S, Saladino C, MacKay C, et al. Fadraciclib (CYC065), a novel CDK inhibitor, targets key pro-survival and oncogenic pathways in cancer. PLOS One 2020;15:e0234103.
- Le Tourneau C, Faivre S, Laurence V, et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur J Cancer 2010;46:3243–50.
- Sielecki TM, Johnson TL, Liu J, et al. Quinazolines as cyclin dependent kinase inhibitors. Bioorg Med Chem Lett 2001;11:1157–60.
- Shewchuk L, Hassell A, Wisely B, et al. Binding mode of the 4-anilinoquinazoline class of protein kinase inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-dependent kinase 2 and p38 kinase. J Med Chem 2000;43:133–8.
- Abdel Gawad NM, Georgey HH, Youssef RM, et al. Synthesis and antitumor activity of some 2,3-disubstituted quinazolin-4(3H)-ones and 4,6-disubstituted-1,2,3,4-tetrahydroquinazolin-2H-ones. Eur J Med Chem 2010;45:6058–67.
- Gatadi S, Pulivendala G, Gour J, et al. Synthesis and evaluation of new 4(3H)-quinazolinone derivatives as potential anticancer agents. J Mol Struct 2020;1200:127097.
- ShaguftaAhmad I. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. Medchemcomm 2017;8:871–85.
- Abbas SE, Barsoum FF, Georgey HH, et al. Synthesis and antitumor activity of certain 2,3,6-trisubstituted quinazolin-4(3H)-one derivatives. Bull Fac Pharm Cairo Univ 2013;51:273–82.
- El Sayed NA, Eissa AA, El Masry GF, et al. Discovery of novel quinazolinones and their acyclic analogues as multi-kinase inhibitors: design, synthesis, SAR analysis and biological evaluation. RSC Adv 2016;6:111767–86.
- Yang Y, Shang P, Cheng C, et al. Novel N-phenyl dichloroacetamide derivatives as anticancer reagents: design, synthesis and biological evaluation. Eur J Med Chem 2010;45:4300–6.
- Alqasoumi SI, Al-Taweel AM, Alafeefy AM, et al. Novel quinolines and pyrimido[4,5-b]quinolines bearing biologically active sulfonamide moiety as a new class of antitumor agents. Eur J Med Chem 2010;45:738–44.
- Guo W, Miao Z, Sheng C, et al. Synthesis and evaluation of 9-benzylideneamino derivatives of homocamptothecin as potent inhibitors of DNA topoisomerase I. Eur J Med Chem 2010;45:2223–8.
- Xu Y, Liang P, Rashid H. u, et al. Design, synthesis, and biological evaluation of matrine derivatives possessing piperazine moiety as antitumor agents. Med Chem Res 2019;28:1618–27.
- Aaglawe J, Dhule S, Bahekar SS, et al. Synthesis and antibacterial activity of some oxazolone derivatives. J Korean Chem Soc 2003;47:133–136.
- Abbas S, Gawad N, Georgey HH, et al. New quinazolinone derivatives: synthesis, anti-inflammatory and antitumor activities. IntJ ChemTech Res 2010;2:1560–1578.
- Georgey H, Abdel-Gawad N, Abbas S. Synthesis and anticonvulsant activity of some quinazolin-4-(3H)-one derivatives. Molecules 2008;13:2557–69.
- Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–12.
- Boyd MR, Paull KD. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Develop Res 1995;34:91–109.
- Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst 1991;83:757–66.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63.
- Scudiero DA, Shoemaker RH, Paull KD, et al. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 1988;48:4827–33.
- Pollack A, Ciancio G, Chapter 3: cell cycle phase-specific analysis of cell viability using Hoechst 33342 and propidium iodide after ethanol preservation. In: Darzynkiewicz Z, Crissman HA, editors. Methods in cell biology. San Diego: Academic Press; 1990:19–24.
- Vermes I, Haanen C, Steffens-Nakken H, et al. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995;184:39–51.
- Hu X, Zhao H, Wang Y, et al. Synthesis and biological evaluation of novel 5,6-dihydropyrimido[4,5-f]quinazoline derivatives as potent CDK2 inhibitors. Bioorg Med Chem Lett 2018;28:3385–90.
- Available from: https://www.rcsb.org/ [last accessed 1 Aug 2021].
- SwissADME. Available from: http://www.swissadme.ch/index.php [last accessed 15 Jun 2021].
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017;7:42717.
- Daina A, Michielin O, Zoete V. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model 2014;54:3284–301.
- NIH Developmental Therapeutics Program. Available from: http://dtp.cancer.gov [last accessed 6 Jun 2020].
- Chohan TA, Qian H, Pan Y, et al. Cyclin-dependent kinase-2 as a target for cancer therapy: progress in the development of CDK2 inhibitors as anti-cancer agents. Curr Med Chem 2015;22:237–63.
- Faber AC, Chiles TC. Inhibition of cyclin-dependent kinase-2 induces apoptosis in human diffuse large B-cell lymphomas. Cell Cycle 2007;6:2982–9.
- Vulpetti A, Pevarello P. An analysis of the binding modes of ATP-competitive CDK2 inhibitors as revealed by X-ray structures of protein-inhibitor complexes. Curr Med Chem Anticancer Agents 2005;5:561–73.
- Martin MP, Endicott JA, Noble MEM. Structure-based discovery of cyclin-dependent protein kinase inhibitors. Essays Biochem 2017;61:439–52.
- Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 2008;9:105–27.