
Abstract
A series of novel 1,2,3-triazole-linked ciprofloxacin-chalcones 4a-j were synthesised as potential anticancer agents. Hybrids 4a-j exhibited remarkable anti-proliferative activity against colon cancer cells. Compounds 4a-j displayed IC50s ranged from 2.53-8.67 µM, 8.67–62.47 µM, and 4.19–24.37 µM for HCT116, HT29, and Caco-2 cells; respectively, whereas the doxorubicin, showed IC50 values of 1.22, 0.88, and 4.15 µM. Compounds 4a, 4b, 4e, 4i, and 4j were the most potent against HCT116 with IC50 values of 3.57, 4.81, 4.32, 4.87, and 2.53 µM, respectively, compared to doxorubicin (IC50 = 1.22 µM). Also, hybrids 4a, 4b, 4e, 4i, and 4j exhibited remarkable inhibitory activities against topoisomerase I, II, and tubulin polymerisation. They increased the protein expression level of γH2AX, indicating DNA damage, and arrested HCT116 in G2/M phase, possibly through the ATR/CHK1/Cdc25C pathway. Thus, the novel ciprofloxacin hybrids could be exploited as potential leads for further investigation as novel anticancer medicines to fight colorectal carcinoma.
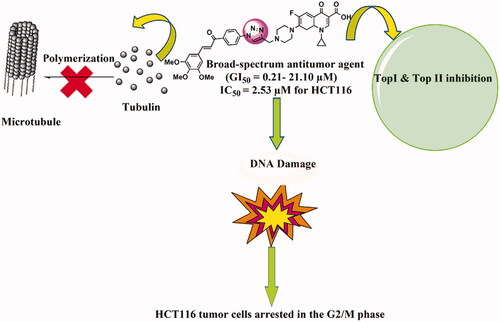
Graphical Abstract
1. Introduction
Cancer is a complicated pathological condition characterised by uncontrolled cell growthCitation1. As the 2nd leading cause of mortality, cancer accounts for 21% of annual deaths globallyCitation2–4. By 2040, the number of new cases is estimated to reach 29.4 million per year, compared to 18.1 million in 2018Citation5. Colorectal Cancer (CRC) was the second most deadly cancer and the third most common malignancy in 2020, which caused 1.9 million incidence cases and 0.9 million deaths globally. The Predictable number of new CRC cases worldwide in 2040 will be 3.2 million casesCitation6. Surgery, chemotherapy, and radiotherapy are currently available cancer treatment optionsCitation7. Although single target chemotherapy achieves some therapeutic benefits, its use is limited by poor efficacy, serious toxic effects and the development of resistanceCitation8,Citation9. Two strategies for multi-target therapeutics are being used in preclinical or clinical practice to overcome such problemsCitation10. The first one is combination therapy; the clinical use of this approach is widely utilised for the treatment of various types of cancer. Still, some combinations may be limited due to undesirable drug-drug interactionsCitation11. The second strategy is based on using smart hybrids or multi-target directed ligands (MTDLs). Two or more pharmacophore fragments are linked together to form a single molecule that can simultaneously modulate multiple oncoproteinsCitation12,Citation13. Thus, the use of multi-target directed ligands can improve efficacy, reduce serious adverse effects, and prevent the development of drug resistanceCitation14,Citation15. Understanding various molecular pathways utilised in the development and formation of tumours can aid in the discovery of novel antitumor agentsCitation16. Ciprofloxacin (CP) is a broad-spectrum antibacterial agent that acts by poisoning bacterial DNA gyrase (topoisomerase II) and topoisomerase IVCitation17,Citation18, which are fundamental enzymes that substantially contribute to the process of DNA replicationCitation19–21. CP has recently gained considerable attention as an anti-proliferative agentCitation21,Citation22. It was reported that poisoning topoisomerase II, arresting cell cycle arrest, and inducing apoptosis are the mechanisms responsible for the anti-proliferative activity of CPCitation23–25. Accordingly, shifting the biological profile of CP from antibacterial to anticancer candidate appears to be possible due to its profound apoptotic and anti-proliferative potentialCitation26. In this regard, the structural modifications required for anticancer activity of CP have been clearly discussedCitation1,Citation27. Bio-isosteric replacement of the carboxylic group at C-3 of CP with tetrazole heterocyclic ring markedly improves its anti-proliferative activityCitation1,Citation28. Also, one key modification was carried out by substituting its N-4 piperazine heterocycle moiety with an aryl or heteroaryl bulky groupCitation1,Citation27. Introducing a substituent on the N-4-piperazinyl moiety of CP affects the physicochemical characteristics and/or significantly improves the anti-proliferative activityCitation29–31. The nature of the N-4-piperazinyl substituent remarkably affects the capacity of CP derivatives to suppress the activity of DNA topoisomerasesCitation32. In addition, the potential selectivity of CP derivatives to human topoisomerases was found to be significantly improved via modification of the N-4-piperazine heterocycleCitation31. Consequently, several CP derivatives have been prepared, characterised, and widely explored for their antitumor propertiesCitation33,Citation34. Recent studies have revealed that the presence of different chalcones on the N-4-piperazine moiety of CP gave rise to new CP hybrids with potent anti-proliferative activities targeting multiple oncoproteins ()Citation16,Citation21. Furthermore, the introduction of the aryl sulfamoyl group into the N-4 piperazine of CP gave rise to new fluoroquinolone derivatives with anticancer activityCitation35. Substitution of N-4 piperazine of CP with 4-chlorophenylcarbamoyl led to a new CP derivative with potent anti-proliferative activity against non-small cell lung cancer ()Citation36. On the other hand, natural and synthetic chalcones represent a vital and central component of various compounds with diverse biological propertiesCitation37. Chalcone is a familiar scaffold in medicinal chemistry that is widely used for the development of a large number of molecules with potential anticancer activitiesCitation38,Citation39. The anticancer potential of chalcone-containing compounds was attributed to apoptosis induction, angiogenesis inhibition, DNA damage, kinases inhibition, and tubulin inhibitionCitation40,Citation41. Chalcones have gained considerable attention from drug hunters due to their ease of synthesis, exploring novel scaffolds with potential biological activitiesCitation42,Citation43.
Figure 1. Chemical structures of several examples of some reported N-4 piperazinyl ciprofloxacin derivatives with anti-proliferative activities.

Furthermore, 1,2,3-Triazoles are essential scaffolds in medicinal chemistry that are widely used in many drug-design protocols as bio-isosteres of ester, amide, and other heterocyclesCitation44,Citation45. Compounds containing 1,2,3-triazole heterocycle can form different non-covalent interactions such as hydrogen bonds, dipole-dipole bonds, hydrophobic interactions, and van der Waals forces with various biological targets, thus they possess various biological effects (e.g., anticancerCitation46–49, antibacterialCitation50,Citation51, antifungalCitation52,Citation53, antiviralCitation54,Citation55, antimalarialCitation56,Citation57, and antitubercularCitation58,Citation59 activities). Additionally, 1,2,3-Triazole is a favourable basic unit in the finding of new anticancer agents, and some of its derivatives have now been in clinics or under clinical trials for combating against cancersCitation45. 1,2,3‐Triazoles exhibited their anticancer effects via different modes of action. They exerted their anti-proliferative effects through Tubulin polymerisation inhibition or inhibition of some kinases such as epidermal growth factor receptor, c-Met Kinase, and vascular endothelial growth factor. They also caused inhibition of vital enzymes such as aromatase, tryptophan 2,3‐dioxygenase, carbonic anhydrases, and thymidylate synthaseCitation60. Also, many drugs have 1,2,3-triazole moiety, such as cefatrizine IV, seviteronel V, carboxyamidotriazole VI, and mubritinib VII entered active clinical trials in 2021 ()Citation61.
Figure 2. Potential anticancer drugs based on 1,2,3-triazole nucleus in active clinical trials.

Moreover, several 1,2,3-triazole chalcone hybrids were reported as anticancer agents aiming for synergistic activity in the last decade. For example, 1,2,3-triazole-chalcone hybrid VIII exhibited nanomolar IC50 values against six cancer cell lines, HCT-116 colon, RPMI-8226 leukaemia, MCF7 breast, SR leukaemia, M14 melanoma, and K-562 leukaemia cancer cell lines, and sensitive cell lines. It caused induction of apoptosis in a dose-dependent manner through activation of caspases 3, 7 & 9 and Bax/Bcl-2 ratio elevationCitation62. Additionally, Chalcone/1,2,3-triazole hybrid IX was reported as an orally tubulin polymerisation inhibitor. Compound IX inhibited proliferation and migration in HepG2 cells growth in a dose-dependent mannerCitation63. Furthermore, 1,2,3-triazole tethered chalcone hybrid X showed potent cytotoxic activity on MCF-7 cancer cell lines with IC50 of 1.27 µM and 0.02 µM at 24 and 48 h; respectivelyCitation64 ().
Figure 3. Some recently reported anti-proliferative 1,2,3-triazole/chalcone hybrids.

Inspired by the above-mentioned aspects, the purpose of our study was to develop a new series of 1,2,3-triazole linked CP-chalcone hybrids 4a-j as possible multi-target-directed ligands with potential anti-proliferative activities. Their in-vitro cytotoxic activity was evaluated using National Cancer Institute (NCI) aganist59 cancer cell lines. Additionally, the IC50s of the new CP derivatives 4a-j against three different colon cancer cell lines (Caco-2, HT29, and HCT116) were determined using the MTT assay. Furthermore, as possible molecular targets, the ability of the most active hybrids to inhibit tubulin polymerisation and the catalytic activities of both topoisomerases I and topoisomerases II were examined. Also, the ability of the most promising hybrids to induce DNA double strand breaks and cell cycle arrest in HCT116 cancer cells in addition to their effect on the protein expression levels of γH2AX was evaluated. Moreover, the docking studies of new hybrids were investigated at the TopI (1T8I) and Top II (6ZY7) active sites. To the best of our knowledge, this is the first report that gathers these three important pharmacophores; Ciprofloxacin, Chalcone and 1,2,3-triazole in one compact unit aiming for synergistic anticancer activity through acting on different molecular targets .
Figure 4. Pharmacophore of target compounds 4a-j.

2. Results and discussion
2.1. Chemistry
Both new compounds and their intermediates were synthesised as drawn in Scheme 1. In particular, the intermediate 1-(4-azidophenyl)ethanone 1 was prepared following the previously published proceduresCitation65,Citation66. Chalcone derivatives 2a-j were synthesised by condensation of 1-(4-azidophenyl)ethanone 1 with the appropriate aromatic aldehyde in ethanol in presence of sodium hydroxideCitation65,Citation66. Compound 3 was prepared by alkylation of CP with propargyl bromide in dimethylformamide (DMF) in the presence of NaHCO3Citation67. The final compounds 4a-j were synthesised by coupling chalcone derivatives 2a-j with CP derivative 3 in DMF using sodium ascorbate and copper sulphate as a catalyst. Structure elucidation of our target compounds was carried out via IR, 1H NMR, 13 C NMR, elemental analysis, and mass spectrometry. The IR spectra of compounds 4a, 4e, and 4i as representative examples showed a broad stretching bands at 3200–3048 cm−1 assigned to OH of the carboxylic group, in addition to three carbonyl stretching bands appeared at 1719–1715 cm−1, 1658–1657 cm−1, and 1625–1622 cm−1 assigned to C = O of the carboxylic, carbonyl of the chalcone and the 4-keto functionality, respectively. The pattern of parent CP and chalcone scaffolds was confirmed by 1H NMR. Chemical shifts (δ) at 1.19–1.35 ppm and 3.45–3.85 ppm (two multiplet signals) represent the protons of cyclopropyl and piperazine moieties, respectively. Also, aromatic protons (3) and carboxylic proton of CP appeared at δ = 7.56–8.66 ppm and 15.05–15.17 ppm, respectively. The two chalcone protons appeared at δ = 7.58–8.03 ppm. One characteristic singlet for the triazolyl H appeared at δ 8.83–8.93 ppm. Moreover, 13 C NMR showed a characteristic signal at δ = 186.77–188.90 ppm which corresponds to the carbonyl group of the chalcone scaffold.
Scheme 1. Reagents and conditions: (a) Conc. H2SO4, NaNO2, NaN3, 0–5 °C; (b) Proper aromatic aldehydes, 60% sodium hydroxide, ethanol, stirred overnight at 0–5 °C; (c) Propargyl bromide, NaHCO3, DMF, stirred overnight at 80 °C; (d) CuSO4·5H2O, Sodium ascorbate, DMF, CH2Cl2, MeOH, H2O (3:2:1:1) rt stirring 24 h.

2.2. Biological investigations
2.2.1. Cytotoxic studies
2.2.1.1. In-vitro single-dose experiment
For screening of anticancer properties, all the developed compounds were chosen by the (NCI) for in-vitro antitumor evaluation. The assay was carried out using 59 human cancer cell lines obtained from nine tumour subpanels (breast, ovarian, melanoma, leukaemia, renal, CNS, lung, prostate, and colon cancer cell lines). Compounds were assessed at a 10 µM concentration. The results of anticancer screening (as shown in Table 1 in Supplementary Data) indicated that all the synthesised CP derivatives produced remarkable anti-proliferative effects against leukaemia (RPMI-8226 and SR) and colon (HCT116) tumour cells with a growth percent ranged from 47.83 to −53.36%. Among the tested compounds, CP hybrid 4j exhibited a strong and wide range of anti-proliferative activities in the majority of cancer cell lines with a growth percent ranged from 67.46 to −39.69%. Also, hybrids 4a, 4b, 4e, 4i and 4j produced significant anti-proliferative effect in several cancer cells, particularly in leukaemia (RPMI-8226) and colon (HCT116) cells with growth percent of 47.87, −14.59, −24.56, −24.42, −15.84, −9.74, −53.36, −10.26, −31.29, and −39.51%, respectively.
2.2.1.2 In-vitro five-dose experiment
Showing promising results in the single-dose assay, compound 4j was advanced for further investigation against a panel of 59 human cancer cell-lines at a five-dose levels. Those cell lines represent nine different tumours, and they were all incubated with our compound at 5 increasing concentrations (0.01, 0.1, 1, 10 and 100 µM). The In-vitro five-dose assay results were listed in , as GI50 which is the concentration causes 50% inhibition of cell proliferation and it was expressed in µM. Moreover, Total growth inhibition concentration (TGI) in µM was calculated and listed in . Results showed that compound 4j displayed high activity and a wide range of antitumor effects with no selectivity towards all cancer cell lines with GI50 values between 0.21 µM and 21.10 µM with selectivity ratio from 0.67 to 1.69 µM at GI50 level, as shown in . Furthermore, compound 4j displayed the capacity to achieve total growth inhibition (TGI) against thirty-one cell lines with TGI values between 0.54 and 45.20 µM.
Table 1. GI50 in µM and total growth inhibition (TGI) in µM of compound 4j against 59 cell lines of nine cancer panels evaluated using NCI's in-vitro 5-dose antitumor assay.
2.2.1.3. In-vitro anti-proliferative study using HCT116, HT29, and Caco-2 cells
As the one dose experiment showed remarkable growth inhibition of the synthesised compounds against the colon cancer cells, we determined the IC50s of the target hybrid compounds 4a-j against three human colon cancer cell lines (Caco-2, HT-29, and HCT116) using the MTT assay after 24 h incubation period. Results in showed that the new CP hybrids 4a-j had stronger anti-proliferative effects in HCT116 cells (IC50 range 2.53–8.67 µM) than H29 cells (IC50 range 8.67–62.47 µM) and Caco-2 cells (IC50 range 4.19–24.37 µM). Compounds 4a, 4b, 4e, 4i, and 4j displayed noticeable anti-proliferative effects against the colon cancer cells. Among the tested compounds, the unsubstituted (4a) and 3,4,5-trimethoxy (4j) derivatives exhibited the strongest anti-proliferative effect. Also, the results indicated that biological activity is not affected by the electronic effect of the substituents. However, the 4j showed the highest cytotoxic effect against HCT116 cells, we noticed difference in the IC50 values of 4j between using the SRB assay (IC50 = 0.2 µM) and the MTT assay (IC50 = 2.53 µM). We assume this difference is due to the SRB assay is more sensitive than the MTT assay as previously described in the literatureCitation68,Citation69. Simple structure-activity relationship (SAR) based on HCT-116 cytotoxicity data showed that the absence of substituents on the aromatic ring is favourable to any substituted except 3,4,5-trimethoxy substituents (compound 4j). For monosubstituted derivatives, the fluorine atom in position 4 is optimal for activity over other donating groups or halogens at the same position. Additionally, the anti-proliferative activity increased with decreasing the halogen size as follows 4-Br < 4-Cl < 4-F.
Table 2: Cytotoxic effects of CP hybrids, 4a-j and doxorubicin against 3 colon cancer cell lines (Caco-2, HT-29, and HCT116).
2.2.1.4. Effect of ciprofloxacin-chalcone hybrids on non-cancerous cells
To evaluate the selectivity and safety profile of the synthesised CP hybrids, a cytotoxicity experiment of compounds 4a, 4b, 4e, 4i, and 4j was performed using normal Human Embryonic Kidney (HEK) 293 cells. Results obtained from cytotoxicity assay showed that CP hybrids 4a, 4b, 4e, 4i, and 4j caused cytotoxic effects in HEK293 cells to different extents after 72 h incubation period with IC50s ranging from 0.6523 µM to10.1600 µM, but their cytotoxicity were much lower than that of doxorubicin cytotoxicity in HEK293 which had IC50 equal to 0.0475 µM (). CP hybrid 4j had the steepest concentration-cell viability curve of any other ciprofloxacin-chalcone hybrids against HEK293 cells, suggesting that the 4j compound had stronger cytotoxic activity than other ciprofloxacin-chalcone hybrids. However, the concentration-cell viability curve of 4j was shallower than the dose-response curve of doxorubicin (positive control). This demonstrates that our synthesised CP hybrids are less cytotoxic against non-cancerous cells (HEK293) when compared to the commonly used chemotherapy (doxorubicin). Further analysis of the cytotoxicity data revealed that CP hybrids (4a, 4b, 4e, and 4i) had significantly larger IC50 values (p ≤ 0.0001, ). Also, the 4j compound had a higher IC50 value than doxorubicin albeit not significantly different. These findings were further supported by microscopic images of cells treated with different concentrations of these compounds (). The cytotoxic effects of CP hybrids in HEK293 may be due to the long incubation period compared to the incubation period of HCT116 cells. Furthermore, compound 4j, a more cytotoxic hybrid, had a selectivity index six-fold better than that of the drug Doxorubicin.
Figure 5. Cytotoxic effects of CP hybrids (4a, 4b, 4e, 4i, and 4j) and doxorubicin against non-cancerous cells (HEK293) after 72 h incubation period. (A) Concentration-cell viability curves of CP hybrids and doxorubicin treated HEK293 cells using different concentrations (0.01–100 µM, n = 3) and 72 h incubation period. (B) IC50 values were estimated by fitting the cytotoxicity data into a linear regression model using GraphPad Prism software. Data were statistically compared by one-way ANOVA followed by Tuckey’s multiple comparisons test (****p < 0.0001, ns = not significant). (C and D). Microscopic images of HEK293 cells treated with different concentrations of CP hybrids (4a, 4b, 4e, 4i, and 4j) and doxorubicin for 72 h (20× objective lens, total magnification = 200×). Scale bar = 400 µM.

2.2.2. Effects of compounds 4a, 4b, 4e, 4i, and 4j on topoisomerase I/II inhibitory activity
Based on our design, we hypothesised that the new CP hybrids 4a-j could act as multi-target ligands that can block the catalytic activity of topoisomerases I (Topo-I), topoisomerases (Topo-II), and inhibit tubulin polymerisation as possible targets for the anti-proliferative activity. First, the Topo-I activity was tested by the DNA relaxation assay. The supercoiled plasmid has been utilised as a substrate in the relaxation assay to study the inhibition of DNA Topo-I activity. The relaxed isomers migrate in the gel more slowly than the supercoiled isomers; hence they can be distinguished from each other. If the synthesised compounds have an inhibitory effect on the catalytic activity of Topo-I, the DNA molecules will remain in the supercoiled form, a faster moving single band. As shown in (), we determined that 4a, 4b, 4e, 4i, and 4j could inhibit Topo-I catalytic activity, with 4a the most potent Topo-I inhibitor.
Figure 6. The effect of IC50 values of CP hybrids 4a, 4b, 4e, 4i, 4j and camptothecin (CPT, positive control in Topo-I assay) on the activity of human topoisomerase I. (A) DNA relaxation assay. (B) in-vitro enzyme assay. Data are plotted as means ± SEM of 3 experiments. ∗∗ p < 0.01 and ∗∗∗ p < 0.001 refer to the statistically significant differences in comparison to DMSO (vehicle, negative control treatment).

Moreover, the Topo-I enzymatic activity inhibition by the synthesised compounds was confirmed using the Human DNA Topoisomerase I Assay Kit (ProFoldin, Hudson, MA, USA) (). The results of Human DNA Topoisomerase I Assay as shown in () indicated that compound 4a had the highest Top-I inhibitory activity which was slightly higher than that of the reference camptothecin. On the other hand, compound 4j was the less potent Top-1 inhibitor.
The remarkable Top-I inhibitory activities of the synthesised hybrids; 4a, 4b, 4e, 4i, and 4j are in agreement with our hypotheses of these hybrid as Top-I inhibitors.
Second, the inhibitory activity of Topo IIα was evaluated for compounds 4a, 4b, 4e, 4i, and 4j utilising Human Topoisomerase II assay kit (TG1001, Topogen Inc.). Etoposide, known inhibitor of Topo IIα, was used as reference. Top-II assay results indicated that the synthesised CP hybrids exhibited strong Topo-II inhibitory activity when compared to the positive control, etoposide (ETO) (). Based on the analysis of band intensity using ImageJ software, it was found that all CP hybrids inhibited Topo-II to a similar magnitude in comparison to ETO when 100 µM of each compound was used (). However, when a lower concentration (10 µM) was tested, it was found that three ciprofloxacin hybrids (4a, 4e, and 4j) had stronger inhibitory activity than 4b, 4i, and ETO. The above results are compatible with our design of these hybrids as Top-II inhibitors. According to these results, CP hybrid 4a had the strongest inhibitory activity on both Topo-I and Topo-II enzymes.
Figure 7. The effect of ciprofloxacin hybrids 4a, 4b, 4e, 4i, 4j, and etoposide (ETO, positive control in Topo-II assay) on the activity of human topoisomerase II. (A). Agarose gel electrophoresis of human topoisomerase II assay. (B). Topo-II inhibitory activity of ciprofloxacin hybrids.

2.2.3. Effects of compounds 4a, 4b, 4e, 4i, and 4j on tubulin polymerisation
The process of tubulin polymerisation in the cells is important for the completion of mitosis and is targeted for cancer treatment. It has been reported that the suppression of tubulin polymerisation will arrest the cells at the G2/M and it eventually leads to cell death. To test the tubulin-disruptive effect of the CP hybrids, HCT116 cells were analysed by immunocytochemistry. HCT116 cells were incubated for 36 h with IC50 of the CP hybrids (4a, 4b, 4e, 4i, and 4j), nocodazole (a well-known tubulin polymerisation inhibitor, as a positive control), DMSO or Paclitaxel (a well-known tubulin polymerisation inducer, as a negative control), and tubulin was visualised by immunofluorescence microscopy. An antibody for α-tubulin was used. Untreated and Paclitaxel treated HCT116 cells showed a spider-web-like structure (. Morphological changes of HCT116 cells treated with either CP hybrids (4a, 4b, 4e, 4i, and 4j) or nocodazole showed a significant tubulin polymerisation inhibition (). Moreover, a Western blot was performed to verify the tubulin polymerisation inhibition ability of the synthesised compounds (). The level of polymerised tubulin was decreased in HCT116 cells treated with IC50 values of CP hybrids 4a, 4b, 4e, 4i, 4j, or nocodazole. Additionally, compounds 4i and 4j showed potent inhibition of tubulin polymerisation () and a similar weak effect on human topoisomerase I activity which may suggest that the tubulin inhibition mechanism is the primary mechanism of action for these compounds. The above-mentioned findings indicated that the synthesised compounds could act as multi-targets; Topo-I, TopII, and tubulin polymerisation inhibitors as intended by our design.
Figure 8. Effect of ciprofloxacin derivatives on tubulin polymerisation. HCT116 cells were untreated (control) and treated with IC50 values of compounds 4a, 4b, 4e, 4i, 4j, paclitaxel or nocodazole for 36 h. (A) Morphological changes of HCT116 cells visualised by immunofluorescence microscopy. Tubulin were shown in green and the nuclei were in blue. (B) Western blotting analysis.

2.2.4. DNA damage induction upon treatment with compounds 4a, 4b, 4e, 4i, and 4j
DNA damage is a major reason for inhibiting proliferation and inducing cytotoxicity in cancer cells as well as the main downstream cascade for the Topo-I, Top-II, and tubulin polymerisation inhibition. The phosphorylation of the histone H2AX always occurs after DNA damage forming double-stranded breaks (DSBs). Therefore, we tested the capability of the potent prepared compounds to trigger DNA double strand breaks through detection of the phosphorylation of γH2AX using two different techniques, Immunofluorescence, and western blotting. Results in () indicated that treatment of HCT116 cells with the IC50 of the potent compounds (4a, 4b, 4e, 4i, and 4j) induced γH2AX foci formation. Also, more γH2AX-positive cells were observed in comparison to the control cells. Similarly, the western blotting analysis () showed increases in the γH2AX protein expression in cells treated with the IC50 of the target compounds (4a, 4b, 4e, 4i, and 4j) in comparison with the control cells. These results clearly show that the 1,2,3-Triazole linked CP hybrids induce DNA damage in colon cancer cells.
Figure 9. CP hybrids induce DNA damage. (A) γH2AX staining in HCT116 cells treated with IC50 values of CP hybrids 4a, 4b, 4e, 4i, 4j, or DMSO (negative control). (B) γH2AX-positive nuclei %. (C) Western blot revealed that levels of γH2AX were increased in HCT116 cells treated with CP hybrids 4a, 4b, 4e, 4i, 4j, DMSO (negative control), and GAPDH (loading control). (D) The relative protein level of γ-H2AX, in comparison to GAPDH was measured by ImageJ. Data are plotted as means ± SEM of 3 experiments. ∗ p < 0.05, ∗∗ p < 0.01, and ∗∗∗ p < 0.001 refer to the statistically significant differences in comparison to DMSO.

2.2.5. Effects of compounds 4a, 4b, 4e, 4i, and 4j on cell cycle analysis
Generally, the mammalian cells counteract the DNA damage by different mechanisms including the cell cycle arrest to allow the cell to repair the damage. There are 2 checkpoints that control DNA damage. The first checkpoint is at the G1/S transition while the other checkpoint is at the G2/M transition. To cover the mechanisms of the synthesised compounds in inhibiting cell proliferation, we analysed the distribution of cell cycle of HCT116 cells following the treatment with different doses of 4a, 4b, 4e, 4i, and 4j. The result showed that 4a, 4b, 4e, 4i, and 4j induced G2/M phase cell cycle arrest in a concentration-dependent manner (. Our results suggest that 1,2,3-Triazole linked CP-Chalcone hybrids arrest the cell cycle of colon tumour cells in G2/M phase.
Figure 10. Analysis of cell cycle. HCT116 cells were treated with different concentrations of ciprofloxacin hybrids 4a, 4b, 4e, 4i, and 4j for 24 h. Doxorubicin, camptothecin, and etoposide were used as assay controls. The X-axis displays the fluorescence intensity corresponding to the DNA content per HCT116 cell. DNA histograms were analysed by ModFit LT software.

2.2.6. Effects of compounds 4a, 4b, 4e, 4i, and 4j on activation of the ATR-Chk1-Cdc25C Cascade
To clarify the molecular mechanism responsible for inducing G2/M phase arrest in HCT116 following the treatment with the synthesised compounds, the signalling in the checkpoint pathway that regulates the G2/M transition was investigated. The mammalian cells respond to the DNA damage through 2 different kinase-signalling cascades (ATR-Chk1 and ATM-Chk2). First, the effect of the synthesised compounds on ATM and ATR phosphorylation was tested. It was found that the ATM phosphorylation on Ser-1981 was unchanged in the comparison with the control (data not shown) after treatment with the synthesised compounds, while p-ATR at Ser-428 markedly increased (). Then, the phosphorylation of downstream effectors of ATM/ATR, phospho-Chk1-S269, and phospho-Chk2-T68 kinases was explored. It was observed that the synthesised compounds upregulated the phosphorylation of Chk1 at S269 () without affecting the phosphorylation of Chk2 at Thr-68 (data not shown). It is well known that in vitro activation of Chk1 phosphorylates Cdc25C on serine-216. Therefore, the impact of the synthesised compounds on the Cdc25C phosphorylation at Ser-216 was investigated. Results showed that the levels of Cdc25C phosphorylation at Ser-216 were substantially elevated in triazole-treated HCT116 cells (). Overall, these results show that the synthesised hybrids induce G2/M cell cycle arrest possibly via ATR/CHK1/Cdc25C pathway.
Figure 11. Ciprofloxacin hybrids 4a, 4b, 4e, 4i, and 4j induce G2/M cell cycle arrest possibly via ATR/CHK1/Cdc25C pathway A. Western blot revealed that expressions of p-ATR, p-CHK1, and p-Cdc25C were increased in HCT116 cells treated with Ciprofloxacin hybrids 4a, 4b, 4e, 4i, 4j, DMSO (negative control), and GAPDH (loading control) B. The relative protein levels of p-ATR, p-CHK1, and p-Cdc25C, compared to their total form ATR, CHK1, and Cdc25C, respectively, were measured by ImageJ. Data are plotted as means ± SEM of 3 experiments. * p < 0.05, ** p < 0.01, and *** p < 0.001 refer to the statistically significant differences in comparison to DMSO.

2.3. Molecular docking studies of ciprofloxacin hybrids 4a, 4b, 4e, 4i, and 4j on Top-I and Top-II active sites
2.3.1. Docking Studies of ciprofloxacin hybrids 4a, 4b, 4e, 4i, and 4j in human Topo I enzyme (PDB ID: 1T8I)
Based on the potent inhibitory Top-I activities of the synthesised hybrids, we study in silico molecular docking simulations using MOE software for the most active compounds 4a, 4b, 4e, 4i, and 4j into human Top-I enzyme (PDB ID: 1T8I) in comparison to camptothecin to examine their binding modes. The validation of the docking method was made by re-docking of the co-crystallized ligand and calculation of RMSD. The RMSD was 0.4626 which means the validity of the docking method. All the tested hybrids had a strong binding affinity to the enzyme as the binding free energy (ΔG) values ranged from −10.905 to −13.267 Kcal/mole which were higher than that of camptothecin (ΔG = −9.761 Kcal/mole), ().
Table 3. ΔG values of the tested compounds 4a, 4b, 4e, 4i, 4j, and camptothecin in the active binding site of Topo I enzyme (PDB ID: 1T8I).
The Docking results (as shown in Table 2 and Figures 1–3, in Supplementary Data) are in agreement with Top-1 inhibitory activity results and indicate that compounds 4a, 4e, and 4i have comparable binding interactions with Top-I enzyme (PDB ID: 1T8I) to that of camptothecin which forms 3 H-bond interactions with Asp533, Lys532, and Arg364 and four π…π interactions with DA 113, DC 112, and TGP 111, (. On the contrast, compounds 4b and 4j have fewer binding interactions with Top-I enzyme (PDB ID: 1T8I) than that of camptothecin. Concerning compound 4a which had the most potent Top-I inhibitory activity even higher than that of camptothecin, its docking pose shows one H-bonding interaction with Trp 416, one cation…π interaction with Lys 425 and three π…π interactions with DT 10, TGP 111, and DA 113, as shown in (. Additionally, compound 4e, which had Topo-I inhibitory activity comparable to camptothecin, has two H-bonding interactions with Arg 488 & His 632 and three π…cation interactions with Trp416, DT 10, and DG 12. On the other side, compound 4j which was the less potent Top-1 inhibitor among the tested hybrids, it has only one cation…π interaction with Glu 356 and one π…π interaction with DC112. See Supplementary Data. The above docking results indicated that although some of the new hybrids have comparable binding interactions as camptothecin, none of these new compounds have H-bond interactions with the same amino acid residues or have three H-bonds like camptothecin, Additionally, none of them have the same number of π…π interactions with the same DNA residues of Top-I enzyme such as camptothecin even the hybrid 4a which was more potent as Top-1 inhibitor than camptothecin which means these hybrids could have a slight distinct interactions with Top-I enzyme.
2.3.2. Docking studies of ciprofloxacin hybrids 4a, 4b, 4e, 4i, and 4j in human Topo II enzyme (PDB ID: 6ZY7)
Upon relying on the results of the Top II assay, docking of the most active hybrids; 4a, 4b, 4e, 4i, and 4j, and the positive control Etoposide; was performed with the active binding site of into human Topo II enzyme (PDB ID: 6ZY7) to elucidate their effective binding modes and concentrate on their similarity to the standard ligand binding modes. The docking method validation was examined by re-docking of the co-crystallized Etoposide and calculation of RMSD. The RMSD was 0.4063 which means the docking method is valid. All the tested hybrids had a strong binding affinity to the enzyme as the binding free energy (ΔG) values ranged from −9.511 to −9.906 Kcal/mole which were lower than that of Etoposide (ΔG = −10.162 Kcal/mole), ().
Table.4. ΔG values of the tested compounds 4a, 4b, 4e, 4i, 4j, and Etoposide in the active binding site of Topo II enzyme (PDB ID: 6ZY7).
From the docking, results (as shown in Table 3 and Figures 4–6, in Supplementary Data), we can say that the tested hybrids form different interactions than that of Etoposide which forms four interaction; two hydrogen bonds with DG 13 & DG 5 residues, one π…cation interaction with DG13, and one π…π interaction with DT 12, (. Despite of compounds 4a, 4e, and 4j were more potent as Top-II inhibitors than etoposide at 10 µM concentrations. They interact with the 6ZY7 binding site in a different manner with different amino acid residues than that of etoposide. For example, compound 4a forms only two hydrogen bonds with Ala 465 and Ser 464, (. Additionally, compound 4e docking pose shows two hydrogen bonds with Ala 801 & Ser 802 and two π…cation interactions with DT 12 and DG 13. Moreover, 4j forms only three interactions with the 6ZY7 binding site; two hydrogen bonds with Ser 800 and DC 3 residue and one π…cation interaction with DC 1 residue. On the other hand, compound 4b which was less potent Top-II inhibitor among the tested compound at 10 µM forms only one hydrogen bond with Lys- 489.
Figure 12. Binding mode and H-bonds interactions of compound 4a (left) and camptothecin (right) within 1T8I active site: (a) 3 D structure and (b) 2D interactions.

Figure 13. Binding mode and H-bonds interactions of compound 4a (left) and etoposide (right) within 6ZY7 active site: (a) 3 D structure and (b) 2 D interactions.

3. Conclusions
We were successfully able to synthesise novel triazole-linked CP chalcone hybrids 4a-j with remarkable anti-proliferative activity against leukaemia (RPMI-8226 and SR) and colon (HCT116) tumour cells with a growth percent ranged from 47.87 to −53.36%. Among the tested compounds, CP hybrid 4j displayed potent and wide-range anti-proliferative effects in the majority of cancer lines with a growth percent range of 67.46 to −39.69%. Also, hybrids 4a, 4b, 4e, 4i, and 4j induced significant anti-proliferative effect in several cancer cells, particularly in leukaemia (RPMI-8226) and colon (HCT116) cell lines with a growth percent values 47.87, −14.59, −24.56, −24.42, −15.84, −9.74, −53.36, −10.26, −31.29, and −39.51%, respectively. Generally, the anti-proliferative activity of hybrids 4a-j was more potent against colon cancer (HCT116) than leukaemia SR (RPMI-8226 and SR) cell lines. Compounds 4a, 4b, 4e, 4i, and 4j displayed the strongest anti-proliferative effect in HCT116 cells with IC50 = 3.57, 4.81, 4.32, 4.87 and 2.53 µM; respectively compared to doxorubicin (IC50 = 1.22 µM). Additionally, Compounds 4a, 4b, 4e, 4i, and 4j exhibited much lower toxicity than that of doxorubicin against normal Human Embryonic Kidney (HEK) 293 cells after 72 h incubation period. Also, hybrids 4a, 4b, 4e, 4i, and 4j exhibited remarkable inhibitory activities against Topo-I, Top-II, and tubulin polymerisation compared with the used references. They increased the protein expression level of γH2AX indicating DNA damage (double strand breaks). Furthermore, they arrested the HCT116 cell cycle at the G2/M phase possibly through ATR/CHK1/Cdc25C pathways as a result of DNA damage. Thus, the new CP hybrids have the capacity to be used as promising leads and advanced for further investigation as novel multi-target anticancer agents to combat colorectal carcinoma.
4. Experimental section
4.1. Chemistry
Thin-layer chromatography (Merck Grade-9385 precoated aluminium TLC plate silica gel 60, size 5 × 20 cm with 0.2 mm thickness) was used to monitor the chemical reaction. Plates were exposed to UV light at a wavelength of 254 nm to detect the spots. In addition, Stuart Electrothermal Melting Point Apparatus was used to determine the melting points (values were un-corrected). Also, 100 MHz for 13 C and 400 MHz for 1H NMR spectra were obtained on Bruker AM400 spectrometer using DMSO-d6 (solvent) and tetramethylsilane (internal standard). In this work, δ (chemical shifts) and J (coupling constants) were reported in ppm and Hz, respectively. The following abbreviations were used to describe the NMR peak multiplicity: singlet = s, doublet = d, doublet of doublets = dd, triplet = t, quartette = q, multiplet = m, broad signal, brs. Shimadzu’s GC/MS-QP5050A (Regional Centre for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt) was used for elemental analyses while Advion’s compact mass spectrometer (Nawah Scientific Centre for Research, Almokattam, Cairo, Egypt) was used to obtain mass spectra (m/z).
4.1.1. Synthesis of the intermediates; 1, 2a-j, and 3
The intermediate 1Citation65,Citation66, 2a-jCitation65,Citation66,Citation70, 3Citation67 were prepared according to the reported methods in the previous studies.
4.1.2. Synthesis of compounds 4a-j
Our target compounds 4a-j were synthesised by the reaction between Ciprofloxacin alkyne derivative 3 (0.5 g, 1.35 mmol.) and the appropriate azide 2a-j (0.37 g, 1.48 mmol.) under the catalysis of sodium ascorbate (0.4 eq) and copper (II) sulphate pentahydrate (0.2 eq) as reported procedureCitation71. See Supplementary Appendix A
4.1.2.1. (E) 7-(4-((1–(4-Cinnamoylphenyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 4a
Yellowish white powder; (0.53 g, 63.85% yield); mp: 240–242 °C; IR (KBr) νmax = broad band 3223-3074 cm−1 (OH of COOH), 1715 cm−1 (C = O of COOH), 1657 cm−1 (C = O of CH = CH–C = O) and 1622 (C = O of 4-keto gp of CP); 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.16–1.22 (2H, m, cyclopropyl-H), 1.28–1.36 (2H, m, cyclopropyl-H), 2.70-2.80 (4H, m, piperazinyl-H), 3.36-3.40 (4H, m, piperazinyl-H), 3.77–3.85 (3H, m, N–CH2 and cyclopropyl-1H), 7.45–7.60 (4H, m, C8-H and Ar-H), 7.79 (1H, d, Jtrans = 16 Hz, =CH), 7.83 (1H, d, JHFortho = 16 Hz, C5-H), 7.90–7.92 (2H, m, Ar-H), 7.99 (1H, d, Jtrans = 16 Hz, =CH), 8.15 (2H, d, J = 8 Hz, Ar-H), 8.35 (2H, d, J = 8 Hz, Ar-H), 8.62 (1H, s, C2-H), 8.93 (1H, s, triazole H), 15.14 (1H, brs, COOH); 13 C NMR (100 MHz, CDCl3) δ ppm: 8.21, 35.26, 49.51, 51.42, 52.48, 52.90, 104.91, 108.23, 112.48 (C-5, d, 2JCFortho = 23 Hz, C-F), 120.12, 121.43, 126.64, 128.57, 129.06, 130.29, 130.91, 134.64, 138.18, 139.08, 140.92, 145.58, 145.81, 146.38, 147.42, 148.43, 153.66 (C-6, d, 1JCFipso = 250 Hz, C-F), 166.89, 177.09, 188.84; ESI-MS (m/z): Calcd. 618.24, found 641.60 [M + Na]+. Anal. Calcd. For C35H31FN6O4: C, 67.95%; H, 5.05%; N, 13.58%. Found: C, 67.84%; H, 5.21%; N, 13.74%.
4.1.2.2. (E)-7-(4-((1–(4-(3-(3-chlorophenyl)acryloyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl) piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 4b
Yellow powder; (0.59 g, 66.91% yield); mp: 254–247 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.18–1.24 (2H, m, cyclopropyl-H), 1.31–1.35 (2H, m, cyclopropyl-H), 2.67–2.73 (4H, m, piperazinyl-H), 3.35–3.39 (4H, m, piperazinyl-H), 3.79–3.85 (3H, m, N–CH2 and cyclopropyl-1H), 7.53–7.60 (2H, m, Ar-H and C8-H), 7.77 (1H, d, Jtrans = 16 Hz, =CH), 7.84–7.91 (2H, m, Ar-H and C5-H), 8.09-8.18 (3H, m, =CH and, Ar-H), 8.17 (2H, d, J = 8 Hz, Ar-H), 8.42 (2H, d, J = 8 Hz, Ar-H), 8.65 (1H, s, C2-H), 8.92 (1H, s, triazole H), 15.17 (1H, brs, COOH); 13 C NMR (100 MHz, CDCl3) δ ppm: 7.22, 34.31, 48.34, 50.44, 51.43, 51.85, 103.99, 107.17, 111.43 (C-5, d, 2JCFortho = 23 Hz, C-F), 119.17, 121.59, 125.94, 127.03, 129.33, 129.70, 134.14, 135.49, 136.87, 138.08, 138.91, 139.33, 139.84, 142.98, 144.58, 144.76, 146.43, 147.55, 152.64 (C-6, d, 1JCFipso = 250 Hz, C-F), 165.88, 176.05, 187.40; ESI-MS (m/z): Calcd. 652.20, found 675.80 [M + Na]+. Anal. Calcd. For C35H30ClFN6O4: C, 64.37%; H, 4.63%; N, 12.87%. Found: C, 64.53%; H, 4.80%; N, 13.09%.
4.1.2.3. (E)-7-(4-((1-(4-(3-(4-chlorophenyl)acryloyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl) piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 4c
Yellow powder; (0.58 g, 65.89% yield); mp: 257–259 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.17–1.22 (2H, m, cyclopropyl-H), 1.30–1.35 (2H, m, cyclopropyl-H), 2.70–2.76 (4H, m, piperazinyl-H), 3.36–3.40 (4H, m, piperazinyl-H), 3.80–3.82 (3H, m, N-CH2 and cyclopropyl-1H), 7.55 (2H, d, J = 8 Hz, Ar-H), 7.57(1H, d, JHFmeta = 8 Hz, C8-H), 7.79 (1H, d, Jtrans = 16 Hz, =CH), 7.90 (1H, d, JHFortho = 12 Hz, C5-H), 7.97 (2H, d, J = 8 Hz, Ar-H), 8.03 (1H, d, Jtrans = 16 Hz, =CH), 8.16 (2H, d, J = 8 Hz, Ar-H), 8.39 (2H, d, J = 8 Hz, Ar-H), 8.66 (1H, s, C2-H), 8.94 (1H, s, triazole H), 15.17 (1H, brs, COOH); 13 C NMR (100 MHz, CDCl3) δ ppm: 8.20, 35.28, 49.44, 52.45, 52.88, 104.92, 108.18, 112.42 (C-5, d, 2JCFortho = 24 Hz, C-F), 120.12, 121.18, 121.79, 126.33, 129.36, 129.70, 130.27, 133.12, 136.88, 137.97, 139.06, 139.85, 144.22, 144.51, 145.59, 147.40, 153.62 (C-6, d, 1JCFipso = 252 Hz, C-F), 166.85, 177.04, 188.50; ESI-MS (m/z): Calcd. 652.20, found 675.80 [M + Na]+. Anal. Calcd. For C35H30ClFN6O4: C, 64.37%; H, 4.63%; N, 12.87%. Found: C, 64.51%; H, 4.89%; N, 13.04%.
4.1.2.4. (E)-7-(4-((1-(4-(3-(4-bromophenyl)acryloyl)phenyl)-1H-1,2,3-triazol-4-yl) methyl)piperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 4d
Pale yellow powder; (0.57 g, 60.50% yield); mp: 247–249 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.17–1.21 (2H, m, cyclopropyl-H), 1.31–1.35 (2H, m, cyclopropyl-H), 2.69–2.75 (4H, m, piperazinyl-H), 3.38–3.43 (4H, m, piperazinyl-H), 3.81–3.83 (3H, m, N-CH2 and cyclopropyl-1H), 7.57(1H, d, JHFmeta = 8 Hz, C8-H), 7.68 (2H, d, J = 8 Hz, Ar-H), 7.76 (1H, d, Jtrans = 16 Hz, =CH), 7.88 (2H, d, J = 8 Hz, Ar-H), 7.91 (1H, d, JHFortho = 12 Hz, C5-H), 8.02 (1H, d, Jtrans = 16 Hz, =CH), 8.15 (2H, d, J = 8 Hz, Ar-H), 8.38 (2H, d, J = 8 Hz, Ar-H), 8.66 (1H, s, C2-H), 8.91 (1H, s, triazole H), 15.05 (1H, brs, COOH); 13 C NMR (100 MHz, CDCl3) δ ppm: 8.22, 35.30, 49.62, 51.41, 52.22, 104.86, 105.18, 108.18, 112.42 (C-5, d, 2JCFortho = 23 Hz, C-F), 120.22, 121.89, 125.24, 129.90, 130.70, 131.08, 131.54, 132.33, 133.55, 138.06, 139.05, 139.74, 144.31, 145.15, 145.81, 153.61 (C-6, d, 1JCFipso = 260 Hz, C-F), 166.90, 177.02, 188.50; ESI-MS (m/z): Calcd. 696.15, found 719.50 [M + Na]+. Anal. Calcd. For C35H30BrFN6O4: C, 60.26%; H, 4.34%; N, 12.05%. Found: C, 60.53%; H, 4.52%; N, 12.31%.
4.1.2.5. (E)-1-cyclopropyl-6-fluoro-7–(4-((1–(4-(3-(4-fluorophenyl)acryloyl) phenyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 4e
Yellow powder; (0.40 g, 46.56% yield); mp: 203–205 °C; IR (KBr) νmax= broad band 3283–3066 cm−1 (OH of COOH), 1718 cm−1 (C = O of COOH), 1658 cm−1 (C = O of CH = CH–C = O) and 1624 (C = O of 4-keto gp of CP); 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.17–1.21 (2H, m, cyclopropyl-H), 1.31–1.34 (2H, m, cyclopropyl-H), 2.75–2.80 (4H, m, piperazinyl-H), 3.39–3.45 (4H, m, piperazinyl-H), 3.82–3.87 (3H, m, N-CH2 and cyclopropyl-1H), 7.30 (2H, t, JHF = 8 Hz, Ar-H), 7.57(1H, d, JHFmeta = 8 Hz, C8-H), 7.79 (1H, d, Jtrans = 16 Hz, =CH), 7.86–7.93 (2H, m, =CH and C5-H), 7.97 (2H, d, J = 8 Hz, Ar-H), 8.13 (2H, d, J = 8 Hz, Ar-H), 8.35 (2H, d, J = 8 Hz, Ar-H), 8.66 (1H, s, C2-H), 8.86 (1H, s, triazole H), 15.05 (1H, brs, COOH); 13 C NMR (100 MHz, CDCl3) δ ppm: 6.41, 33.50, 47.43, 50.58, 50.98, 103.22, 106.42, 110.64 (C-5, d, 2JCFortho = 23 Hz, C-F), 113.67, 114.46 (CAr, d, 2JCFortho = 23 Hz, C-F), 118.35, 119.38, 119.74, 128.43, 128.67, 128.75, 129.14, 136.36, 137.27, 137.97, 142.62, 143.64, 145.62, 151.82 (C-6, d, 1JCFipso = 251 Hz, C-F), 162.53 (CAr, d, 1JCFipso = 252 Hz, C-F), 165.01, 175.23, 186.77; ESI-MS (m/z): Calcd. 636.23, found 659.03 [M + Na]+. Anal. Calcd. For C35H30F2N6O4: C, 66.03%; H, 4.75%; N, 13.20%. Found: C, 65.94%; H, 4.89%; N, 12.98%.
4.1.2.6. (E)-1-cyclopropyl-6-fluoro-4-oxo-7-(4-((1-(4-(3-(p-tolyl)acryloyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid 4f
Yellow white powder; (0.47 g, 55.03% yield); mp: 259–261 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.15–1.23 (2H, m, cyclopropyl-H), 1.29–1.34 (2H, m, cyclopropyl-H), 2.37 (3H, s, Ar-CH3), 2.71–2.75 (4H, m, piperazinyl-H), 3.39-3.46 (4H, m, piperazinyl-H), 3.79–3.83 (3H, m, N-CH2 and cyclopropyl-1H), 7.30 (2H, d, J = 8 Hz, Ar-H), 7.56 (1H, d, JHFmeta = 8 Hz, C8-H), 7.75–7.82 (3H, m, =CH and Ar-H), 7.89–7.99 (2H, m, C5-H and = CH), 8.14 (2H, d, J = 8 Hz, Ar-H), 8.35 (2H, d, J = 8 Hz, Ar-H), 8.66 (1H, s, C2-H), 8.96 (1H, s, triazole H), 15.05 (1H, brs, COOH); 13 C NMR (100 MHz, CDCl3) δ ppm: 8.20, 21.50, 35.26, 49.38, 50.46, 52.41, 52.83, 104.96, 108.27, 112.48 (C-5, d, 2JCFortho = 23 Hz, C-F), 120.11, 120.47, 124.77, 128.60, 129.03, 129.79, 130.21, 131.94, 138.41, 139.07, 139.70, 141.55, 145.48, 145.89, 147.41, 148.32, 153.64 (C-6, d, 1JCFipso = 252 Hz, C-F), 166.79, 177.06, 188.90; ESI-MS (m/z): Calcd. 632.25, found 655.40 [M + Na]+. Anal. Calcd. For C36H33FN6O4: C, 68.34%; H, 5.26%; N, 13.28%. Found: C, 68.35%; H, 5.40%; N, 13.41%.
4.1.2.7. (E)-1-Cyclopropyl-6-fluoro-7-(4-((1-(4-(3-(4-methoxyphenyl)acryloyl) phenyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 4 g
Yellow powder; (0.47 g, 53.67% yield); mp: 254–257 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.18–1.24 (2H, m, cyclopropyl-H), 1.29–1.34 (2H, m, cyclopropyl-H), 2.70–2.75 (4H, m, piperazinyl-H), 3.35–3.40 (4H, m, piperazinyl-H), 3.76 (3H, s, OCH3), 3.81–3.85 (3H, m, N-CH2, cyclopropyl-H), 7.04 (2H, d, J = 8 Hz, Ar-H), 7.56 (1H, d, JHFmeta = 8 Hz, C8-H), 7.77 (1H, d, Jtrans = 16 Hz, =CH), 7.85–7.90 (4H, m, =CH, Ar-H and C5-H), 8.14 (2H, d, J = 8 Hz, Ar-H), 8.36 (2H, d, J = 8 Hz, Ar-H), 8.66 (1H, s, C2-H), 8.93 (1H, s, triazole-H), 15.16 (1H, brs, COOH); 13 C NMR (100 MHz, CDCl3) δ ppm: 8.21, 35.25, 49.45, 52.47, 52.90, 55.45, 104.91, 108.30, 112.56 (C-5, d, 2JCFortho = 23 Hz, C-F), 113.73, 114.58, 119.07, 120.07, 122.97, 127.38, 130.17, 130.42, 131.87, 138.58, 139.07, 139.58, 141.40, 145.69, 147.45, 153.66 (C-6, d, 1JCFipso = 252 Hz, C-F), 162.09, 166.88, 177.12, 188.83; ESI-MS (m/z): Calcd. 648.25, found 671.60 [M + Na]+. Anal. Calcd. For C36H33FN6O5: C, 66.66%; H, 5.13%; N, 12.96%. Found: C, 66.39%; H, 5.36%; N, 13.18%.
4.1.2.8. (E)-1-cyclopropyl-7–(4-((1–(4-(3-(4-(dimethylamino)phenyl)acryloyl) phenyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 4h
Pale yellow powder; (0.39 g, 43.65% yield); mp: 177–179 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.17–1.21 (2H, m, cyclopropyl-H), 1.31–1.35 (2H, m, cyclopropyl-H), 2.74–2.78 (4H, m, piperazinyl-H), 3.04 (6H, s, N(CH3)2), 3.39–3.45 (4H, m, piperazinyl-H), 3.82–3.87 (3H, m, N–CH2 and cyclopropyl-1H), 6.79 (2H, d, J = 8 Hz, Ar-H), 7.58–7.65 (2H, m, C8-H and = CH), 7.70–7.76 (3H, m, Ar-H and = CH), 7.91 (1H, d, JHFortho = 12 Hz, C5-H), 8.10 (2H, d, J = 8 Hz, Ar-H), 8.30 (2H, d, J = 8 Hz, Ar-H), 8.67 (1H, s, C2-H), 8.83 (1H, s, triazole H), 15.07 (1H, brs, COOH); 13 C NMR (100 MHz, CDCl3) δ ppm: 8.19, 35.28, 40.06, 49.57, 52.49, 52.97, 104.89, 108.15, 111.86, 112.38 (C-5, d, 2JCFortho = 23 Hz, C-F), 116.02, 119.82, 119.97, 121.04, 122.34, 130.00, 130.65, 139.07, 139.13, 139.31, 144.77, 145.63, 145.72, 146.79, 147.37, 153.63 (C-6, d, 1JCFipso = 252 Hz, C-F), 166.91, 177.06, 188.79; ESI-MS (m/z): Calcd. 661.28, found 684.50 [M + Na]+. Anal. Calcd. For C37H36FN7O4: C, 67.16%; H, 5.48%; N, 14.82%. Found: C, 67.43%; H, 5.41%; N, 14.70%.
4.1.2.9. (E)-1-cyclopropyl-7-(4-((1-(4-(3–(3,4-dimethoxyphenyl)acryloyl) phenyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid 4i
Yellow powder; (0.52 g, 56.77% yield); mp: 167–169 °C; IR (KBr) νmax= broad band 3234–3067 cm−1 (OH of COOH), 1719 cm−1 (C = O of COOH), 1658 cm−1 (C = O of CH = CH–C = O) and 1625 (C = O of 4-keto gp of CP); 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.18–1.22 (2H, m, cyclopropyl-H), 1.29–1.32 (2H, m, cyclopropyl-H), 2.70-2.75 (4H, m, piperazinyl-H), 3.35–3.42 (4H, m, piperazinyl-H), 3.81–3.84 (6H, m, N-CH2, cyclopropyl-H, OCH3), 3.88 (3H, s, OCH3), 7.04–7.08 (1H, m, Ar-H), 7.43-7.45 (1H, m, Ar-H), 7.55–7.60 (2H, m, Ar-H and C8-H), 7.76 (1H, d, Jtrans = 16 Hz, =CH), 7.86–7.93 (2H, m, C5-H and = CH), 8.15 (2H, d, J = 8 Hz, Ar-H), 8.37 (2H, d, J = 8 Hz, Ar-H), 8.66 (1H, s, C2-H), 8.94 (1H, s, triazole-H), 15.17 (1H, brs, COOH); 13 C-NMR (100 MHz, CDCl3) δ ppm: 8.20, 35.29, 49.48, 52.45, 52.89, 55.89, 56.06, 104.92, 108.13, 110.48, 111.32, 112.36 (C-5, d, 2JCFortho = 23 Hz, C-F), 119.34, 120.07, 121.18, 123.45, 127.62, 130.19, 138.45, 139.06, 139.63, 145.58, 145.98, 147.38, 148.45, 149.44, 151.78, 151.91, 153.62 (C-6, d, 1JCFipso = 252 Hz, C-F), 166.87, 177.02, 188.87; ESI-MS (m/z): Calcd. 678.26, found 701.50 [M + Na]+. Anal. Calcd. For C37H35FN6O6: C, 65.48%; H, 5.20%; N, 12.38%. Found: C, 65.69%; H, 5.37%; N, 12.54%.
4.1.2.10. (E)-1-cyclopropyl-6-fluoro-4-oxo-7–(4-((1–(4-(3-(3,4,5-trimethoxyphenyl) acryloyl)phenyl)-1H-1,2,3-triazol-4-yl)methyl)piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid 4j
Yellow powder; (0.49 g, 51.21% yield); mp: 247–249 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.18–1.22 (2H, m, cyclopropyl-H), 1.27–1.32 (2H, m, cyclopropyl-H), 2.69–2.76 (4H, m, piperazinyl-H), 3.35–3.45 (4H, m, piperazinyl-H), 3.74 (3H, s, OCH3), 3.78–3.85 (3H, m, N-CH2, cyclopropyl-H), 3.89 (6H, s, 2X OCH3), 7.26 (2H, s, Ar-H), 7.57 (1H, d, JHFmeta = 8 Hz, C8-H), 7.75 (1H, d, Jtrans = 16 Hz, =CH), 7.87 (1H, d, JHFortho = 12 Hz, C5-H), 7.94 (1H, d, Jtrans = 16 Hz, =CH), 8.15 (2H, d, J = 8 Hz, Ar-H), 8.37 (2H, d, J = 8 Hz, Ar-H), 8.65 (1H, s, C2-H), 8.94 (1H, s, triazole-H), 15.16 (1H, brs, COOH); 13 C NMR (100 MHz, CDCl3) δ ppm: 8.22, 35.27, 49.15, 49.44, 52.42, 56.35, 61.00, 104.95, 106.11, 107.52, 108.23, 112.46 (C-5, d, 2JCFortho = 23 Hz, C-F), 120.14, 120.76, 121.29, 130.02, 130.30, 137.38, 138.28, 139.07, 140.51, 141.07, 144.76, 146.00, 147.42, 153.21 (C-6, d, 1JCFipso = 252 Hz, C-F), 153.62, 166.85, 177.07, 188.87; ESI-MS (m/z): Calcd. 707.28, found 730.26 [M + Na]+. Anal. Calcd. For C38H37FN6O7: C, 64.40%; H, 5.26%; N, 11.86%. Found: C, 64.53%; H, 5.49%; N, 12.03%. HPLC analysis: 94.39%.
4.2. Biological investigations
4.2.1. Cell culture experiments and reagents
Colo-rectal cancer cell lines (Caco-2, HT29, and HCT116) were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured and maintained in appropriate media supplemented with foetal bovine serum (10%) (Sigma-Aldrich, St. Louis, MO, USA), streptomycin (100 μg/mL), and penicillin (100 U/mL) (Life Technologies) and incubated at 37 °C with CO2 (5%). Ciprofloxacin derivatives were chemically synthesised (4a-j) while doxorubicin was obtained from Sigma-Aldrich. Analytical or cell culture grade chemicals were used throughout this study.
4.2.2. Proliferation assay
The method description for NCI anticancer screening is provided in detail in the standard NCI-60 testing protocolCitation72.
4.2.3. 3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
The proliferation of colorectal cancer cells (HCT116, HT29 and Caco-2) was assessed using MTT assay as previously reportedCitation73. Cells were treated with increasing concentrations of 1, 5, 10, 20, 40, 80 or 100 µM of CP hybrids 4a-j, doxorubicin, or DMSO. See Supplementary Appendix A.
4.2.4. Cytotoxicity assay with non-cancerous cells
To evaluate the selectivity and safety profile of the synthesised ciprofloxacin-chalcone hybrids, a cytotoxicity experiment was performed using a non-cancerous cell line. Briefly, Human Embryonic Kidney (HEK) 293 cells (ATCC, Manassas, VA, USA) were cultured in a Dulbecco’s Modified Eagle Medium (DEMEM) supplemented with 1 mM sodium pyruvate, 10 mM HEPES buffer solution, 2 mM GlutaMAX (Gibco, Grand Island, NY, USA), 50 µg/mL gentamycin sulphate (Dubuque, IA, USA), and 10% foetal bovine serum (ThermoFisher Scientific, USA). Cells were seeded in 96-well plates and incubated overnight in a humidified atmosphere with 5% CO2 at 37 °C. After 24 h, cells were treated with different concentrations (0.01–100 µM) of ciprofloxacin-chalcone hybrids. Also, DMSO and doxorubicin were used as negative and positive controls, respectively. After 72 h, old medium containing treatments were removed, and fresh medium and MTS reagents (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega, Madison, WI, USA) were added. The plates were then incubated at 37 °C. After 2 h, the absorbance was measured at 490 nm using a Spectra Max plus 384 Microplate reader (Molecular Devices, Sunnyvale, CA, USA) Citation74. Also, before adding the MTS reagents, cells were examined under a cell imaging system (EVOS FL Digital Microscope using a 20x objective lens, total magnification = 200×).
4.2.5. Topoisomerase I inhibition assay
Two different assays were used to examine the inhibitory activity of IC50 of the CP hybrids against Human topoisomerase I. Firstly; Gel electrophoresis-based assay (DNA relaxation assay) was performed following the manufacturer's protocol (TG-1015, Topogen Inc., USA). Secondly, the inhibitory activity of compounds 4a, 4b, 4e, 4i, 4j, and camptothecin to human topoisomerase I was analysed by Human DNA Topoisomerase I Assay Kit (ProFoldin, Hudson, MA, USA) with recombinant human DNA topoisomerase I (Sigma-Aldrich) according to the manufacturer’s protocol.
4.2.6. Topoisomerase II inhibition assay
The inhibitory activity of the selected ciprofloxacin hybrids on human Top-II was tested using a Human Topoisomerase II assay kit (TG1001, Topogen Inc.). In this assay, 1% agarose gel was used, and the compounds were tested at two different concentrations, 10 and 100 µM. Also, etoposide was used as a positive control in this experiment. The assay was performed according to the manufacturer’s protocol. The image of the electrophoretic gel was captured using an imaging system (iBright-CL1500, Thermo Fisher Scientific).
4.2.7. Immunofluorescence microscopy
HCT116 cells were cultured and treated with the IC50 of the potent compounds (4a, 4b, 4e, 4i, and 4j) or DMSO (control), fixed, permeabilized, and they were incubated with either γH2AX or α-tubulin antibody as the reported procedure in the literatureCitation75. Confocal images were captured using a Confocal Microscope with a 63X objective. All images were processed using ImageJ software. See Supplementary Appendix A.
4.2.8. Analysis of cell cycle distribution
The effects of the most potent CP hybrids; 4a, 4b, 4e, 4i, and 4j, and assay controls; Doxorubicin, camptothecin, and etoposide on cell cycle progression were analysed through propidium iodide staining and subsequent flow cytometry according to the reported procedureCitation76. See Supplementary Appendix A.
4.2.9. Western blotting
Western blotting analysis for HCT116 cells was performed according to the reported procedure in the literatureCitation77–79 to determine the effects of CP hybrids; 4a, 4b, 4e, 4i, and 4j on the expression of polymerised tubulin, γH2AX, p-ATR, p-CHK1, and p-Cdc25C by using the appropriate antibody. See Supplementary Appendix A.
4.3. Docking studies
Compounds 4a, 4b, 4e, 4i, and 4j were drawn and docked in the human Topo-I in complex with DNA and camptothecin (PDB ID: 1T8I) and in the human Top II in complex with DNA and Etoposide (PDB ID: 6ZY7) using Molecular Operating Environment (MOE 2019.01) program as reported in the literature. See Supplementary Appendix A.
Supplemental Material
Download PDF (3.9 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Abdel-Aal MAA, Abdel-Aziz SA, Shaykoon MSA, Abuo-Rahma GE-DA. Towards anticancer fluoroquinolones: a review article. Arch Pharm 2019;352:e1800376.
- Kaushik CP, Sangwan J, Luxmi R, et al. Design, synthesis, anticancer and antioxidant activities of amide linked 1,4-disubstituted 1,2,3-triazoles. J. Mol. Struct 2021;1226:129255.
- Chekkara R, Gorla VR, Susithra E, et al. Design, synthesis and anticancer evaluation of 2-amino pyrimidine linked 7-azaindzole derivatives. Chem. Data Collect 2020;29:100513.
- El-Damasy AK, Haque MM, Park JW, et al. 2-Anilinoquinoline based arylamides as broad spectrum anticancer agents with B-RAFV600E/C-RAF kinase inhibitory effects: design, synthesis, in vitro cell-based and oncogenic kinase assessments. Eur J Med Chem 2020;208:112756.
- Baglini E, Salerno S, Barresi E, et al. Multiple topoisomerase I (TopoI), topoisomerase II (TopoII) and Tyrosyl-DNA phosphodiesterase (TDP) inhibitors in the development of anticancer drugs. Eur J Pharm Sci 2021;156:105594.
- Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Transl Oncol 2021;14:101174.
- Rodriguez-Wallberg KA. Principles of cancer treatment: impact on reproduction. Adv Exp Med Biol 2012;732:1–8.
- Dong Y, Hu H, Sun Y, et al. Design, synthesis and biological evaluation of novel c-Met/HDAC dual inhibitors. Bioorg Med Chem Lett 2020;30:127610.
- Chen J, Li D, Li W, et al. Design, synthesis and anticancer evaluation of acridine hydroxamic acid derivatives as dual Topo and HDAC inhibitors. Bioorg Med Chem 2018;26:3958–66.
- Zimmermann GR, Lehár J, Keith CT. Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov Today 2007;12:34–42.
- Anighoro A, Bajorath J, Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem 2014;57:7874–87.
- Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. j Med Chem 2005;48:6523–43.
- Bass AKA, El-Zoghbi MS, Nageeb E-SM, et al. Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors. Eur. J. Med. Chem 2021; 209: 112904.
- Proschak E, Stark H, Merk D. Polypharmacology by design: a medicinal chemist's perspective on multitargeting compounds. J Med Chem 2019;62:420–44.
- Raghavendra NM, Pingili D, Kadasi S, et al. Dual or multi-targeting inhibitors: the next generation anticancer agents. Eur J Med Chem 2018;143:1277–300.
- Mohammed HHH, Abbas SH, Hayallah AM, et al. Novel urea linked ciprofloxacin-chalcone hybrids having antiproliferative topoisomerases I/II inhibitory activities and caspases-mediated apoptosis. Bioorganic Chem 2021;106:104422.
- Drlica K. Mechanism of fluoroquinolone action. Curr Opin Microbiol 1999;2:504–8.
- Shen LL, Baranowski J, Pernet AG. Mechanism of inhibition of DNA gyrase by quinolone antibacterials: specificity and cooperativity of drug binding to DNA. Biochemistry 1989;28:3879–85.
- Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol 2002;3:430–40.
- Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 2001;70:369–413.
- Abdel-Aziz M, Park S-E, Abuo-Rahma GE-DAA, et al. Novel N-4-piperazinyl-ciprofloxacin-chalcone hybrids: synthesis, physicochemical properties, anticancer and topoisomerase I and II inhibitory activity. Eur J Med Chem 2013;69:427–38.
- Azéma J, Guidetti B, Dewelle J, et al. 7-((4-Substituted)piperazin-1-yl) derivatives of ciprofloxacin: synthesis and in vitro biological evaluation as potential antitumor agents. Bioorg Med Chem 2009;17:5396–407.
- Sissi C, Palumbo M. The quinolone family: from antibacterial to anticancer agents. Curr Med Chem Anticancer Agents 2003;3:439–50.
- Reuveni D, Halperin D, Shalit I, et al. Quinolones as enhancers of camptothecin-induced cytotoxic and anti-topoisomerase I effects. Biochem Pharmacol 2008;75:1272–81.
- Yadav V, Varshney P, Sultana S, et al. Moxifloxacin and ciprofloxacin induces S-phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation. BMC Cancer 2015;15:581.
- Repositioning of fluoroquinolones from antibiotic to anti-cancer agents: an underestimated truth. Biomed. Pharmacother 2019;111:934–46.
- Mohammed HHH, Abuo-Rahma GE-DAA, Abbas SH, Abdelhafez E-SMN. Current trends and future directions of fluoroquinolones. Curr Med Chem 2019;26:3132–49.
- Ahadi H, Emami S. Modification of 7-piperazinylquinolone antibacterials to promising anticancer lead compounds: synthesis and in vitro studies. Eur J Med Chem 2020;187:111970.
- Yogeeswari P, Sriram D, Kavya R, Tiwari S. Synthesis and in-vitro cytotoxicity evaluation of gatifloxacin Mannich bases. Biomed Pharmacother 2005;59:501–10.
- Nieto MJ, Alovero FL, Manzo RH, Mazzieri MR. Benzenesulfonamide analogs of fluoroquinolones. Antibacterial activity and QSAR studies. Eur J Med Chem 2005;40:361–9.
- Gootz TD, McGuirk PR, Moynihan MS, Haskell SL. Placement of alkyl substituents on the C-7 piperazine ring of fluoroquinolones: dramatic differential effects on mammalian topoisomerase II and DNA gyrase. Antimicrob Agents Chemother 1994;38:130–3.
- Foroumadi A, Emami S, Rajabalian S, et al. N-Substituted piperazinyl quinolones as potential cytotoxic agents: structure-activity relationships study. Biomed Pharmacother 2009;63:216–20.
- Azéma J, Guidetti B, Korolyov A, et al. Synthesis of lipophilic dimeric C-7/C-7-linked ciprofloxacin and C-6/C-6-linked levofloxacin derivatives. Versatile in vitro biological evaluations of monomeric and dimeric fluoroquinolone derivatives as potential antitumor, antibacterial or antimycobacterial agents. Eur J Med Chem 2011;46:6025–38.
- Suresh N, Nagesh HN, Sekhar KVG, et al. Synthesis of novel ciprofloxacin analogues and evaluation of their anti-proliferative effect on human cancer cell lines. Bioorg Med Chem Lett 2013;23:6292–5.
- Abdel-Aziz AA-M, El-Azab AS, Alanazi AM, et al. Synthesis and potential antitumor activity of 7-(4-substituted piperazin-1-yl)-4-oxoquinolines based on ciprofloxacin and norfloxacin scaffolds: in silico studies. J Enzyme Inhib Med Chem 2016;31:796–809.
- Mohammed HHH, Abd El-Hafeez AA, Abbas SH, et al. New antiproliferative 7-(4-(N-substituted carbamoylmethyl)piperazin-1-yl) derivatives of ciprofloxacin induce cell cycle arrest at G2/M phase. Bioorg Med Chem 2016;24:4636–46.
- Wang G, Liu W, Gong Z, et al. Design, synthesis, biological evaluation and molecular docking studies of new chalcone derivatives containing diaryl ether moiety as potential anticancer agents and tubulin polymerization inhibitors. Bioorg Chem 2020;95:103565.
- Zhang E-H, Wang R-F, Guo S-Z, Liu B. An update on antitumor activity of naturally occurring chalcones, Evid Based Complement Alternat Med 2013;2013:815621.
- Das M, Manna K. Chalcone scaffold in anticancer armamentarium: a molecular insight. J Toxicol 2016;2016:7651047.
- Mirzaei H, Shokrzadeh M, Modanloo M, et al. New indole-based chalconoids as tubulin-targeting antiproliferative agents. Bioorg Chem 2017;75:86–98.
- Yadav VR, Prasad S, Sung B, Aggarwal BB. The role of chalcones in suppression of NF-κB-mediated inflammation and cancer. Int Immunopharmacol 2011;11:295–309.
- Singh P, Anand A, Kumar V. Recent developments in biological activities of chalcones: a mini review. Eur J Med Chem 2014;85:758–77.
- Batovska DI, Todorova IT. Trends in utilization of the pharmacological potential of chalcones. Curr Clin Pharmacol 2010;5:1–29.
- Bonandi E, Christodoulou MS, Fumagalli G, et al. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov Today 2017;22:1572–81.
- Xu Z, Zhao S-J, Liu Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: current developments, action mechanisms and structure-activity relationships. Eur J Med Chem 2019;183:111700.
- Akhtar J, Khan AA, Ali Z, et al. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur J Med Chem 2017;125:143–89.
- Lal K, Yadav P. Recent advancements in 1,4-disubstituted 1H-1,2,3-triazoles as potential anticancer agents. Anticancer Agents Med Chem 2018;18:21–37.
- Rashdan HRM, Gomha SM, El-Gendey MS, et al. Eco-friendly one-pot synthesis of some new pyrazolo[1,2-b]phthalazinediones with antiproliferative efficacy on human hepatic cancer cell lines. Green Chem. Lett. Rev 2018;11:264–74.
- Gomha SM, Ahmed SA, Abdelhamid AO. Synthesis and Cytotoxicity evaluation of some novel thiazoles, thiadiazoles, and pyrido[2,3-d][1,2,4]triazolo[4,3-a]pyrimidin-5(1H)-ones incorporating triazole moiety. Molecules 2015;20:1357–76.
- Dheer D, Singh V, Shankar R. Medicinal attributes of 1,2,3-triazoles: current developments. Bioorg Chem 2017;71:30–54.
- Zhang B. Comprehensive review on the anti-bacterial activity of 1,2,3-triazole hybrids. Eur J Med Chem 2019;168:357–72.
- Victoria Castelli M, Gabriel Derita M, Noelí López S. Novel antifungal agents: a patent review (2013 –present). Expert Opin Ther Pat 2017;27:415–26.
- Emami S, Ghobadi E, Saednia S, Hashemi SM. Current advances of triazole alcohols derived from fluconazole: design, in vitro and in silico studies. Eur J Med Chem 2019;170:173–94.
- Tian Y, Liu Z, Liu J, et al. Targeting the entrance channel of NNIBP: discovery of diarylnicotinamide 1,4-disubstituted 1,2,3-triazoles as novel HIV-1 NNRTIs with high potency against wild-type and E138K mutant virus. Eur J Med Chem 2018;151:339–50.
- Kaoukabi H, Kabri Y, Curti C, et al. Dihydropyrimidinone/1,2,3-triazole hybrid molecules: synthesis and anti-varicella-zoster virus (VZV) evaluation. Eur J Med Chem 2018;155:772–81.
- Chu X-M, Wang C, Wang W-L, et al. Triazole derivatives and their antiplasmodial and antimalarial activities. Eur J Med Chem 2019;166:206–23.
- Kalaria PN, Karad SC, Raval DK. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur J Med Chem 2018;158:917–36.
- Keri RS, Patil SA, Budagumpi S, Nagaraja BM. Triazole: a promising antitubercular agent. Chem Biol Drug Des 2015;86:410–23.
- Zhang S, Xu Z, Gao C, et al. Triazole derivatives and their anti-tubercular activity. Eur J Med Chem 2017;138:501–13.
- M. M A. 1,2,3‐Triazole hybrids as anticancer agents: a review. Arch. Pharm 2022;355:2100158.
- Oubella A, Bimoussa A, Oussidi AN, et al. New 1,2,3-triazoles from(R)-carvone: synthesis, DFTMechanistic study and in vitro cytotoxic evaluation. Molecules 2022;27:769.
- Ashour HF, Abou-zeid LA, El-Sayed MA-A, Selim KB. 1,2,3-Triazole-chalcone hybrids: synthesis, in vitro cytotoxic activity and mechanistic investigation of apoptosis induction in multiple myeloma RPMI-8226. Eur J Med Chem 2020;189:112062.
- Yan W, Xiangyu C, Ya L, et al. An orally antitumor chalcone hybrid inhibited HepG2 cells growthand migration as the tubulin binding agent. Invest New Drugs 2019;37:784–90.
- Gurrapu N, Kumar EP, Kolluri PK, et al. Synthesis, biological evaluation and molecular docking studies of novel 1,2,3-triazole tethered chalcone hybrids as potential anticancer agents. J. Mol. Struct 2020;1217:128356.
- Rahulan KM, Balamurugan S, Meena KS, et al. Synthesis and nonlinear optical absorption of novel chalcone derivative compounds. Opt. Laser Technol 2014;56:142–5.
- Balamurugan S, Nithyanandan S, Selvarasu C, et al. Photophysical and photochemical investigations on triazole ring linked chalcone containing polymethacrylates. Polymer 2012;53:4104–11.
- McPherson JC, Runner R, Buxton TB, et al. Synthesis of osteotropic hydroxybisphosphonate derivatives of fluoroquinolone antibacterials. Eur J Med Chem 2012;47:615–8.
- Keepers YP, Pizao PE, Peters GJ, et al. Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur J Cancer Clin Oncol 1991;27:897–900.
- Perez RP, Godwin AK, Handel LM, Hamilton TC. A comparison of clonogenic, microtetrazolium and sulforhodamine B assays for determination of cisplatin cytotoxicity in human ovarian carcinoma cell lines. Eur J Cancer 1993;29:395–9.
- Iqbal H, Prabhakar V, Sangith A, et al. Synthesis, anti- inflammatory and antioxidant activity of ring-A-monosubstituted chalcone derivatives. Med. Chem. Res 2014;23:4383–94.
- Worrell BT, Malik JA, Fokin VV. Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science 2013;340:457–60.
- https://dtp.cancer.gov.
- Marwa Ali A. Fathi AA, Abd El-Hafeez DA, Abbas SH, Montano MM. Mohamed Abdel-Aziz, 1,3,4-oxadiazole/chalcone hybrids: Design, synthesis, and inhibition of leukemia cell growth and EGFR, Src, IL-6 and STAT3 activities. Bioorganic Chem 2019;84:153–60.
- Mekkawy AI, Naguib YW, Alhaj-Suliman SO, et al. Paclitaxel anticancer activity is enhanced by the MEK 1/2 inhibitor PD98059 in vitro and by PD98059-loaded nanoparticles in BRAFV600E melanoma-bearing mice. Int J Pharm 2021;606:120876.
- Popp HD, Brend S, Hofmann W-K, Fabarius A. Immunofluorescence microscopy of γH2AX and 53BP1 for analyzing the formation and repair of DNA double-strand breaks. J Vis Exp 2017;129:e56617.
- Ryu MJ, Chung HS. [10]-Gingerol induces mitochondrial apoptosis through activation of MAPK pathway in HCT116 human colon cancer cells. In Vitro Cell Dev Biol Anim 2015;51:92–101.
- Ahmed FF, Abd El-Hafeez AA, Abbas SH, et al. New 1,2,4-triazole-chalcone hybrids induce caspase-3 dependent apoptosis in A549 human lung adenocarcinoma cells. Eur J Med Chem 2018;151:705–22.
- P. Giannakakou DL, Sackett Y-K, Kang Z, et al. Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem 1997;272:17118–25.
- Sinha S, Field JJ, Miller JH. Use of substitute Nonidet P-40 nonionic detergents in intracellular tubulin polymerization assays for screening of microtubule targeting agents. Biochem Cell Biol 2017;95:379–84.