
Abstract
The poly (ADP-ribose) polymerase (PARP) inhibitors play a crucial role in cancer therapy. However, most approved PARP inhibitors cannot cross the blood-brain barrier, thus limiting their application in the central nervous system. Here, 55 benzodiazepines were designed and synthesised to screen brain penetrating PARP-1 inhibitors. All target compounds were evaluated for their PARP-1 inhibition activity, and compounds with better activity were selected for further assays in vitro. Among them, compounds H34, H42, H48, and H52 displayed acceptable inhibition effects on breast cancer cells. Also, computational prediction together with the permeability assays in vitro and in vivo proved that the benzodiazepine PARP-1 inhibitors we synthesised were brain permeable. Compound H52 exhibited a B/P ratio of 40 times higher than that of Rucaparib and would be selected to develop its potential use in neurodegenerative diseases. Our study provided potential lead compounds and design strategies for the development of brain penetrating PARP-1 inhibitors.
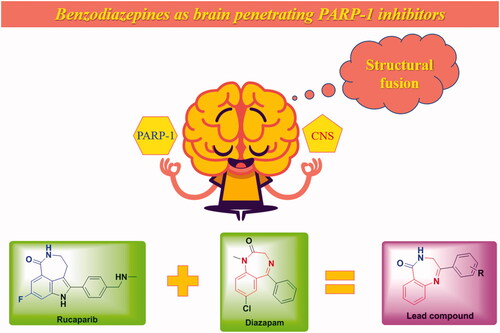
Structural fusion was used to screen brain penetrating PARP-1 inhibitors.
55 benzodiazepines were evaluated for their PARP-1 inhibition activity.
Four compounds displayed acceptable inhibition effects on breast cancer cells.
The benzodiazepine PARP-1 inhibitors were proved to be brain permeable.
HIGHLIGHTS
Graphical Abstract
1. Introduction
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear protein that creates poly(ADP-ribose) (PAR) covalently attached to target proteins to mediate genomic stability, DNA damage repair, and apoptosisCitation1. And PARP-1 is responsible for repairing DNA single-strand breaks (SSBs)Citation2, whereas breast cancer susceptibility gene 1/2 (BRCA1/2) are key components involved in the repair of DNA double-strand breaks(DSBs) by homologous recombination (HR)Citation3. Cancer cells with BRCA1/2 mutations treated with PARP inhibitors lead to cell cycle arrest and apoptosis through synthetic lethalityCitation4,Citation5, thus the PARP-1 protein has become an attractive target for the treatment of cancers in recent yearsCitation6. Since OlaparibCitation7 was approved as the first PARP-1 inhibitor to treat advanced ovarian cancer associated with defective BRCA mutations in 2014, several PARP inhibitors were approved subsequently, including RucaparibCitation8, NiraparibCitation9, TalazoparibCitation10, FluzoparibCitation11,Citation12, and PamiparibCitation13,Citation14 ().
Figure 1. Approved PARP-1/2 inhibitors and representative benzodiazepine drugs.

Although PARP inhibitors play a crucial role in cancer therapy, effective therapy against the primary malignancy facilitates a more resilient systemic disease that leads to central nervous system (CNS) metastatic infiltrationCitation15. Breast cancer is the second-leading cause of metastatic disease in the CNSCitation16. Patients with metastatic HER2+ primary cancer have a risk of 35–50% to develop metastatic brain diseaseCitation17 and patients with adjuvant trastuzumab treatment have a risk of 2.5% to relapse in CNS firstlyCitation18. In addition, the risk of CNS involvement in metastatic triple-negative breast cancer (TNBC) is as high as 46%Citation19. Similarly, primary CNS tumours, such as neurofibromatosisCitation20, diffuse gliomas, medulloblastomas, and glioblastomaCitation21, are also difficult to overcome because of the existence of the blood-brain barrier. However, several well-known PARP-1 inhibitors have been reported to be not CNS penetrant, including OlaparibCitation22,Citation23, RucaparibCitation24,Citation25, and TalazoparibCitation26. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumour, which is recently approved in ChinaCitation13,Citation14. Nevertheless, more brain penetrating PARP-1 inhibitors are needed to meet the huge market.
PARP-1 is also known to participate in a range of important cellular processes including regulation of apoptosis, cell division and differentiation, transcriptional regulation, chromosome stability, and metabolic regulationCitation27,Citation28, and it has become an attractive therapeutic target for other diseases, such as neuroinflammationCitation29, neurodegenerationCitation30, cardiovascular diseaseCitation31, and drug addictionCitation32,Citation33. To deal with these diseases, the factor of the blood–brain barrier (BBB) should also be consideredCitation34. In addition, the synergism between PARP-1 and various DNA damage response (DDR) targets is also being studied to trigger synthetic lethality artificiallyCitation35. Lallo et al.Citation36 combined Olaparib and the WEE1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Mak et al.Citation37 reported that synergism between ataxia-telangiectasia mutated (ATM) and PARP-1 inhibition involves DNA damage and abrogating the G2 DNA damage checkpoint. Li et al.Citation38 proposed that PARP inhibitors could enhance the priming and tumour-killing activities of T cells, boost the whole cancer-immunity cycle, and thereby improve the response to immune checkpoint blockade. The above-combined strategies show that PARP-1 inhibitors have a strong potential for the treatment of tumours, and the development of a new generation of PARP-1 inhibitors with high selectivity or brain penetration is helpful to make better use of its therapeutic potential. Based on the above facts, this paper is devoted to the study of CNS penetrant PARP-1 inhibitors, which are expected to meet the diverse needs of clinical treatment in the future.
2. Materials and methods
2.1. Compound design
PARP inhibitors have been developed for several years and the efforts of predecessors have led to a good understanding of key pharmacophores for the binding of small molecules to PARP-1. Most PARP inhibitors are designed to mimic the nicotinamide moiety of natural substrate NAD+ and bind competitively with NAD+ at the NI site of the PARP-1 catalytic domainCitation6. Key hydrogen bonding interactions between the inhibitors and residues SER904 and GLY863 of PARP-1 are crucial for activity retentionCitation39 (). The π–π stacking formed between the ligand and the nearby Tyr residue is also essential. The strategies of restraint in amide rotation are designed either by constraining the amide into a cyclic amide, such as Rucaparib and Olaparib, or by forming a pseudo cyclic amide via an intramolecular H-bond as in NiraparibCitation40. To our delight, benzodiazepines have been widely concerned by pharmaceutical chemists because of their effective penetration of the blood-brain barrier and significant inhibition of the central nervous systemCitation41, such as Diazepam and Clonazepam, which are all classical benzodiazepines (). Accordingly, the pharmacophore of Rucaparib and benzodiazepine structure were fused in this paper to find brain penetrant PARP inhibitors (), and preliminary compounds H1, H2, H3, and H4 were obtained. As shown in , compound H4 exhibited similar interactions with known PARP-1 inhibitors and was used for subsequent structural optimisation.
Figure 2. (A) Binding mode of PARP-1 inhibitors. (B) Drug design strategy.

Figure 3. (A) Predicted binding mode of H4 with PARP-1. (B) 2D diagram of H4 interacting with PARP-1. The catalytic domain structure of PARP-1 is coloured in cyan (crystal structure: PARP-1 PDB code 4zzz). Compounds and the key residues within the binding pocket are shown as sticks and coloured in yellow and green, respectively. Hydrogen bonds are shown as yellow and the distances (Å) of H-bonds are also labelled. The molecular docking was accomplished using Glide flexible docking. The 3D images were prepared with PyMOL, and the 2D picture was drawn in Discovery Studio.

2.2. Chemistry
The syntheses of all target compounds were accomplished according to the synthetic routes outlined in Schemes 1–3. The first series of 2-phenyl-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one derivatives were synthesised in three steps as shown in Scheme 1. Firstly, a variety of 2-bromo-1-phenylethanone derivatives (X-001) were used as starting materials to react with urotropine, and then the quaternary ammonium salt was hydrolysed in acid to synthesise primary amine hydrochloride (X-002). Then, the key intermediates (X-003) were synthesised in good yields by the reaction of these primary amine hydrochlorides with isatoic anhydride at room temperature. Finally, H4–H19 were obtained by refluxing the above intermediates in xylene. After reduction with NaBH4 in methanol at 0 °C, compounds H20–H27 were prepared. As shown in Scheme 2, compounds H28 and H29 were prepared by different methylation conditions. Sodium diformylamide was used to get H30-002, then compound H30 and H31 were also obtained by the above similar reactions. With H19 and H27 in hand, the target compound H32–H55 were synthesised through Suzuki Coupling or Buchwald-Hartwig Coupling as shown in Scheme 3.
Scheme 1. Synthetic route of compounds H4–H27.

Scheme 2. Synthetic route of compounds H28–H31.

Scheme 3. Synthetic route of compounds H32–H55.

3. Results and discussion
3.1. Enzyme inhibition activity against PARP-1 and structure-activity relationship
All target compounds were evaluated for PARP-1 enzyme inhibition activity at the concentration of 50 nM determined by previous pre-experiments.
3.1.1. Inhibitory activities of compounds H1–H19 against PARP-1
As shown in , the results of compound H1–H4 show that the phenyl substituent improves the activity, which may form π–π stacking similar to that of Rucaparib. Because of its preliminary activity, H4 was selected for structural optimisation. Then, H5–H11 were obtained by introducing methyl, methoxy, fluorine, and chlorine into the benzene ring of H4, and they all showed slightly increased PARP-1 inhibitory activity except H11. For compounds H6–H9, the activities of meta-substitution were higher than para-substitution, while chlorine substituted H10–H11 showed the opposite, indicating that para-substitution was more beneficial to the activity when the volume of substituents is larger. H12 and H13 were obtained by introducing polar cyano and hydroxyl groups into the para-position. H12 dramatically decreased enzyme inhibition potency, while H13 maintained the enzyme activity. Considering that fluorine, methoxy, and chlorine substitutions help to improve the activity and lipophilicity of the compounds, the corresponding disubstituted compounds H14–H17 were synthesised. However, compounds H14, H15, and H17 maintained parallel activity, while H16 exhibited decreased potency. The benzene ring was replaced by naphthalene ring to obtain H18, which showed similar activity, indicating that the introduction of large substituents would not cause the decrease of activity. To introduce diverse substituents through the classical coupling reactions, compound H19 containing bromine substitution was synthesised with an inhibition rate of 38.4%.
Table 1. The chemical structures and inhibitory activities of compounds H1–H19.
3.1.2. Inhibitory activities of compounds H20–H31 against PARP-1
To obtain better lead compounds, we investigated the structure-activity relationship of different variants of benzodiazepine structure. The inhibition rates are shown in . Compounds H20–H27 were obtained by the reduction of the imines in compounds H6–H9, H14–H16, and H19. Compared with the corresponding unreduced compounds, the activities of H20–H24 decreased slightly, while the activities of compounds H25 and H27 increased. Compound H26 regained acceptable activity, which may be due to the increase of molecular flexibility after reduction. Compounds H28 and H29 were synthesised from compound H27 by nitrogen methylation reaction, and the decreased inhibition indicated that the hydrogen of amide was necessary for PARP-1 inhibitory activity, which was consistent with the previously reported structure-activity relationship. When methyl was introduced into the ortho position of amide nitrogen in compound H19, the obtained compound H30 showed no obvious change in activity. However, the activity of the reduction product H31 decreased obviously, which may be due to the excessive flexibility of the seven-membered ring. For this series of compounds, the conclusions are as follows: (i) the reduction of imine structure has little effect on the activity and increases the flexibility of the compound; (ii) hydrogen on amide nitrogen is necessary for activity, and the introduction of substituents leads to the decrease of activity; (iii) the introduction of methyl into the ortho position of amide nitrogen has no effect on the activity on the rigid ring but has a greater effect on the flexible ring. Based on the above results, compounds H19 and H27 display the highest activity, and their bromine substitution is conducive to the introduction of various substituents through coupling reactions, which can be used as new lead compounds for follow-up optimisation.
Table 2. The chemical structures and inhibitory activities of compounds H20–H31.
3.1.3. Inhibitory activities of compounds H32–H46 against PARP-1
According to our previous studyCitation42, three key amino acids in PARP-1 (GLN759, GLU763, ASP766) were found, which formed an acidic cavity and specifically contained the hydrophilic group. Therefore, compounds containing exposed amino groups were synthesised to match the acidic domain of the PARP-1 active pocket (). With H19 in hand, compounds H32–H35 were obtained by coupling with pyrazole and its derivatives, and their inhibition rates were significantly higher than that of H19 at the same concentration, among which compound H34 reached 56.7%. The activities of H36 and H37 synthesised by replacing pyrazole with aniline were only slightly improved. Increasing the number of heteroatoms on the aniline to get H38–H40, which exhibited decreased enzyme inhibition potency. It is considered that the increased number of heteroatoms will increase the polar surface area of the compound, which is not conducive to crossing the blood-brain barrier. Thus, compounds H41–H44 were obtained by replacing the amino group on the benzene ring with methoxy and trifluoromethyl to improve the lipophilicity of the compound. The results show that the position of the methoxy group has a great influence on the activity, and the inhibition rate of para-substituted compound H42 was 51.2%. On the contrary, trifluoromethyl substituted compounds showed similar activity. The results of H36–H37 and H41–H44 show that the activity of para-substitution is generally better than that of meta-substitution with the increase of length. Compound H45 was obtained by replacing benzene with 1-methyl-1,2,3,6-tetrahydropyridine, and the activity was not significantly improved. H46 was obtained by the introduction of carbazole, and the loss of activity indicated that the size of substituents needs to be controlled to avoid the decrease of activity caused by steric hindrance.
Table 3. The chemical structures and inhibitory activities of compounds H32–H46.
3.1.4. Inhibitory activities of compounds H47–H55 against PARP-1
The activity of H27 is higher than that of H19, and its imine structure is more stable after reduction. Therefore, a series of compounds were synthesised based on H27, the results were shown in . The compounds H47–H52 were obtained by coupling amino, methoxy and trifluoromethyl substituted benzene with H27, and their inhibition rates were higher than that of unreduced H36–H37 and H41–H44. In addition, it was also concluded that para-substitution was better than meta-substitution, and the inhibition rates of H48, H50, and H52 were all more than 50%. Unexpectedly, the activities of compounds H53–H55 substituted by 1-isopropylpiperazine, 1-acetylpiperazine, and (2S, 6R)-2,6-dimethylmorpholine were lower than that of their lead compound H27, indicating that saturated heterocycles were not conducive to maintaining activity.
Table 4. The chemical structures and inhibitory activities of compounds H47–H55.
To sum up, through the study of structure–activity relationship, five compounds (H34 56.7%, H42 51.2%, H48 55.5%, H50 52.3%, H52 57.7%) were obtained with an inhibition rate of more than 50%. Then, the IC50 values of compounds H34, H42, H48, H50, and H52 against PARP-1 were determined, as shown in . Although their enzyme activity is weaker than that of the control drug Rucaparib, we think it is necessary to make a further evaluation, especially brain permeability, which is the focus of this article. And lower activity may also be allowed when such inhibitors are used as a combination strategy to treat tumours or alleviate neurological diseases. PARP-2 is the closest homolog of PARP-1, which also resides in the nucleus and functions similarly to PARP-1. The IC50 values of the compounds against PARP-2 were also determined (). In addition, compounds H19 and H27 have preliminary PARP-1 inhibitory activity and active reaction groups, and further structural optimisation of the compounds or analogues as lead compounds to find more active PARP-1 inhibitors will also be carried out.
Table 5. IC50 values of compounds H34, H42, H48, H50, and H52 against PARP-1
3.2. Molecular docking
To rationalise the observed moderate activity of compounds H34, H42, H48, H50, and H52, we next obtained the possible binding patterns from molecular docking results. As shown in , the three key hydrogen bonding interactions were formed between the five compounds and SER904 and GLY863 of PARP-1, which were necessary for activity and consistent with the binding patterns of Rucaparib. In addition, the pyrazole group of H34 formed an additional hydrogen bond with GLN759, the phenolic group of H42 and H50 also form additional hydrogen bonds with GLN759. However, the aniline of H48 formed an additional hydrogen bond with GLY888, while the trifluoromethyl of H52 stayed in the cavity formed by ASP766, GLU763, and GLN759. These interactions mentioned above theoretically explained the moderate PARP-1 inhibitory activity of the benzodiazepines synthesised in this paper.
Figure 4. Predicted binding poses of compounds with PARP-1 (A: H34, B: H42, C: H48, D: H50, E: H52). The catalytic domain structure of PARP-1 is coloured in cyan (crystal structure: PARP-1 PDB code 4zzz). Compounds and the key residues within the binding pocket are shown as sticks and coloured in yellow and green, respectively. Hydrogen bonds are shown as yellow and the distances (Å) of H-bonds are also labelled. The molecular docking was accomplished using Glide flexible docking. The 3D images were prepared with PyMOL.

3.3. Inhibition of cellular proliferation
Due to their acceptable inhibition rates (more than 50%) at the concentration of 50 nM, compounds H34, H42, H48, H50, and H52 were selected to evaluate the proliferation inhibition activity on several cancer cells, including BRCA1 mutant tumour cells (MX-1) and BRCA1 wild type tumour cells (MCF7 and A549). Rucaparib was used as the reference molecule and the results were outlined in . The results showed that all the five compounds showed a certain inhibitory effect on proliferation, and were weaker than those of Rucaparib, especially H50. In addition, the inhibitory effect of H34, H42, H48, and H52 on breast cancer cells was stronger than that of lung cancer cells, and the compounds showed slightly stronger effects on MX-1 cells than MCF7 cells.
Table 6. Antiproliferation activity of compounds H34, H42, H48, H50, and H52 in tumour cell lines.
3.4. Prediction of ADME properties
The computational prediction of absorption, distribution, metabolism, and excretion (ADME) was used in evaluating the brain permeability of synthesised compounds. Several parameters of four compounds (H34, H42, H48, and H52) were displayed in , which were often used in the evaluation of blood-brain barrier and permeability. These include polar surface area (PSA), octanol/water partition coefficient (QPlogPo/w), central nervous system activity (CNS), brain/blood partition coefficient (QPlogBB), apparent MDCK cell permeability (QPPMDCK), apparent Caco-2 cell permeability (QPPCaco). Based on the above parameters, the prediction results show that compounds H42 and H52 have good brain permeability, while H34, H48, and Rucaparib are difficult to pass through the blood-brain barrier.
Table 7. Predicted ADME properties of compounds H34, H42, H48, and H52.
3.5. MDCK cell permeability
The MDCK cells monolayer model was also used to determine whether the four compounds were capable of crossing the blood-brain barrierCitation43,Citation44. The cell culture was carried out in accordance with routine operations or instructions. The MDCK monolayer model was established after 5 days of culture, and then the trans-epithelial electrical resistance (TEER) was measured using an EVOM epithelial volt-ohmmeter with STX2 electrodes to evaluate the integrity of the membrane. The tested compounds (10 µM) of 100 µL were added to the apical compartment of the transwell cell culture devices (Millipore), and then the devices were incubated at 37 °C for 3 h. The concentrations of tested compounds in an apical compartment and basolateral compartment were detected by HPLC. The apparent permeability in an apical compartment to the basolateral compartment (Papp (A→B)) was then calculatedCitation45, and the results are listed in . Rucaparib was not detected in the basolateral compartment, which was consistent with the literature that it could not pass through the blood-brain barrierCitation24,Citation25. The four compounds we synthesised all showed the permeability of the monolayer model, and the Papp (A→B) value of compound H52 was the highest, reaching 19.7 cm/s × 10−6.
Table 8. MDCK cell permeability of compounds H34, H42, H48, and H52.
3.6. Brain/plasma pharmacokinetic studies in vivo
Further, the brain permeability of representative compounds was evaluated in ICR mice. After tail vein injection of a 1.5 mg/kg dose, the concentrations of the compounds in the brain and plasma were measured after 5 min and 60 min of administration, and the brain/plasma (B/P) ratio was calculated. As shown in , compounds H34 and H48 exhibited low B/P ratios in 5 min and 60 min, which were similar to that of Rucaparib. On the contrary, compounds H42 and H52 performed high B/P ratios, which were consistent with our initial expectations. Among them, the B/P ratio of H42 reached balance rapidly in vivo and maintained above 3, while the proportion of H52 in the brain increased gradually and reached 4.41 at 60 min. In addition, H42 showed a low concentration in plasma and brain after administration of 60 min, while the concentration of H52 was still high, indicating that H52 was more likely to accumulate in vivo.
Table 9. Brain penetration study for compounds H34, H42, H48, and H52a.
Based on the above results, compounds H42, H52, and Rucaparib were then selected to evaluate their antiproliferation activity in combination with TMZ on glioblastoma (U87) cells. As shown in , both H42 and H52 showed significant sensitisation effects in U87 cells.
Figure 5. The cytotoxicity of H42 and H52 in combination with TMZ in glioblastoma (U87) cells. TMZ: Temozolomide; RU: Rucaparib.

4. Conclusions
Blood–brain barrier protects the central nervous system from the interruption and damage of xenobiotics. However, it prevents potential drugs aimed at the central nervous system, thus becomes an obstruction for the development of central nervous system drugs. The PARP inhibitors have played a crucial role in cancer therapy. However, most approved PARP inhibitors cannot cross the blood-brain barrier, thus limiting their application in the central nervous system. In this study, 55 benzodiazepines were designed and synthesised to screen brain penetrating PARP-1 inhibitors. All target compounds were evaluated for their PARP-1 inhibition activity, and compounds with high inhibition rates at 50 nM were selected to assess for cellular assays in vitro. Among them, compounds H34, H42, H48, and H52 displayed acceptable inhibition effects on breast cancer cells. Also, computational prediction and the permeability assays in vitro and in vivo proved that the benzodiazepine PARP-1 inhibitors we synthesised are brain permeable. Compound H52 exhibited acceptable PARP-1 inhibition and good brain penetration, and its B/P ratio was more than 40 times higher than that of Rucaparib. It has been reported in Science that strategies aimed at inhibiting PARP-1 activation could hold promise as a disease-modifying therapy to prevent the loss of dopamine neurons in Parkinson’s disease and related a-synucleinopathiesCitation46. Therefore, in addition to improving the anti-tumour activity of these benzodiazepine PARP-1 inhibitors, compound H52 would be selected to develop its potential use in neurodegenerative diseases. Overall, our study provided potential lead compounds and design strategies for the development of brain penetrating PARP-1 inhibitors.
5. Experimental section
5.1. Chemistry
5.1.1. General methods
All reagents and solvents were obtained from commercial suppliers and used without further purification. All reactions were performed in glassware containing a Teflon coated stir bar. The reactions were monitored by thin-layer chromatography (TLC) using silica gel GF254 and spots were visualised by UV light at 254 or 365 nm. Flash column chromatography on silica gel (200–300 mesh) was used for the routine purification of reaction products. Since the reactions were carried out at the millimole level, all yields have not been accurately calculated. The purity of the target compounds determined by high-performance liquid chromatography was all higher than 95%. The intermediates and the products were fully characterised by spectroscopic data. 1H and 13C NMR spectra were recorded with Bruker BioSpin GmbH at 400 and 100 MHz, respectively. DMSO-d6 and CDCl3 were selected as deuterated solvents. Chemical shifts were expressed in δ (ppm) and coupling constants were given in Hz. Low-resolution mass spectra were obtained from Waters 3100.
5.1.2. Synthesis procedure for X-002
5.1.2.1. 2-Amino-1-phenylethanone hydrochloride (H4-002)
2-Bromo-1-phenylethanone (H4-001) (8 mmol, 1 equiv.) and urotropine (8 mmol, 1 equiv.) were dissolved in 40 ml of dry chloroform. The reaction mixture was stirred at 50 °C for 2 h. The precipitate was isolated by filtration and thoroughly washed with chloroform and ethanol. Then the urotropine salt was dissolved in 40 ml of absolute ethanol and 4 ml of concentrated hydrochloric acid. The mixture was heated for another 2 h. After the completion of reaction monitored by TLC, the solid was removed by filtration and washed with ethanol. The filtrate was evaporated under reduced pressure to yield the title compound (H4-002) as a white solid. 1H NMR (400 MHz, DMSO) δ 8.57 (s, 3H), 8.01 (d, J = 7.3 Hz, 2H), 7.72 (t, J = 7.4 Hz, 1H), 7.57 (t, J = 7.7 Hz, 2H), 4.60 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 193.29, 134.94, 134.13, 129.46, 128.67, 45.23; ESI-MS m/z = 136.03 [M + H]+.
5.1.2.2. 2-Amino-1-(p-tolyl)ethanone hydrochloride (H5-002)
The title compound (H5-002) was synthesised from H5-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.53 (s, 3H), 7.92 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 8.1 Hz, 2H), 4.54 (s, 2H), 2.39 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 192.78, 145.58, 131.72, 129.99, 128.76, 45.05, 21.77. ESI-MS m/z = 150.11 [M + H]+.
5.1.2.3. 2-Amino-1-(4-methoxyphenyl)ethanone hydrochloride (H6-002)
The title compound (H6-002) was synthesised from H6-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.41 (s, 3H), 8.00 (d, J = 8.9 Hz, 2H), 7.10 (d, J = 8.9 Hz, 2H), 4.53 (d, J = 5.3 Hz, 2H), 3.86 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 191.56, 164.54, 131.12, 127.06, 114.71, 56.24, 44.83. ESI-MS m/z = 166.13 [M + H]+.
5.1.2.4. 2-Amino-1-(3-methoxyphenyl)ethanone hydrochloride (H7-002)
The title compound (H7-002) was synthesised from H7-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.51 (s, 3H), 7.64–7.59 (m, 1H), 7.53–7.47 (m, 3H), 4.59 (d, J = 5.2 Hz, 2H), 3.84 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 193.21, 159.95, 135.49, 130.70, 121.10, 120.99, 113.10, 56.01, 45.35. ESI-MS m/z = 166.13 [M + H]+.
5.1.2.5. 2-Amino-1-(4-fluorophenyl)ethanone hydrochloride (H8-002)
The title compound (H8-002) was synthesised from H8-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.48 (s, 3H), 8.19–8.07 (m, 2H), 7.44 (t, J = 8.8 Hz, 2H), 4.60 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 192.07, 166.15 (d, 1JC-F = 235.7 Hz), 131.87 (d, 3JC-F = 9.79 Hz, 2 C), 130.97 (d, 4JC-F = 2.63 Hz), 116.61 (d, 2JC-F = 22.11 Hz, 2 C), 45.22. ESI-MS m/z = 154.18 [M + H]+.
5.1.2.6. 2-Amino-1-(3-fluorophenyl)ethanone hydrochloride (H9-002)
The title compound (H9-002) was synthesised from H9-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.54 (s, 3H), 7.93–7.80 (m, 2H), 7.71–7.56 (m, 2H), 4.62 (d, J = 4.4 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 192.60 (d, 4JC-F = 2.22 Hz), 162.63 (d, 1JC-F = 246.74 Hz), 136.26 (d, 3JC-F = 6.67 Hz), 131.78 (d, 3JC-F = 7.88 Hz), 124.95 (d, 4JC-F = 2.83 Hz), 121.87 (d, 2JC-F = 21.41 Hz), 115.30 (d, 2JC-F = 22.62 Hz), 45.47. ESI-MS m/z = 154.12 [M + H]+.
5.1.2.7. 2-Amino-1-(4-chlorophenyl)ethanone hydrochloride (H10-002)
The title compound (H10-002) was synthesised from H10-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.56 (s, 3H), 8.04 (d, J = 8.6 Hz, 2H), 7.67 (d, J = 8.6 Hz, 2H), 4.59 (d, J = 3.9 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 192.50, 139.81, 132.87, 130.63, 129.60, 45.27. ESI-MS m/z = 170.13 [M + H]+.
5.1.2.8. 2-Amino-1-(3-chlorophenyl)ethanone hydrochloride (H11-002)
The title compound (H11-002) was synthesised from H11-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.55 (s, 3H), 8.08–8.03 (m, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.84–7.78 (m, 1H), 7.63 (t, J = 7.9 Hz, 1H), 4.63 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 192.63, 135.97, 134.60, 134.35, 131.49, 128.40, 127.32, 45.42. ESI-MS m/z = 170.09 [M + H]+.
5.1.2.9. 4-(2-Aminoacetyl)benzonitrile hydrochloride (H12-002)
The title compound (H12-002) was synthesised from H12-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.63 (s, 3H), 8.18 (d, J = 8.6 Hz, 2H), 8.08 (d, J = 8.5 Hz, 2H), 4.66 (d, J = 4.5 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 193.02, 137.29, 133.45, 129.34, 118.47, 116.62, 45.61. ESI-MS m/z = 161.13 [M + H]+.
5.1.2.10. 2-Amino-1-(4-hydroxyphenyl)ethanone hydrochloride (H13-002)
The title compound (H13-002) was synthesised from H13-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 10.92 (s, 1H), 8.43 (s, 3H), 7.88 (d, J = 8.8 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 4.45 (d, J = 5.3 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 191.12, 163.88, 131.26, 125.56, 116.07, 44.55. ESI-MS m/z = 152.09 [M + H]+.
5.1.2.11. 2-Amino-1-(2,4-difluorophenyl)ethanone hydrochloride (H14-002)
The title compound (H14-002) was synthesised from H14-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.58 (s, 3H), 8.13–8.01 (m, 1H), 7.56–7.50 (m, 1H), 7.38–7.33 (m, 1H), 4.40 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 189.63, 189.59, 167.72, 165.05, 164.47, 161.90, 133.08, 133.01, 119.72, 119.60, 113.47, 113.22, 106.23, 105.97, 105.70, 48.05, 47.94. ESI-MS m/z = 172.10 [M + H]+.
5.1.2.12. 2-Amino-1-(3,5-difluorophenyl)ethanone hydrochloride (H15-002)
The title compound (H15-002) was synthesised from H15-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.63 (s, 3H), 7.80–7.67 (m, 3H), 4.61 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 191.71, 164.25, 164.13, 161.78, 161.66, 137.16, 112.25, 112.17, 112.06, 111.98, 110.26, 110.00, 45.58. ESI-MS m/z = 172.11 [M + H]+.
5.1.2.13. 2-Amino-1-(3,4-dimethoxyphenyl)ethanone hydrochloride (H16-002)
The title compound (H16-002) was synthesised from H16-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.48 (s, 3H), 7.69 (dd, J = 8.5, 1.9 Hz, 1H), 7.48 (d, J = 1.9 Hz, 1H), 7.13 (d, J = 8.5 Hz, 1H), 4.54 (d, J = 4.9 Hz, 2H), 3.85 (d, J = 10.2 Hz, 6H); 13 C NMR (101 MHz, DMSO) δ 191.62, 154.48, 149.17, 126.98, 123.64, 111.57, 110.64, 56.39, 56.14, 44.82. ESI-MS m/z = 196.16 [M + H]+.
5.1.2.14. 2-Amino-1-(3,4-dichlorophenyl)ethanone hydrochloride (H17-002)
The title compound (H17-002) was synthesised from H17-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.63 (s, 3H), 8.26 (d, J = 1.9 Hz, 1H), 7.98 (dd, J = 8.4, 2.0 Hz, 1H), 7.88 (d, J = 8.4 Hz, 1H), 4.62 (d, J = 3.8 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 191.92, 137.68, 134.32, 132.51, 131.81, 130.71, 128.67, 45.40. ESI-MS m/z = 204.04 [M + H]+.
5.1.2.15. 2-Amino-1-(naphthalen-2-yl)ethanone hydrochloride (H18-002)
The title compound (H18-002) was synthesised from H18-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.81 (s, 1H), 8.56 (s, 3H), 8.17 (d, J = 8.0 Hz, 1H), 8.11–7.99 (m, 3H), 7.75– 7.62 (m, 2H), 4.73 (d, J = 5.1 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 193.28, 135.99, 132.49, 131.44, 131.32, 130.21, 129.77, 129.12, 128.28, 127.75, 123.52, 45.29. ESI-MS m/z = 136.03 [M + H]+. ESI-MS m/z = 186.08 [M + H]+.
5.1.2.16. 2-Amino-1-(4-bromophenyl)ethanone hydrochloride (H19-002)
The title compound (H19-002) was synthesised from H19-001 using the same method as the preparation of compound H4-002; 1H NMR (400 MHz, DMSO) δ 8.65 (s, 3H), 7.95 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H), 4.58 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 192.73, 133.19, 132.53, 130.67, 129.11, 45.22. ESI-MS m/z = 213.90, 215.92 [M + H]+.
5.1.2.17. 2-Amino-1-(4-bromophenyl)propan-1-one hydrochloride (H30-002)
Sodium diformylamide (9.6 mmol, 1.2 eq) was suspended in 30 ml acetonitrile. 2-bromo-1-(4-bromophenyl)propan-1-one (H30-001) (8 mmol, 1 equiv.) was added drop-wise and with stirring. The mixture was then stirred at 75 °C for 4 h. The mixture was hot filtered and the solid was washed twice with acetonitrile. The combined organics were evaporated and the residue was dissolved in 20 ml water. 2 ml 10 N hydrochloric acid was added and the mixture was stirred at reflux for 1 h. After completed, the solvent was removed under reduced pressure. The residue was washed twice with ethyl acetate and the solid was collected by filtration and dried to give H30-002; 1H NMR (400 MHz, DMSO) δ 8.66 (s, 3H), 8.01 (d, J = 8.5 Hz, 2H), 7.81 (d, J = 8.5 Hz, 2H), 5.18–5.04 (m, 1H), 1.42 (d, J = 7.1 Hz, 3H); 13 C NMR (101 MHz, DMSO) δ 196.41, 132.73, 132.37, 131.24, 129.21, 51.28, 17.39. ESI-MS m/z = 227.92, 229.91 [M + H]+.
5.1.3. Synthesis procedure for X-003
5.1.3.1. 2-Amino-N-(2-oxo-2-phenylethyl)benzamide (H4-003)
To a solution of 2-amino-1-phenylethanone hydrochloride (H4-002) (6 mmol, 1 equiv.) in 20 ml water, sodium carbonate (3.72 mmol, 0.62 eq) were added. After being stirred at room temperature for 10 min, isatoic anhydride (6 mmol, 1 equiv.) was dissolved in 10 ml water and added dropwise to reaction mixture. The solution was stirred at room temperature for 3 h and then monitored by TLC. After completion was indicated, the resultant solution was extracted with ethyl acetate and then the organic layer dried over anhydrous Na2SO4. After filtration and concentration, the crude product was obtained and purified with column chromatography to provide H4-003; 1H NMR (400 MHz, DMSO) δ 8.60 (t, J = 5.4 Hz, 1H), 8.05 (d, J = 7.8 Hz, 2H), 7.69 (t, J = 7.4 Hz, 1H), 7.63–7.51 (m, 3H), 7.17 (t, J = 7.6 Hz, 1H), 6.72 (d, J = 8.2 Hz, 1H), 6.55 (t, J = 7.5 Hz, 1H), 6.45 (s, 2H), 4.72 (d, J = 5.6 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 196.12, 169.59, 150.27, 135.61, 134.00, 132.41, 129.31, 128.64, 128.32, 116.90, 115.06, 114.43, 46.61. ESI-MS m/z = 255.24 [M + H]+, 277.70 [M + Na]+.
5.1.3.2. 2-Amino-N-(2-oxo-2-(p-tolyl)ethyl)benzamide (H5-003)
Compound H5-003 was prepared by following a similar procedure to that for H4-003; 1H NMR (400 MHz, DMSO) δ 8.57 (t, J = 5.5 Hz, 1H), 7.95 (d, J = 8.1 Hz, 2H), 7.60 (d, J = 7.1 Hz, 1H), 7.37 (d, J = 8.0 Hz, 2H), 7.22–7.12 (m, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.55 (t, J = 7.5 Hz, 1H), 6.45 (s, 2H), 4.69 (d, J = 5.6 Hz, 2H), 2.40 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 195.56, 169.56, 150.26, 144.40, 133.10, 132.39, 129.84, 128.63, 128.42, 116.90, 115.05, 114.47, 46.47, 21.69. ESI-MS m/z = 269.13 [M + H]+, 291.28 [M + Na]+.
5.1.3.3. 2-Amino-N-(2-(4-methoxyphenyl)-2-oxoethyl)benzamide (H6-003)
Compound H6-003 was prepared by following a similar procedure to that for H4-003; 1H NMR (400 MHz, DMSO) δ 8.54 (t, J = 5.6 Hz, 1H), 8.03 (d, J = 8.9 Hz, 2H), 7.59 (d, J = 7.1 Hz, 1H), 7.21–7.14 (m, 1H), 7.08 (d, J = 8.9 Hz, 2H), 6.72 (d, J = 8.2 Hz, 1H), 6.55 (t, J = 7.5 Hz, 1H), 6.45 (s, 2H), 4.67 (d, J = 5.6 Hz, 2H), 3.86 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 194.36, 169.55, 163.80, 150.22, 132.36, 130.64, 128.63, 128.48, 116.89, 115.06, 114.58, 114.49, 56.03, 46.21. ESI-MS m/z = 285.16 [M + H]+, 307.15 [M + Na]+.
5.1.3.4. 2-Amino-N-(2-(3-methoxyphenyl)-2-oxoethyl)benzamide (H7-003)
Compound H7-003 was prepared by following a similar procedure to that for H4-003; 1H NMR (400 MHz, DMSO) δ 8.61 (t, J = 5.5 Hz, 1H), 7.68–7,57 (m, 2H), 7.54–7.45 (m, 2H), 7.28–7.22 (m, 1H), 7.20–7.14 (m, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.55 (t, J = 7.3 Hz, 1H), 6.45 (s, 2H), 4.71 (d, J = 5.6 Hz, 2H), 3.84 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 195.94, 169.58, 159.91, 150.27, 136.97, 132.41, 130.49, 128.64, 120.76, 120.02, 116.90, 115.06, 114.42, 112.81, 55.84, 46.76. ESI-MS m/z = 285.30 [M + H]+, 307.55 [M + Na]+.
5.1.3.5. 2-Amino-N-(2-(4-fluorophenyl)-2-oxoethyl)benzamide (H8-003)
Compound H8-003 was prepared by following a similar procedure to that for H4-003; 1H NMR (400 MHz, DMSO) δ 8.61 (t, J = 5.4 Hz, 1H), 8.20–8.08 (m, 2H), 7.59 (d, J = 7.9 Hz, 1H), 7.40 (t, J = 8.8 Hz, 2H), 7.21–7.13 (m, 1H), 6.71 (d, J = 8.3 Hz, 1H), 6.55 (t, J = 7.5 Hz, 1H), 6.45 (s, 2H), 4.71 (d, J = 5.6 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 194.80, 169.59, 168.43, 166.77, 150.27, 146.75, 133.24, 133.21, 132.42, 132.34, 132.10, 131.42, 131.33, 131.00, 130.91, 130.34, 128.63, 127.06, 126.97, 126.18, 116.90, 116.45, 116.41, 116.24, 116.20, 115.06, 114.37, 46.57. ESI-MS m/z = 273.38 [M + H]+, 295.13 [M + Na]+.
5.1.3.6. 2-Amino-N-(2-(3-fluorophenyl)-2-oxoethyl)benzamide (H9-003)
Compound H9-003 was prepared by following a similar procedure to that for H4-003; 1H NMR (400 MHz, DMSO) δ 8.64 (t, J = 5.4 Hz, 1H), 7.93–7.80 (m, 2H), 7.67–7.52 (m, 3H), 7.22–7.13 (m, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.58–6.52 (m, 1H), 6.45 (s, 2H), 4.71 (d, J = 5.6 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 195.28, 169.61, 168.33, 166.85, 163.89, 161.44, 150.30, 146.49, 139.18, 137.79, 137.73, 132.45, 132.15, 131.58, 131.51, 131.42, 131.34, 130.37, 128.64, 127.14, 127.04, 126.48, 124.59, 124.57, 121.00, 120.79, 118.79, 118.58, 116.92, 115.05, 114.80, 114.28, 46.85. ESI-MS m/z = 273.11 [M + H]+, 295.15 [M + Na]+.
5.1.3.7. 2-Amino-N-(2-(4-chlorophenyl)-2-oxoethyl)benzamide (H10-003)
Compound H10-003 was prepared by following a similar procedure to that for H4-003; 1H NMR (400 MHz, DMSO) δ 8.62 (t, J = 5.4 Hz, 1H), 8.06 (d, J = 8.5 Hz, 2H), 7.61 (dd, J = 22.5, 7.8 Hz, 3H), 7.22–7.12 (m, 1H), 6.71 (d, J = 8.0 Hz, 1H), 6.55 (t, J = 7.4 Hz, 1H), 6.44 (s, 2H), 4.70 (d, J = 5.5 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 195.32, 169.59, 150.28, 138.86, 134.29, 132.45, 130.29, 129.43, 128.63, 116.91, 115.05, 114.29, 46.66. ESI-MS m/z = 289.15 [M + H]+.
5.1.3.8. 2-Amino-N-(2-(3-chlorophenyl)-2-oxoethyl)benzamide (H11-003)
Compound H11-003 was prepared by following a similar procedure to that for H4-003; 1H NMR (400 MHz, DMSO) δ 8.65 (t, J = 5.4 Hz, 1H), 8.05 (s, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.79–7.71 (m, 1H), 7.66–7.54 (m, 2H), 7.23–7.11 (m, 1H), 6.71 (d, J = 8.0 Hz, 1H), 6.55 (t, J = 7.4 Hz, 1H), 6.45 (s, 2H), 4.71 (d, J = 5.5 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 195.38, 169.60, 150.31, 137.44, 134.23, 133.68, 132.47, 131.31, 128.64, 128.03, 127.04, 116.92, 115.05, 114.23, 46.82. ESI-MS m/z = 289.04 [M + H]+, 311.29 [M + Na]+.
5.1.3.9. 2-Amino-N-(2-(4-cyanophenyl)-2-oxoethyl)benzamide (H12-003)
Compound H12-003 was prepared by following a similar procedure to that for H4-003. The crude product was used for the follow-up reaction directly without further purification. ESI-MS m/z = 280.26 [M + H]+, 302.11 [M + Na]+.
5.1.3.10. 2-Amino-N-(2-(4-hydroxyphenyl)-2-oxoethyl)benzamide (H13-003)
Compound H13-003 was prepared by following a similar procedure to that for H4-003. 1H NMR (400 MHz, DMSO) δ 10.45 (s, 1H), 8.49 (t, J = 5.6 Hz, 1H), 7.93 (d, J = 8.7 Hz, 2H), 7.59 (d, J = 7.9 Hz, 1H), 7.21–7.12 (m, 1H), 6.89 (d, J = 8.7 Hz, 2H), 6.71 (d, J = 8.2 Hz, 1H), 6.55 (t, J = 7.5 Hz, 1H), 6.43 (s, 2H), 4.63 (d, J = 5.6 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 194.00, 169.53, 162.76, 150.19, 132.33, 130.85, 128.62, 127.14, 116.87, 115.82, 115.06, 114.66, 46.00. ESI-MS m/z = 271.11 [M + H]+, 293.58 [M + Na]+.
5.1.3.11. 2-Amino-N-(2-(2,4-difluorophenyl)-2-oxoethyl)benzamide (H14-003)
Compound H14-003 was prepared by following a similar procedure to that for H4-003. 1H NMR (400 MHz, DMSO) δ 8.66–8.57 (m, 1H), 7.98 (dd, J = 15.3, 8.5 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.48–7.36 (m, 1H), 7.24 (t, J = 8.4 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.54 (t, J = 7.5 Hz, 1H), 6.42 (s, 2H), 4.54 (dd, J = 5.2, 2.8 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 193.20, 193.15, 169.62, 168.35, 167.00, 166.88, 165.67, 164.75, 164.71, 164.47, 164.35, 163.87, 163.74, 163.04, 162.77, 162.64, 161.32, 161.19, 160.25, 160.12, 150.27, 146.01, 132.99, 132.95, 132.88, 132.84, 132.39, 132.11, 130.35, 128.62, 127.17, 127.06, 126.62, 121.16, 121.13, 121.02, 120.99, 116.90, 115.02, 114.22, 113.09, 113.06, 112.88, 112.85, 112.71, 112.67, 105.87, 105.60, 105.34, 105.30, 79.72, 79.39, 79.06, 49.74, 49.65. ESI-MS m/z = 291.20 [M + H]+, 313.16 [M + Na]+.
5.1.3.12. 2-Amino-N-(2-(3,5-difluorophenyl)-2-oxoethyl)benzamide (H15-003)
Compound H15-003 was prepared by following a similar procedure to that for H4-003. 1H NMR (400 MHz, DMSO) δ 8.67 (t, J = 5.4 Hz, 1H), 7.80–7.71 (m, 2H), 7.68–7.56 (m, 2H), 7.21–7.13 (m, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.55 (t, J = 7.5 Hz, 1H), 6.45 (s, 2H), 4.70 (d, J = 5.5 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 194.41, 169.62, 168.21, 165.93, 164.37, 164.24, 161.91, 161.79, 150.32, 146.13, 140.31, 132.50, 132.20, 130.40, 128.63, 127.18, 127.08, 126.79, 116.92, 115.04, 114.11, 111.82, 111.67, 111.41, 107.41, 107.15, 106.89, 46.97. ESI-MS m/z = 291.35 [M + H]+, 313.16 [M + Na]+.
5.1.3.13. 2-Amino-N-(2-(3,4-dimethoxyphenyl)-2-oxoethyl)benzamide (H16-003)
Compound H16-003 was prepared by following a similar procedure to that for H4-003. The crude product was used for the follow-up reaction directly without further purification. ESI-MS m/z = 315.29 [M + H]+, 337.25 [M + Na]+.
5.1.3.14. 2-Amino-N-(2-(3,4-dichlorophenyl)-2-oxoethyl)benzamide (H17-003)
Compound H17-003 was prepared by following a similar procedure to that for H4-003. The crude product was used for the follow-up reaction directly without further purification. ESI-MS m/z = 323.06 [M + H]+.
5.1.3.15. 2-Amino-N-(2-(naphthalen-2-yl)-2-oxoethyl)benzamide (H18-003)
Compound H18-003 was prepared by following a similar procedure to that for H4-003. The crude product was used for the follow-up reaction directly without further purification.
5.1.3.16. 2-Amino-N-(2-(4-bromophenyl)-2-oxoethyl)benzamide (H19-003)
Compound H19-003 was prepared by following a similar procedure to that for H4-003. 1H NMR (400 MHz, DMSO) δ 8.68–8.56 (m, 1H), 8.01–7.95 (m, 2H), 7.84–7.75 (m, 2H), 7.62–7.55 (m, 1H), 7.25–7.11 (m, 1H), 6.77–6.67 (m, 1H), 6.61–6.51 (m, 1H), 6.44 (s, 2H), 4.68 (d, J = 5.6 Hz, 2H). ESI-MS m/z = 333.18, 335.11 [M + H]+, 354.99 [M + Na]+.
5.1.3.17. 2-Amino-N-(1-(4-bromophenyl)-1-oxopropan-2-yl)benzamide (H30-003)
Compound H30-003 was prepared by following a similar procedure to that for H4-003. 1H NMR (400 MHz, DMSO) δ 8.74–8.58 (m, 1H), 7.94 (d, J = 8.2 Hz, 2H), 7.75 (d, J = 8.2 Hz, 2H), 7.59–7.47 (m, 1H), 7.14 (t, J = 7.0 Hz, 1H), 6.72–6.62 (m, 1H), 6.51 (t, J = 7.0 Hz, 1H), 6.37 (s, 2H), 5.44–5.27 (m, 1H), 1.37 (d, J = 6.8 Hz, 3H); 13 C NMR (101 MHz, DMSO) δ 199.26, 168.97, 150.25, 134.67, 132.46, 132.24, 130.67, 128.92, 127.63, 116.77, 114.92, 114.12, 50.52, 16.83. ESI-MS m/z = 347.08, 349.38 [M + H]+, 370.26 [M + Na]+.
5.1.4. Synthesis procedure for compounds H4–H19
5.1.4.1. 2-Phenyl-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H4)
2-Amino-N-(2-oxo-2-phenylethyl)benzamide (H4-003) (2 mmol) was dissolved in xylene (10 ml), and the reaction was heated and refluxed in nitrogen atmosphere for 3 h. The solvent xylene was evaporated to dryness, and the crude product was purified and crystallised from ethyl acetate to afford compound H4; 1H NMR (400 MHz, DMSO) δ 8.61 (t, J = 5.8 Hz, 1H), 8.09 (dd, J = 7.7, 1.6 Hz, 2H), 7.88 (d, J = 7.4 Hz, 1H), 7.65–7.52 (m, 4H), 7.42–7.31 (m, 2H), 3.96 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 168.46, 167.87, 146.86, 136.67, 132.09, 131.86, 130.34, 129.29, 128.37, 127.09, 126.99, 126.15, 38.99. ESI-MS m/z = 237.02 [M + H]+, 259.13 [M + Na]+.
5.1.4.2. 2-(p-Tolyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H5)
Starting from H5-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H5; 1H NMR (400 MHz, DMSO) δ 8.59 (t, J = 5.9 Hz, 1H), 7.99 (d, J = 8.2 Hz, 2H), 7.90–7.83 (m, 1H), 7.64–7.57 (m, 1H), 7.40–7.30 (m, 4H), 3.93 (s, 2H), 2.40 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 168.51, 167.60, 146.99, 141.95, 133.95, 132.05, 130.32, 129.88, 128.40, 127.09, 126.95, 125.95, 38.88, 21.51. ESI-MS m/z = 251.06 [M + H]+, 273.31 [M + Na]+.
5.1.4.3. 2-(4-Methoxyphenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H6)
Starting from H6-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H6; 1H NMR (400 MHz, DMSO) δ 8.65–8.44 (m, 1H), 8.06 (d, J = 7.5 Hz, 2H), 7.86 (d, J = 7.0 Hz, 1H), 7.66–7.50 (m, 1H), 7.33 (d, J = 7.0 Hz, 2H), 7.10 (d, J = 7.5 Hz, 2H), 3.93 (s, 2H), 3.86 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 168.60, 166.93, 162.39, 147.17, 132.01, 130.28, 129.02, 127.02, 126.94, 125.68, 114.66, 55.95, 38.73. ESI-MS m/z = 267.33 [M + H]+, 289.01 [M + Na]+.
5.1.4.4. 2-(3-Methoxyphenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H7)
Starting from H7-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H7; 1H NMR (400 MHz, DMSO) δ 8.60 (t, J = 5.9 Hz, 1H), 7.87 (dd, J = 7.7, 1.0 Hz, 1H), 7.70–7.56 (m, 3H), 7.47 (t, J = 7.9 Hz, 1H), 7.40–7.33 (m, 2H), 7.16 (dd, J = 8.1, 2.0 Hz, 1H), 3.94 (s, 2H), 3.85 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 168.40, 167.71, 160.02, 146.77, 138.14, 132.07, 130.38, 130.32, 127.10, 126.99, 126.19, 120.83, 117.72, 113.29, 55.81, 39.17. ESI-MS m/z = 267.15 [M + H]+, 289.33 [M + Na]+.
5.1.4.5. 2-(4-Fluorophenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H8)
Starting from H8-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H8; 1H NMR (400 MHz, DMSO) δ 8.58 (t, J = 5.9 Hz, 1H), 8.15 (dd, J = 8.8, 5.6 Hz, 2H), 7.87 (dd, J = 8.1, 1.4 Hz, 1H), 7.66–7.59 (m, 1H), 7.44–7.33 (m, 4H), 3.96 (d, J = 3.3 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.43, 166.76, 165.79, 163.30, 146.75, 133.24, 133.21, 132.10, 131.00, 130.91, 130.34, 127.06, 126.98, 126.18, 116.41, 116.20, 38.95. ESI-MS m/z = 255.24 [M + H]+, 277.13 [M + Na]+.
5.1.4.6. 2-(3-Fluorophenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H9)
Starting from H9-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H9; 1H NMR (400 MHz, DMSO) δ 8.63–8.49 (m, 1H), 7.97–7.82 (m, 3H), 7.69–7.55 (m, 2H), 7.50–7.32 (m, 3H), 3.96 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 168.33, 166.85, 166.82, 164.08, 161.66, 146.49, 139.18, 139.11, 132.14, 131.41, 131.33, 130.36, 127.14, 127.03, 126.47, 124.56, 124.53, 118.78, 118.57, 115.00, 114.78, 49.07, 39.02. ESI-MS m/z = 255.09 [M + H]+, 277.25 [M + Na]+.
5.1.4.7. 2-(4-Chlorophenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H10)
Starting from H10-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H10; 1H NMR (400 MHz, DMSO) δ 8.59 (t, J = 5.7 Hz, 1H), 8.10 (d, J = 8.6 Hz, 2H), 7.87 (dd, J = 8.0, 1.1 Hz, 1H), 7.67–7.59 (m, 3H), 7.40–7.33 (m, 2H), 3.94 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 168.37, 166.88, 146.63, 136.73, 135.46, 132.13, 130.37, 130.18, 129.37, 127.11, 127.00, 126.35, 38.85. ESI-MS m/z = 271.26 [M + H]+, 293.54 [M + Na]+.
5.1.4.8. 2-(3-Chlorophenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H11)
Starting from H11-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H11; 1H NMR (400 MHz, DMSO) δ 8.56 (t, J = 5.8 Hz, 1H), 8.13 (s, 1H), 8.04 (d, J = 7.7 Hz, 1H), 7.88 (dd, J = 7.7, 1.0 Hz, 1H), 7.70–7.55 (m, 3H), 7.44–7.34 (m, 2H), 3.95 (d, J = 4.1 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.29, 166.76, 146.46, 138.77, 134.23, 132.15, 131.55, 131.21, 130.36, 128.00, 127.15, 127.02, 126.52, 38.98. ESI-MS m/z = 271.29 [M + H]+, 293.00 [M + Na]+.
5.1.4.9. 4-(5-Oxo-4,5-dihydro-3H-benzo[e][1,4]diazepin-2-yl)benzonitrile (H12)
Starting from H12-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H12; 1H NMR (400 MHz, DMSO) δ 8.60 (t, J = 5.9 Hz, 1H), 8.24 (d, J = 8.5 Hz, 2H), 8.05 (d, J = 8.5 Hz, 2H), 7.89 (dd, J = 8.1, 1.4 Hz, 1H), 7.69–7.61 (m, 1H), 7.43–7.36 (m, 2H), 3.98 (d, J = 5.0 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.25, 166.90, 146.32, 140.72, 133.24, 132.22, 130.43, 129.04, 127.14, 126.82, 118.94, 113.87, 38.91. ESI-MS m/z = 262.26 [M + H]+, 284.15 [M + Na]+.
5.1.4.10. 2-(4-Hydroxyphenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H13)
Starting from H13-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H13; 1H NMR (400 MHz, DMSO) δ 10.21 (s, 1H), 8.53 (t, J = 5.9 Hz, 1H), 7.96 (d, J = 8.7 Hz, 2H), 7.84 (dd, J = 8.0, 1.4 Hz, 1H), 7.62–7.54 (m, 1H), 7.34–7.26 (m, 2H), 6.91 (d, J = 8.7 Hz, 2H), 3.90 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 170.80, 146.90, 143.78, 132.27, 131.56, 129.58, 128.77, 127.55, 127.27, 120.52, 119.19, 117.06, 45.97. ESI-MS m/z = 253.08 [M + H]+.
5.1.4.11. 2-(2,4-Difluorophenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H14)
Starting from H14-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H14; 1H NMR (400 MHz, DMSO) δ 8.69 (t, J = 5.6 Hz, 1H), 8.00–7.85 (m, 2H), 7.68–7.60 (m, 1H), 7.53–7.44 (m, 1H), 7.43–7.35 (m, 2H), 7.31–7.24 (m, 1H), 3.85 (d, J = 5.4 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.30, 165.53, 164.84, 163.15, 162.74, 162.61, 160.22, 160.09, 146.01, 133.04, 133.00, 132.94, 132.90, 132.21, 130.39, 127.20, 127.07, 126.72, 122.89, 122.77, 113.02, 112.80, 105.68, 105.42, 105.16, 41.66, 41.59. ESI-MS m/z = 273.13 [M + H]+, 295.06 [M + Na]+.
5.1.4.12. 2-(3,5-Difluorophenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H15)
Starting from H15-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H15; 1H NMR (400 MHz, DMSO) δ 8.49 (t, J = 5.6 Hz, 1H), 7.91–7.84 (m, 1H), 7.83–7.73 (m, 2H), 7.68–7.59 (m, 1H), 7.56–7.47 (m, 1H), 7.43–7.34 (m, 2H), 3.95 (d, J = 5.1 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.20, 165.94, 164.37, 164.24, 161.92, 161.79, 146.13, 140.31, 132.20, 130.40, 127.19, 127.08, 126.79, 111.68, 111.42, 107.42, 107.16, 38.96. ESI-MS m/z = 273.35 [M + H]+, 295.31 [M + Na]+.
5.1.4.13. 2-(3,4-Dimethoxyphenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H16)
Starting from H16-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H16; 1H NMR (400 MHz, DMSO) δ 8.57 (t, J = 5.9 Hz, 1H), 7.85 (dd, J = 7.8, 1.2 Hz, 1H), 7.72–7.63 (m, 2H), 7.63–7.56 (m, 1H), 7.38–7.28 (m, 2H), 7.11 (d, J = 8.4 Hz, 1H), 3.94 (s, 2H), 3.86 (s, 6H); 13 C NMR (101 MHz, DMSO) δ 168.59, 166.98, 152.32, 149.33, 147.15, 131.99, 130.29, 129.08, 127.02, 126.95, 125.68, 122.42, 111.60, 110.75, 56.16, 56.01, 38.73. ESI-MS m/z = 297.25 [M + H]+.
5.1.4.14. 2-(3,4-Dichlorophenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H17)
Starting from H17-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H17; 1H NMR (400 MHz, DMSO) δ 8.53 (t, J = 5.9 Hz, 1H), 8.30 (d, J = 1.9 Hz, 1H), 8.05 (dd, J = 8.5, 2.0 Hz, 1H), 7.90–7.80 (m, 2H), 7.64 (td, J = 7.8, 1.4 Hz, 1H), 7.43–7.34 (m, 2H), 3.95 (d, J = 4.5 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.25, 165.96, 146.31, 137.18, 134.53, 132.29, 132.18, 131.55, 130.39, 130.11, 128.39, 127.16, 127.05, 126.65, 38.81. ESI-MS m/z = 305.03 [M + H]+.
5.1.4.15. 2-(Naphthalen-2-yl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H18)
Starting from H18-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H18; 1H NMR (400 MHz, DMSO) δ 8.71–8.62 (m, 2H), 8.27 (dd, J = 8.7, 1.5 Hz, 1H), 8.10–7.98 (m, 3H), 7.90 (dd, J = 7.8, 1.2 Hz, 1H), 7.69–7.59 (m, 3H), 7.45–7.34 (m, 2H), 4.12 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 168.53, 167.68, 146.97, 134.66, 133.91, 133.01, 132.12, 130.37, 129.43, 128.92, 128.44, 128.18, 127.41, 127.08, 126.20, 124.77, 38.84. ESI-MS m/z = 287.75 [M + H]+.
5.1.4.16. 2-(4-Bromophenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H19)
Starting from H19-003 (2 mmol), and following the procedure similar to that of preparation of H4 to give H19; 1H NMR (400 MHz, DMSO) δ 8.56 (t, J = 5.9 Hz, 1H), 8.02 (d, J = 8.6 Hz, 2H), 7.87 (dd, J = 8.0, 1.3 Hz, 1H), 7.76 (d, J = 8.6 Hz, 2H), 7.65–7.58 (m, 1H), 7.40–7.32 (m, 2H), 3.93 (d, J = 5.2 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.37, 167.02, 146.63, 135.83, 132.30, 132.13, 130.37, 130.35, 127.11, 126.99, 126.36, 125.72, 38.82. ESI-MS m/z = 315.12, 317.01 [M + H]+.
5.1.5. Synthesis procedure for compound H20–H27
5.1.5.1. 2-(4-Methoxyphenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H20)
The solution of 2-(4-methoxyphenyl)-3H-benzo[e][1,4]diazepin-5(4H)-one (H6) (1 mmol, 1eq) in methanol was placed in an ice bath, then the sodium borohydride (4 mmol, 4 equiv.) was added to the reaction bottle in batches maintaining the temperature below 10 °C. The reaction was monitored by TLC. After completed, the solvent methanol was removed under reduced pressure. The residue was extracted twice with ethyl acetate and water, the combined organic layer was washed with brine, dried over anhydrous Na2SO4. After filtration and concentration, the crude product was obtained and purified with column chromatography to afford the compound H20; 1H NMR (400 MHz, DMSO) δ 7.89 (t, J = 5.9 Hz, 1H), 7.62 (dd, J = 7.9, 1.4 Hz, 1H), 7.28–7.17 (m, 3H), 6.92 (d, J = 8.7 Hz, 2H), 6.86 (d, J = 8.0 Hz, 1H), 6.67 (t, J = 7.4 Hz, 1H), 6.48 (d, J = 3.4 Hz, 1H), 4.73–4.65 (m, 1H), 3.74 (s, 3H), 3.40–3.34 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.78, 158.90, 146.88, 135.70, 132.32, 132.24, 128.32, 119.36, 119.20, 116.99, 114.16, 62.25, 55.56, 46.22. ESI-MS m/z = 291.06 [M + Na]+.
5.1.5.2. 2-(3-Methoxyphenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H21)
Starting from H7 (1 mmol), and following the procedure similar to that of preparation of H20 to give H21; 1H NMR (400 MHz, DMSO) δ 7.90 (t, J = 5.8 Hz, 1H), 7.61 (dd, J = 7.9, 1.4 Hz, 1H), 7.31–7.17 (m, 2H), 6.93–6.81 (m, 4H), 6.68 (t, J = 7.1 Hz, 1H), 6.54 (d, J = 3.7 Hz, 1H), 4.78–4.69 (m, 1H), 3.74 (s, 3H), 3.44–3.37 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.76, 159.74, 146.84, 145.40, 132.36, 132.25, 129.84, 119.48, 119.44, 119.17, 117.10, 113.07, 112.75, 62.63, 55.44, 45.90. ESI-MS m/z = 269.19 [M + H]+, 291.05 [M + Na]+.
5.1.5.3. 2-(4-Fluorophenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H22)
Starting from H8 (1 mmol), and following the procedure similar to that of preparation of H20 to give H22;1H NMR (400 MHz, DMSO) δ 7.89 (t, J = 5.8 Hz, 1H), 7.62 (dd, J = 7.9, 1.2 Hz, 1H), 7.40–7.31 (m, 2H), 7.26–7.15 (m, 3H), 6.86 (d, J = 8.1 Hz, 1H), 6.69 (t, J = 7.4 Hz, 1H), 6.57 (d, J = 4.1 Hz, 1H), 4.82–4.75 (m, 1H), 3.46–3.32 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.80, 163.02, 160.61, 146.76, 139.98, 139.95, 132.39, 132.25, 129.23, 129.15, 119.52, 119.17, 117.20, 115.53, 115.32, 61.94, 45.79. ESI-MS m/z = 257.12 [M + H]+, 279.08 [M + Na]+.
5.1.5.4. 2-(3-Fluorophenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H23)
Starting from H9 (1 mmol), and following the procedure similar to that of preparation of H20 to give H23;1H NMR (400 MHz, DMSO) δ 7.90 (t, J = 5.9 Hz, 1H), 7.62 (dd, J = 7.9, 1.4 Hz, 1H), 7.45–7.36 (m, 1H), 7.28–7.21 (m, 1H), 7.20–7.06 (m, 3H), 6.87 (d, J = 8.0 Hz, 1H), 6.70 (t, J = 7.1 Hz, 1H), 6.63 (d, J = 4.5 Hz, 1H), 4.86–4.79 (m, 1H), 3.48–3.41 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.78, 163.89, 161.47, 146.95, 146.88, 146.68, 132.43, 132.26, 130.71, 130.63, 123.35, 123.33, 119.55, 119.16, 117.31, 114.32, 114.17, 114.11, 113.96, 62.01, 45.42. ESI-MS m/z = 257.18 [M + H]+, 279.14 [M + Na]+.
5.1.5.5. 2-(2,4-Difluorophenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H24)
Starting from H14 (1 mmol), and following the procedure similar to that of preparation of H20 to give H24; 1H NMR (400 MHz, DMSO) δ 7.89 (t, J = 5.5 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.39–7.21 (m, 3H), 7.10 (t, J = 8.5 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.71 (t, J = 7.4 Hz, 1H), 6.57 (d, J = 4.6 Hz, 1H), 5.02–4.94 (m, 1H), 3.54–3.33 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.72, 163.25, 163.12, 160.81, 160.69, 160.57, 158.24, 158.12, 146.59, 132.47, 132.23, 131.08, 126.72, 126.62, 119.55, 119.20, 117.45, 111.81, 111.57, 104.45, 104.19, 103.94, 60.23, 43.85. ESI-MS m/z = 296.96 [M + H]+.
5.1.5.6. 2-(3,5-Difluorophenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H25)
Starting from H15 (1 mmol), and following the procedure similar to that of preparation of H20 to give H25; 1H NMR (400 MHz, DMSO) δ 7.88 (t, J = 5.9 Hz, 1H), 7.60 (dd, J = 7.9, 1.2 Hz, 1H), 7.29–7.20 (m, 1H), 7.18–7.09 (m, 1H), 7.07–6.98 (m, 2H), 6.85 (d, J = 8.1 Hz, 1H), 6.71 (t, J = 7.4 Hz, 1H), 6.65 (d, J = 5.0 Hz, 1H), 4.89–4.77 (m, 1H), 3.51–3.40 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.72, 164.07, 163.94, 161.63, 161.50, 148.93, 148.85, 148.77, 146.44, 132.48, 132.23, 119.68, 119.14, 117.55, 110.55, 110.49, 110.30, 103.05, 102.79, 102.53, 61.69, 44.91. ESI-MS m/z = 297.11 [M + H]+.
5.1.5.7. 2-(3,4-Dimethoxyphenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H26)
Starting from H16 (1 mmol), and following the procedure similar to that of preparation of H20 to give H26; 1H NMR (400 MHz, DMSO) δ 7.91 (t, J = 5.9 Hz, 1H), 7.61 (dd, J = 7.9, 1.4 Hz, 1H), 7.25–7.18 (m, 1H), 6.95–6.90 (m, 2H), 6.88 (d, J = 8.0 Hz, 1H), 6.83 (dd, J = 8.3, 1.5 Hz, 1H), 6.67 (t, J = 7.2 Hz, 1H), 6.45 (d, J = 3.4 Hz, 1H), 4.71–4.64 (m, 1H), 3.73 (d, J = 2.7 Hz, 6H), 3.42 − 3.35 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.77, 149.07, 148.43, 146.85, 136.12, 132.30, 132.20, 119.55, 119.21, 117.06, 112.10, 111.01, 62.57, 56.04, 55.88, 46.21. ESI-MS m/z = 321.16 [M + H]+.
5.1.5.8. 2-(4-Bromophenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H27)
Starting from H19 (1 mmol), and following the procedure similar to that of preparation of H20 to give H27; 1H NMR (400 MHz, DMSO) δ 7.86–7.74 (m, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.55 (d, J = 7.6 Hz, 2H), 7.27 (d, J = 7.7 Hz, 2H), 7.22 (t, J = 7.6 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.69 (t, J = 7.3 Hz, 1H), 6.55 (s, 1H), 4.83–4.71 (m, 1H), 3.48–3.33 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.75, 146.69, 143.26, 132.38, 132.27, 131.56, 129.58, 120.52, 119.51, 119.14, 117.25, 61.94, 45.46. ESI-MS m/z = 317.23, 319.05 [M + H]+, 341.02 [M + Na]+.
5.1.6. Synthesis procedure for compounds H28–H31
5.1.6.1. 2-(4-Bromophenyl)-4-methyl-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H28)
Compound H27 (317 mg, 1 mmol) and 10 ml acetonitrile were added to 50 ml reaction flask to obtain uniform turbid solution. Then NaH (80 mg, 2 mmol) was added in batches in an ice bath and slowly warmed to room temperature. The acetonitrile solution of dimethyl sulphate (252 mg, 2 mmol) was then slowly dripped into the reaction bottle and reacted at room temperature for about 2 h. The progress of the reaction was monitored by TLC. After the reaction was completed, the inorganic salts were removed by filtration, and the residue was washed twice with ethyl acetate. The combined organic layers were concentrated and filtered to afford the desired compound. 1H NMR (400 MHz, DMSO) δ 7.60–7.52 (m, 3H), 7.33 (d, J = 8.4 Hz, 2H), 7.27–7.20 (m, 1H), 6.86 (d, J = 7.9 Hz, 1H), 6.74 (t, J = 7.4 Hz, 1H), 6.45 (d, J = 4.2 Hz, 1H), 4.95–4.89 (m, 1H), 3.75–3.56 (m, 2H), 2.63 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 169.29, 146.33, 142.84, 132.20, 131.98, 131.64, 129.42, 121.44, 120.68, 119.17, 117.91, 61.98, 53.57, 36.52. ESI-MS m/z = 331.23, 333.07 [M + H]+, 355.39 [M + Na]+.
5.1.6.2. 2-(4-Bromophenyl)-1,4-dimethyl-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H29)
Formic acid (88%, 5 mmol) was added to the aqueous solution of H27 (1 mmol) in an ice bath, and the mixture was slowly warmed until solution was obtained. Then 1.2 mmol CH2O (37%) was added at room temperature and the mixture was heated at 100 °C until the generation of CO2 ceased. The solution was cooled, acidified with concentrated HCl and evaporated. The residue was dissolved in H2O and alkalified with NaOH and extracted three times with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4. After filtration and concentration, the crude product was obtained and purified with column chromatography to afford the desired compound. 1H NMR (400 MHz, DMSO) δ 7.59–7.52 (m, 3H), 7.50–7.45 (m, 1H), 7.13 (d, J = 8.2 Hz, 2H), 7.05 (t, J = 7.4 Hz, 1H), 6.95 (d, J = 8.1 Hz, 1H), 5.03 (d, J = 9.8 Hz, 1H), 4.64–4.55 (m, 1H), 3.57–3.50 (m, 1H), 3.23 (s, 3H), 2.58 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 170.34, 146.53, 139.12, 132.34, 131.52, 130.25, 129.26, 128.49, 121.01, 120.89, 119.12, 76.65, 70.00, 50.94, 38.18. ESI-MS m/z = 345.08, 347.05 [M + H]+.
5.1.6.3. 2-(4-Bromophenyl)-3-methyl-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H30)
Starting from H30-003 (1 mmol), and following the procedure similar to that of preparation of H4 to give H30; 1H NMR (400 MHz, DMSO) δ 8.70 (d, J = 7.3 Hz, 1H), 7.87 (d, J = 7.6 Hz, 3H), 7.75 (d, J = 8.5 Hz, 2H), 7.66–7.59 (m, 1H), 7.42–7.31 (m, 2H), 4.53 (s, 1H), 1.10 (d, J = 6.9 Hz, 3H); 13 C NMR (101 MHz, DMSO) δ 169.76, 167.34, 145.93, 136.79, 132.46, 132.11, 130.66, 130.30, 127.06, 126.36, 125.06, 46.21, 15.91. ESI-MS m/z = 329.19, 331.77 [M + H]+, 351.02 [M + Na]+.
5.1.6.4. 2-(4-Bromophenyl)-3-methyl-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H31)
Starting from H30 (1 mmol), and following the procedure similar to that of preparation of H20 to give H31; 1H NMR (400 MHz, DMSO) δ 7.64 (d, J = 5.5 Hz, 1H), 7.61–7.53 (m, 3H), 7.28–7.17 (m, 3H), 6.81 (d, J = 8.0 Hz, 1H), 6.71 (t, J = 7.4 Hz, 1H), 6.59 (d, J = 5.5 Hz, 1H), 4.58 (d, J = 3.8 Hz, 1H), 3.91–3.78 (m, 1H), 0.99 (d, J = 6.9 Hz, 3H); 13 C NMR (101 MHz, DMSO) δ 169.70, 146.61, 141.33, 132.35, 131.84, 131.23, 130.97, 120.83, 120.37, 119.04, 117.56, 66.84, 49.08, 17.12. ESI-MS m/z = 331.23, 333.05 [M + H]+, 353.02, 355.00 [M + Na]+.
5.1.7. Synthesis procedure for compounds H32–H55
5.1.7.1. 2-(4-(1H-pyrazol-4-yl)phenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H32)
4-Pyrazole-pinacol borate (1.1 mmol, 1.1 eq), H19 (1 mmol, 1 equiv.), Na2CO3 (2 mmol, 2 equiv.) and 10 ml dioxane were placed into a 50 ml three-necked bottle, the reaction system was evacuated and backfilled with argon three times. Pd(dppf)Cl2 (0.05 mmol, 0.05 eq) was added to the reaction bottle under argon flow. The resulting solution was heated to 90 °C and stirred at this temperature for 2 h. The reaction was monitored by TLC. The mixture was diluted with water and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated to give a crude product, which was purified by column chromatography to give compound H32; 1H NMR (400 MHz, DMSO) δ 13.11 (s, 1H), 8.60 (t, J = 5.9 Hz, 1H), 8.37 (s, 1H), 8.08 (d, J = 8.4 Hz, 3H), 7.87 (dd, J = 7.8, 1.2 Hz, 1H), 7.81 (d, J = 8.4 Hz, 2H), 7.66–7.58 (m, 1H), 7.40–7.31 (m, 2H), 3.97 (s, 2H). 13 C NMR (101 MHz, DMSO) δ 168.54, 167.34, 147.06, 136.57, 133.82, 132.05, 130.33, 129.04, 128.63, 127.10, 127.01, 125.92, 125.62, 125.46, 120.85, 38.79. ESI-MS m/z = 303.13 [M + H]+.
5.1.7.2. 2-(4-(1-Methyl-1H-pyrazol-4-yl)phenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H33)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 1-methyl-4-pyrazole-pinacol borate (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.59 (t, J = 6.0 Hz, 1H), 8.31 (s, 1H), 8.08 (d, J = 8.5 Hz, 2H), 8.02 (s, 1H), 7.87 (dd, J = 7.8, 1.4 Hz, 1H), 7.75 (d, J = 8.5 Hz, 2H), 7.66–7.58 (m, 1H), 7.40–7.30 (m, 2H), 3.96 (s, 2H), 3.89 (s, 3H). 13 C NMR (101 MHz, DMSO) δ 168.54, 167.31, 147.05, 137.02, 136.21, 133.90, 132.05, 130.33, 129.09, 127.10, 127.00, 125.93, 125.43, 121.54, 38.78. ESI-MS m/z = 317.34 [M + H]+.
5.1.7.3. 2-(4-(3,5-Dimethyl-1H-pyrazol-4-yl)phenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H34)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 3,5-dimethyl-4-pyrazole-pinacol borate (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 12.46 (s, 1H), 8.61 (t, J = 5.9 Hz, 1H), 8.12 (d, J = 8.3 Hz, 2H), 7.91–7.84 (m, 1H), 7.66–7.58 (m, 1H), 7.48 (d, J = 8.3 Hz, 2H), 7.41–7.31 (m, 2H), 3.98 (s, 2H), 2.26 (s, 6H). 13 C NMR (101 MHz, DMSO) δ 168.52, 167.50, 147.05, 137.91, 133.77, 132.06, 130.34, 129.22, 128.60, 127.09, 127.01, 125.98, 116.63, 38.86, 26.81, 25.85. ESI-MS m/z = 331.10 [M + H]+.
5.1.7.4. 2-(4-(1-Methyl-1H-pyrazol-5-yl)phenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H35)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 1-methyl-5-pyrazole boric acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.63 (t, J = 5.9 Hz, 1H), 8.19 (d, J = 8.3 Hz, 2H), 7.92–7.85 (m, 1H), 7.74 (d, J = 8.3 Hz, 2H), 7.64 (t, J = 7.6 Hz, 1H), 7.53 (d, J = 1.7 Hz, 1H), 7.43–7.34 (m, 2H), 6.55 (d, J = 1.7 Hz, 1H), 4.00 (s, 2H), 3.93 (s, 3H). 13 C NMR (101 MHz, DMSO) δ 168.44, 167.33, 146.80, 142.33, 138.56, 136.26, 133.25, 132.13, 130.38, 129.12, 128.78, 127.12, 127.06, 126.29, 106.89, 38.93, 38.29. ESI-MS m/z = 317.11 [M + H]+.
5.1.7.5. 2-(3'-Amino-[1,1'-biphenyl]-4-yl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H36)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 3-aminophenylboronic acid pinacol ester (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.62 (t, J = 5.9 Hz, 1H), 8.15 (d, J = 8.4 Hz, 2H), 7.89 (dd, J = 7.8, 1.2 Hz, 1H), 7.75 (d, J = 8.4 Hz, 2H), 7.67–7.58 (m, 1H), 7.43–7.32 (m, 2H), 7.15 (t, J = 7.8 Hz, 1H), 6.98–6.93 (m, 1H), 6.89 (d, J = 7.6 Hz, 1H), 6.63 (dd, J = 7.9, 1.3 Hz, 1H), 5.26 (s, 2H), 3.99 (s, 2H); 13 C NMR (101 MHz, DMSO) δ 168.51, 167.45, 149.75, 146.98, 144.27, 140.20, 135.23, 132.09, 130.36, 130.05, 128.95, 127.17, 127.11, 127.03, 126.09, 114.92, 114.33, 112.53, 60.24, 38.89, 21.24, 14.56. ESI-MS m/z = 328.36 [M + H]+.
5.1.7.6. 2-(4′-Amino-[1,1′-biphenyl]-4-yl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H37)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 4-aminophenylboronic acid pinacol ester (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.56 (t, J = 5.9 Hz, 1H), 8.08 (d, J = 8.4 Hz, 2H), 7.87 (dd, J = 7.8, 1.2 Hz, 1H), 7.73 (d, J = 8.5 Hz, 2H), 7.64–7.57 (m, 1H), 7.50 (d, J = 8.5 Hz, 2H), 7.40–7.29 (m, 2H), 6.68 (d, J = 8.5 Hz, 2H), 5.39 (s, 2H), 3.96 (d, J = 4.6 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.56, 167.36, 149.72, 147.13, 143.91, 133.59, 132.03, 130.33, 128.98, 127.95, 127.10, 127.02, 126.31, 125.87, 125.68, 114.67, 38.81. ESI-MS m/z = 328.29 [M + H]+.
5.1.7.7. 2-(4-(6-Aminopyridin-3-yl)phenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H38)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 2-aminopyridine-5-borate pinacol ester (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.71–8.52 (m, 1H), 8.41 (s, 1H), 8.11 (d, J = 6.9 Hz, 2H), 7.93–7.71 (m, 4H), 7.68–7.54 (m, 1H), 7.43–7.26 (m, 2H), 6.57 (d, J = 7.5 Hz, 1H), 6.26 (s, 2H), 3.97 (s, 2H). 13 C NMR (101 MHz, DMSO) δ 168.53, 167.36, 160.18, 147.04, 146.74, 141.42, 135.84, 134.22, 132.07, 130.34, 129.08, 127.11, 127.03, 125.98, 125.71, 122.98, 108.50, 38.83. ESI-MS m/z = 329.01 [M + H]+.
5.1.7.8. 2-(4-(2-Aminopyrimidin-5-yl)phenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H39)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 2-aminopyrimidine-5-borate pinacol ester (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.72 (s, 2H), 8.62 (t, J = 5.9 Hz, 1H), 8.14 (d, J = 8.4 Hz, 2H), 7.90–7.81 (m, 3H), 7.67–7.58 (m, 1H), 7.41–7.31 (m, 2H), 6.98 (s, 2H), 3.98 (s, 2H). 13 C NMR (101 MHz, DMSO) δ 168.50, 167.34, 163.66, 156.70, 146.96, 138.60, 134.87, 132.09, 130.35, 129.13, 127.11, 127.04, 126.07, 125.64, 121.29, 38.86. ESI-MS m/z = 330.19 [M + H]+.
5.1.7.9. 2-(4-(5-Amino-6-methoxypyridin-3-yl)phenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H40)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 3-amino-2-methoxypyridine-5-borate pinacol ester (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.62 (t, J = 5.9 Hz, 1H), 8.15 (d, J = 8.5 Hz, 2H), 7.88 (dd, J = 7.8, 1.3 Hz, 1H), 7.79 (d, J = 2.2 Hz, 1H), 7.75 (d, J = 8.5 Hz, 2H), 7.66–7.60 (m, 1H), 7.41–7.33 (m, 2H), 7.25 (d, J = 2.2 Hz, 1H), 5.17 (s, 2H), 3.98 (s, 2H), 3.92 (s, 3H). 13 C NMR (101 MHz, DMSO) δ 168.50, 167.39, 152.51, 146.96, 141.38, 135.11, 133.12, 132.09, 131.06, 130.36, 129.38, 129.06, 127.11, 127.03, 126.81, 126.08, 117.15, 53.55, 38.88. ESI-MS m/z = 359.03 [M + H]+.
5.1.7.10. 2-(3′-Methoxy-[1,1′-biphenyl]-4-yl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H41)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 3-methoxyphenylboronic acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.62–8.54 (m, 1H), 8.16 (d, J = 7.9 Hz, 2H), 7.92–7.82 (m, 3H), 7.62 (t, J = 7.5 Hz, 1H), 7.46–7.26 (m, 5H), 7.00 (d, J = 8.0 Hz, 1H), 3.99 (d, J = 4.1 Hz, 2H), 3.85 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 168.49, 167.42, 160.33, 146.94, 143.16, 141.02, 135.72, 132.07, 130.61, 130.36, 128.98, 127.56, 127.14, 127.05, 126.13, 119.66, 114.21, 112.86, 55.69, 38.95. ESI-MS m/z = 343.18 [M + H]+.
5.1.7.11. 2-(4′-Methoxy-[1,1′-biphenyl]-4-yl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H42)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 4-methoxyphenylboronic acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.58 (t, J = 5.9 Hz, 1H), 8.14 (d, J = 8.3 Hz, 2H), 7.88 (d, J = 7.6 Hz, 1H), 7.81 (d, J = 8.3 Hz, 2H), 7.73 (d, J = 8.6 Hz, 2H), 7.65–7.59 (m, 1H), 7.40–7.32 (m, 2H), 7.06 (d, J = 8.7 Hz, 2H), 3.98 (d, J = 5.0 Hz, 2H), 3.81 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 168.51, 167.40, 159.99, 147.00, 142.95, 134.81, 132.07, 131.74, 130.36, 129.03, 128.50, 127.12, 127.04, 126.77, 126.04, 114.99, 55.72, 38.89. ESI-MS m/z = 343.14 [M + H]+.
5.1.7.12. 2-(3′-(Trifluoromethyl)-[1,1′-biphenyl]-4-yl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H43)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 3-(trifluoromethyl)phenylboronic acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.60 (t, J = 5.9 Hz, 1H), 8.21 (d, J = 8.4 Hz, 2H), 8.13–8.07 (m, 2H), 7.97 (d, J = 8.4 Hz, 2H), 7.92–7.87 (m, 1H), 7.82–7.72 (m, 2H), 7.67–7.61 (m, 1H), 7.43–7.34 (m, 2H), 4.01 (d, J = 5.3 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.45, 167.38, 146.86, 141.54, 140.60, 136.35, 132.09, 131.48, 130.66, 130.37, 129.13, 127.82, 127.14, 127.06, 126.23, 125.20, 123.77, 38.97. ESI-MS m/z = 381.11 [M + H]+.
5.1.7.13. 2-(4′-(Trifluoromethyl)-[1,1′-biphenyl]-4-yl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H44)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 4-(trifluoromethyl)phenylboronic acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.59 (t, J = 5.9 Hz, 1H), 8.21 (d, J = 8.4 Hz, 2H), 8.00 (d, J = 8.2 Hz, 2H), 7.96–7.82 (m, 5H), 7.67–7.58 (m, 1H), 7.42–7.32 (m, 2H), 4.00 (d, J = 5.2 Hz, 2H); 13 C NMR (101 MHz, DMSO) δ 168.45, 167.36, 146.84, 143.52, 141.57, 136.54, 132.09, 130.37, 129.14, 128.16, 127.88, 127.72, 127.58, 127.15, 127.06, 126.36, 126.32, 38.96. ESI-MS m/z = 381.30 [M + H]+.
5.1.7.14. 2-(4-(1-Methyl-1,2,3,6-tetrahydropyridin-4-yl)phenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H45)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 1-methyl-1, 2, 3, 6-tetrahydropyridine-4-borate pinacol ester (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.63 (t, J = 5.6 Hz, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.90–7.83 (m, 1H), 7.65–7.56 (m, 3H), 7.39–7.30 (m, 2H), 6.36 (s, 1H), 3.97 (d, J = 22.2 Hz, 2H), 3.05 (d, J = 2.6 Hz, 2H), 2.61–2.55 (m, 2H), 2.54–2.48 (m, 2H), 2.28 (s, 3H). 13 C NMR (101 MHz, DMSO) δ 168.49, 167.36, 146.97, 143.27, 134.98, 133.40, 132.06, 130.34, 128.54, 127.10, 127.01, 126.03, 125.23, 124.58, 55.13, 52.16, 45.87, 38.81, 27.68. ESI-MS m/z = 332.09 [M + H]+.
5.1.7.15. 2-(4-(9H-carbazol-3-yl)phenyl)-3,4-dihydro-5H-benzo[e][1,4]diazepin-5-one (H46)
This compound was synthesised by employing the procedure for H32 using H19 (1 mmol, 1 equiv.) and 9H-carbazole-3-boronic acid pinacol ester (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 11.44 (s, 1H), 8.66 (t, J = 6.0 Hz, 1H), 8.61 (d, J = 1.3 Hz, 1H), 8.27 (d, J = 7.7 Hz, 1H), 8.20 (d, J = 8.5 Hz, 2H), 7.99 (d, J = 8.5 Hz, 2H), 7.93–7.81 (m, 2H), 7.67–7.58 (m, 2H), 7.53 (d, J = 8.1 Hz, 1H), 7.47–7.33 (m, 3H), 7.21 (t, J = 7.4 Hz, 1H), 4.06–3.96 (m, 2H). 13 C NMR (101 MHz, DMSO) δ 168.56, 167.47, 147.07, 144.51, 140.74, 140.21, 134.39, 132.09, 130.37, 130.05, 129.07, 127.17, 127.13, 127.06, 126.39, 126.01, 125.16, 123.66, 123.06, 121.05, 119.30, 119.12, 111.94, 111.65, 38.89. ESI-MS m/z = 402.46 [M + H]+.
5.1.7.16. 2-(3′-Amino-[1,1′-biphenyl]-4-yl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H47)
This compound was synthesised by employing the procedure for H32 using H27 (1 mmol, 1 equiv.) and (3-aminophenyl)boronic acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 7.92 (t, J = 5.9 Hz, 1H), 7.64 (dd, J = 7.9, 1.3 Hz, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 7.27–7.19 (m, 1H), 7.09 (t, J = 7.8 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.83 (s, 1H), 6.77 (d, J = 7.7 Hz, 1H), 6.69 (t, J = 7.2 Hz, 1H), 6.60–6.52 (m, 2H), 5.15 (s, 2H), 4.83–4.76 (m, 1H), 3.50–3.39 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.83, 149.59, 146.89, 142.60, 141.16, 140.41, 132.38, 132.30, 129.88, 127.72, 126.83, 119.40, 119.20, 117.08, 114.75, 113.56, 112.48, 62.51, 45.91. ESI-MS m/z = 330.30 [M + H]+.
5.1.7.17. 2-(4′-Amino-[1,1′-biphenyl]-4-yl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H48)
This compound was synthesised by employing the procedure for H32 using H27 (1 mmol, 1 equiv.) and 4-aminophenylboronic acid pinacol ester (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 7.95–7.85(m, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.51 (d, J = 7.1 Hz, 2H), 7.40–7.26 (m, 4H), 7.22 (t, J = 7.5 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.73–6.59 (m, 3H), 6.53 (s, 1H), 5.21 (s, 2H), 4.81 − 4.69(m, 1H), 3.50–3.38 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.82, 148.81, 146.92, 141.03, 140.10, 132.36, 132.29, 127.67, 127.55, 125.72, 119.36, 119.20, 117.02, 114.70, 62.53, 46.03. ESI-MS m/z = 330.49 [M + H]+.
5.1.7.18. 2-(3′-Methoxy-[1,1′-biphenyl]-4-yl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H49)
This compound was synthesised by employing the procedure for H32 using H27 (1 mmol, 1 equiv.) and 3-methoxyphenylboronic acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 7.91 (t, J = 5.9 Hz, 1H), 7.70–7.61 (m, 3H), 7.43–7.34 (m, 3H), 7.28–7.15 (m, 3H), 6.96–6.86 (m, 2H), 6.69 (t, J = 7.4 Hz, 1H), 6.58 (d, J = 3.9 Hz, 1H), 4.86–4.77 (m, 1H), 3.82 (s, 3H), 3.53–3.39 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.82, 160.22, 146.89, 143.16, 141.95, 139.36, 132.40, 132.31, 130.48, 127.85, 127.17, 119.43, 119.39, 119.20, 117.11, 113.33, 112.63, 62.40, 55.57, 45.85. ESI-MS m/z = 345.35 [M + H]+.
5.1.7.19. 2-(4′-Methoxy-[1,1′-biphenyl]-4-yl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H50)
This compound was synthesised by employing the procedure for H32 using H27 (1 mmol, 1 equiv.) and 4-methoxyphenylboronic acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 7.90 (t, J = 5.8 Hz, 1H), 7.66–7.56 (m, 5H), 7.36 (d, J = 8.1 Hz, 2H), 7.26–7.18 (m, 1H), 7.02 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.2 Hz, 1H), 6.69 (t, J = 7.4 Hz, 1H), 6.56 (d, J = 3.8 Hz, 1H), 4.84–4.75 (m, 1H), 3.79 (s, 3H), 3.48–3.40 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.83, 159.32, 146.90, 142.21, 139.15, 132.78, 132.38, 132.30, 128.12, 127.83, 126.54, 119.40, 119.20, 117.07, 114.85, 62.43, 55.63, 45.91. ESI-MS m/z = 345.35 [M + H]+.
5.1.7.20. 2-(3′-(Trifluoromethyl)-[1,1′-biphenyl]-4-yl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H51)
This compound was synthesised by employing the procedure for H32 using H27 (1 mmol, 1 equiv.) and 3-(trifluoromethyl)phenylboronic acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 8.02–7.93 (m, 2H), 7.90 (t, J = 5.4 Hz, 1H), 7.80–7.67 (m, 4H), 7.64 (d, J = 7.8 Hz, 1H), 7.45 (d, J = 7.9 Hz, 2H), 7.24 (t, J = 7.6 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.69 (t, J = 7.4 Hz, 1H), 6.64–6.56 (m, 1H), 4.90–4.79 (m, 1H), 3.53–3.41 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.82, 146.86, 143.96, 141.45, 137.78, 132.40, 132.31, 131.16, 130.57, 128.11, 127.36, 123.36, 119.47, 119.20, 117.16, 62.33, 45.74. ESI-MS m/z = 383.24 [M + H]+.
5.1.7.21. 2-(4′-(Trifluoromethyl)-[1,1′-biphenyl]-4-yl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H52)
This compound was synthesised by employing the procedure for H32 using H27 (1 mmol, 1 equiv.) and 4-(trifluoromethyl)phenylboronic acid (1.1 mmol, 1.1 eq) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 7.97–7.86 (m, 3H), 7.81 (d, J = 8.1 Hz, 2H), 7.74 (d, J = 7.8 Hz, 2H), 7.66 (d, J = 7.8 Hz, 1H), 7.46 (d, J = 7.9 Hz, 2H), 7.25 (t, J = 7.6 Hz, 1H), 6.91 (d, J = 8.2 Hz, 1H), 6.70 (t, J = 7.4 Hz, 1H), 6.62 (d, J = 3.7 Hz, 1H), 4.92–4.80 (m, 1H), 3.54–3.42 (m, 2H); 13 C NMR (101 MHz, DMSO) δ 170.86, 146.86, 144.39, 144.19, 137.81, 132.41, 132.32, 128.38, 128.12, 127.80, 127.43, 126.28, 126.25, 126.21, 119.45, 119.20, 117.16, 62.34, 45.71. ESI-MS m/z = 383.20 [M + H]+.
5.1.7.22. 2-(4-(4-Isopropylpiperazin-1-yl)phenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H53)
H27 (1 mmol, 1 equiv.), NaOtBu (2 mmol, 2 equiv.), Pd(OAc)2 (0.05 mmol, 0.05 eq) and RuPhos (0.1 mmol, 0.11 eq) were placed into a 10 ml Schlenk tube. The tube was evacuated and backfilled with argon three times, then 1-isopropylpiperazine (2 mmol, 2 equiv.) and dioxane (4 ml) were added via syringe under argon flow . The reaction mixture was heated at 100 °C for 24 h under vigorous stirring. The cooled solution was diluted with ethyl acetate, washed with brine. The organic phase was dried over anhydrous Na2SO4, concentrated in vacuo, and purified by column chromatography to afford the corresponding compound 7 m; 1H NMR (400 MHz, DMSO) δ 7.90 (t, J = 5.4 Hz, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.24–7.08 (m, 3H), 6.97–6.79 (m, 3H), 6.65 (t, J = 7.3 Hz, 1H), 6.46–6.35 (m, 1H), 4.70–4.55 (m, 1H), 3.48–3.32 (m, 2H), 3.18–2.99 (m, 4H), 2.75–2.63 (m, 1H), 2.62–2.51 (m, 4H), 1.00 (d, J = 6.4 Hz, 6H); 13 C NMR (101 MHz, DMSO) δ 170.76, 150.86, 146.93, 133.89, 132.28, 132.24, 127.79, 119.26, 119.20, 116.87, 115.71, 62.43, 54.21, 49.19, 48.54, 46.35, 18.68. ESI-MS m/z = 365.24 [M + H]+.
5.1.7.23. 2-(4-(4-Acetylpiperazin-1-yl)phenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H54)
This compound was synthesised by employing the procedure for H53 using H27 (1 mmol, 1 equiv.) and 1-(piperazin-1-yl)ethanone (2 mmol, 2 equiv.) as the starting reagents; 1H NMR (400 MHz, DMSO) δ 7.90 (t, J = 5.6 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.25–7.12 (m, 3H), 6.96 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.1 Hz, 1H), 6.66 (t, J = 7.4 Hz, 1H), 6.44 (d, J = 2.8 Hz, 1H), 4.72–4.58 (m, 1H), 3.64–3.50 (m, 4H), 3.35–3.27 (m, 2H), 3.16–3.01 (m, 4H), 2.04 (s, 3H); 13 C NMR (101 MHz, DMSO) δ 170.77, 168.70, 150.57, 146.91, 134.57, 132.30, 132.25, 127.89, 119.27, 119.19, 116.91, 116.33, 62.35, 49.39, 49.03, 46.27, 45.94, 41.12, 21.68. ESI-MS m/z = 387.20 [M + H]+.
5.1.7.24. 2-(4-((2S,6R)-2,6-dimethylmorpholino)phenyl)-1,2,3,4-tetrahydro-5H-benzo[e][1,4]diazepin-5-one (H55)
This compound was synthesised by employing the procedure for H53 using H27 (1 mmol, 1 equiv.) and (2S,6R)-2,6-dimethylmorpholine (2 mmol, 2 equiv.) as the starting reagents; 1H NMR (400 MHz, CDCl3) δ 7.90–7.82 (m, 1H), 7.33–7.20 (m, 4H), 6.94–6.84 (m, 3H), 6.66 (d, J = 8.0 Hz, 1H), 6.60 (t, J = 5.8 Hz, 1H), 4.74 (t, J = 4.7 Hz, 1H), 4.16 (s, 1H), 3.86–3.74 (m, 2H), 3.52 (t, J = 5.6 Hz, 2H), 3.48–3.40 (m, 1H), 2.41 (t, J = 11.1 Hz, 2H), 1.27 (d, J = 6.3 Hz, 6H); 13 C NMR (101 MHz, CDCl3) δ 172.04, 150.84, 145.29, 133.16, 132.91, 132.23, 127.55, 120.27, 119.27, 115.80, 71.60, 64.22, 54.61, 54.55, 47.04, 19.09. ESI-MS m/z = 352.23 [M + H]+, 374.34 [M + Na]+.
5.2. Virtual computational studies
5.2.1. Molecular docking
The X-ray cocrystal structure of PARP-1 (PDB code: 4zzz) was obtained from the RCSB Protein Data Bank. The protein was prepared by deleting waters and adding hydrogen atoms using BIOVIA Discovery Studio 2017 R2 (DS 2017). The preparation of ligands was conducted by ChemDraw 19.0, DS 2017 and LigPrep (Schrödinger @2018). After the preliminary protein preparation and receptor grid generation were completed, the docking calculations were completed through Glide flexible docking. The other parameters were set to the default values. There were 20 docking conformations per docking run, and the results were ranked based on the value of their docking scores. Finally, the graphical display was processed by PyMOL.
5.2.2. Prediction of ADME parameters
The preparation of ligands and the prediction of ADME parameters of compounds were carried out with LigPrep and QikProp, respectively (Schrodinger Suite 2018). The LigPrep process consists of a series of steps that perform conversions, apply corrections to the structures, generate variations on the structures, eliminate unwanted structures, and optimise the structures. Many of the steps are optional and are controlled by selecting options in the LigPrep panel or by specifying command-line options. QikProp can efficiently evaluate a widely applicable set of physically significant descriptors and pharmaceutically relevant properties, making it an indispensable tool for applying ADME principles in lead discovery and optimisation. We output all the parameters according to the user's manual and the parameters related to BBB were displayed.
5.3. Biological evaluation
5.3.1. PARP-1 enzyme inhibition assay
The ability of the compounds to inhibit PARP-1 enzyme activity was assessed using BPS Bioscience's PARP Colorimetric Assay Kit (BPS Bioscience, Catalog # 80580). PARP-1 is known to catalyse the NAD-dependent addition of poly(ADP-ribose) to histones. The PARP-1 assay kit comes in a convenient 96-well format, with purified PARP-1 enzyme, histone mixture, activated DNA, and PARP-1 assay buffer. The key to the PARP-1 Colorimetric Activity Assay is the biotinylated substrate. With this kit, only three simple steps are required for PARP-1 reactions. First, histone proteins are coated on a 96-well plate. Next, the PARP-1 biotinylated substrate is incubated with an assay buffer that contains the PARP-1 enzyme. Finally, the plate is treated with streptavidin-HRP followed by the addition of the colorimetric HRP substrate to produce colour that can then be measured using a UV/Vis spectrophotometer microplate reader. IC50 values were calculated using GraphPad Prism5 software.
5.3.2. Cellular inhibition assay
The MTT assay was performed to detect the sensitivity of cells to anticancer drugs in vitro. The human A549, MCF7, and U87 tumour cells, as well as human BRCA mutant tumour cells (MX-1), were tested in this study. The cell culture was carried out in accordance with routine operations or instructions. Briefly, 5,000 cells were seeded in each well of a 96-well plate. After cell attachment to the bottom of the well, different concentrations of tested compounds were added into designated wells. After 48 h of incubation, 20 µL of MTT solution (5 mg/mL) were added to each well for cell staining. After 4 h, the media were removed and 150 µL of DMSO were added to each well. Finally, the absorbance was determined at 570 nm using a multifunctional microplate reader (TECAN).
5.3.3. MDCK cell permeability assay
The MDCK cells were cultured in accordance with the instructions, and the integrity of the membrane was evaluated by measuring the trans-epithelial electrical resistance (TEER) using an EVOM epithelial volt-ohmmeter with STX2 electrodes. The tested compounds (10 µM) of 100 µL were added to the apical compartment of the transwell cell culture devices (Millipore), and then the devices were incubated at 37 °C for 3 h. The concentrations of tested compounds in an apical compartment and basolateral compartment were then detected by HPLC, and the apparent permeability was then calculated. The calculation was Papp (A→B) = (ΔQ/Δt)/(A·C0) where ΔQ is the amount of compound in the basolateral compartment (µg), Δt is incubation time (s), A is the surface area of transwell in cm2 (0.33 cm2), and C0 is starting concentration (10 µM).
5.3.4. Brain/plasma pharmacokinetic studies in vivo
All animal experiments were complied with the ARRIVE guidelines and were carried out in accordance with the U.K. Animals Act, 1986 and associated guidelines. The ICR mice were used in plasma–brain distribution assay of compounds. After tail vein injection of a 1.5 mg/kg dose, the concentrations of the compounds in the brain and plasma were measured after 5 min and 60 min of administration, and the brain/plasma (B/P) ratio was calculated. The samples of plasma and brain tissue were prepared as previously reportedCitation47. An established LC-MS/MS method was used for determining the concentration of representative compounds in plasma and brain tissue.
Supplemental Material
Download PDF (6.2 MB)Disclosure statement
The authors declare no conflicts of interest. We declare that we have no financial and personal relationships with other people or organisations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service, and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled.
Additional information
Funding
References
- Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 2010;39:8–24.
- Jagtap P, Szabo C. Poly(adp-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 2005;4:421–40.
- Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002;108:171–82.
- Goldberg MS, Xing D, Ren Y, et al. Nanoparticle-mediated delivery of siRNA targeting parp1 extends survival of mice bearing tumors derived from brca1-deficient ovarian cancer cells. Proc Natl Acad Sci USA 2011;108:745–50.
- Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(adp-ribose) polymerase. Nature 2005;434:913–7.
- Wang YQ, Wang PY, Wang YT, et al. An update on poly(adp-ribose)polymerase-1 (PARP-1) inhibitors: opportunities and challenges in cancer therapy. J Med Chem 2016;59:9575–98.
- Deeks ED. Olaparib: first global approval. Drugs 2015;75:231–40.
- Syed YY. Rucaparib: first global approval. Drugs 2017;77:585–92.
- Scott LJ. Niraparib: first global approval. Drugs 2017;77:1029–34.
- Hoy SM. Talazoparib: first global approval. Drugs 2018;78:1939–46.
- Wang L, Yang C, Xie C, et al. Pharmacologic characterization of fluzoparib, a novel poly(adp-ribose) polymerase inhibitor undergoing clinical trials. Cancer Sci 2019;110:1064–75.
- Li N, Bu H, Liu J, et al. An open-label, multicenter, single-arm, phase ii study of fluzoparib in patients with germline brca1/2 mutation and platinum-sensitive recurrent ovarian cancer. Clin Cancer Res 2021;27:2452–8.
- Wang H, Ren B, Liu Y, et al. Discovery of pamiparib (bgb-290), a potent and selective poly (adp-ribose) polymerase (PARP) inhibitor in clinical development. J Med Chem 2020;63:15541–63.
- Xiong Y, Guo Y, Liu Y, et al. Pamiparib is a potent and selective PARP inhibitor with unique potential for the treatment of brain tumor. Neoplasia 2020;22:431–40.
- Mateo J, Lord CJ, Serra V, Tutt A, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol 2019;30:1437–47.
- Costa R, Carneiro BA, Wainwright DA, et al. Developmental therapeutics for patients with breast cancer and central nervous system metastasis: current landscape and future perspectives. Ann Oncol 2017;28:44–56.
- Brufsky AM, Mayer M, Rugo HS, et al. Central nervous system metastases in patients with her2-positive metastatic breast cancer: incidence, treatment, and survival in patients from registHER. Clin Cancer Res 2011;17:4834–43.
- Olson EM, Abdel-Rasoul M, Maly J, et al. Incidence and risk of central nervous system metastases as site of first recurrence in patients with her2-positive breast cancer treated with adjuvant trastuzumab. Ann Oncol 2013;24:1526–33.
- Lin NU, Claus E, Sohl J, et al. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer 2008;113:2638–45.
- Campian J, Gutmann DH. CNS tumors in neurofibromatosis. J Clin Oncol 2017;35:2378–85.
- Louis DN, Perry A, Reifenberger G, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:803–20.
- Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of brca1-deficient mammary tumors to the PARP inhibitor azd2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA 2008;105:17079–84.
- Lawlor D, Martin P, Busschots S, et al. PARP inhibitors as p-glyoprotein substrates. J Pharm Sci 2014;103:1913–20.
- Durmus S, Sparidans RW, van Esch A, et al. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699). Pharm Res 2015;32:37–46.
- Parrish KE, Cen L, Murray J, et al. Efficacy of PARP inhibitor rucaparib in orthotopic glioblastoma xenografts is limited by ineffective drug penetration into the central nervous system. Mol Cancer Ther 2015;14:2735–43.
- Kizilbash SH, Gupta SK, Chang K, Kawashima R, et al. Restricted delivery of talazoparib across the blood–brain barrier limits the sensitizing effects of PARP inhibition on temozolomide therapy in glioblastoma. Mol Cancer Ther 2017;16:2735–46.
- Weaver AN, Yang ES. Beyond DNA repair: additional functions of PARP-1 in cancer. Front Oncol 2013;3:290.
- Bai P. Biology of poly(adp-ribose) polymerases: the factotums of cell maintenance. Mol Cell 2015;58:947–58.
- Martinez-Zamudio RI, Ha HC. PARP1 enhances inflammatory cytokine expression by alteration of promoter chromatin structure in microglia. Brain Behav 2014;4:552–65.
- Martire S, Mosca L, d'Erme M. PARP-1 involvement in neurodegeneration: a focus on Alzheimer's and Parkinson's diseases. Mech Ageing Dev 2015;146–148:53–64.
- Pacher P, Szabó C. Role of poly(adp-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: The therapeutic potential of PARP inhibitors. Cardiovasc Drug Rev 2007;25:235–60.
- Scobie KN, Damez-Werno D, Sun H, et al. Essential role of poly(adp-ribosyl)ation in cocaine action. Proc Natl Acad Sci USA 2014;111:2005–10.
- Dash S, Balasubramaniam M, Rana T, et al. Poly (adp-ribose) polymerase-1 (PARP-1) induction by cocaine is post-transcriptionally regulated by mir-125b. eNeuro 2017;4:ENEURO.0089-17.2017.
- Reilly SW, Puentes LN, Schmitz A, et al. Synthesis and evaluation of an azd2461[18F]PET probe in non-human primates reveals the PARP-1 inhibitor to be non-blood-brain barrier penetrant. Bioorg Chem 2019;83:242–9.
- Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov 2017;7:20–37.
- Lallo A, Frese KK, Morrow CJ, et al. The combination of the PARP inhibitor Olaparib and the WEE1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin Cancer Res 2018;24:5153–64.
- Mak JPY, Ma HT, Poon RYC. Synergism between atm and PARP1 inhibition involves DNA damage and abrogating the G2 DNA damage checkpoint. Mol Cancer Ther 2020;19:123–34.
- Li A, Yi M, Qin S, et al. Prospects for combining immune checkpoint blockade with PARP inhibition. J Hematol Oncol 2019;12:98.
- Jones P, Wilcoxen K, Rowley M, Toniatti C. Niraparib: a poly(adp-ribose) polymerase (PARP) inhibitor for the treatment of tumors with defective homologous recombination. J Med Chem 2015;58:3302–14.
- Zhou J, Ji M, Zhu Z, et al. Discovery of 2-substituted 1h-benzo[d]immidazole-4-carboxamide derivatives as novel poly(adp-ribose)polymerase-1 inhibitors with in vivo anti-tumor activity. Eur J Med Chem 2017;132:26–41.
- Verma R, Bhatia R, Singh G, et al. Design, synthesis and neuropharmacological evaluation of new 2,4-disubstituted-1,5-benzodiazepines as CNS active agents. Bioorg Chem 2020;101:104010.
- Yu J, Luo L, Hu T, et al. Structure-based design, synthesis, and evaluation of inhibitors with high selectivity for PARP-1 over PARP-2. Eur J Med Chem 2022;227:113898.
- Garberg P, Ball M, Borg N, et al. In vitro models for the blood-brain barrier. Toxicol In Vitro 2005;19:299–334.
- Mensch J, Oyarzabal J, Mackie C, Augustijns P. In vivo, in vitro and in silico methods for small molecule transfer across the BBB. J Pharma Sci 2009;98:4429–68.
- Wang Y, Zhao H, Brewer JT, et al. De novo design, synthesis, and biological evaluation of 3,4-disubstituted pyrrolidine sulfonamides as potent and selective glycine transporter 1 competitive inhibitors. J Med Chem 2018;61:7486–502.
- Kam TI, Mao X, Park H, et al. Poly(adp-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson's disease. Science 2018;362:1–10.
- Guo Z, Zhang Z, Zhang Y, et al. Design, synthesis and biological evaluation of brain penetrant benzazepine-based histone deacetylase 6 inhibitors for alleviating stroke-induced brain infarction. Eur J Med Chem 2021;218:113383.