
Abstract
As regioisomers/bioisosteres of 1a, a 4-phenylbenzamide tranylcypromine (TCP) derivative previously disclosed by us, we report here the synthesis and biological evaluation of some (hetero)arylbenzoylamino TCP derivatives 1b-6, in which the 4-phenyl moiety of 1a was shifted at the benzamide C3 position or replaced by 2- or 3-furyl, 2- or 3-thienyl, or 4-pyridyl group, all at the benzamide C4 or C3 position. In anti-LSD1-CoREST assay, all the meta derivatives were more effective than the para analogues, with the meta thienyl analogs 4b and 5b being the most potent (IC50 values = 0.015 and 0.005 μM) and the most selective over MAO-B (selectivity indexes: 24.4 and 164). When tested in U937 AML and prostate cancer LNCaP cells, selected compounds 1a,b, 2b, 3b, 4b, and 5a,b displayed cell growth arrest mainly in LNCaP cells. Western blot analyses showed increased levels of H3K4me2 and/or H3K9me2 confirming the involvement of LSD1 inhibition in these assays.
Graphical Abstract
1. Introduction
Protein post-translational modifications (PTMs), such as ac(et)ylation, methylation, and phosphorylation, play an important role in regulating many protein functions. Among such PTMs, reversible methylation/demethylation of lysine residues is typically involved in regulating the chromatin environment and eukaryotic gene expressionCitation1,Citation2.
In mammals there are two families of lysine demethylases (KDMs): the lysine-specific demethylases (LSDs, including LSD1 and −2, also called KDM1A and -B) and the jumonij-containing demethylases, called KDM2-8Citation2. LSD1 is a flavoenzyme that specifically removes the methyl groups of H3K4me1/2 and H3K9me1/2 in complexes with various chromatin-associated factors including CoRESTCitation3–7. LSD2 also demethylates histone H3, mainly to mono- and dimethylated Lys4, although its activity is weaker than that of LSD1 in vitroCitation8.
LSD1 plays a vital role in a broad spectrum of biological processes, including embryonic development, stem cell maintenance and differentiation, and is also a key player in oncogenic processes, including cancer cell growth and metastasis. LSD1 has been reported to be overexpressed in a variety of cancers, and its inactivation or downregulation of its expression inhibits the development of cancer cellsCitation2,Citation9. LSD1 regulates cellular signalling pathways in various cancerous entities such as breast cancer, where it interacts in a multiprotein complex containing deacetylase (NuRD)Citation10. In prostate cancer, high levels of LSD1 are associated with cancer progression and metastasis and therefore LSD1 levels could be a useful biomarkerCitation11,Citation12. LSD1 and LSD2 share a similar catalytic domain (45% sequence identity) that is structurally homologous with the amine oxidases, a class of flavin-dependent enzymes that act on biogenic aminesCitation5,Citation13. Among these proteins, human monoamine oxidases (MAOs) A and B have been the subject of more than 50 years of research that has led to the development of a multitude of inhibitors including antidepressant and antiparkinson drugsCitation14. Their similarity in the catalytic and structural properties prompted the investigation of anti-MAO drugs as potential LSD1 inhibitorsCitation15. Among them, tranylcypromine (TCP) is one of the most reliable molecules able to inhibit LSD1 irreversibly via a covalent interaction with the FAD cofactor and has been widely used as a fragment to create valuable LSD1 inhibitorsCitation16.
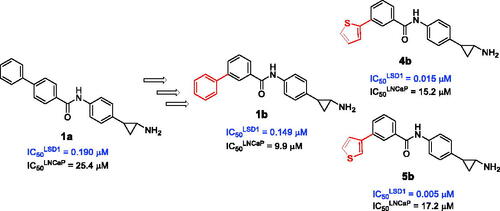
In the past, we have described numerous TCP-containing compounds as covalent LSD1 inhibitorsCitation17–26, and among them, MC2580Citation17,Citation26,Citation27 and MC2584Citation17,Citation18 () were the most effective prototypes at enzyme level. MC2580 showed a quite good selectivity for LSD1 over MAO-B (not MAO-A), and it strongly enhanced the efficacy of retinoic acid on growth and differentiation of acute promyelocytic leukaemia (APL) cells, including primary murine APL blastsCitation17. The two enantiomers of MC2584 displayed at 0.25 μM strong (64%, 1 R,2S, and 83.4%, 1S,2R) inhibition on colony formation in the same murine APL blastsCitation18. The introduction of a phenyl ring at the para position of the MC2584 benzoylamino moiety led to MC2652 (1a) (), less potent in LSD1 inhibition but more selective towards MAOs, and able to arrest MV4-11 acute myeloid leukaemia (AML) and NB4 APL cell growth with IC50 values at low, single-digit micromolar levelsCitation25.
Figure 1. TCP-based LSD1 inhibitors disclosed by us.

Based on these data, we designed and synthesised new analogs of 1a as potential inhibitors of LSD1, by shifting the 4-phenyl ring from the 4- to the 3-position of the benzoylamino moiety (1b) () and replacing this 4- or 3-phenyl ring with different heterocyclic moieties such as furan, thiophene, and pyridine (2–6) (), aiming to improve their potency and LSD1 selectivity. Selected compounds were tested against U937 AML and prostate LNCaP cancer cells to assess their antiproliferative potential.
Figure 2. Novel TCP-based analogs as LSD1 inhibitors 1 b-6.

2. Materials and methods
2.1. Chemistry
Melting points were determined on a Buchi 530 melting point apparatus and are uncorrected. 1H and 13 C NMR spectra were recorded at 400 MHz and 100 MHz, respectively, on a Bruker AC 400 spectrometer; chemical shifts are reported in δ (ppm) units relative to the internal reference tetramethylsilane (Me4Si). EIMS spectra were recorded with a Thermo MSQ Plus spectrometer; only molecular ions (M+) and base peaks are given. All the compounds were routinely checked by TLC, 1H NMR and 13 C NMR spectra. TLC was performed on aluminum-backed silica gel plates (Merck DC, Alufolien Kieselgel 60 F254) with spots visualised by UV light. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. The concentration of solutions after reactions and extractions involved the use of a rotary evaporator operating at reduced pressure of ca. 20 Torr. Organic solutions were dried over anhydrous sodium sulphate. Elemental analysis has been used to determine the purity of the described compounds, that is >95%. Analytical results are within ± 0.40% of the theoretical values. All chemicals were purchased from Sigma Aldrich, Milan (Italy), or from TCI Europe, Zwjindrecht (Belgium), and were of the highest purity.
2.2. General procedure for the synthesis of the tert-butyl (2-(4-(4- or 3-(phenyl/heteroaryl)benzamido)phenyl)cyclopropyl)carbamates 14–19 example:
tert-butyl (2-(4-([1,1′-biphenyl]-3-carboxamido)phenyl)cyclopropyl)carbamate (14b). Triethylamine (1.05 mmol, 0.15 ml) and benzotriazol-1-yloxytripyrrolidino-phosphonium hexafluorophosphate (PyBOP) (0.24 mmol, 0.126 g) were added to a solution of [1,1′-biphenyl]-3-carboxylic acid (0.20 mmol, 0.04 g) in anhydrous N,N-dimethylformamide (3 ml) under a nitrogen atmosphere. The resulting mixture was stirred for 45 min at room temperature followed by the addition of tert-butyl (2-(4-aminophenyl)cyclopropyl)carbamate 13Citation17 (0.24 mmol, 0.06 g) under a nitrogen atmosphere, and the reaction was stirred overnight. The reaction was quenched with brine (50 ml) and the precipitated solid was filtered off and washed with water (3 × 5 ml). The residue was purified by chromatographic column on silica gel eluting with ethyl acetate/chloroform 1:3 to afford the pure 14b. Chemical and physical data: m.p., 212–214 °C; yield, 64%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.06–1.09 (m, 2H, CH2 cyclopropane), 1.39 (s, 9H, (CH3)3), 1.86–1.89 (m, 1H, CHNH2 cyclopropane), 2.62 (bs, 1H, PhCH cyclopropane), 7.09– 7.11 (d, 2H, benzene protons, J = 8 Hz), 7.24 (bs, 1H, benzene protons), 7.42–7.44 (t, 1H, benzene proton, J = 8 Hz), 7.49–7.46 (t, 2H, benzene protons, J = 12 Hz), 7.61–7.64 (t, 1H, benzene proton, J = 12 Hz), 7.67–7.69 (d, 2H, benzene protons, J = 8 Hz), 7.71–7.73 (d, 2H, benzene protons, J = 8 Hz), 7.87–7.89 (m, 1H, benzene proton), 7.93–7.95 (d, 1H, benzene proton, J = 8 Hz), 8.22 (bs, 1H, CONH), 10.28 (bs, 1H, CONH), ppm; 13 C (100 MHz, DMSO-d6) δ 166.9, 155.44, 140.6, 139.7, 139.1, 137.9, 133.4, 130.9, 130.2, 129.1, 128.1, 127.9, 127.7 (3 C), 127.3, 127.1 (3 C), 121.2 (3 C), 79.7, 33.8, 28.3, 26.3, 18.1; MS (EI) m/z: 428.21 [M]+.
tert-Butyl (2-(4-(4-(furan-2-yl)benzamido)phenyl)cyclopropyl)carbamate (15a). Chemical and physical data: m.p., 223–225 °C; yield, 49%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.09–1.12 (m, 2H, CH2 cyclopropane), 1.39 (s, 9H, (CH3)3), 1.98 (bs, 1H, CHNH2 cyclopropane), 2.63 (bs, 1H, PhCH cyclopropane), 4.81 (bs, 1H, CONH), 6.45–6.46 (m, 1H, furan proton), 6.71–6.72 (d, 1H, furan proton, J = 4 Hz), 7.07–7.09 (d, 2H, benzene protons, J = 8 Hz), 7.46–7.49 (m, 3H, furan and benzene protons), 7.69–7.71 (d, 2H, benzene protons, J = 8 Hz), 7.76 (bs, 1H, CONH), 7.81–7.83 (d, 2H, benzene protons, J = 8 Hz) ppm; 13 C (100 MHz, DMSO-d6) δ 168.5, 155.4, 153.2, 142.4 (2 C), 139.1, 137.9, 137.2, 134.2, 128.9 (2 C), 127.3 (2 C), 125.1 (2 C), 121.2 (2 C), 111.0, 107.7, 79.7 (2 C), 33.8, 28.3, 26.3, 18.1; MS (EI) m/z: 418.19 [M]+.
tert-Butyl (2-(4-(3-(furan-2-yl)benzamido)phenyl)cyclopropyl)carbamate (15b). Chemical and physical data: m.p., 197–200 °C; yield, 77%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.28–1.29 (m, 2H, CH2 cyclopropane), 1.51 (s, 9H, (CH3)3), 1.96–198 (m, 1H, CHNH2 cyclopropane), 2.64 (bs, 1H, PhCH cyclopropane), 4.85 (bs, 1H, CONH), 6.43–6.45 (d, 1H, furan proton, J = 8 Hz), 6.69–6.71 (d, 1H, furan proton, J = 8 Hz), 7.10–7.12 (d, 2H, benzene protons, J = 8 Hz), 7.44–7.45 (m, 2H, benzene protons), 7.48–7.50 (d, 2H, benzene protons, J = 8 Hz), 7.65–7.67 (d, 1H, furan proton, J = 8 Hz), 7.75–7.79 (m, 2H, benzene protons), 8.08 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 166.7, 155.4, 154.2, 142.3 (2 C), 139.1, 137.9, 133.3, 130.7 (2 C), 128.3, 128.1, 127.3 (2 C), 126.5, 121.2 (2 C), 111.5, 107.7, 79.7 (2 C), 33.8, 28.3, 26.3, 18.1; MS (EI) m/z: 418.19 [M]+.
tert-Butyl (2-(4-(4-(furan-3-yl)benzamido)phenyl)cyclopropyl)carbamate (16a). Chemical and physical data: m.p., 232–234 °C; yield, 49%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.08–1.09 (m, 2H, CH2 cyclopropane), 1.39 (s, 9H, (CH3)3), 1.97–1.98 (m, 1H, CHNH2 cyclopropane), 2.63 (bs, 1H, PhCH cyclopropane), 4.80 (bs, 1H, CONH), 6.68 (bs, 1H, furan proton), 7.07–7.09 (d, 2H, benzene protons, J = 8 Hz), 7.43–7.46 (m, 3H, furan and benzene protons), 7.51–7.53 (d, 2H, benzene protons, J = 8 Hz), 7.71–7.73 (m, 1H, furan protons), 7.75 (bs, 1H, CONH), 7.79–7.81 (d, 2H, benzene protons, J = 8 Hz) ppm; 13 C (100 MHz, DMSO-d6) δ 168.2, 155.5, 143.0, 140.77, 139.1 (2 C), 138.4, 137.8, 136.6, 130.1 (2 C), 127.3 (2 C), 127.2, 126.6 (2 C), 121.1 (2 C), 108.8, 79.7 (2 C), 33.9, 28.3, 26.3, 18.5; MS (EI) m/z: 418.19 [M]+.
tert-Butyl (2-(4-(3-(furan-3-yl)benzamido)phenyl)cyclopropyl)carbamate (16b). Chemical and physical data: m.p., 196–198 °C; yield, 41%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.08–1.10 (m, 2H, CH2 cyclopropane), 1.39 (s, 9H, (CH3)3), 1.97–1.99 (m, 1H, CHNH2 cyclopropane), 2.64 (bs, 1H, PhCH cyclopropane), 4.80 (bs, 1H, CONH), 6.69 (bs, 1H, furan proton), 7.08–7.10 (d, 2H, benzene protons, J = 8 Hz), 7.41–7.47 (m, 4H, furan and benzene protons), 7.57–7.62 (m, 2H, benzene protons), 7.74 (bs, 2H, furan and benzene protons), 7.92 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 166.4, 155.5, 141.9, 140.7, 139.1, 138.4, 133.9 (2 C), 133.9, 130.3, 129.9, 128.5, 127.4, 127.3 (2 C), 126.8 (2 C), 121.1 (2 C), 109.5, 79.7, 33.9, 28.3, 26.3, 18.5; MS (EI) m/z: 418.19 [M]+.
tert-Butyl (2-(4-(4-(thiophen-2-yl)benzamido)phenyl)cyclopropyl)carbamate (17a). Chemical and physical data: m.p., 226–228 °C; yield, 35%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.18–1.21 (m, 2H, CH2 cyclopropane), 1.39 (s, 9H, (CH3)3), 1.95–1.97 (m, 1H, CHNH2 cyclopropane), 2.64 (bs, 1H, PhCH cyclopropane), 4.78 (bs, 1H, CONH), 7.05–7.09 (m, 3H, thiophene protons), 7.29–7.30 (m, 1H, benzene proton), 7.34– 7.36 (m, 1H, benzene protons), 7.47–7.49 (d, 2H, benzene protons, J = 8 Hz), 7.64–7.66 (d, 2H, benzene protons, J = 8 Hz), 7.70 (bs, 1H, CONH), 7.79–7.82 (d, 2H, benzene protons, J = 12 Hz), ppm; 13 C (100 MHz, DMSO-d6) δ 168.2, 155.5 (2 C), 143.0, 139.2, 139.1, 138.4, 136.9, 128.6 (2 C), 127.9, 127.3 (2 C), 126.9, 126.8 (2 C), 124.7, 121.1 (2 C), 79.7, 33.9 (2 C), 28.3, 26.3, 18.5; MS (EI) m/z: 434.17 [M]+.
tert-Butyl (2-(4-(3-(thiophen-2-yl)benzamido)phenyl)cyclopropyl)carbamate (17b). Chemical and physical data: m.p., 189–191 °C; yield, 55%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.12–1.15 (m, 2H, CH2 cyclopropane), 1.45 (s, 9H, (CH3)3), 2.02–2.04 (m, 1H, CHNH2 cyclopropane), 2.70 (bs, 1H, PhCH cyclopropane), 4.82 (bs, 1H, CONH), 7.09–7.12 (t, 1H, thiophene proton, J = 12 Hz), 7.15–7.17 (d, 2H, benzene proton, J = 8 Hz), 7.32– 7.34 (d, 1H, thiophene proton, J = 8 Hz), 7.39–7.40 (d, 1H, benzene proton, J = 4 Hz), 7.48–7.51 (m, 1H, thiophene proton), 7.53–7.56 (d, 2H, benzene protons, J = 12 Hz), 7.70–7.78 (m, 3H, benzene protons), 8.09 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 165.9, 155.5 (2 C), 141.70, 139.1, 138.4, 135.9 (2 C), 132.8, 130.3, 128.5, 128.3, 128.1, 127.8, 127.3 (2 C), 126.5, 124.4, 121.1 (2 C), 79.7, 33.9, 28.3, 26.3, 18.5; MS (EI) m/z: 434.17 [M]+.
tert-Butyl (2-(4-(4-(thiophen-3-yl)benzamido)phenyl)cyclopropyl)carbamate (18a). Chemical and physical data: m.p., > 287 °C; yield, 40%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.09–1.11 (m, 2H, CH2 cyclopropane), 1.39 (s, 9H, (CH3)3), 1.97–1.98 (m, 1H, CHNH2 cyclopropane), 2.64 (bs, 1H, PhCH cyclopropane), 4.78 (bs, 1H, CONH), 7.08–7.10 (m, 2H, thiophene protons), 7.37 (bs, 2H, benzene protons), 7.48–7.51 (m, 3H, thiophene and benzene protons), 7.61–7.63 (d, 2H, benzene protons, J = 8 Hz), 7.71 (bs, 1H, CONH), 7.81–7.83 (d, 2H, benzene protons, J = 8 Hz) ppm; 13 C (100 MHz, DMSO-d6) δ 168.5, 155.4, 141.7, 140.5, 139.1, 137.9, 136.8, 129.7 (3 C), 127.7, 127.3, 127.2 (3 C), 127.1, 122.4, 121.2 (3 C), 79.7, 33.8, 28.3, 26.3, 18.1; MS (EI) m/z: 434.17 [M]+.
tert-Butyl (2-(4-(3-(thiophen-3-yl)benzamido)phenyl)cyclopropyl)carbamate (18b). Chemical and physica data: m.p., 214–216 °C; yield, 56%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.09–1.11 (m, 2H, CH2 cyclopropane), 1.39 (s, 9H, (CH3)3), 1.98–2.02 (m, 1H, CHNH2 cyclopropane), 2.64 (bs, 1H, PhCH cyclopropane), 4.81 (bs, 1H, CONH), 7.07–7.09 (d, 2H, benzene protons, J = 8 Hz), 7.36–7.38 (m, 2H, thiophene and benzene protons), 7.41–7.47 (m, 4H, thiophene and benzene protons), 7.65–7.67 (t, 2H, thiophene and benzene protons, J = 8 Hz), 7.78 (bs, 1H, benzene protons), 8.03 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 166.9, 155.4, 140.8, 139.1, 138.6, 137.9, 133.6, 130.3, 130.2, 128.4, 127.4, 127.3 (3 C), 127.1, 127.1, 122.6, 121.2 (3 C), 79.7, 33.8, 28.3, 26.3, 18.1; MS (EI) m/z: 434.17 [M]+.
tert-Butyl (2-(4-(4-(pyridin-4-yl)benzamido)phenyl)cyclopropyl)carbamate (19a). Chemical and physical data: m.p., 198–200 °C; yield, 40%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.09–1.11 (m, 2H, CH2 cyclopropane), 1.44 (s, 9H, (CH3)3), 1.98–1.99 (m, 1H, CHNH2 cyclopropane), 2.64 (bs, 1H, PhCH cyclopropane), 4.81 (bs, 1H, CONH), 7.09–7.11 (m, 2H, benzene protons), 7.47–7.49 (m, 4H, pyridine and benzene protons), 7.67–7.69 (d, 2H, benzene protons, J = 8 Hz), 7.80 (bs, 1H, CONH), 7.91–7.93 (d, 2H, benzene protons, J = 8 Hz), 8.63–8.65 (m, 2H, pyridine protons) ppm; 13 C (100 MHz, DMSO-d6) δ 168.5, 155.4, 148.2 (2 C), 147.5, 141.8, 139.1, 137.9, 136.8 (2 C), 129.6 (2 C), 128.4 (2 C), 127.3 (2 C), 121.6 (2 C), 121.2 (2 C), 79.7, 33.8, 28.3, 26.3, 18.1 (2 C); MS (EI) m/z: 429.21 [M]+.
tert-Butyl (2-(4-(3-(pyridin-4-yl)benzamido)phenyl)cyclopropyl)carbamate (19b). Chemical and physical data: m.p., 158–160 °C; yield, 70%. 1H NMR (CDCl3, 400 MHz, δ; ppm) δ 1.15–1.16 (m, 2H, CH2 cyclopropane), 1.44 (s, 9H, (CH3)3), 2.03–2.04 (m, 1H, CHNH2 cyclopropane), 2.63 (bs, 1H, PhCH cyclopropane), 5.12 (bs, 1H, CONH), 7.13–7.15 (m, 2H, benzene protons), 7.55–7.62 (m, 6H, pyridine and benzene protons), 7.79–7.81 (d, 1H, benzene protons, J = 8 Hz), 7.93–7.95 (d, 1H, benzene protons, J = 8 Hz), 8.19 (bs, 1H, CONH), 8.64 (bs, 2H, pyridine protons) ppm; 13 C (100 MHz, DMSO-d6) δ 166.9, 155.4, 150.2 (2 C), 146.2, 139.3, 139.1, 137.9, 133.4, 131.5, 130.2, 128.1, 128.0, 127.3 (2 C), 121.7 (3 C), 121.2 (3 C), 79.7, 33.8, 28.3, 26.3, 18.1; MS (EI) m/z: 429.21 [M]+.
2.3. General procedure for the synthesis of the N-(4-(2-aminocyclopropyl)phenyl)-4- or 3-(phenyl/heteroaryl)benzamides 1–6 as hydrochloride salts example
N-(4-(2-Aminocyclopropyl)phenyl)-[1,1′-biphenyl]-3-carboxamide hydrochloride (1b). A solution of tert-butyl (2-(4-([1,1′-biphenyl]-3-carboxamido)phenyl)cyclopropyl)carbamate (14b) (0.12 mmol, 0.05 g) in dry THF (8 ml) was treated with 4 N HCl in dioxane (1.20 mmol, 0.3 ml) at 0 °C, and the resulting reaction mixture was left under stirring at rt for 24 h. After the end of the reaction, the resulting precipitate was isolated by filtration and washed with Et2O to give the pure 1b as hydrochloride salt. Chemical and physical data: m.p., >250 °C (methanol); yield, 57%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.19–1.21 (m, 1H, CHH cyclopropane), 1.21–1.28 (m, 1H, CHH cyclopropane), 2.31–2.39 (m, 1H, CHNH2 cyclopropane), 2.77–2.81 (m, 1H, PhCH cyclopropane), 7.16–7.18 (d, 2H, benzene protons, J = 8 Hz), 7.41–7.45 (t, 1H, benzene proton, J = 16 Hz), 7.50–7.55 (t, 2H, benzene protons, J = 20 Hz), 7.60–7.65 (t, 1H, benzene protons, J = 20 Hz), 7.72–7.80 (m, 4H, benzene protons), 7.87–7.90 (d, 1H, benzene protons, J = 12 Hz), 7.92–7.97 (d, 1H, benzene protons, J = 20 Hz), 8.21–8.24 (m, 1H, benzene protons), 8.52 (bs, 3H, NH3+), 10.37 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 166.9, 140.6, 139.7, 137.8, 137.7, 133.4, 130.9, 130.2, 129.1 (2 C), 128.3 (2 C), 128.1, 127.9 (2 C), 127.7, 127.1, 121.3 (2 C), 34.7, 25.3, 19.6; MS (EI) m/z: 364.13 [M]+. Anal. (C22H21ClN2O) Calcd. (%): C, 72.42; H, 5.80; Cl, 9.72; N, 7.68. Found (%) C, 72.68; H, 5.94; Cl, 9.41; N, 7.35.
N-(4-(2-Aminocyclopropyl)phenyl)-4-(furan-2-yl)benzamide hydrochloride (2a). Chemical and physical data: m.p., >250 °C (acetonitrile/methanol); yield, 55%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.19–1.21 (m, 1H, CHH cyclopropane), 1.37–1.39 (m, 1H, CHH cyclopropane), 2.32–2.33 (m, 1H, CHNH2 cyclopropane), 2.79–2.80 (m, 1H, PhCH cyclopropane), 6.68–6.70 (m, 1H, furan proton), 7.14–7.16 (m, 3H, furan and benzene protons), 7.72–7.74 (d, 2H, benzene protons, J = 8 Hz), 7.84–7.86 (m, 3H, furan and benzene protons), 8.02–8.04 (d, 2H, benzene protons, J = 8 Hz), 8.49 (bs, 3H, NH3+), 10.30 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 168.2, 153.6, 142.3, 138.6, 138.4, 137.0, 134.4, 128.7 (2 C), 128.2 (2 C), 125.2 (2 C), 121.2 (2 C), 111.4, 107.7, 34.6, 25.3, 19.3; MS (EI) m/z: 354.11 [M]+. Anal. (C20H19ClN2O2) Calcd. (%): C, 67.70; H, 5.40; Cl, 9.99; N, 7.89. Found (%) C, 67.39; H, 5.27; Cl, 10.12; N, 7.94.
N-(4-(2-Aminocyclopropyl)phenyl)-3-(furan-2-yl)benzamide hydrochloride (2b). Chemical and physical data: m.p., 208–210 °C (acetonitrile/methanol); yield, 61%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.20–1.24 (m, 1H, CHH cyclopropane), 1.35–1.40 (m, 1H, CHH cyclopropane), 2.28–2.33 (m, 1H, CHNH2 cyclopropane), 2.80–2.82 (m, 1H, PhCH cyclopropane), 6.65–6.66 (m, 1H, furan proton), 7.09–7.10 (d, 1H, furan proton, J = 4 Hz), 7.15–7.18 (d, 2H, benzene protons, J = 12 Hz), 7.57–7.60 (t, 1H, furan proton, J = 12 Hz), 7.69–7.74 (m, 2H, benzene protons), 7.74–7.79 (m, 2H, benzene protons), 7.94–7.96 (m, 1H, benzene proton), 8.25 (s, 1H, benzene protons), 8.39 (bs, 3H, NH3+), 10.37 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 166.2, 154.0, 142.2, 138.6, 138.4, 133.1, 130.8, 130.7, 128.2, 128.2 (2 C), 128.0, 126.4, 121.2 (2 C), 111.5, 107.3, 34.6, 25.3, 19.3; MS (EI) m/z: 354.11 [M]+. Anal. (C20H19ClN2O2) Calcd. (%): C, 67.70; H, 5.40; Cl, 9.99; N, 7.89. Found (%) C, 67.52; H, 5.30; Cl, 10.07; N, 7.92.
N-(4-(2-Aminocyclopropyl)phenyl)-4-(furan-3-yl)benzamide hydrochloride (3a). Chemical and physical data: m.p., >250 °C (acetonitrile/methanol); yield, 75%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.19–1.21 (m, 1H, CHH cyclopropane), 1.38–1.39 (m, 1H, CHH cyclopropane), 2.29–2.31 (m, 1H, CHNH2 cyclopropane), 2.80 (bs, 1H, PhCH cyclopropane), 7.09 (s, 1H, furan proton), 7.14–7.16 (d, 2H, benzene proton, J = 8 Hz), 7.72–7.73 (d, 2H, benzene proton, J = 4 Hz), 7.79–7.81 (m, 3H, furan and benzene proton), 7.96–7.98 (d, 2H, benzene proton, J = 8 Hz), 8.36 (s, 1H, benzene proton), 8.47 (bs, 3H, NH3+), 10.26 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 168.2, 143.0, 140.8, 138.6, 138.4, 137.8, 136.6, 130.1 (2 C), 128.2 (2 C), 127.2, 126.6 (2 C), 121.2 (2 C), 108.8, 34.6, 25.3, 19.3; MS (EI) m/z: 354.11 [M]+. Anal. (C20H19ClN2O2) Calcd. (%): C, 67.70; H, 5.40; Cl, 9.99; N, 7.89. Found (%) C, 67.98; H, 5.41; Cl, 9.79; N, 7.54.
N-(4-(2-Aminocyclopropyl)phenyl)-3-(furan-3-yl)benzamide hydrochloride (3b). Chemical and physical data: m.p., 235–238 °C (acetonitrile/methanol); yield, 71%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.20–1.21 (m, 1H, CHH cyclopropane), 1.38–1.39 (m, 1H, CHH cyclopropane), 2.29–2.31 (m, 1H, CHNH2 cyclopropane), 2.79–2.81 (m, 1H, PhCH cyclopropane), 7.08 (s, 1H, furan proton), 7.15–7.17 (d, 2H, benzene protons, J = 8 Hz), 7.53–7.55 (t, 1H, furan proton, J = 8 Hz), 7.71–7.73 (d, 2H, benzene proton, J = 8 Hz), 7.80–7.81 (m, 3H, furan and benzene proton), 8.17 (s, 1H, benzene proton), 8.32 (s, 1H, benzene proton), 8.45 (bs, 3H, NH3+), 10.33 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 166.4, 141.9, 140.7, 138.6, 138.4, 133.9, 133.9, 130.3, 129.9, 128.5, 128.2 (2 C), 127.4, 126.8, 121.2 (2 C), 109.5, 34.6, 25.3, 19.3; MS (EI) m/z: 354.11 [M]+. Anal. (C20H19ClN2O2) Calcd. (%): C, 67.70; H, 5.40; Cl, 9.99; N, 7.89. Found (%) C, 67.65; H, 5.31; Cl, 10.06; N, 7.99.
N-(4-(2-Aminocyclopropyl)phenyl)-4-(thiophen-2-yl)benzamide hydrochloride (4a). Chemical and physical data: m.p., >250 °C (methanol); yield, 82%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.17–1.24 (m, 1H, CHH cyclopropane), 1.36–1.41 (m, 1H, CHH cyclopropane), 2.30–2.35 (m, 1H, CHNH2 cyclopropane), 2.77–2.81 (m, 1H, PhCH cyclopropane), 7.14–7.17 (m, 2H, thiophen protons), 7.19–7.21 (m, 1H, thiophen proton), 7.65–7.67 (m, 1H, benzene protons), 7.68–7.69 (m, 1H, benzene protons), 7.72–7.74 (d, 2H, benzene protons, J = 8 Hz), 7.81–7.83 (d, 2H, benzene protons, J = 8 Hz), 8.01–8.03 (d, 2H, benzene protons, J = 8 Hz), 8.42 (bs, 3H, NH3+), 10.23 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 168.2, 143.0, 139.2, 138.6, 138.4, 136.9, 128.6 (2 C), 128.2 (2 C), 127.9, 126.9, 126.8 (2 C), 124.7, 121.2 (2 C), 34.6, 25.3, 19.3; MS (EI) m/z: 370.09 [M]+. Anal. (C20H19ClN2OS) Calcd. (%): C, 64.77; H, 5.16; Cl, 9.56; N, 7.55; S, 8.64. Found (%) C, 64.43; H, 5.09; Cl, 9.68; N, 7.84; S, 8.49.
N-(4-(2-Aminocyclopropyl)phenyl)-3-(thiophen-2-yl)benzamide hydrochloride (4b). Chemical and physical data: m.p., 245–248 °C (methanol); yield, 67%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.17–1.23 (m, 1H, CHH cyclopropane), 1.35–1.40 (m, 1H, CHH cyclopropane), 2.29–2.34 (m, 1H, CHNH2 cyclopropane), 2.79–2.81 (m, 1H, PhCH cyclopropane), 7.16–7.21 (m, 3H, thiophene and benzene protons), 7.56–7.60 (t, 1H, thiophene protons, J = 16 Hz), 7.62–7.66 (m, 2H, thiophene and benzene protons), 7.72–7.74 (d, 2H, benzene protons, J = 8 Hz), 7.86–7.89 (d, 2H, benzene protons, J = 12 Hz), 8.18–8.20 (m, 1H, benzene protons), 8.37 (bs, 3H, NH3+), 10.37 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 165.9, 141.7, 138.6, 138.4, 135.9, 132.8, 130.3, 128.5, 128.3, 128.2 (2 C), 128.1, 127.8, 126.5, 124.4, 121.2 (2 C), 34.6, 25.3, 19.3; MS (EI) m/z: 370.109 [M]+. Anal. (C20H19ClN2OS) Calcd. (%): C, 64.77; H, 5.16; Cl, 9.56; N, 7.55; S, 8.64. Found (%) C, 64.94; H, 5.28; Cl, 9.45; N, 7.42; S, 8.77.
N-(4-(2-Aminocyclopropyl)phenyl)-4-(thiophen-3-yl)benzamide hydrochloride (5a). Chemical and physical data: m.p., 222–225 °C (methanol); yield, 81%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.18–1.20 (m, 1H, CHH cyclopropane), 1.21–1.23 (m, 1H, CHH cyclopropane), 2.28–2.33 (m, 1H, CHNH2 cyclopropane), 2.80 (bs, 1H, PhCH cyclopropane), 7.15–7.19 (d, 2H, benzene protons, J = 16 Hz), 7.67–7.75 (m, 4H, thiophene and benzene protons), 7.89–7.94 (d, 2H, benzene protons, J = 20 Hz), 8.05–8.09 (d, 2H, benzene protons, J = 16 Hz), 8.14–8.16 (m, 1H, thiophene proton), 8.40 (bs, 3H, NH3+), 10.28 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 168.5, 155.4, 141.7, 140.5, 139.1, 137.9, 136.8, 129.7 (2 C), 127.7, 127.3, 127.2, 127.1, 122.4, 121.2, 79.7, 33.8, 28.3, 26.3, 18.1; MS (EI) m/z: 370.09 [M]+. Anal. (C20H19ClN2OS) Calcd. (%): C, 64.77; H, 5.16; Cl, 9.56; N, 7.55; S, 8.64. Found (%) C, 65.07; H, 5.24; Cl, 9.39; N, 7.22; S, 8.78.
N-(4-(2-Aminocyclopropyl)phenyl)-3-(thiophen-3-yl)benzamide hydrochloride (5b). Chemical and physical data: m.p., >250 °C (methanol); yield, 85%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.20–1.21 (m, 1H, CHH cyclopropane), 1.35–1.38 (m, 1H, CHH cyclopropane), 2.29–2.32 (m, 1H, CHNH2 cyclopropane), 2.81 (bs, 1H, PhCH cyclopropane), 7.17–7.18 (d, 2H, thiophene proton, J = 4 Hz), 7.49–7.51 (t, 1H, thiophene proton, J = 8 Hz), 7.67–7.69 (m, 1H, benzene protons), 7.70–7.73 (m, 3H, thiophene and benzene protons), 7.84– 7.86 (d, 1H, benzene proton, J = 8 Hz), 7.90–7.92 (d, 1H, benzene proton, J = 8 Hz), 8.02 (bs, 1H, benzene proton), 8.26 (s, 1H, benzene proton), 8.40 (bs, 3H, NH3+), 10.34 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 166.9, 140.8, 138.6, 137.8, 137.7, 133.6, 130.3, 130.2, 128.4, 128.3 (2 C), 127.4, 127.1, 127.1, 122.6, 121.3 (2 C), 34.7, 25.3, 19.6; MS (EI) m/z: 370.09 [M]+. Anal. (C20H19ClN2OS) Calcd. (%): C, 64.77; H, 5.16; Cl, 9.56; N, 7.55; S, 8.64. Found (%) C, 64.92; H, 5.23; Cl, 9.47; N, 7.69; S, 8.47.
N-(4-(2-Aminocyclopropyl)phenyl)-4-(pyridin-4-yl)benzamide hydrochloride (6a). Chemical and physical data: m.p., >250 °C (methanol); yield, 48%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.17–1.22 (m, 1H, CHH cyclopropane), 1.39–1.44 (m, 1H, CHH cyclopropane), 2.33–2.39 (m, 1H, CHNH2 cyclopropane), 2.77–2.80 (m, 1H, PhCH cyclopropane), 7.16–7.18 (d, 2H, benzene protons, J = 8 Hz), 7.75–7.77 (d, 2H, benzene protons, J = 8 Hz), 8.14–8.20 (m, 4H, benzene protons), 8.30–8.32 (d, 2H, pyridine protons, J = 8 Hz), 8.58 (bs, 3H, NH3+), 8.93–8.95 (d, 2H, pyridine protons, J = 8 Hz), 10.49 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 168.5, 148.2 (2 C), 147.5, 141.8, 137.9, 137.7, 136.8, 129.6 (2 C), 128.4 (2 C), 128.3 (2 C), 121.6 (2 C), 121.3 (2 C), 34.7, 25.3, 19.6; MS (EI) m/z: 365.13 [M]+. Anal. (C21H20ClN3O) Calcd. (%): C, 68.94; H, 5.51; Cl, 9.69; N, 11.49. Found (%) C, 69.17; H, 5.64; Cl, 9.49; N, 11.38.
N-(4-(2-Aminocyclopropyl)phenyl)-3-(pyridin-4-yl)benzamide hydrochloride (6b). Chemical and physical data: m.p., >250 °C (methanol); yield, 63%. 1H NMR (DMSO-d6, 400 MHz, δ; ppm) δ 1.18–1.23 (m, 1H, CHH cyclopropane), 1.37–1.42 (m, 1H, CHH cyclopropane), 2.32–2.35 (m, 1H, CHNH2 cyclopropane), 2.79–2.82 (m, 1H, PhCH cyclopropane), 7.17–7.19 (d, 2H, benzene protons, J = 8 Hz), 7.76–7.79 (t, 3H, benzene protons, J = 12 Hz), 8.15–8.21 (m, 2H, pyridine protons), 8.39–8.40 (d, 2H, benzene protons, J = 4 Hz), 8.50 (bs, 3H, NH3+), 8.55 (s, 1H, benzene protons), 8.95–8.97 (d, 2H, pyridine protons, J = 8 Hz), 10.58 (bs, 1H, CONH) ppm; 13 C (100 MHz, DMSO-d6) δ 166.9, 150.2 (2 C), 146.2, 139.3, 137.8, 137.7, 133.4, 131.5, 130.2, 128.3, 128.1 (2 C), 128.0, 121.8 (2 C), 121.3 (2 C), 34.7, 25.3, 19.6; MS (EI) m/z: 365.13 [M]+. Anal. (C21H20ClN3O) Calcd. (%): C, 68.94; H, 5.51; Cl, 9.69; N, 11.49. Found (%) C, 69.22; H, 5.68; Cl, 9.54; N, 11.31.
2.4. Lsd1-CoREST binding and inhibition assays
Thermal stability, activity on histone H3K4me peptide, and inhibition of human LSD1-CoREST were measured using established protocolsCitation17,Citation28.
The complex of human recombinant LSD1/CoREST protein was produced in E. coli as separate proteins and co-purified following previously reported proceduresCitation13,Citation29. The experiments were performed in 96 well half area white plates (cat. 3693, Corning, Corning, NY) using a di-methylated H3K4 peptide containing 21 amino acids (custom synthesis done by Thermo Scientific) as substrate in 100 μL volume of 50 mM HEPES, pH 7.5. The peptide purity was >95% as checked by analytical high-pressure liquid chromatography and mass spectrometry. The demethylase activity was estimated under aerobic conditions at room temperature by measuring the release of H2O2 produced during the catalytic process by the Amplex UltraRed detection system coupled with horseradish peroxidase (HRP). Briefly, 300 nM of LSD1/CoREST complex was incubated at room temperature for 10 min in the absence and/or the presence of various concentrations of the inhibitors, 50 mM Amplex UltraRed (Life Technologies) and 0.023 mM HRP (Sigma-Aldrich, Schnellendorf, Germany) in 50 mM HEPES pH 7.5 and 0.05 mg/mL BSA. The inhibitors were tested twice in duplicates at each concentration. Tranylcypromine (Sigma) was used as a control. After preincubation of the enzyme with the inhibitor, the reaction was initiated by adding 10 μМ of the dimethylated H3K4 peptide. The conversion of the Amplex UltraRed reagent to resurfin was monitored by fluorescence (excitation at 510 nm, emission at 595 nm). Arbitrary units were used to measure the level of H2O2 produced in the absence and/or in the presence of inhibition. The maximum demethylase activity of LSD1/CoREST was obtained in the absence of inhibitors and corrected for background fluorescence in the absence of the substrate. The IC50 values were calculated using GraphPad Prism version 4.0 (GraphPad Software, San Diego, CA).
2.5. Anti-MAO assays
The tested compounds were dissolved in DMSO (Sigma-Aldrich, Schnellendorf, Germany) to prepare 10 mM stock solutions which were kept for storage at −20 °C. The percentage of DMSO used in the experiments was never higher than 1%. Clorgyline and selegiline, used as reference inhibitors, have been acquired from Sigma-Aldrich, Schnellendorf, Germany. Human recombinant MAO isoforms, used in the experiments, was purchased from Sigma-Aldrich. Resorufin sodium salt, p-tyramine hydrochloride, sodium phosphate buffer, horseradish peroxidase, and Amplex Red reagent have been supplied in the assay kit of Amplex Red MAO Molecular Probes (Molecular Probes Inc., Eugene, OR). Determination of MAO isoforms enzymatic activity: Briefly, 0.1 ml of sodium phosphate buffer (0.05 M, pH 7.4) containing different concentrations of the test drugs (new compounds or reference inhibitors) in various concentrations and adequate amounts of recombinant hMAO-A or hMAO-B required and adjusted to obtain in our experimental conditions the same reaction velocity, that is, to oxidise (in the control group) the same concentration of substrate: 165 pmol of p-tyramine/min (hMAO-A: 1.1 μg protein; specific activity: 150 nmol of p-tyramine oxidised to p-hydroxyphenylacetaldehyde/min/mg protein; hMAO-B: 7.5 μg protein; specific activity: 22 nmol of p-tyramine transformed/min/mg protein) were incubated for 15 min at 37 °C in a flat-black-bottom 96-well microtest plate, placed in the dark fluorimeter chamber. After this incubation period, the reaction was started by adding (final concentrations) 200 μM Amplex Red reagent, 1 U/mL horseradish peroxidase, and 1 mM p-tyramine. The production of H2O2 and, consequently, of resorufin was quantified at 37 °C in a multidetection microplate fluorescence reader (FLX800, Bio-Tek Instruments Inc., Winooski, VT) based on the fluorescence generated (excitation, 545 nm, emission, 590 nm) over a 15 min period, in which the fluorescence increased linearly. Control experiments were carried out simultaneously by replacing the tested drugs (new compounds and reference inhibitors) with appropriate dilutions of the vehicles. In addition, the possible capacity of the above test drugs to modify the fluorescence generated in the reaction mixture due to non-enzymatic inhibition (e.g. for directly reacting with Amplex Red reagent) was determined by adding these drugs to solutions containing only the Amplex Red reagent in a sodium phosphate buffer. To determine the kinetic parameters of hMAO-A and hMAO-B (Km and Vmax), the corresponding enzymatic activity of both isoforms was evaluated (under the experimental conditions described above) in the presence of a wide range of p-tyramine concentrations. The specific fluorescence emission (used to obtain the final results) was calculated after subtraction of the background activity, which was determined from wells containing all components except the hMAO isoforms, which were replaced by a sodium phosphate buffer solution. In our experimental conditions, this background activity was practically negligible. MAO activity of the test compounds and reference inhibitors is expressed as IC50, i.e. the concentration of each drug required to result in a 50% decrease in respect to the control value activity of the relative MAO isoforms. The corresponding IC50 values were calculated by using the Origin 5.0 software (Microcal Software Inc., Northampton, MA).
2.6. Cellular assays
2.6.1. Chemicals
The tested compounds have been dissolved in DMSO at 50 μM as stock concentration. All compounds have been tested at 10 μM and 50 μM as final concentrations for WB analysis. All compounds have been used at 0.1 μM, 5 μM, 10 μM, 25 μM, 50 μM as final concentration for proliferation assays. ORY-1001 (Selleck Catalog No. S7795) was used as reference compound at the concentration of 25 μM.
2.6.2. Cell cultures
Cell culture LNCaP and U937 were purchased from ATCC (Milan, Italy). LNCaP were grown in Roswell Park Memorial Institute culture medium (RPMI; EuroClone, Milan, Italy), supplemented with 10% heat-inactivated foetal bovine serum (FBS; Sigma-Aldrich, Schnellendorf, Germany), antimicrobials (100 U/mL penicillin, 100 µg/mL streptomycin, 250 ng/mL amphotericin-B), 2 mM L-glutamine (EuroClone, Milan, Italy), and 1% essential amino acids solution (MEM; EuroClone, Milan, Italy). U937 were cultured in Dulbecco’s Modified Eagle Medium (DMEM; EuroClone, Milan, Italy), supplemented with 10% heat-inactivated foetal bovine serum (FBS; Sigma-Aldrich, Schnellendorf, Germany), antimicrobials (100 U/mL penicillin, 100 µg/mL streptomycin, 250 ng/mL amphotericin-B), 2 mM L-glutamine (EuroClone). Cells have grown in standard incubator conditions at 37 °C and 5% CO2.
2.6.3. Histone extraction, Western blot analysis and protein extraction
After stimulation for 48 h with the compounds at 10 μM and 50 μM, ORY-1001 (Selleck Catalog No. S7795), commercially available LSD1 inhibitor, was used as positive controls. ORY-1001 was used at the final concentration of 25 μM. Cells were collected and washed 2 times with PBS then processed for histone extraction. Pellets were resuspended in triton extraction buffer [TEB; PBS containing 0.5% Triton X 100 (v/v), 2 mmol/L PMSF, 0.02% (w/v) NaN3], and the lysis was performed for 10 min at 4 °C. The samples were centrifuged at 2000× g for 10 min at 4 °C and pellets were washed in TEB (half volume). Samples were then resuspended in 0.2 N HCl, and acid histone extraction was carried out overnight at 4 °C. The supernatants were recovered, and protein concentration was quantified by Bradford assay (Bio-Rad). For each sample, 4 μg of proteins were loaded on 15% polyacrylamide gels. The nitrocellulose filters were stained with Ponceau red (Sigma-Aldrich, Schnellendorf, Germany) as an additional control for equal loading. H3K4me2, H3K9me2 (Diagenode, Ougrée, Belgium; pAB-035–050, pAb-060–050), and H4 (Cell Signalling #2592) were used according to the manufacturer’s instructions.
2.6.4. Statistical analysis and software tool analysis
Raw data were acquired by using ImageJ software and a semi-quantitative analysis was performed. Relative intensities were normalised on the control and reported above images, in .
2.6.5. Proliferative assay
Cell viability was determined on LNCaP and U937 cell lines using Thiazolyl Blue Tetrazolium Bromide [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (MTT; Sigma-Aldrich, Schnellendorf, Germany) assay, following manufacturer’s instructions. A total of 5 × 103 cells/well were plated in a 96-well plate and treated, in duplicates, and repeated for three times, with the tested compounds at different concentrations 0.1 μM, 5 μM, 10 μM, 25 μM, 50 μM as final concentrations for 48 h, and 72 h. Tranylcypromine (TCP) and ORY-1001 were used as positive controls (data not shown). TCP was purchased from Sigma-Aldrich (St Louis, MO) and used at a final concentration of 100 μM and ORY-1001 at 25 μM. Absorbance was read at a wavelength of 570 nm with a TECAN M − 200 reader (Tecan, Männedorf, Switzerland).
3. Results and discussion
3.1. Chemistry
The tert-butyl carbamates 14–19, key intermediates for the synthesis of the final compounds 1–6, were prepared by coupling the tert-butyl (2-(4-aminophenyl)cyclopropyl)carbamate) 13Citation17 with the appropriate 4- or 3-substituted benzoic acids 7–12, all commercially available, in the presence of benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) and triethylamine, using dry N,N-dimethylformamide as the solvent. The subsequent cleavage of the Boc protection from the carbamates 14–19 with 4 N HCl in dioxane and dry THF afforded the N-(4-(2-aminocyclopropyl)phenyl)-(hetero)aryl-4- and 3-carboxamides 1–6 as hydrochloride salts (Scheme 1).
Scheme 1. Synthesis of the N-(4-(2-aminocyclopropyl)phenyl)-(hetero)aryl-4- and 3-carboxamides 1 b-6. Reagents and conditions: (a) PyBop, Et3N, dry DMF, N2 atmosphere, overnight, rt. (b) 4 N HCl, dry dioxane/THF, overnight, rt.

3.2. LSD1 and MAO enzymes evaluation assays
First, the new TCP-based compounds 1b-6 were tested to detect their binding and inhibition of LSD1. ThermoFAD assayCitation28 was used to detect the effects of 1b-6 on the thermal stability of the LSD1-CoREST complex, and the largest shifts in the Tm of the enzyme usually indicate the strongest LSD1 binding abilities (). Representative curves relative to ThermoFAD assay and FAD spectral bleaching using LSD1-CoREST (LC305) with selected compounds have been depicted in , respectively.
Figure 3. Representative samples of thermal shifts by ThermoFAD assay (A) and FAD spectral bleaching (B) for selected compounds. LC305 is the code for the LSD1-CoREST complex.

Table 1. ThermoFAD assay and inhibition values (IC50 values) of 1 b-6 vs LSD1-CoREST complex.
The inhibition of LSD1 was assessed by protein expression in Escherichia coli and co-purification of LSD1Δ124-CoREST1Δ305 (LSD1-CoREST) and using H3K4me1 (residues 1–21) peptide as the substrate, as previously describedCitation17. The IC50 values of 1b-6 against the LSD1-CoREST complex are reported in , and the relative curves are depicted in . TCP and 1a were used as reference compounds.
Figure 4. Inhibition curves of 1 b-6 against LSD1-CoREST. Compound 1a has been reported for comparison purpose.

The shift of the 4-phenyl ring of the benzoylamino moiety of 1a from the para to meta position led to slightly increased potency (1.3-fold) against LSD1 (compare 1b with 1a). This trend of inhibition potency is confirmed for all the following derivatives, in which the phenyl ring has been replaced by a 2-furyl (2a,b), 3-furyl (3a,b), 2-thienyl (4a,b), 3-thienyl (5a,b), and 4-pyridyl (6a,b) group. In general, the meta regioisomers 2b, 3b, 4b, 5b and 6b displayed 10.6- to 71.8-fold higher potency against LSD1 than the para counterparts. When compared to the phenyl-containing 1a and 1b, the 2-furyl (2a,b) compounds decreased the inhibitory potency when placed in para (2a), and gave little improvement when inserted in meta (2b). Differently, the replacement with a 3-furyl (3a,b) ring was detrimental for the anti-LSD1 activity regardless of the para or meta position. The introduction of the 2- or 3-thienyl (4a,b and 5a,b) ring led to a severe (1.8- to 5.7-fold) drop of potency when the heterocycle was placed in para (compare 4a and 5a with 1a), whereas the meta corresponding analogs 4b and 5b showed 10- to 30-fold improved potency respect to 1b in the anti-LSD1 assay. At last, the 4-pyridyl (6a,b) ring abated the inhibitory potency from 73 (6a) to 4 (6b) times when compared with 1a and 1b, respectively.
Since 1b-6 contain in their structure the TCP fragment, they were tested against MAOs to assess their eventual LSD1 selectivity. The IC50 values of 1b-6 against MAO-A and MAO-B have been summarised in . Clorgyline and R-(-)-deprenyl were used as reference MAO-A and -B selective inhibitors, respectively.
Table 2. Anti-MAO activities of 1 b-6.a
The tested compounds displayed IC50 values against MAO-A in the range 0.024–1.120 μM, and against MAO-B they were often less potent, with a range of inhibition between 0.032 and 10.42 μM. Among the para and meta (a and b) substituted analogs, the first generally were more potent than the latter against the two MAO isoforms, reversing the behaviour observed with LSD1. Exceptions to this rule are the thienyl derivatives (4a,b and 5a,b) versus MAO-A: indeed, against this MAO isoform, the 2-thienyl derivatives 4a and 4b showed the same inhibitory activity, and the 3-thienyl meta derivative 5b was 1.5-fold more potent than the corresponding para analog 5a.
Comparing the activities of 1b-6 against LSD1 and MAOs, all the meta-substituted derivatives showed from 2.2- to 164-fold selectivity for LSD1 vs MAO-B, and some of them (3b-5b) also vs MAO-A (). The meta 2- and 3-thienyl compounds 4b and 5b emerged as the most potent against LSD1 (IC50 values = 0.015 (4b) and 0.005 (5b) μM) and the most LSD1-selective respect to MAO-B (selectivity indexes = 24.4 (4b) and 164 (5b)).
Table 3. LSD1 selectivity over MAOs.
3.3. Effects of 1a,b, 2b, 3b, 4b, and 5a,b in U937 AML and prostate cancer LNCaP cells
Selected compounds among the most potent against LSD1 at enzyme level (1a,b, 2b, 3b, 4b, and 5a,b, with IC50 values in the range 0.005–0.341 μM) were tested at 0.1, 5, 10, 25, and 50 μM for 48 and 72 h in U937 AML and prostate cancer LNCaP cells to determine their effects on cell viability. and show the dose-dependent decrease of cell viability observed after treatment in U937 and LNCaP cells, respectively.
Table 4. Effects of 1a,b, 2 b, 3 b, 4 b, and 5a,b on cell viability in U937 cells after 48 and 72 h of treatment.
Table 5. Effects of 1a,b, 2 b, 3 b, 4 b, and 5a,b on cell viability in LNCaP cells after 48 and 72 h of treatment.
In U937 cells the new LSD1 inhibitors showed moderate activities, with 1b, 3b, 5a, and 5b being the most potent and 1a, 2b, and 4b almost ineffective at both time points. In this cell line, the meta phenyl and 3-thienyl derivatives 1b and 5b exerted the highest potency at 48 and 72 h. LNCaP cells were more sensitive than U937 cells to these inhibitors, likely due to the ability of the tested LSD1 inhibitors to also inhibit MAO-A, that is known to play a role in prostate cancer cell proliferationCitation30–33. In LNCaP cells, all the tested compounds exhibited dose-dependent decreased cell viability. After 48 h of treatment 3b was the most potent, with IC50 = 12.4 μM, and after 72 h all the tested inhibitors displayed IC50 values comprised between 9.9 and 25.4 μM, with 1b and 3b being the most effective ().
Table 6. IC50 values of 1a,b, 2 b, 3 b, 4 b, and 5a,b in LNCaP cells. For comparison, the IC50 (72 h) of the selective MAO-A inhibitor clorgyline has been added.
To link the reduction of cell viability to the inhibition of LSD1, western blot analyses have been performed on the new TCP-based inhibitors at 10 and 50 μM after 48 h of treatment to determine the changes in H3K4me2 (U937) or H3K4me2 and H3K9me2 (LNCaP) levels, typical marks for LSD1 demethylase activity (). In LNCaP cells we also detected the levels of H3K9me2 because LSD1 is known to interact with the androgen receptor and to promote androgen-dependent transcription of target genes by ligand-induced demethylation of H3K9me1/2Citation4,Citation34. ORY-1001 (iadademstat)Citation35, a known covalent LSD1 inhibitor, was used as a reference drug (at 25 μM).
Figure 5. Western blot analyses of the H3K4me2 levels in U937 cells (A) and of H3K4me2 and H3K9me2 levels in LNCaP cells (B).

Western blot analyses in both cell lines confirmed the capability of the tested compounds to inhibit LSD1 in cellular contexts, showing a moderate to huge increase of methylation levels of all the investigated marks.
4. Conclusions
The known anti-MAO drug TCP is the most reliable fragment to design potent, covalent LSD1 inhibitors, active in cancer cells as antiproliferative agentsCitation16. In our previous works, we identified MC2580Citation17 and MC2584Citation18 as TCP analogs with higher potency against LSD1. The insertion of a phenyl ring at the para position of the benzoylamino moiety of MC2584 led to 1a, which retained potency against LSD1 and displayed high effects in MV4-11 and NB4 leukaemia cellsCitation25. Starting from these findings, we designed and prepared new 1a analogs by shifting the phenyl ring from para to meta position (1b), and by inserting 5- or 6-membered heterocyclic rings either at para or meta positions (2a,b-6a,b). Compounds 1b-6 were tested against LSD1: the inhibition assay showed the meta derivatives (b) to be more potent than the corresponding para counterparts (a), with the meta 2- and 3-thienyl analogs 4b and 5b being the most potent (IC50 values = 0.015 (4b) and 0.005 (5b) μM). When tested against MAOs, 1b-6 were found active at submicromolar/nanomolar levels mainly against MAO-A. The comparison between the inhibitory activities of 1b-6 against the two families of flavoenzymes (LSD1 and MAOs) showed an LSD1-selective behaviour for all the meta regioisomers over MAO-B and in some cases respect to MAO-A, with the 2- and 3-thienyl derivatives 4b and 5b being the most LSD1-selective respect to MAO-B (selectivity indexes: 24.4 and 164, respectively).
Selected compounds 1a,b, 2b, 3b, 4b, and 5a,b were tested in U937 AML and prostate cancer LNCaP cells to detect their antiproliferative activities after 48 and 72 h of treatment. LNCaP cells turned out to be more sensitive than U937 to these LSD1 inhibitors because all the tested compounds displayed a dose-dependent decrease of cell viability with 1b and 3b being the most effective. In U937 cells, only 1b, 3b, 5a, and 5b gave an effect, while the remaining compounds were less active or even inactive. Western blot analyses, performed in both the cell lines after 48 h of treatment with the same compounds to detect the levels of H3K4me2 (U937 and LNCaP) and H3K9me2 (LNCaP), confirmed the involvement of LSD1 inhibition in these cellular assays showing a general increase of the methylation levels.
Further studies will be performed with these compounds in different cancer contexts to assess their anticancer potential.
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Bhat KP, Umit Kaniskan H, Jin J, Gozani O. Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat Rev Drug Discov 2021;20:265–86.
- Sterling J, Menezes SV, Abbassi RH, Munoz L. Histone lysine demethylases and their functions in cancer. Int J Cancer 2021;148:2375–2388.
- Laurent B, Ruitu L, Murn J, et al. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol Cell 2015;57:957–70.
- Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005;437:436–9.
- Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004;119:941–53.
- Shi YJ, Matson C, Lan F, et al. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 2005;19:857–64.
- Wada T, Koyama D, Kikuchi J, et al. Overexpression of the shortest isoform of histone demethylase LSD1 primes hematopoietic stem cells for malignant transformation. Blood 2015;125:3731–46.
- Karytinos A, Forneris F, Profumo A, et al. A novel mammalian flavin-dependent histone demethylase. J Biol Chem 2009;284:17775–82.
- Yang GJ, Lei PM, Wong SY, et al. Pharmacological Inhibition of LSD1 for Cancer Treatment. Molecules 2018;23:3194.
- Wang Y, Zhang H, Chen Y, et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009;138:660–72.
- Wang M, Liu X, Jiang G, et al. Relationship between LSD1 expression and E-cadherin expression in prostate cancer. Int Urol Nephrol 2015;47:485–90.
- Chen L, Xu Y, Xu B, et al. Over-expression of lysine-specific demethylase 1 predicts tumor progression and poor prognosis in human esophageal cancer. Int J Clin Exp Pathol 2014;7:8929–34.
- Forneris F, Binda C, Battaglioli E, Mattevi A. LSD1: oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem Sci 2008;33:181–9.
- Edmondson DE, Binda C, Wang J, et al. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 2009;48:4220–30.
- Yang M, Culhane JC, Szewczuk LM, et al. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat Struct Mol Biol 2007;14:535–9.
- Dai XJ, Liu Y, Xiong XP, et al. Tranylcypromine based lysine-specific demethylase 1 inhibitor: summary and perspective. J Med Chem 2020;63:14197–215.
- Binda C, Valente S, Romanenghi M, et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J Am Chem Soc 2010;132:6827–33.
- Valente S, Rodriguez V, Mercurio C, et al. Pure enantiomers of benzoylamino-tranylcypromine: LSD1 inhibition, gene modulation in human leukemia cells and effects on clonogenic potential of murine promyelocytic blasts. Eur J Med Chem 2015;94:163–74.
- Valente S, Rodriguez V, Mercurio C, et al. Pure diastereomers of a tranylcypromine-based LSD1 inhibitor: enzyme selectivity and in-cell studies. ACS Med Chem Lett 2015;6:173–7.
- Vianello P, Botrugno OA, Cappa A, et al. Synthesis, biological activity and mechanistic insights of 1-substituted cyclopropylamine derivatives: a novel class of irreversible inhibitors of histone demethylase KDM1A. Eur J Med Chem 2014;86:352–63.
- Vianello P, Botrugno OA, Cappa A, et al. Discovery of a novel inhibitor of histone lysine-specific demethylase 1A (KDM1A/LSD1) as orally active antitumor agent. J Med Chem 2016;59:1501–17.
- Rodriguez V, Valente S, Rovida S, et al. Pyrrole- and indole-containing tranylcypromine derivatives as novel lysine-specific demethylase 1 inhibitors active on cancer cells. MedChemComm 2015;6:665–70.
- Rotili D, Tomassi S, Conte M, et al. Pan-histone demethylase inhibitors simultaneously targeting Jumonji C and lysine-specific demethylases display high anticancer activities. J Med Chem 2014;57:42–55.
- Kalin JH, Wu M, Gomez AV, et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat Commun 2018;9:53.
- Fioravanti R, Romanelli A, Mautone N, et al. Tranylcypromine-based LSD1 inhibitors: structure-activity relationships, antiproliferative effects in leukemia, and gene target modulation. ChemMedChem 2020;15:643–58.
- Ravasio R, Ceccacci E, Nicosia L, et al. Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid-induced differentiation. Sci Adv 2020;6:eaax2746.
- Nicosia L, Boffo FL, Ceccacci E, et al. Pharmacological inhibition of LSD1 triggers myeloid differentiation by targeting GSE1 oncogenic functions in AML. Oncogene 2022;41:878–894.
- Forneris F, Orru R, Bonivento D, et al. ThermoFAD, a thermofluor-adapted flavin ad hoc detection system for protein folding and ligand binding. Febs J 2009;276:2833–40.
- Forneris F, Binda C, Adamo A, et al. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J Biol Chem 2007;282:20070–4.
- Gaur S, Gross ME, Liao CP, et al. Effect of monoamine oxidase A (MAOA) inhibitors on androgen-sensitive and castration-resistant prostate cancer cells. Prostate 2019;79:667–77.
- Wu JB, Shao C, Li X, et al. Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. J Clin Invest 2014;124:2891–908.
- Wu JB, Lin TP, Gallagher JD, et al. Monoamine oxidase A inhibitor-near-infrared dye conjugate reduces prostate tumor growth. J Am Chem Soc 2015;137:2366–74.
- Wang K, Luo J, Yeh S, et al. The MAO inhibitors phenelzine and clorgyline revert enzalutamide resistance in castration resistant prostate cancer. Nat Commun 2020;11:2689.
- Wissmann M, Yin N, Muller JM, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 2007;9:347–53.
- Maes T, Mascaro C, Tirapu I, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 2018;33:495–511 e12.