
Abstract
Histone deacetylases (HDACs) are a family of enzymes responsible for regulating DNA transcription by modulating its binding to histone proteins. HDACs are overexpressed in several types of cancers and are recognised as drug targets. Vorinostat, or suberanilohydroxamic acid (SAHA), is an histone deacetylase (HDAC) inhibitor with a hydroxamic acid as a zinc-binding group (ZBG), and it has been FDA approved for the treatment of T-cell lymphoma. In this work, phosphorus-based SAHA analogues were synthesised to assess their zinc-binding effectiveness compared to the hydroxamic acid of SAHA. Specifically, we examined phosphate, phosphoramidate and phosphorothiolate groups as isosteres of the canonical hydroxamic acid motif of conventional HDAC inhibitors. The compounds were screened for binding to HDAC enzymes from HeLa cell lysate. The most potent derivatives were then screened against HDAC3 and HDAC8 isoforms. HDAC inhibition assays demonstrated that these phosphorus-based SAHA analogs exhibited slow binding to HDACs but with greater potency than phosphonate SAHA analogs examined previously. All compounds inhibited HDACs, the most potent having an IC50 of 50 µM.
Graphical Abstract
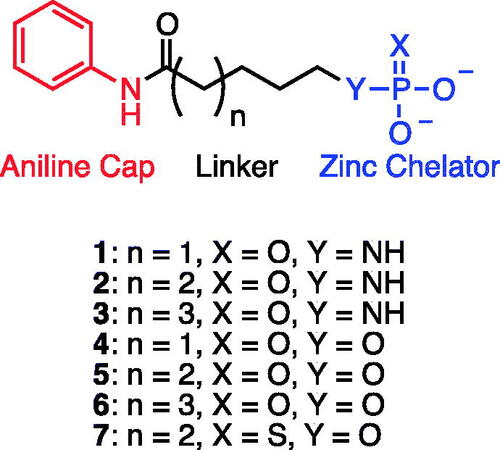
Introduction
Chromatin is a protein–DNA complex that consist of segments of DNA wrapped around a histone octamer which are then woven into fibresCitation1,Citation2. These chromatin fibres condense the vast amounts of DNA into compact dense structuresCitation3,Citation4. Histones proteins are modified via acetylation or deacetylation by histone acetyltransferase HAT and histone deacetylase HDAC enzymes, respectivelyCitation5,Citation6 to regulate DNA transcription by affecting how tightly DNA strands are bound to histone proteinsCitation7. HDACs inhibit transcription by removing N-acetyl modifications on histone lysine residues allowing the histone to carry a positive charge and thereby strengthening its electrostatic interactions with DNACitation8,Citation9.
The HDAC family of zinc metalloproteinases contains 11 members and are conserved across all eukaryotesCitation10. With the exception of NAD+-dependent class III HDACS, all HDAC family enzymes share a common catalytic mechanism. In brief, a zinc (II) ion in the active site functions to simultaneously coordinate a water molecule and act as a Lewis acid towards substrate acetyl groupsCitation11. This coordination serves to lower the pKa of the water molecule and polarise the carbonyl group, thus increasing the nucleophilicity and electrophilicity of each, respectivelyCitation12. Nucleophilic addition of water to the carbonyl centre of the substrate acetyl leads to a tetrahedral intermediateCitation13, which, once collapsed, releases the lysine amine and acetic acid.
HDACs have served as drug targets for many diseases including various cancers, interstitial fibrosis, autoimmune and inflammatory diseases, and metabolic disordersCitation14. Indeed, considerable efforts have been made to develop HDAC inhibitors (HDACi). Vorinostat, or SAHA, is a broad spectrum HDACi (IC50 = 13 nM) has been FDA approved to treat cutaneous T-cell lymphomaCitation15,Citation16. This molecule utilises a hydroxamic acid as a zinc-binding groupCitation17 as do Belinostat and PanobinostatCitation18, while a thiol serves as the zinc-binding group in RomidepsinCitation19. Because none of the known HDAC inhibitors are specific for a single HDAC, off-target effects remain an issue.
In recent work, much attention has been towards increasing the potency of HDACi molecules and improving selectivity for certain isoenzymes. Negmeldin et al. modified the C2 position of SAHA with a n-hexyl to exploit a wider active site entrance of HDAC6/8. This compound resulted in a 49- to 300-fold HDAC6/8 (IC50 = 0.6 and 2.0 µM) selectivity over HDAC1-3Citation20. Procainamide-SAHA fused inhibitors proposed by Nardella et al. targeted post translational modifications in the malaria parasite plasmodium falciparum. This compound combines SAHA, a potent pan-HDAC inhibitor with a DNA methyltransferase inhibitor procainamide. The lead SAHA/procainamide fusion molecule was fully active in drug resistant plasmodium falciparum isolates (IC50 = 41 nM) and human HDAC6 (IC50 = 14 nM)Citation21. Another strategy for optimisation of SAHA derivatives is replacing the anilide with different hydrophobic functional groups. Huang et al. synthesised and evaluated SAHA derivatives with osthole fused to the aliphatic hydroxamate core. Their best compound showed potency and selectivity similar to SAHA with moderate selectivity towards HDAC6 (IC50 = 14 nM)Citation22.
Kapustin et al.Citation23 demonstrated the utility of phosphonamidate, phosphonate and phosphinate analogs of SAHA () as HDAC inhibitors. The most potent of these was a monobasic phosphoramidate-based compound PA1 () with an IC50 of 570 µM against HeLa cell lysates. It also exhibited a slow binding mode of inhibition, requiring a 10-h preincubation time. The focus of this study was to expand upon the Kapustin study by examining dibasic phosphoryl motifs as zinc-binding groups in the context of HDAC inhibitors.
Figure 1. Phosphorus containing SAHA analogues discovered by Kapustin et al. IC50 values reported for Hela cell lysate and 10 h incubation time.

Our inhibitor selectivity experiments focussed on the Class-I HDACs (HDAC1, 2, 3, and 8). The HDAC isoforms 3 and 8 were chosen from this class due to the differences in their sequence and structure. Both of these enzymes are present in the cell nucleus and use zinc as a cofactor for catalytic activity. There are 4 key differences in the active site amino acid sequence suggesting that there is a selectivity towards substratesCitation24. HDAC8 contains a flexible L1 loop made up of 7 amino acids that form a hydrophobic secondary pocket adjacent to the active siteCitation25. This pocket has been exploited for HDAC8-specific inhibitor research and has led to “L shaped” molecules with improved activity against HDAC8Citation26. These HDACs are clinically relevant due to HDAC 8 being overexpressed in T-Cell leukaemia and NeuroblastomaCitation27,Citation28. HDAC3, however, is associated with neurodegenerative diseases such as Alzheimer’s disease.
The time course enzyme inhibition assay using compound 2 showed optimal inhibition at 8 h for HeLa cell Lysate and HDAC 8, and 4 h for HDAC 3. These results suggest that our series of phosphoryl compounds are slow binding inhibitors. These types of inhibitors also express tight binding qualities such that the molecules have low dissociation rates and long drug target residence timeCitation29. In vitro, this strong binding quality can disrupt cell viability due to the inhibitors ability to shutdown the enzyme for a long period of time. If enzyme synthesis time in targeted cells cannot overcome the inhibition time, cell viability can be affectedCitation30. The advantage of slow and tight binding inhibitors for in vivo biomedical purposes stems from the decreased off target toxicity of the compound. Because of the decreased systemic circulation time and increased inhibitor residence time a lower concentration of the compound is available in the blood stream to bind to non-targeted proteinCitation31. There are several known examples of slow binding as FDA-approved drugsCitation32. There are also several know types of HDAC inhibitors that exhibit slow binding kineticsCitation33.
Results and discussion
Amino aniline amidesCitation14–16 were synthesised from commercially available Boc-protected amino acids by a HBTU coupling reaction with aniline followed by deprotection with HCl (Scheme 1). PhosphoramidatesCitation17–19 were synthesised by an Atherton-Todd reaction with amino aniline amides and dibenzylphosphite. The resulting dibenzyl protected phosphoramidates were deprotected by catalytic hydrogenation in the presence of potassium bicarbonate to provide productsCitation1–3. Hydroxy aniline amidesCitation27–29 were synthesised either starting from the commercially available bromo-alkyl ester or corresponding lactone. Ethyl 7-bromoheptanoate 20 was hydrolysed using HBr in acetic acid to provide 7-bromoheptanoic acid 22. Both bromoheptanoic acid 22 and commercially available bromopentanoic acid 21 were coupled with aniline using DCC. An O-acetyl group was installed by reaction with bromo alkyl acid 23 and 24 and potassium acetate. Saponification with NaOH was preformed to provide the alcohols 27 and 29. ε-Caprolactone was hydrolysed and TBDMS protected following an established literature procedure (ref). TBDMS protected alkyl acid was coupled to aniline using HBTU followed by deprotection of the silyl ester using aqueous acid to provide alcohol 28 (Citation34). The alcohols were treated with dibenzyl diisopropylphosphoramidite and oxidised with tert-butyl hydrogen peroxide to give the dibenzyl phosphatesCitation30–32. Deprotection was accomplished by catalytic hydrogenation in the presence of potassium bicarbonate to provide productsCitation4–6. Phosphorothioates were synthesised by reacting 6-hydroxy-N-phenylhexamadmide 28 with bis(2-cyanoethyl) diisopropylphosphoramidite then oxidising with elemental sulphur. The cyanoethyl esters were removed by treating with an excess of sodium hydroxide to give product 7.
Once prepared, the compounds were screened for inhibition of HDAC activity from the cell lysates of HeLa cells and the recombinant isoforms HDAC3 and HDAC8. Linker lengths of 5–7 atoms (including O or NH) were chosen for these structures to compare to the analogous structure of SAHA with its 6-atom linker between the hydrophobic analide cap and hydroxamate ZBG. While incubation times less than 1 h resulted in little inhibition of HDAC activity, the compounds exhibited significant inhibition when pre-incubated with HeLa cell lysates, for 8 h (), which was consistent with phosphonyl-based HDAC inhibitors (PA1-3). Recombinant HDAC3 and HDAC8 were also tested for slow binding inhibition using compound 2.
Figure 2. Time-dependent inhibition of HDACs from HeLa cell lysates, HDAC8 and HDAC3 with inhibitor 2 (100 μM).

The screening results () showed evidence of concentration dependent inhibition for each compound. The most potent compounds were the phosphoramidate 2 and phosphate 5, both possessing a 6-atom linker. Based on these results, we further expanded the library to include the thiophosphate 7, while maintaining a 6-atom linker. Interestingly, the inhibitory potency of compounds 2, 5, and 7 were similar, suggesting little difference in the zinc-binding of these the three motifs; phosphoramidate, phosphate, and thiophosphate. Therefore, we tested these compounds 2, 5, and 7 using recombinant HDAC3 and HDAC8 (). The assay results showed that compound 2 had the highest potency for the cell lysate (IC50 = 70 ± 8 µM) but was more selective for HDAC8 (IC50 = 129 ± 23 µM) over HDAC3 (IC50 = 240 ± 34 µM). Similarly compound 5 also had the highest potency for cell lysate (IC50 = 60 ± 9 µM), and was more selective for HDAC8 (IC50 = 179 ± 34 µM) over HDAC3 (IC50 = 690 ± 60 µM). Compound 7 showed similar potency between cell lysate (IC50 = 50 ± 13 µM) and HDAC8 (IC50 = 49 ± 8 µM) but was less selective for HDAC3 (IC50 = 103 ± 20 µM).
Table 1. IC50 values for inhibitors 1–7 and SAHA.
Conclusion
In summary, we synthesised a library of 7 phosphoryl-based analogs of SAHA. These inhibitors were designed to contain the canonical hydrophobic anilide cap and aliphatic linker, but present an alternative phosphoryl-based ZBG. While each of these motifs could provide multidentate interactions with the HDAC active-site zinc ion, this was not an advantage with respect to inhibitory potency against HDACs. However, while these compounds were considerably less potent than SAHA (0.2 µM) against HDACs from HeLa cell lysates or HDAC3 (0.24 µM) and HDAC8 (1 µM), they exhibited greater potency compared to previously reported phopshonyl-based HDAC inhibitors (Citation23). Investigations into the specificity of inhibitory potency against individual HDACs and cancer cells by these compounds will be forthcoming.
Supplemental Material
Download PDF (125.3 KB)Acknowledgements
The authors extend their gratitude for technical assistance to Dr. G. Helms and Dr. W. Hiscox at the WSU Center for NMR Spectroscopy, Dr. G. Munske at the WSU Molecular Biology and Genomics Core, and to both Dr. A. Berim and Dr. B. M Lange at the WSU Institute of Biological Chemistry for assistance with high resolution mass spectrometric analysis.
Disclosure statement
The authors declare no competing financial or personal interest.
Scheme 1. Synthesis of HDAC inhibitors 1–7. a) HBTU, DIPEA, aniline, DMF b) 4 N HCl in dioxane c) dibenzyl phosphite, BrCCl3, Et3N, CH3CN d) H2, Pd/C, MeOH, KHCO3 e) 33% HBr in AcOH f) DCC, DMAP, aniline, DMF g) KOAc, DMF, 70 °C h) NaOH, MeOH i) NaOH, H2O j) imidazole, TBDMS-Cl, DCM k) HBTU, DIPEA, aniline, DMF l) 4 N HCl in dioxane m) Dibenzyl N,N-diisopropylphosphoramidite, 5-(Ethylthio)-1H-tetrazole, ACN, tert-butyl hydrogen peroxide n) H2, Pd/C, MeOH, KHCO3 o) Bis(2-cyanoethyl)-N,N-diisopropylphosphoramidite, Sulphur, ACN p) KOH, MeOH.

Additional information
Funding
References
- Bruce Alberts AJ, J, Lewis, M, Raff, et al. Chromosomal DNA and its packaging in the chromatin fiber, In: Molecular Biology of the Cell. New York: Garland Science; 2002.
- Morales V, Giamarchi C, Chailleux C, et al. Chromatin structure and dynamics: Functional implications. Biochimie 2001;83:1029–39.
- Nair N, Shoaib M, Sørensen CS. Chromatin dynamics in genome stability: roles in suppressing endogenous DNA damage and facilitating DNA repair. Int J Mol Sci 2017;18:1486.
- Koltover I, Wagner K, Safinya CR. DNA condensation in two dimensions. Proc Natl Acad Sci U S A 2000;97:14046–51.
- Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007;26:5310–8.
- Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol 2011;2011:371832.
- Bowman GD, Poirier MG. Post-translational modifications of histones that influence nucleosome dynamics. Chem Rev 2015;115:2274–95.
- Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 2014;6:a018713.
- Bassett SA, Barnett MPG. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 2014;6:4273–301.
- Milazzo G, Mercatelli D, Di Muzio G, et al. Histone deacetylases (HDACs): evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes (Basel) 2020;11:556.
- Fernandes HS, Teixeira CSS, Sousa SF, Cerqueira NMFSA. Formation of unstable and very reactive chemical species catalyzed by metalloenzymes: a mechanistic overview. Molecules 2019;24:2462.
- Porter NJ, Christianson DW. Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Curr Opin Struct Biol 2019;59:9–18.
- Hai Y, Christianson DW. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol 2016;12:741–7.
- Tang J, Yan H, Zhuang S. Histone deacetylases as targets for treatment of multiple diseases. Clin Sci (Lond) 2013;124:651–62.
- Beckers T, Burkhardt C, Wieland H, et al. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int J Cancer 2007;121:1138–48.
- Al-Yacoub N, Fecker LF, Möbs M, et al. Apoptosis induction by SAHA in cutaneous T-cell lymphoma cells is related to downregulation of c-FLIP and enhanced TRAIL signaling. J Invest Dermatol 2012;132:2263–74.
- Vannini A, Volpari C, Filocamo G, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U. S. A. 2004;101:15064–9.
- Zhang L, Zhang J, Jiang Q, et al. Zinc binding groups for histone deacetylase inhibitors. J Enzyme Inhib Med Chem 2018;33:714–21.
- Gong W, Wu R, Zhang Y. Thiol versus hydroxamate as zinc binding group in HDAC inhibition: An ab initio QM/MM molecular dynamics study. J Comput Chem 2015;36:2228–35.
- Negmeldin AT, Padige G, Bieliauskas AV, Pflum MKH. Structural requirements of HDAC inhibitors: SAHA analogues modified at the C2 position display HDAC6/8 selectivity. ACS Med Chem Lett 2017;8:281–6.
- Nardella F, Halby L, Dobrescu I, et al. Procainamide-SAHA fused inhibitors of hHDAC6 tackle multidrug-resistant malaria parasites. J Med Chem 2021;64:10403–17.
- Huang W-J, Chen C-C, Chao S-W, et al. Synthesis and evaluation of aliphatic-chain hydroxamates capped with osthole derivatives as histone deacetylase inhibitors. Eur J Med Chem 2011;46:4042–9.
- Kapustin GV, Fejér G, Gronlund JL, et al. Phosphorus-based SAHA analogues as histone deacetylase inhibitors. Org Lett 2003;5:3053–6.
- Yang F, Zhao N, Ge D, Chen Y. Next-generation of selective histone deacetylase inhibitors. RSC Advances 2019;9:19571–83.
- Chakrabarti A, Oehme I, Witt O, et al. HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol Sci 2015;36:481–92.
- Marek M, Shaik TB, Heimburg T, et al. Characterization of histone deacetylase 8 (HDAC8) selective inhibition reveals specific active site structural and functional determinants. J Med Chem 2018;61:10000–16.
- Spreafico M, Gruszka AM, Valli D, et al. HDAC8: a promising therapeutic target for acute myeloid leukemia. Front Cell Develop Biol 2020;8:844.
- Rettig I, Koeneke E, Trippel F, et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis 2015;6:e1657.
- Robers MB, Dart ML, Woodroofe CC, et al. Target engagement and drug residence time can be observed in living cells with BRET. Nature Communications 2015;6:10091.
- (2013) Slow Binding Inhibitors, in Evaluation of Enzyme Inhibitors in Drug Discovery pp 203–244.
- Walkup GK, You Z, Ross PL, et al. Translating slow-binding inhibition kinetics into cellular and in vivo effects. Nat Chem Biol 2015;11:416–23.
- Lu H, Tonge PJ. Drug-target residence time: critical information for lead optimization. Curr Opin Chem Biol 2010;14:467–74.
- Chou CJ, Herman D, Gottesfeld JM. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases *. J Biol Chem 2008;283:35402–9.
- Stals PJM, Phan TNT, Gigmes D, et al. Nitroxide-mediated controlled radical polymerizations of styrene derivatives. J. Polym. Sci. A: Polym. Chem 2012;50:780–91.