
Abstract
A novel series of aminotrimethylpyridinol and aminodimethylpyrimidinol derivatives were designed and synthesised for FGFR4 inhibitors. Structure-activity relationship on the FGFR4 inhibitory activity of the new compounds was clearly elucidated by an intensive molecular docking study. Anti-cancer activity of the compounds was evaluated using hepatocellular carcinoma (HCC) cell lines and a chick chorioallantoic membrane (CAM) tumour model. Compound 6O showed FGFR4 inhibitory activity over FGFR1 − 3. Compared to the positive control BLU9931, compound 6O exhibited at least 8 times higher FGFR4 selectivity. Strong anti-proliferative activity of compound 6O was observed against Hep3B, an HCC cell line which was a much more sensitive cell line to BLU9931. In vivo anti-tumour activity of compound 6O against Hep3B-xenografted CAM tumour model was almost similar to BLU9931. Overall, compound 6O, a novel derivative of aminodimethylpyrimidinol, was a selective FGFR4 kinase inhibitor blocking HCC tumour growth.
Graphical abstract
A series of methylated aminopyri(mi)dine compounds were designed and synthesised as FGFR4 inhibitors.
Compound 6O showed a selective FGFR4 inhibitory activity over FGFR1 ∼ 3.
Dimethyl groups at the pyrimidine ring of compound 6O prohibited a proper conformation for covalent bonding to FGFR1 ∼ 3.
The smaller size of fluorine at the dimethoxyphenyl ring in compound 6O gives suitable conformation for strong binding interaction with FGFR4.
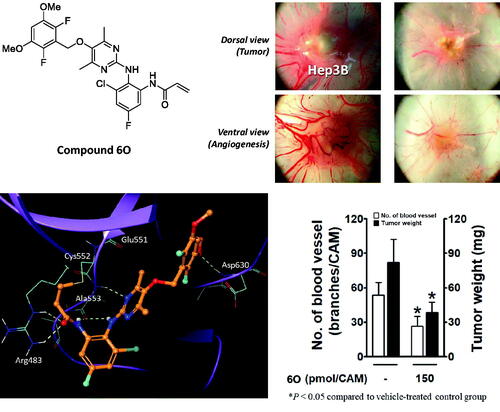
Compound 6O inhibited Hep3B tumour growth xenografted on chick chorioallantoic membrane (CAM) tumour model
Highlights
1. Introduction
Hepatocellular carcinoma (HCC) has been increasingly diagnosed over the past few decades and has become the third leading cause of cancer-related mortality worldwideCitation1. After sorafenib (a multi-tyrosine kinase inhibitor) was authorised for the first-line treatment option of HCC treatmentCitation2, several multi-tyrosine kinase inhibitors including cabozantinib have been approved for patients with advanced-stage HCCCitation3. In addition, PD-1 immune checkpoint inhibitors, such as nivolumab and pembrolizumab, have been added as therapeutic options in HCC treatmentCitation3. Despite these drugs being approved as the first-line or second-line therapy, tumour regression by immune checkpoint blockade only occurs in a limited proportion of patientsCitation4.
Fibroblast growth factor receptor 4 (FGFR4), one of the families of fibroblast growth factor receptors, is a tyrosine kinase receptor with a distinct 802 amino acid sequence. Normally, FGFR4 expression level is high during foetal development and drastically reduced thereafterCitation5. Most of FGF family members, except FGF11 subfamily, are ligands of FGFR4Citation6. FGF19, which is produced from the ileum as a postprandial hormone, has a more specific selective affinity to FGFR4 than the other FGF ligands. In contrast to the normal production and action of FGF19 as an endocrine hormone, FGF19 is overexpressed and co-expressed with FGFR4 in various cancers of liver, breast, lung, bladder, head, and neckCitation7–10, indicating FGF19 as a driver oncogene. Similarly, amplification of FGFR4 gene is the predominant type and accounts for 78% of all FGFR4 gene alterations in various cancersCitation11–14. In addition, a significant correlation between overexpression of FGFR4 in tumour tissues and patients’ poor survival rate indicates FGFR4 is also acting as an oncogeneCitation15,Citation16.
Numbers of research for the discovery of selective FGFR4 inhibitors have underlined the covalent interaction between Cys552 residue in the middle-hinge domain of the ATP-binding site of FGFR4 and acrylamide moiety of small molecule compounds, which was supported by X-ray co-crystal structures. BLU9931 (1) was discovered as a notable compound to possess potent FGFR4 inhibitory activity with good selectivity over FGFR1 ∼ 3. After that, a number of analogues of BLU9931 were designed and prepared, where fisogatinib (BLU554) (2) with better physicochemical properties has been evaluated in a clinical trial for the treatment of HCC. In addition, other Cys552 − acrylamide inhibitors such as H3B-6527 (3), aminopyrimidines (4), and aminopyridines (5) are undergoing either preclinical or clinical studies in recent years () Citation17–26. Multiple pan-FGFR inhibitors have shown weak efficacy against FGFR4 together with toxicity (hyperphosphatemia and soft tissue mineralization) due to activity against FGFR1 ∼ 3, which indicates a strong need for the discovery of FGFR4-selective inhibitorsCitation27.
Figure 1. Representative selective and irreversible FGFR4 inhibitors (1 ∼ 5) and general structure (6) of novel compounds prepared in this study.

We recently reported a string of anti-HCC compounds where bioisosterism was employed as the main strategy for the design of new compoundsCitation28,Citation29. Bioisosterism is one of the efficient and practical strategies of new drug discovery. Replacement of a functional group of a lead compound may improve efficacy by changes in binding mode to molecular target, metabolic stability by modifying structure of labile sites, or physicochemical properties, etcCitation30,Citation31. In our previous studies, we designed and prepared a series of bioisosteres of sorafenib and cabozantinib for the discovery of anti-HCC agents, respectivelyCitation28,Citation29. As part of our on-going project of the discovery of new drug by bioisosteric replacement of a benzene, pyridine, or pyrimidine core ring of well-known drugs or promising compounds in the development stage with methyl-containing pyridine/pyrimidine rings, the present study was focussed on the synthesis and evaluation of a new series of aminotrimethylpyridinol- and aminodimethylpyrimidinol-derived FGFR4 inhibitors which are depicted as a general structure (6) in . In addition, a molecular docking study was done in-depth for an explanation of the difference in FGFR4 inhibitory activity with structural change.
2. Results and discussion
2.1. Synthesis
shows the structures of twenty-eight new compounds for this study. They were designed based on common aminopyridine/aminopyrimidine backbones of about hundred compounds such as compounds 4 and 5 in introduced in the literatureCitation21–23. The pyridine/pyrimidine centred rings were replaced by trimethylpyridine/dimethylpyrimidine, which was the major structural variation. Fluorine and chlorine were employed at the halogen position in the dimethoxy ring part and seven kinds of acryl amide-containing aromatic rings were combined.
Figure 2. New compounds designed and synthesised for this study.

We thought that, among several synthetic methods shown in the literature on aminopyri(mi)dine FGFR4 inhibitors, the synthetic strategies described in the Eisai's patent specification of Eisai Co., LtdCitation21. were most suitable for preparing our new compounds (6). All twenty-eight final target compounds (6A ∼ 6BB) were prepared by the synthetic method of Scheme 1 depicted as general structures for convenience. In brief, 3,5-dimethoxy-2,6-dihalobenzyl bromides (7, 8) were combined with 6-bromo-2,4,5-trimethylpyridin-3-ol (9) or 2-bromo-4,6-dimethylpyrimidin-5-ol (10) to afford the four common intermediates 11 ∼ 14. Combination of these four bromo-intermediates (11 ∼ 14) with seven aromatic amines (15 ∼21) under the palladium-catalyzed amination reaction conditions afforded twenty-eight coupled compounds (22A∼22BB). The nitro groups in compounds 22 were reduced to the corresponding amines 23, and finally, the acryloyl group was introduced to the resulting amine groups producing the final compounds (6A ∼ 6BB).
Scheme 1. General synthetic scheme for 6A∼6BB. Reagents and Conditions: (a) K2CO3, DMF, 80 °C; (b) Pd2(dba)3, BINAP, NaOtBu, PhMe, reflux or Pd2(dba)3, xantphos, Cs2CO3, PhMe, reflux; (c) SnCl2·2H2O, EtOH, reflux or Zn, AcOH, DCM, r.t.; (d) acrylic acid, T3P, Et3N, DMF, 100 °C or acryloyl chloride, Et3N, DCM, 0 °C to r.t.

Preparation of the required substrates 7 ∼ 10 is shown in Scheme 2. First, the fluoro-containing benzyl bromide 7 was readily prepared by a simple bromination from commercially available alcohol 22Citation21. For the synthesis of the chloro-version substrate 8, we put the ring-chlorination step at the final stage. Reduction of methyl 3,5-dimethoxybenzoate (23) to primary alcohol 24 followed by bromination afforded the corresponding bromide 25Citation32. Lastly, chlorination of the ring with sulphuryl chloride gave the chloro-substrate 8. The centred ring, 2,4,5-trimethylpyridin-3-ol (9), was prepared from pyridoxine (26) by the known procedure developed by usCitation33. The two hydroxymethyl groups of 26 were chlorinated to give 27 which was then reductively cleaved to afford methyl compound 28. Ring bromination of 28 finally gave the substrate 9. Another centred ring, 2-bromo-4,6-dimethylpyrimidin-5-ol (10), was obtained by a three-step sequence. 3-Chloropentane-2,4-dione (29) was reacted with formamide in formic acid to give an oxazole intermediate 30 which was then treated with ammonia water to afford 4,6-dimethylpyrimidin-5-ol (31)Citation34. The pyrimidine substrate 10 was finally obtained by bromination of 31.
Scheme 2. Preparation of the substrates (7 ∼ 10). Reagents and Conditions: (a) PBr3, CH2Cl2, r.t., 15 min, quant.; (b) LiAlH4, THF, −20 °C to r.t., 12 h, 97%; (c) PBr3, CH2Cl2, r.t., 15 min; (d) SO2Cl2, CH3CN-CH2Cl2, 0 °C to r.t., 42 h, 90% (for 2 steps); (e) SOCl2, DMF, ClCH2CH2Cl, reflux, 5 h, 93%; (f) Zn, AcOH, reflux, 3 h, 80%; (g) DBDMH, THF, r.t., 3 h, 80%; (h) HCO2NH2, HCO2H, 140 °C, 12 h; (i) NH4OH, 120 °C, 5 h, 34% (for 2 steps); (j) DBDMH, THF, r.t., 5 h, 63%.

Scheme 3 shows the preparation of substrates (15 ∼ 21) for the third part constituting the target compounds. They were synthesised from commercially available materials (32, 34, 35, 37, 38, and 40) by a one- or three-step procedure. The compounds 15, 16, 17, 18, and 20 were prepared by a similar synthetic sequence, i.e., N-acetylation, nitration, and N-deacetylationCitation21,Citation23,Citation35,Citation36. For compound 19, methylation of 2-amino-3-nitrophenol (37) was used, and replacement of fluoro of 5-fluoro-2-nitroaniline (40) with 1-ethylpiperazine afforded compound 21, where the two materials, 37 and 40, were purchasable.
Scheme 3. Preparation of the nitroamine compounds (1 5 ∼21). Reagents and Conditions: (a) AcCl, THF, 0 °C to r.t., 2 h, quant.; (b) HNO3, H2SO4, 0 °C to r.t., 15 h, 51%; (c) i) Ac2O, 0 °C, 2 h, ii) HNO3, H2SO4, AcOH, 0 °C, 3 h then r.t., 12 h, iii) HCl, H2O, reflux, 5 h, 16 (35%), 17 (23%); (d) AcCl, THF, 0 °C to r.t., 2 h, quant.; (e) i) HNO3, H2SO4, AcOH, 0 °C, 3 h then r.t., 12 h, ii) HCl, H2O, reflux, 5 h, 59%; (f) MeI, K2CO3, acetone, r.t., 20 h, 95%; (g) AcCl, THF, 0 °C to r.t., 0.5 h, quant.; (h) i) HNO3, H2SO4, AcOH, 0 °C, 1.5 h then r.t., 2 d, ii) HCl, H2O, reflux, 3 h, 13%; (i) 1-ethylpiperazine, iPr2NEt, NMP, 130 °C, 5 h, 98%.

2.2. Inhibitory activity of new compounds against FGFR kinases
The new compounds (6A∼6BB) were subjected to cell-free kinase assay to examine their inhibitory activities against FGFR4 kinase at a fixed concentration (1 µM) along with BLU9931 (1), the positive control. Most compounds generally showed low inhibitory activity against FGFR4 (). Roughly, the inhibitory activity of the difluoro-containing compounds was better than the corresponding dichloro analgoues. The inhibitory activity of compounds 6A, 6O, and 6S was notable, and, in particular, compound 6O showed an excellent inhibition comparable to that of BLU9931 at the 1 µM concentration tested.
Figure 3. Inhibitory activities of the new compounds (6A∼6BB) along with BLU9931 (1) as the positive control at a fixed concentration (1 μM) against FGFR4 kinase. The numbers in the bar-graphs are mean values of % inhibition obtained from two independent experiments performed by the Reaction Biology Corp. (Malvern, PA, USA).

A follow-up kinase assay of selected compounds, 6A and 6O, with 7 different concentrations, showed inhibition of the target kinase FGFR4 with IC50 values of 190 nM and 75.3 nM, respectively (). Compound 6A inhibited FGFR1 ∼ 3 with IC50 1565, 1149, and 277 nM, respectively, which corresponds to 1.5 ∼ 8 fold selectivity of FGFR4 over FGFR1 ∼ 3. Surprisingly, compound 6O showed a marked FGFR4 selectivity of 398 ∼ 664 fold over FGFR1 ∼ 3 IC50 values of >50,000, 35,482, and >30,000 nM. This result indicates that compound 6O has at least 8 times better FGFR4 selectivity than BLU9931 (1) ().
Table 1. IC50 values of the selected compounds against FGFR1 ∼ 4 kinases and selectivity.
2.3. Structure-activity relationship (SAR) based on molecular docking
As shown in , most of the trimethylpyridine- or dimethylpyrimidine-containing new compounds generally have low inhibitory activity against FGFR4 except for compounds 6A, 6O, and 6S, which have fluorine in common at X position in . We thought that a combination of fluorine substitution at the dimethoxyphenyl ring and the introduction of methyl groups to the pyridine or pyrimidine ring may contribute to the higher FGFR4 activity and selectivity of 6A and 6O as shown in . To clarify these substituent effects, we focussed on compounds 6E and 6G. These compounds are methylated analogs of known FGFR4 inhibitors 41 and 42, which have bare pyridine and pyrimidine at the centreCitation21. The FGFR4 percent inhibition data at 0.01 µM of these compounds are shown in . It is clear that the substitution of methyl groups at the pyridine and pyrimidine rings is detrimental to the activity. In order to further investigate the substitution effects of fluorines and methyl groups on the FGFR4 activity and selectivity, we performed covalent docking studies on relevant compounds.
Table 2. Percentages of inhibition of compounds at 0.01 μM concentration against FGFR4 kinase.
2.3.1. Effect of methyl groups and halogen substituents on SAR
Covalent docking studies were carried out using Flare from Cresset according to the methods described in detail in the experimental section. The LF Rank score and LF VSscore were used to identify correct binding pose and rank compounds in a virtual screening context, respectively. Introducing three methyl groups into the pyridine core (compound 41 → 6E) cost LF VSscore of 0.908. As shown in , this is a direct result of losing hydrogen bonds from Ala553 to the pyridine N and anilyl NH. Due to steric bulkiness of three methyl groups in compound 6E, the pyridine ring plane is twisted by 90° and thus the hydrogen bonds are destroyed. On the other hand, introducing only two methyl groups into the pyrimidine ring is generally tolerated (compound 6O). Unlike compound 6E, it does not cause a twist of the pyrimidine ring plane (). However, replacing the fluorine atoms of the dimethoxyphenyl ring with chlorine (6O → 6P) abolished the FGFR4 inhibitory activity (). To find out the origin of the activity change, we replaced hydrogens of the pyrimidine ring with methyl in the crystal structure of FGFR4 − ligand complex (PDB 7DTZ) and examined the binding interactions (). It turned out that the methyl group would clash with the peptide carbonyl oxygen of Glu551 and thus the ideal binding pose cannot be maintained as shown in . LF VSscore also changed from −12.23 to −10.81 in agreement with the observed FGFR4 activities of 6O and 6P. Interestingly, removing methyl groups from the pyrimidine ring of 6P restores the FGFR4 activity, i.e., it is known that compound 6P-1 exhibits an FGFR4 activity of IC50 2.6 nMCitation37. shows the X-ray co-crystal structure of FGFR4−6P-1 complex (PDB 6NVI), in which, unlike 6P (), one of the pyrimidine nitrogen atoms forms a hydrogen bond to Ala553. Therefore it can be concluded that both the small size of fluorine at the dimethoxyphenyl ring and the bulkiness of methyl groups at the pyrimidine ring amount to the observed FGFR4 activity and selectivity of compound 6O.
Figure 4. Effect of methyl groups and halogen substituents on binding to FGFR4. (a) Binding pose of FGFR4−41 complex (LF VSscore= −11.88). (b) Binding pose of FGFR4−6E complex (LF VSscore= −10.97). Hydrogen bonds to Ala553 are hindered by the methyl groups of the pyridine ring. (c) Binding pose of FGFR4−6O complex (LF VSscore= −12.23). (d) Binding pose of FGFR4−6P complex (LF VSscore= −10.81). Hydrogen bonds to Ala553 are lost. (e) Introduction of methyl groups into the pyrimidine ring in FGFR4 − ligand complex crystal structure (PDB 7DTZ). Methyl groups in the pyrimidine ring would bump into the Glu551 peptide bond, forcing the ring plane to twist. (f) X-ray crystal structure of FGFR4−6P-1 complex (PDB 6NVI).

2.3.2. Selective inhibition of FGFR4 by compound 6O over FGFR1 − 3
Compound 6O showed an excellent selectivity towards FGFR4 over other FGFR subtypes (FGFR1 ∼ 3), whereas compound 6A had a marginal selectivity (). Although detailed explanation for the selectivity profile of BLU554 for the hinge cysteine of FGFR4 has been reported recentlyCitation37, more rationales are needed to address the exceptionally high selectivity profile of compound 6O. We further conducted a molecular docking study to provide an answer to such a high degree of selectivity by 6O. FGFR4 has a cysteine residue available for covalent binding of ligand in the hinge region, while other FGFR subtypes have one on the P-loop. X-ray crystal structures are found for FGFR − ligand complexes with a covalent bond at both the hinge and P-loop cysteine residues. It was proposed that the FGFR selectivity of BLU554 mainly comes from the rotational energy barriers of the ligand in solution and steric clash between the tetrahydropyran ring and the P-loop of FGFR1, specifically Leu484Citation37. Although compound 6O does not have a tetrahydropyran-like fragment that would clash with the P-loop of FGFR1, the methyl groups in the central pyrimidine ring would be too close to Glu562 at the hinge, hindering the binding (). Therefore we propose that the outstanding FGFR4 selectivity of 6O originates from the steric clash of the pyrimidine methyl groups to the hinge loop.
Figure 5. Introduction of methyl groups into the pyrimidyl ring in FGFR1 − ligand complex crystal structure (PDB 6NVL). Methyl groups in the pyrimidine ring would clash with the Glu562 on the hinge loop of FGFR1.

2.4. Anti-hepatocellular carcinoma efficacy evaluation
To examine anti-proliferative activities of compounds 6A and 6O, two HCC cell lines, Hep3B and Huh7 cells, were treated for 48 h with increasing concentrations, and their growth was measured by MTT assay. Hep3B and Huh7 cell lines overexpress FGF19 as a result of FGF19 gene amplification and are suitable model cells to study FGF19–FGFR4 signallingCitation38–40. Treatment of Hep3B cells with compounds 6A and 6O resulted in IC50 values of 25.2 and 4.5 µM, respectively, which was 28 and 5-fold less potent than compound 1 (). The anti-proliferative activity of the positive control compound 1 in Huh7 cells was less effective than in Hep3B, showing a 7-fold difference. Compound 6O showed a 2.8-fold difference of anti-proliferative activity in the two cell lines, whereas compounds 6A did not show a significant difference between the two cell lines. IC50 values of cytotoxicity of the compounds 1, 6A, and 6O measured in H6c7 normal human pancreatic duct epithelial cell line were 13.4, >100, and 18 µM, respectively, indicating compound 6A was fairly safe, and cytotoxicity of 6O and 1 was similar.
Figure 6. IC50 values of the selected compounds on proliferation of human liver cancer cell lines (Hep3B and Huh7) and viability of normal human pancreatic duct epithelial cell line (H6c7). Drugs were treated for 48 h. The values are mean ± SEM of three independent experiments performed in triplicate.

Next, we examined the in vivo antitumor efficacy of compound 6O using a chick chorioallantoic membrane (CAM) tumour model implanted with Hep3B cells. The implanted Hep3B cells in the vehicle-treated control group developed a tumour mass and tumour-induced angiogenesis (). Compound 6O and positive control compound (BLU9931, 1) dose-dependently inhibited the tumour growth. The inhibitory effect of 6O on the tumour weight was slightly weaker than that of compound 1. The tumour-induced angiogenesis was also significantly blocked by compounds, 6O and 1, and the antiangiogenic effect of compound 6O was also slightly weaker than compound 1.
Figure 7. Inhibitory effects of compound 6O and BLU9931 (1) on tumour growth and tumour-induced angiogenesis in Hep3B-xenografted CAM tumour model. Four days after Hep3B xenograft, both tumour growth and tumour-induced angiogenesis on the CAM tissues are shown in (A). The weight of tumour masses isolated from CAMs (B) and the number of new vessel branches formed on CAM were counted using Image J program (C). *P< 0.05 compared to the vehicle-treated group.

3. Conclusion
A series of novel compounds containing aminotrimethylpyri(mi)dinol were designed and synthesised with the aim of the discovery of selective FGFR4 inhibitors. Among them, compound 6O showed a potent and highly selective inhibitory activity against FGFR4 over FGFR1 ∼ 3. Structure-activity relationship study using intensive molecular docking calculations revealed that, by and large, introducing methyl groups at the central pyridine or pyrimidine ring can cause a steric clash with the hinge of FGFR4 kinase domain and thus weaken the binding affinity. However, exceptionally, compound 6O with dimethylpyrimidine core ring and difluoro substituent was observed to maintain its conformation suitable for strong binding interaction. Molecular docking study also suggests that a steric clash of dimethyl groups of compound 6O to a hinge of FGFR1 ∼ 3 interfere with adopting a proper pose for a strong binding between them. This explains the highly selective inhibition of compound 6O against FGFR4 over other families. The anti-proliferative activity of compound 6O on HCC cell lines was a little weaker than BLU9931 at low concentrations, however, the strength of the inhibitory effect was reversed at high concentrations. In addition, compound 6O effectively inhibited both the growth and angiogenesis of HCC, one of the hypervascular tumours. Overall, the antitumor effect of compound 6O was excellent and comparable to that of BLU9931.
4. Experimental section
4.1. Synthesis (Supplemental data)
4.1.1. General
Unless noted otherwise, materials were purchased from commercial suppliers and used without further purification. Air or moisture-sensitive reactions were carried out under argon atmosphere. The reaction progress was monitored by thin layer-chromatography (TLC) using silica gel F254 plates. Products were purified by flash column chromatography using silica gel 60 (70–230 mesh) or by using the Biotage ‘Isolera One’ system with indicated solvents. Melting points were determined using a Fisher–Johns melting point apparatus and were not corrected. Low-resolution mass spectra (LRMS) were obtained using a JMS-700 (JEOL) and recorded either in molecular ion peak mode with an electron ionisation (EI) source or in positive ion mode with fast electron bombardment (FAB) source or using a Waters ZQ 2000 and recorded in a positive ion mode with an electrospray (ESI) source. High-resolution mass spectra (HRMS) were obtained using a JMS-700 (JEOL) and recorded either in molecular ion peak mode with an electron ionisation (EI) source or in positive ion mode with fast electron bombardment (FAB) source. NMR spectra were obtained using a Bruker-250 spectrometer (250 MHz for 1H NMR and 62.5 MHz for 13C NMR) and a Bruker Avance Neo 400 spectrometer (400 MHz for 1H NMR). Chemical shifts (δ) were expressed in ppm using a solvent as an internal standard and the coupling constant (J) in hertz.
4.1.2. 3-(Bromomethyl)-2,4-difluoro-1,5-dimethoxybenzene (7) [CAS RN: 1956324–83-6]
To a solution of (2,6-difluoro-3,5-dimethoxyphenyl)methanol (22) (3.0 g, 14.69 mmol) in DCM (15 ml) was added a solution of PBr3 (1 M in DCM, 15.7 ml, 15.72 mmol) dropwise at 0 °C then stirred at room temperature for 30 min. The reaction mixture was quenched with ice water followed by DCM extraction. The combined organic solution was dried over MgSO4, filtered, concentrated to obtain 7 (3.75 g, 100%) as a grey solid.
4.1.3. 4,6-Dimethylpyrimidin-5-ol (31) [CAS RN: 70345–38-9]
To a solution of 3-chloro-2,4-pentadione (29) (20 ml, 177.17 mmol) in formic acid (30 ml) was added formamide (16 ml, 402.84 mmol) and refluxed for 12 h. The reaction mixture was cooled to room temperature then aq. NH4OH was added dropwise until the basic pH was maintained. After refluxing the mixture for 5 h, it was cooled to room temperature and then concentrated. Acetone was added to the residue to acquire product in acetone. The acetone fraction was concentrated, and the residue was purified by silica gel column chromatography with EtOAc to obtain nd 31 (7.4 g, 34%) as a pale yellow solid.
4.1.4. 2-Bromo-4,6-dimethylpyrimidin-5-ol (10)
To a solution of 4,6-dimethylpyrimidin-5-ol (31) (150 mg, 1.21 mmol) in THF (4 ml) was added DBDMH (242 mg, 0.85 mmol). The resulting mixture was stirred at room temperature for 5 h and then concentrated. The residue was diluted with EtOAc and water, and the aqueous layer was extracted with EtOAc. The combined organic solution was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography with 10% to 40% EtOAc/Hexanes to obtain 10 (156 mg, 63%). Brown solid; TLC Rf 0.55 (EtOAc/Hexanes = 1/1); m.p. 170 °C; 1H NMR (CDCl3) δ 2.47 (s, 6H); 13C NMR (CDCl3) δ 156.4 (2C), 147.2, 141.3, 18.7 (2C); HRMS (FAB) m/z calculated for C6H8BrN2O [M + H]+ 201.9742, found 201.9739.
4.1.5. 2-Bromo-5-((2,6-difluoro-3,5-dimethoxybenzyl)oxy)-4,6-dimethylpyrimidine (13)
To a solution of 2-bromo-4,6-dimethylpyrimidin-5-ol (10) (90 mg, 0.44 mmol) in DMF (3 ml) were added 3-(bromomethyl)-2,4-difluoro-1,5-dimethoxybenzene (7) (94 mg, 0.35 mmol) and K2CO3 (304 mg, 2.20 mmol). The mixture was stirred at 80 °C for 12 h and then cooled to room temperature. It was quenched with ice water and stirred at 0 °C for 15 min. The precipitate formed in the mixture was collected by filtration and washed with ice water to obtain 13 (114 mg, 67%). White solid; TLC Rf 0.63 (EtOAc/Hexanes = 1/2); m.p. 145 °C; 1H NMR (CDCl3) δ 6.70 (t, J= 8.2 Hz, 1H), 4.96 (t, J= 1.8 Hz, 2H), 3.89 (s, 6H), 2.47 (s, 6H); 13 C NMR (CDCl3) δ 164.2 (2C), 149.5, 147.0 (d, J= 5.7 Hz), 145.7, 143.9 (d, J= 5.0 Hz), 143.7 (d, J= 5.0 Hz), 143.1 (d, J= 5.6 Hz), 113.1, 102.4, 62.5, 57.5 (2C), 18.9 (2C); HRMS (EI) m/z calculated for C15H15BrF2N2O3 [M]+ 388.0234, found 388.0239.
4.1.6. N-(2-Chloro-4-fluorophenyl)acetamide (36) [CAS RN: 399–35-9]
To a solution of 2-chloro-4-fluoroaniline (35) (5.0 ml, 41.87 mmol) in THF (10 ml) was added acetyl chloride (5.9 ml, 83.75 mmol) at 0 °C. The mixture was stirred at room temperature for 5 h. The precipitate formed in the reaction mixture was collected to obtain 36 (8.2 g) as a white solid then used in the next step reaction without further purification.
4.1.7. 2-Chloro-4-fluoro-6-nitroaniline (18) [CAS RN: 153505–32-9]
To a mixture of N-(2-chloro-4-fluorophenyl)acetamide (36) (1.0 g, 5.33 mmol) in acetic acid (2 ml) and concentrated H2SO4 (10 ml, 187.60 mmol) was added fuming HNO3 (334 µL, 8.0 mmol) dropwise at 0 °C. The mixture was stirred at 0 °C for 3 h and then it was poured into ice water and stirred at room temperature for 12 h. The mixture was neutralised using a solution of 6 M NaOH followed by EtOAc extraction. The combined organic solution was dried over MgSO4, filtered, and concentrated. After addition of an excessive amount of 1 M HCl, the mixture was refluxed for 5 h and then neutralised using a solution of 6 M NaOH followed by EtOAc extraction. The combined organic solution was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography with 1% to 5% EtOAc/Hexanes to obtain 18 (596 mg, 59%) as a yellow solid.
4.1.8. N-(2-Chloro-4-fluoro-6-nitrophenyl)-5-((2,6-difluoro-3,5-dimethoxybenzyl)oxy)-4,6-dimethylpyrimidin-2-amine (22 O)
To a mixture of 2-bromo-5-((2,6-difluoro-3,5-dimethoxybenzyl)oxy)-4,6-dimethylpyrimidine (13) (150 mg, 0.39 mmol) and 2-chloro-4-fluoro-6-nitroaniline (18) (110 mg, 0.58 mmol) in toluene (3 ml) were added Cs2CO3 (376 mg, 1.16 mmol), Xantphos (67 mg, 0.12 mmol), and Pd2(dba)3 (40 mg, 0.04 mmol). The mixture was refluxed for 20 h. and then diluted with DCM and washed with brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography with 10% to 20% EtOAc/Hexanes to obtain 22O (136 mg, 71%). Yellow solid; TLC Rf 0.32 (EtOAc/Hexanes = 1/2); m.p. 170 °C; 1H NMR (CDCl3) δ 7.64 (dd, J= 7.9, 3.0 Hz, 1H), 7.45 (dd, J= 7.3, 3.0 Hz, 1H), 7.32 (s, 1H), 6.69 (t, J= 8.2 Hz, 1H), 4.86 (t, J= 1.8 Hz, 2H), 3.89 (s, 6H), 2.35 (s, 6H); 13C NMR (CDCl3) δ 161.8, 158.9, 154.9, 153.5, 147.2 (d, J= 5.6 Hz), 144.8, 144.5 (d, J= 8.8 Hz), 143.8 (d, J= 5.0 Hz), 143.7 (d, J= 4.9 Hz), 143.3 (d, J= 5.8 Hz), 130.7 (d, J= 10.0 Hz), 128.0 (d, J= 4.0 Hz), 121.6 (d, J= 25.3 Hz), 113.7 (t, J= 17.9 Hz), 111.8 (d, J= 26.8 Hz), 102.4 (t, J= 2.3 Hz), 62.5 (t, J= 3.6 Hz), 57.6 (2C), 18.6 (2C); HRMS (EI) m/z calculated for C21H18ClF3N4O5 [M]+ 498.0918, found 498.0914.
4.1.9. 6-Chloro-N1-(5-((2,6-difluoro-3,5-dimethoxybenzyl)oxy)-4,6-dimethylpyrimidin-2-yl)-4-fluorobenzene-1,2-diamine (23 O)
To a mixture of N-(2-chloro-4-fluoro-6-nitrophenyl)-5-((2,6-difluoro-3,5-dimethoxybenzyl)oxy)-4,6-dimethylpyrimidin-2-amine (22O) (120 mg, 0.24 mmol) in EtOH (3 ml) was added SnCl2·2H2O (271 mg, 1.20 mmol). The reaction mixture was refluxed for 12 h and then concentrated. The residue was diluted with EtOAc and saturated K2CO3 followed by EtOAc extraction. The combined organic solution was dried over MgSO4, filtered, and concentrated. The residual solid was washed with EtOAc/Hexanes to obtain 23O (101 mg, 89%). Grey solid; TLC Rf 0.48 (DCM/MeOH = 20/1); m.p. 260 °C; 1H NMR (DMSO-d6) δ 8.04 (s, 1H), 7.07 (t, J= 7.9 Hz, 1H), 6.61–6.36 (m, 2H), 5.37 (s, 2H), 4.81 (s, 2H), 3.87 (s, 6H), 2.18 (s, 6H); 13C NMR (DMSO-d6) δ 162.4, 160.2, 158.5, 157.0, 148.8 (d, J= 13.5 Hz), 145.6 (d, J= 6.0 Hz), 143.4 (d, J= 4.6 Hz), 143.2 (d, J= 4.2 Hz), 142.0, 141.7, 134.0 (d, J= 14.6 Hz), 118.7, 113.3 (t, J= 18.3 Hz), 102.7 (d, J= 26.2 Hz), 101.7, 99.3 (d, J= 25.0 Hz), 61.9, 56.9 (2C), 18.3 (2C); HRMS (EI) m/z calculated for C21H20ClF3N4O3 [M]+ 468.1176, found 468.1177.
4.1.10. N-(3-Chloro-2-((5-((2,6-difluoro-3,5-dimethoxybenzyl)oxy)-4,6-dimethylpyrimidin-2-yl)amino)-5-fluorophenyl)acrylamide (6 O)
To a solution of 6-chloro-N1-(5-((2,6-difluoro-3,5-dimethoxybenzyl)oxy)-4,6-dimethylpyrimidin-2-yl)-4-fluorobenzene-1,2-diamine (23O) (30 mg, 0.06 mmol) in DCM (2 ml) were added triethylamine (27 µL, 0.19 mmol) and acryloyl chloride (7.8 µL, 0.10 mmol) dropwise at 0 °C. The mixture was stirred at room temperature for 12 h and then diluted with DCM and saturated NaHCO3 followed by DCM extraction. The combined organic solution was dried over MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography with 30% to 40% EtOAc/Hexanes to obtain 6O (7 mg, 20%). White solid; TLC Rf 0.35 (EtOAc/Hexanes = 1/1); m.p. 160 °C; 1H NMR (CDCl3) δ 9.12 (s, 1H), 8.01 (dd, J= 10.5, 2.7 Hz, 1H), 6.98 (dd, J= 7.7, 2.9 Hz, 1H), 6.70 (t, J= 8.2 Hz, 2H), 6.32 (dd, J= 16.9, 1.1 Hz, 1H), 6.11 (dd, J= 16.9, 10.1 Hz, 1H), 5.71 (dd, J= 10.1, 1.1 Hz, 1H), 4.90 (t, J= 1.7 Hz, 2H), 3.89 (s, 6H), 2.38 (s, 6H); 13C NMR (CDCl3) δ 163.7, 162.4, 162.2, 158.3, 156.4, 147.1 (d, J= 5.8 Hz), 144.2, 143.9 (d, J= 5.0 Hz), 143.7 (d, J= 5.0 Hz), 143.2 (d, J= 5.7 Hz), 136.8 (d, J= 12.9 Hz), 131.6, 131.1 (d, J= 12.7 Hz), 127.9, 124.3 (d, J= 3.4aHz), 113.6 (t, J= 17.8 Hz), 112.4 (d, J= 26.3 Hz), 109.4 (d, J= 27.1 Hz), 102.3 (t, J= 2.5 Hz), 62.6 (t, J= 3.6 Hz), 57.5 (2C), 18.9 (2C); HRMS (EI) m/z calculated for C24H22ClF3N4O4 [M]+ 522.1282, found 522.1285.
4.2. Biological evaluation
4.2.1. FGFR kinase assay
A FGFR4 kinase assay was performed at the Reaction Biology Corporation (Malvern, PA, USA) using a Kinase HotSpotSM assay platform (www.reactionbiology.com, last accessed at 19/10/2020). Briefly, human FGFR4 kinase (5–10 mU), peptide substrate, and poly[Glu:Tyr] (4:1) 0.2 mg/ml were prepared in a reaction buffer with a final volume of 25 µL. The compounds were delivered into the reaction, followed ∼20 min. later by the addition of a mixture of ATP and [γ-33P-ATP] (specific activity approx. 500 cpm/pmol, concentration as required) to a final concentration of 10 µM. After incubation for 40 min. at 25 °C, the reaction was stopped by the addition of a 3% phosphoric acid solution. Then, the reaction was spotted onto a P30 filtermat, and unbound phosphate was removed by washing 3 times for 5 min in 75 mM phosphoric acid and once in methanol prior to drying and scintillation counting. The background counting derived from the control reactions containing the inactive enzyme was subtracted, and specific kinase activity data were expressed as the percentage of remaining kinase activity in the test compounds compared to the vehicle (dimethyl sulfoxide) reactions. IC50 values and curve fits were obtained using GraphPad Prism 5 software.
FGFR1 ∼ 3 kinase assays were performed using the adenosine diphosphate (ADP)-glow kinase assay kit of FGFR1, FGFR2, and FGFR3 kinase enzyme systems (Promega, WI, USA) in accordance with the manufacturer’s instructions. Kinase activity was detected by the addition 50 µM adenosine triphosphate (ATP) to a mixture of 0.2 µg/µL poly (Glu4, Tyr1), test drug (1, 6A, 6O), and corresponding enzyme, FGFR1 (1.5 ng/µL), FGFR2 (3 ng/µL), or FGFR3 (6 ng/µL). The reaction was carried out at 25 °C for 1 h in a total volume of 25 µL. Subsequently, 25 µL of ADP-Glow reagent was added to the mixture, and then incubated for 40 min at 25 °C. After the addition of 50 µL of ADP detection reagents for 30 min at 25 °C, luminescence was measured using a Fluostar Omega microplate reader (BMG LABTECH GmbH, Ortenberg, Germany).
4.2.2. Cell lines and culture
Human HCC cell lines, Hep3B and Huh7 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). An H6c7 human normal pancreatic duct epithelial cell line was purchased from Kerafast (Boston, MA, USA). Hep3B and Huh7 cells were cultured in Dulbecco′s Modified Eagle′s Medium (DMEM) (Hyclone, Logan, UT, USA). The media were supplemented with 10% foetal bovine serum (FBS) (Gibco/ThermoFisher Scientific) and 1% penicillin/streptomycin (Gibco/ThermoFisher Scientific). H6c7 cells were maintained in a keratinocyte serum-free medium supplemented with a recombinant endothelial growth factor (rEGF) and bovine pituitary extract (Gibco/ThermoFisher Scientific). All the cells were incubated at 37 °C under a 5% CO2 atmosphere.
4.2.3. Cell proliferation assay
Cells were seeded at a density of 5000 cells/well in a 96-well plate. After overnight incubation, the cells were serum-starved using 1% FBS for 24 h. The next day, the cells were pre-treated with the different concentrations (0.1, 1, 3, 10, 30, 100 µM) of drugs for 1 h prior to the treatment with serum (10% FBS). After 48 h of incubation, 3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye solution was added and incubated for 4 h. Next, DMSO was added, and after 30 min, the colour intensities were measured using a microplate reader (Versamax, Molecular Devices, Inc., USA) at 490 nm.
4.2.4. Cytotoxicity assay
The cytotoxicity of the compounds was assessed by measuring the cell viability decrease using the MTT staining method. Briefly, H6c7 cells were seeded in a 96-well plate (Falcon, USA) at a density of 4 × 104 cells per well in keratinocyte serum-free medium. After 24 h, Cells were incubated with different concentrations (0.1, 1, 3, 10, 30, and 100) µM of each compound for 48 h, and the cell viability was measured using MTT assay. Optical density was measured at 540 nm using a microplate reader (BMG LABTECH).
4.2.5. Cam (chick chorioallantoic membrane) tumour model
Fertile chicken eggs were purchased from Byeolbichon Farm (Gyeongbuk, South Korea) and incubated at 37 °C and under 55% relative humidity. On the 9th day of egg incubation (post-fertilization), false air sac was generated on the relatively flat side of eggs by a negative pressure technique. A small window (1 cm2) was created on the false air sac surface of the eggs by separating the shell and membrane beneath (technique) using a grinding wheel (Dremel, Racine, WI, USA). Next, Hep3B cells were loaded at a density of 1.5 × 106 cells/CAM with or without compound. After four days of drug treatment, the tumour weight, number of vessel branch points within the tumour region were analysed.
The chick embryo experiments were approved beforehand by the Institutional Animal Care and Use Committee of Yeungnam University and were performed accordingly the guidelines issued by the Institute of Laboratory Animal Resources (1996) and Yeungnam University (The care and use of animals 2009).
4.2.6. Statistical analysis
The results are presented as the mean ± SEM and were analysed using one-way ANOVA followed by the Newman-Keuls comparison method using the GraphPad Prism software (version 5.0) (San Diego, CA, USA). P values less than 0.05 were considered statistically significant.
4.3. Computational study
Flare version 5.0 from Cresset (http://www.cresset-group.com/flare/) was used for covalent docking for this study. The crystal structure of the complex of FGFR4 and a 2-aminopyridine-based inhibitor (PDB ID 7DTZ) was chosen for docking owing to its high crystal resolution (2.01 Å) and the possession of strong hydrogen bonding interactions between the guanidyl group of Arg483 and the carbonyl oxygen of the acrylamide moiety of the ligand, which would help keep the ligands within the binding cavity in a proper orientation during the docking. Docking parameters were set up to use Cys552 as the covalent bonding residue and to use “Very Accurate but Slow” as the calculation method. For post-docking analysis, LF Rank Score was used to identify correct binding pose, while LF VSscore was used to rank compounds in a virtual screening contextCitation41.
Procedures for the synthesis of compound 6O are provided here. Synthetic procedures for all compounds are described in Supplementary Material.
Supplemental Material
Download PDF (11.6 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424.
- Ganten TM, Stauber RE, Schott E, et al. Sorafenib in patients with hepatocellular carcinoma-results of the observational INSIGHT study. Clin Cancer Res 2017;23:5720–8.
- Li D, Sedano S, Allen R, et al. Current treatment landscape for advanced hepatocellular carcinoma: patient outcomes and the impact on quality of life. Cancers (Basel) 2019;11:841.
- Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell 2018;33:581–98.
- Partanen J, Mäkelä TP, Eerola E, et al. FGFR-4, a novel acidic fibroblast growth factor receptor with a distinct expression pattern. Embo J 1991;10:1347–54.
- Prieto-Dominguez N, Shull AY, Teng Y. Making way for suppressing the FGF19/FGFR4 axis in cancer. Future Med Chem 2018;10:2457–70.
- Huang X, Gollin SM, Raja S, Godfrey TE. High-resolution mapping of the 11q13 amplicon and identification of a gene, TAOS1 that is amplified and overexpressed in oral cancer cells. Proc Natl Acad Sci U S A 2002;99:11369–174.
- Sawey ET, Chanrion M, Cai C, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell 2011;19:347–58.
- Tiong KH, Tan BS, Choo HL, et al. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival. Oncotarget 2016;7:57633–50.
- Zhang X, Kong M, Zhang Z, et al. FGF19 genetic amplification as a potential therapeutic target in lung squamous cell carcinomas. Thorac Cancer 2017;8:655–65.
- Ho HK, Pok S, Streit S, et al. Fibroblast growth factor receptor 4 regulates proliferation, anti-apoptosis and alpha-fetoprotein secretion during hepatocellular carcinoma progression and represents a potential target for therapeutic intervention. J Hepatol 2009;50:118–27.
- Penault-Llorca F, Bertucci F, Adélaïde J, et al. Expression of FGF and FGF receptor genes in human breast cancer. Int J Cancer 1995;61:170–6.
- Koole K, van Kempen PM, van Bockel LW, et al. fgfr4 is a potential predictive biomarker in oral and oropharyngeal squamous cell carcinoma. Pathobiology 2015;82:280–9.
- Helsten T, Elkin S, Arthur E, et al. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res 2016;22:259–67.
- Gowardhan B, Douglas DA, Mathers ME, et al. Evaluation of the fibroblast growth factor system as a potential target for therapy in human prostate cancer. Br J Cancer 2005;92:320–7.
- Tang S, Hao Y, Yuan Y, et al. Role of fibroblast growth factor receptor 4 in cancer. Cancer Sci 2018;109:3024–31.
- Liu H, Niu D, Tham Sjin RT, et al. Discovery of selective, covalent FGFR4 inhibitors with antitumor activity in models of hepatocellular carcinoma. ACS Med Chem Lett 2020;11:1899–904.
- Joshi JJ, Coffey H, Corcoran E, et al. H3B-6527 is a potent and selective inhibitor of FGFR4 in FGF19-driven hepatocellular carcinoma. Cancer Res 2017;77:6999–7013.
- Guagnano V, Furet P, Spanka C, et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem 2011;54:7066–83.
- Hagel M, Miduturu C, Sheets M, et al. First selective small molecule inhibitor of fgfr4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov 2015;5:424–37.
- Reynolds D, Hao MH, Wang J, Prajapati S, Satoh T, Selvaraj A. Eisai R&D Management Co., LTD. FGFR4 inhibitors. WO2016164703A1. 2016 Oct 13.
- Wang Y, Chen Z, Dai M, et al. Discovery and optimization of selective FGFR4 inhibitors via scaffold hopping. Bioorg Med Chem Lett 2017;27:2420–3.
- Mo C, Zhang Z, Guise CP, et al. 2-aminopyrimidine derivatives as new selective fibroblast growth factor receptor 4 (FGFR4) inhibitors. ACS Med Chem Lett 2017;8:543–8.
- Weiss A, Adler F, Buhles A, et al. Graus porta D. FGF401, a first-in-class highly selective and potent FGFR4 inhibitor for the treatment of FGF19-driven hepatocellular cancer. Mol Cancer Ther 2019;18:2194–206.
- Kim RD, Sarker D, Meyer T, et al. First-in-human phase I study of fisogatinib (BLU-554) validates aberrant FGF19 signaling as a driver event in hepatocellular carcinoma. Cancer Discov 2019;9:1696–707.
- Rezende Miranda R, Fu Y, Chen X, et al. Development of a potent and specific FGFR4 inhibitor for the treatment of hepatocellular carcinoma. J Med Chem 2020;63:11484–97.
- Packer LM, Pollock PM. Paralog-specific kinase inhibition of FGFR4: adding to the arsenal of anti-FGFR agents. Cancer Discov 2015;5:355–7.
- Awasthi BP, Chaudhary P, Guragain D, et al. Synthesis and anti-hepatocellular carcinoma activity of aminopyridinol-sorafenib hybrids. J Enzyme Inhib Med Chem 2021;36:1884–97.
- Karmacharya U, Guragain D, Chaudhary P, et al. Novel pyridine bioisostere of cabozantinib as a potent c-met kinase inhibitor: synthesis and anti-tumor activity against hepatocellular carcinoma. Int J Mol Sci 2021;22:9685.
- Patani GA, LaVoie EJ. Bioisosterism: a rational approach in drug design. Chem Rev 1996;96:3147–76.
- Lima LM, Barreiro EJ. Bioisosterism: a useful strategy for molecular modification and drug design. Curr Med Chem 2005;12:23–49.
- Zeng D, Mi Q, Sun H, Wang H. A convenient synthesis of 14C-labelled resveratrol. J Label Compd Radiopharm 2004;47:167–74.
- Kim DG, Kang Y, Lee H, et al. 6-Amino-2,4,5-trimethylpyridin-3-ols: a new general synthetic route and antiangiogenic activity. Eur J Med Chem 2014;78:126–39.
- Dornow A, Hell H. Syntheses of nitrogen-containing heterocycles. XXIII. The preparation of 5-acetyloxazoles and their conversion to 5-hydroxypyrimidines. Chem Ber 1960;93:1998–2001.
- Glišić BĐ, Hoffmann M, Warżajtis B, et al. Selectivity of the complexation reactions of four regioisomeric methylcamphorquinoxaline ligands with gold(III): X-ray, NMR and DFT investigations. Polyhedron 2016;105:137–49.
- Garst ME, Dolby LJ, Esfandiari S, Mackenzie VR, Avey AA, Muchmore DC, Cooper GK, Malone TC. Allergan, Inc. Process for preparing isomerically pure prodrugs of proton pump inhibitors such as omeprazole and pantoprazole. US20050038076. 2005 Feb 17.
- Lin X, Yosaatmadja Y, Kalyukina M, et al. Rotational freedom, steric hindrance, and protein dynamics explain BLU554 selectivity for the hinge cysteine of FGFR4. ACS Med Chem Lett 2019;10:1180–6.
- Guagnano V, Kauffmann A, Wöhrle S, et al. Graus-Porta D. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov 2012;2:1118–33.
- Zhao H, Lv F, Liang G, et al. FGF19 promotes epithelial-mesenchymal transition in hepatocellular carcinoma cells by modulating the GSK3β/β- catenin signaling cascade via FGFR4 activation. Oncotarget 2016;7:13575–86.
- Kanzaki H, Chiba T, Ao J, et al. The impact of FGF19/FGFR4 signaling inhibition in antitumor activity of multi-kinase inhibitors in hepatocellular carcinoma. Sci Rep 2021;11:5303.
- Stroganov OV, Novikov FN, Stroylov VS, et al. Lead finder: an approach to improve accuracy of protein-ligand docking, binding energy estimation, and virtual screening. J Chem Inf Model 2008;48:2371–85.