
Abstract
Herein, a series of novel O-alkyl ferulamide derivatives were designed and synthesised through the multi-target-directed ligands (MTDLs) strategy. The biological activities in vitro showed that compounds 5a, 5d, 5e, 5f, and 5h indicated significantly selective MAO-B inhibitory potency (IC50 = 0.32, 0.56, 0.54, 0.73, and 0.86 μM, respectively) and moderate antioxidant activity. Moreover, compounds 5a, 5d, 5e, 5f, and 5h showed potent anti-inflammatory properties, remarkable effects on self-induced Aβ1-42 aggregation, and potent neuroprotective effect on Aβ1-42-induced PC12 cell injury. Furthermore, compounds 5a, 5d, 5e, 5f, and 5h presented good blood–brain barrier permeation in vitro and drug-like properties. More interesting, the PET/CT images with [11C]5f demonstrated that [11C]5f could penetrate the BBB with a high brain uptake and exhibited good brain clearance kinetic property. Therefore, compound 5f would be a promising multi-functional agent for the treatment of AD.
Graphical Abstract
1. Introduction
Alzheimer’s disease (AD) is first described by Dr. Alois Alzheimer in 1906. It is an irreversible and severe progressive neurodegenerative brain diseaseCitation1. Currently, the approved drugs for treating AD include the N-methyl-D-aspartate receptor antagonist (memantine) and the cholinesterase inhibitors (rivastigmine, doneipezil, and galantamine), however, these drugs cannot effectively prevent or reverse the progression of AD, but they do provide short-term symptomatic reliefCitation2,Citation3.
The pathogenesis of AD is not clear completely, the characterised pathologic brain changes include amyloid-β (Aβ) deposits, neurofibrillary tangles, and low levels of acetylcholine (ACh). Accumulated evidence shows that over 100 targets relate to AD, and many hypotheses are proposed, such as the cholinergic hypothesis, amyloid cascade hypothesis, tau hypothesis, oxidative stress hypothesis, inflammation hypothesis, metal ion hypothesis, and mutation hypothesisCitation4.
Monoamine oxidases A and B (MAO-A and MAO-B) are important FAD-dependent enzymes (flavoenzymes) responsible for the metabolism of neurotransmitters, such as dopamine, serotonin, adrenaline, and noradrenaline and for the inactivation of exogenous arylalkylamines. Increasing evidence suggests that levels of monoamine oxidase-B (MAO-B) activity could increase up to 3-fold in the temporal, parietal, and frontal cortex of AD patients. The excess MAO-B produces hydroxyl radicals, which could increase the former of Aβ plaquesCitation5. Thus, a selective MAO-B inhibitor would be a potential strategy for treating AD. Fortunately, selective MAO-B inhibitor rasagiline has been performed in a clinical trial in people with mild-to-moderate ADCitation6.
The inflammatory hypothesis states that inflammation is considered to be a major factor in AD whereby the beta-amyloid plaques and neurofibrillary tangles co-localize with astrocytes and microglia as well as local immune cells in the brain. In AD, the primary driver of activation of microglia is the accumulation of Aβ. Activated microglia respond to Aβ resulting in migration to the plaques and phagocytosis of Aβ. This results in an accumulation of Aβ and sustained pro-inflammatory cytokine singling beginning to damage neuronsCitation7. In addition, the excessive accumulation of reactive oxygen species (ROS) in patients with AD induces oxidative stress and further aggravates the production and aggregation of Aβ oligomerCitation8. Thus, modulation of inflammation or preventing the formation of the free radicals has been one of the most dynamic areas in the search for new therapeutic targets for AD.
Given the multifactorial aetiology of AD, the development of multi-target-directed ligands (MTDLs) has been considered the best pharmacological option for addressing the progression of ADCitation9–11. These MTDLs possess two or more complementary biological activities, and several candidates with disease-modifying potential are now in the pipeline and have completed clinical trials, such as masitinib mesylate (Angiogenesis inhibitors/Lyn kinase inhibitors/Fibroblast growth factor receptor 3 inhibitors/Signal transduction modulators/KIT inhibitors/Tyrosine kinase inhibitors), leuco methylthioninium (microtubule-associated protein tau aggregation inhibitors/MAO inhibitors/Nitric oxide (NO) production inhibitors), blarcamesine hydrochloride (Signal transduction modulators/Muscarinic M1 receptor agonists/sigma non-opioid intracellular receptor 1 agonists/Drugs acting on NMDA receptors/Drugs acting on sodium channels/Lipid peroxidation inhibitors) and troriluzole hydrochloride (Glutamate release inhibitors/Voltage-gated sodium channel blockers/signal transduction modulators/K(V) 4.3 channel blockers) have reached a testing stage in clinical phase II/III trials ()Citation12.
Figure 1. Candidates have reached testing stage in clinical phase II/III trials.

Ferulic acid (FA) is a polyphenol that is abundant in vegetables and maize bran. Several lines of evidence have displayed that FA would be hopeful for treating AD because of its scavenging free radicals, Aβ aggregation inhibition properties, anti-inflammatory properties, and neuroprotective effectsCitation13,Citation14. However, the low bioavailability of FA limits its clinical uses in AD. Based on this, many groups have developed the FA derivative to treat ADCitation14. Rasagiline, a selective MAO-B inhibitor, which has been approved for treating symptoms of Parkinson’s disease, is in Phase II clinical trial with mild to moderate AD, the data showed that Rasagiline improved blood flow in a separate small study of 11 people with AD treated for a median of 1.7 years with 1 mg per day, along with donepezilCitation6. The propargyl group has been confirmed as the pharmacophore of rasagiline. In addition, evidences show that introducing benzyl derivatives and alkyl fragment into the skeleton could increase MAO-B inhibitory potencyCitation15. Thus, we plan to introduce the propargyl, benzyl, and alkyl fragment into the FA skeleton based on the MTDLs strategy and create novel O-alkyl ferulamide derivatives as multifunctional agents ().
Figure 2. Design strategy of 2-acetylphenol-O-alkylhydroxyethylamine derivatives.

Herein, a series of novel O-alkyl ferulamide derivatives are designed and synthesised. The in vitro biological activities of target compounds are evaluated through MAO-A/MAO-B inhibition, anti-inflammatory property, anti-Aβ aggregation, and neuroprotective effects. Further, the optimised compound is studied in depth by PET-CT imaging.
2. Results and discussion
2.1. Chemistry
The synthetic of novel O-alkyl ferulamide derivatives 5a–5i was described in Scheme 1, the starting material FA (1) was with secondary amine NR1R2 (2a–2c) in the presence of EDCI and HOBt to get key intermediate 3a–3cCitation16, yield from 40–56%. Finally, compounds 3a–3c were treated with halogenated hydrocarbons (R-X) including propargyl bromide (4a), CH3I (4b), benzyl bromide (4c), cyclohexyl bromide (4d), respectively, to obtain the target compounds 5a–5i, yield from 39–76%. All the target compounds were confirmed by 1H NMR and HR-ESI-MS, and most of them were characterised by 13 C NMR.
Scheme 1. Synthesis of 5a–5i. Reagents and conditions: (i) CH2Cl2, EDCI, HOBT, room temperature, overnight, yield 40–56%. (ii) RX (4a–4d), K2CO3, CH3CN, 65 °C, 6–10 h, yield 39–76%.

2.2. huMAO-A and huMAO-B inhibitory activities
The inhibition potency of compounds 5a–5j towards huMAO-A and huMAO-B were evaluated through fluorescence assayCitation17,Citation18. Rasagiline mesylate was also assessed as a positive compound. As listed in , the positive drug rasagiline mesylate showed excellent huMAO-B inhibitory activity (IC50 = 0.03 μM) and good huMAO-A inhibitory activity (IC50 = 0.63 μM), which were consistent with the references. All the target compounds showed good huMAO-B inhibitory potency with IC50 values ranging from 3.8 μM to 0.32 μM, while displayed weak huMAO-A inhibitory activity. Among them, compound 5a presented the best selective MAO-B inhibitory activity (IC50 = 0.32 μM). The results showed that both the secondary amine NR1R2 and alkyl fragment significantly influenced MAO-B inhibitory activity. To explore the inhibition potency of alkyl fragment, firstly NR1R2 was fixed with benzylpiperidine (2a), compound 5a with propargyl fragment showed good MAO-B inhibitory potency with an IC50 value of 0.32 μM. When changing propargyl fragment of 5a with a methyl group or benzyl fragment to get compounds 5b and 5c, respectively, the MAO-B inhibitory activity decreased to 1.1 and 3.8 μM, respectively, suggesting that the propargyl fragment contributed to the MAO-B inhibitory activity, which was consistent with the previous workCitation19. Then, when changing propargyl fragment of 5a with cyclohexyl group to get compound 5d, the MAO-B inhibitory activity slightly decreased to 0.56 μM. Further, when NR1R2 was fixed with 1,2,3,4-tetrahydroisoquinoline fragment, the similar inhibition tendency of alkyl fragment was also observed, such as compound 5e with propargyl (IC50 = 0.54 μM) > compound 5f with methyl (IC50 = 0.73 μM) > compound 5h with cyclohexyl (IC50 = 0.73 μM) > compound 5g with benzyl (IC50 = 3.1 μM). On the other side, the NR1R2 fragment also affected MAO-B inhibitory activity. When the alkyl fragment was propargyl (4a), compound 5a with benzylpiperidine (2a) fragment showed good MAO-B inhibitory activity with an IC50 value was 0.32 μM. When changing benzylpiperidine (2a) fragment with 1,2,3,4-tetrahydroisoquinoline (2b) to get compound 5e, the MAO-B inhibitory activity decreased to 0.54 μM. Subsequently, when the alkyl fragment was methyl, compound 5b with benzylpiperidine showed good MAO-B inhibitory potency (IC50 = 1.1 μM). When changing benzylpiperidine of 5b with 1,2,3,4-tetrahydroisoquinoline to get compound 5f, the MAO-B inhibitory activity slightly increased to 0.73 μM. When changing benzylpiperidine of 5b with benzylpiperazine to get compound 5i, the MAO-B inhibitory activity decreased to 1.9 μM. Overall speaking, compounds 5a, 5d, 5e, 5f, and 5h presented good selective MAO-B inhibitory potency, deserving further study.
Table 1. Inhibitory activity of MAO-A/MAO-B, antioxidant activity and effects on self-induced Aβ1-42 aggregation by target compounds and positive compounds. 
2.3. Kinetic studies for huMAO-B
Compound 5a was selected to perform the kinetic study against huMAO-B using Lineweaver-Burk plots assayCitation20. As displayed in , the Lineweaver-Burk plots for three concentrations of 5a were linear and intersected at y-axis. These results indicated that 5a was a competitive MAO-B inhibitor.
Figure 3. Linewear-Burk plots resulting from the subvelocity curve of the MAO-B activity different substrate concentration in the absence and presence of 5a (0.15, 0.3, and 0.6 μM).

2.4. Molecular modelling study of MAO-B
From the above data, compound 5a, a good selective MAO-B inhibitor, was selected to further explore the binding mode with MAO-B (PDB code: 2V60) through a molecular dockingCitation21. In 5a-MAO-B complex (), the benzene ring of benzylpiperidine interacted with key residue Tyr435 via Pi-Pi interaction. The Tyr435 residue was a key amino acid in the active site of MAO-B (PDB ID: 2v60), in the structure of human MAO-B in complex with the selective inhibitor 7–(3-chlorobenzyloxy)-4-carboxaldehyde-coumarin from PBD database (), the aldehyde group interacted with residue Tyr435 via one intermolecular hydrogen bonding. In addition, showed that the O atom of the carbonyl group interacted with key residue Tyr326 and Gln206 via one intermolecular hydrogen bonding, respectively. Moreover, the CH = CH fragment interacted with key amino acid Tyr326 via one Sigma-Pi interaction. Besides, there were some hydrophobic interactions that could be observed between the ligand 5a and amino acids Leu164, Pro104, Phe103, Ile199, Gln206, Leu171, Phe 343, and Tyr326. Similarly, the hydrophobic interactions were also observed between the ligand and residues Leu164, Pro104, Ile199, Gln206, Leu171, Phe 343, and Tyr326 in . Thus, the observed interactions offered a rational explanation for the high MAO-B inhibitory activity towards 5a.
Figure 4. (A) Compound 5a (green stick) interacted with residues in the binding site of hMAO-B (PDB code: 2V60). (B) The selective inhibitor 7-(3-chlorobenzyloxy)-4-carboxaldehyde-coumarin interacted with residues in the binding site of hMAO-B (PDB code: 2V60).

2.5. Antioxidant activity assay
The antioxidant activity of synthesised compounds 5a–5i was tested by oxygen radical absorbance capacity fluorescein (ORAC-FL) assay, FA was also tested as a referenced compoundCitation22,Citation23. As presented in , FA showed good antioxidant activity with an ORAC value of 1.5 eq. Correspondingly, compounds 5a–5i displayed moderate to good antioxidant potency with ORAC value of 0.2–0.7 eq. Generally speaking, compound 5i (ORAC = 0.7 eq) with benzylpiperazine fragment showed better antioxidant activity than other derivatives. The benzylpiperidine, 1,2,3,4-tetrahydroisoquinoline, and alkyl fragments did not produce a significant influence on the antioxidant activity of the target compounds.
2.6. Anti-inflammatory property
Based on the results of MAO-B inhibitory activity, compounds 5a, 5d, 5e, 5f, and 5h showed good MAO-B inhibitory potency, which was selected to test the anti-inflammatory potency by measuring the production of inflammatory mediators TNF-α and NO in LPS-induced BV-2 cellsCitation21,Citation24.
2.6.1. Cytotoxicity of compounds 5a, 5d, 5e, 5f, and 5h on BV-2 cells
The cytotoxicity of compounds 5a, 5d, 5e, 5f, and 5h were tested using MTT assay, and FA was also tested for comparison. As shown in , the cell viability did not show obvious change after adding compounds 5a, 5d, 5e, 5f, 5h, and FA (5 μM and 10 μM), showing that compounds 5a, 5d, 5e, 5f, 5h, and FA were non-toxic on BV-2 cells at 5 and 10 μM, respectively.
Figure 5. The cell viability of compounds 5a, 5d, 5e, 5f, 5h, and FA on the BV-2 cells were testing using MTT assay. The data were expressed as the mean ± SD through three independent experiments.

2.6.2. Evaluation of NO and TNF-α in LPS-stimulated BV-2 cells
The inhibition of NO production was tested on LPS-induced BV-2 cells through the Griess reaction methodCitation24. As displayed in , the release volume of NO did not produce obvious change after adding tested compounds (5a, 5d, 5e, 5f, 5h, and FA) at the concentration of 5 and 10 μM, respectively, exhibiting that the tested compounds did not produce an effect on the release of NO in BV-2 cells. When BV-2 cells were treated with LPS (1 μg/mL), the release volume of NO remarkably increased. Moreover, when the BV-2 cells were pre-treated with compounds 5a, 5d, 5e, 5f, 5h, and FA at 5 μM, leading to a remarkable reduction of LPS-induced NO production with 32.8, 38.1, 35.1, 41.2, 40.4, and 51.9% inhibition rate, respectively. And when pre-treatment with compounds 5a, 5d, 5e, 5f, 5h, and FA at 10 μM, the inhibition rate significantly improved to 55.7, 59.5, 57.3, 62.5, 64.1, and 75.6%, respectively. In addition, the effects of compounds 5a, 5d, 5e, 5f, and 5h on LPS-induced TNF-α production in BV-2 cells were determined using the enzyme-linked immunosorbent assay (ELISA). As displayed in , when BV-2 cells were exposed to 1.0 μg/mL LPS, the levels of TNF-α significantly increased to 1880 pg/mL (p < 0.01) compared with the untreated group (140 pg/mL). When treatment with tested compounds (5a, 5d, 5e, 5f, 5h, and FA) at 5 μM, respectively, the levels of TNF-α production decreased to 820 pg/mL (p < 0.05), 780 pg/mL (p < 0.01), 805 pg/mL (p < 0.05), 750 pg/mL (p < 0.01), 740 pg/mL (p < 0.01), and 539 pg/mL (p < 0.01), respectively, and the inhibitory rate were 51.7, 54.1, 52.6, 55.9, 56.5, and 68.3%, respectively. When treatment with tested compounds (5a, 5d, 5e, 5f, 5h, and FA) at 10 μM, respectively, the levels of TNF-α production decreased to 520 pg/mL (p < 0.05), 460 pg/mL (p < 0.01), 550 pg/mL (p < 0.05), 455 pg/mL (p < 0.01), 435 pg/mL (p < 0.01) and 234 pg/mL (p < 0.01), respectively. The above results showed that compounds 5a, 5d, 5e, 5f, and 5h could reduce the release of NO and suppress TNF-α production in LPS-induced BV-2 cells, and presented lower anti-neuroinflammatory potency in vitro than the skeleton FA.
Figure 6. Effects of compounds 5a, 5d, 5e, 5f, 5h, and FA on NO release in BV-2 cells and LPS-stimulated BV-2 cells. Data were expressed as mean ± SD through three independent experiments. con. = control; mod. = model. ##p < 0.01 vs. control; **p < 0.01, *p < 0.05 vs. LPS-induced group.

Figure 7. Effects of compounds 5a, 5d, 5e, 5f, 5h, and FA on TNF-α release in LPS-stimulated BV-2 cells. Data were expressed as mean ± SD through three independent experiments. ##p < 0.01 vs. control; ***p < 0.01, **p < 0.01, *p < 0.05 vs. LPS-induced group.

2.7. Effects on self-induced Aβ1 − 42 aggregation
There were two experiments were performed: inhibition experiments and disaggregation experimentsCitation22,Citation25. Based on the above results, the potent compounds 5a, 5d, 5e, 5f, and 5h were selected to test the inhibition effects against self-induced Aβ1 − 42 aggregation by thioflavin T (ThT) fluorescence assay, as well as FA and curcumin acted as positive compounds. As listed in , compounds 5a, 5d, 5e, 5f, and 5h showed significant inhibitory potency against self-induced Aβ1 − 42 aggregation with 52.3, 59.6, 63.5, and 61.7% inhibition rates at 25 μM, respectively, which were better than FA (30.6%) and curcumin (46.1%). Further, the transmission electron microscopy (TEM) images in also supported the results from the ThT assay.
Figure 8. TEM images of Aβ species. (A) inhibition experiments of self-induced Aβ1 − 42 aggregation. (B) disaggregation experiments of self-induced Aβ1 − 42 aggregation.

For the disaggregation experiment, the obtained data in displayed that compounds 5a, 5d, 5e, 5f, and 5h presented potent disaggregation potency with 40.1, 47.2, 43.8, 45.9% disaggregation rates, respectively, which were also supported by TEM images in .
2.8. Neuroprotective effects against Aβ-induced PC12 cell injury
To consolidate the results obtained from the inhibition studies of self-induced Aβ1-42 aggregation, further study was performed to evaluate the neuroprotective effect of compounds 5a, 5d, 5e, 5f, and 5h against PC12 cell injury induced by Aβ1-42 using MTT assayCitation26. Two different concentrations (5 and 10 μM) of each compound were employed in this experiment. As displayed in , when the PC12 cells were treated with 25 μM Aβ1-42 for 48 h, the cell viability decreased to 47.5% (p < 0.01) compared with the control group. When treated with compound 5a, the cell viability was 58.2 and 63.7%, respectively. Similarly, when treating with compounds 5d, 5e, 5f, and 5h, respectively, the cell viability gradually increased in a dose-dependent manner, showing compounds 5a, 5d, 5e, 5f, and 5h presented a potent neuroprotective effect against Aβ1-42-induced PC12 cell injury.
Figure 9. The cell viability (%) of compounds 5a, 5d, 5e, 5f, and 5h on Aβ1-42-induced PC12 cell injury by MTT assay. Percentages of the cell viability were presented as mean ± SD from three independent experiments. ##p < 0.01 vs. untreated group; **p < 0.01, *p < 0.05 vs. Aβ1-42-induced group.

2.9. In vitro blood − brain barrier permeability
The blood-brain barrier (BBB) permeability is an important factor for the development of anti-CNS drugs, herein the ability of compounds 5a, 5d, 5e, 5f, and 5h to access BBB was evaluated by the parallel artificial membrane permeation assay of the blood − brain barrier (PAMPA-BBB)Citation25,Citation27. 11 commercial drugs were employed to verify this assay (). A plot of the experimental data versus the reported values produced a good linear correlation, Pe (exp) = 0.9163Pe (bibl.) − 0.2247 (r2 = 0.9558) (). From this equation, and considering the limit established by Di et al. for blood − brain barrier permeation, the following ranges of permeability Pe (×10−6 cm/s) were established: Pe > 3.44 for compounds with high BBB permeation; 3.44 > Pe > 1.61 showed uncertain BBB permeation; Pe < 1.61 showed weak BBB permeation. As displayed in , compounds 5a, 5d, 5e, 5f, and 5h presented good BBB permeation with 14.7 × 10−6, 18.2 × 10−6, 12.5 × 10−6, 20.4 × 10−6, and 16.7 × 10−6 cm/s permeability, respectively, as similar with testosterone and diazepam, displaying that compounds 5a, 5d, 5e, 5f, and 5h could cross BBB through passive diffusion and deserving further investigation.
Figure 10. Linear correlation between experimental and reported permeability of commercial drugs using the PAMPA-BBB assay. Pe(exp) = 0.9163, Pe(bibl.) −0.2247 (r2 = 0.9558).

Table 2. Permeability (Pe × 10−6 cm/s) in the PAMPA-BBB assay for 11 commercial drugs used in the experiment validation.
Table 3. The predictive permeation of compounds 5a, 5d, 5e, 5f, and 5h by PAMPA-BBB assay.
2.10. Theoretical prediction of the ADME properties
The drug-like properties of compounds 5a, 5d, 5e, 5f, and 5h were evaluated using the Molinspiration property programCitation28. A widely accepted method to predict ADME properties is the Rule of Five proposed by Lipinski in 1997. The items of Rule of Five were that octanol-water partition coefficient (log P) <5, molecular weight (MW) <500, number of hydrogen-bond donors (n-OHNH) <5, number of hydrogen-bond acceptors (n-ON) <10, number of rotatable bonds ≤10, and the rotatable bonds were single bonds that are not adjacent to triple bonds, do not connect hydrogen or halogen atoms and are not included in rings containing <5 single bonds. Topological Polar Surface Area (TPSA), for the CNS drugs, TPSA ≤90 Å2. As demonstrated in , compounds 5a, 5d, 5e, 5f, and 5h complied with the Rule of Five, deserving further investigations.
Table 4. Theoretical prediction of the druglike properties of compounds 5a, 5d, 5e, 5f, and 5h.
2.11. In vivo PET-CT imaging with [11C]5f in mice
To further explore the BBB permeation of the O-alkyl ferulamide derivatives, compound 5f was selected to evaluate using PET-CT imaging in mice.
2.11.1. Preparation of the precursor and radiosynthesis of [11C]5fCitation29,Citation30
Precursor 3b was used as the [11C]5f preparation (Scheme 2). [11C]5f was obtained from the methylation of 3b by reacting with [11C]CH3I in the presence of sodium hydroxide. The radio-desired product was collected by injecting it into the semi-preparative reversed-phase HPLC and was reformulated by loading it onto solid-phase exchange (SPE) C-18 cartridges. The preparation process of [11C]5f was accomplished in 30–40 min after the end of bombardment (EOB), with an overall non-decay corrected radiochemical yields (12–15%) at the end of synthesis (EOS) and high specific activity of 83.2 GBq/μmol (EOB).
Scheme 2. Radiosynthesis of [11C]5f. Reagents and conditions: NaOH, DMF, 100 °C, 3 min.
![Scheme 2. Radiosynthesis of [11C]5f. Reagents and conditions: NaOH, DMF, 100 °C, 3 min.](/cms/asset/497792a6-9bd6-4022-b4ae-2517d7e68ba5/ienz_a_2073442_sch0002_c.jpg)
2.11.2. In vivo PET-CT imaging with [11C]5f in mice
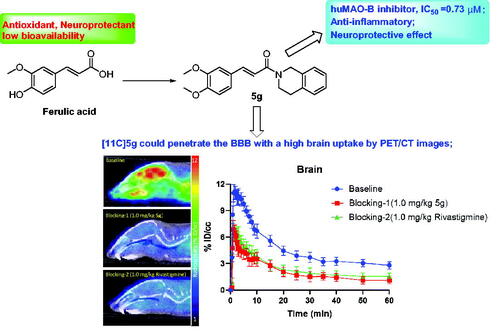
Preliminary rodent PET/CT imaging studies were performed to investigate the in vivo bio-distribution of [11C]5f. 60-min dynamic PET scans were conducted after the tail vein injection of [11C]5f in mice. The radioactivity was presented as the percentage of injected dose per unit volume (% ID/cc). Representative mice brain PET/CT images with [11C]5f are shown in . Encouragingly, the results demonstrated that [11C]5f could penetrate the BBB with a high brain uptake. From the time-activity curve (TAC) analysis in the whole brain, [11C]5f reached a maximum uptake of 11.2%ID/cc in the first few minutes after injection and exhibited good brain clearance kinetic properties during the 60-min scanning time.
Figure 11. PET/CT (baseline and blocking) images in mice brain (20‒60 min) with [11C]5f after intravenous administration (i.v.) and time-activity curve of the whole brain (n = 4).
![Figure 11. PET/CT (baseline and blocking) images in mice brain (20‒60 min) with [11C]5f after intravenous administration (i.v.) and time-activity curve of the whole brain (n = 4).](/cms/asset/16b737c7-c081-4de9-9c0b-da125a11b9e1/ienz_a_2073442_f0011_c.jpg)
Further blocking studies were performed to verify the specific binding of [11C]5f (). The anti-AD drug Rivastigmine approved by FDA was performed. Unlabelled [11C]5f (1.0 mg/kg, self-blocking) and Rivastigmine (1.0 mg/kg) were pre-treated 5 min before the radiotracer administration. Compared with the bassline, a significant radio-uptake decrease (∼35%) in the brain was observed in both 5f and Rivastigmine pre-treated mice, indicating specific binding of [11C]5f.
The in vivo whole-body distribution of [11C]5f was analysed at several scan time points (5, 15, 30, and 60 min). The mean radioactivity uptake in the organs of interest at each time point is presented in the histogram (). The uptake of [11C]5f reached the highest concentration after injection at 5 min in the heart (14.20 ± 0.18%ID/cc), lung (9.5 ± 0.15%ID/cc), and spleen (11.4 ± 0.11%ID/cc) and gradually washed out from these organs. A high accumulation of radioligand was found in the liver and kidney, where the radioactivity steadily increased over time and achieved a maximum of 24.3 ± 0.16%ID/cc and 18.7 ± 0.13%ID/cc, respectively. The relatively high uptake in the liver and kidney indicated that [11C]5f is primarily excreted through the hepatobiliary and urinary pathways.
Figure 12. Left: the PET/CT imaging of the mice with [11C]5f (whole body, 0–60 min); right: biodistribution of [11C]5f in organs of interest at 5, 15, 30, and 60 min after injection of radioligand (n = 4 for each time point). Error bars represent SEM.
![Figure 12. Left: the PET/CT imaging of the mice with [11C]5f (whole body, 0–60 min); right: biodistribution of [11C]5f in organs of interest at 5, 15, 30, and 60 min after injection of radioligand (n = 4 for each time point). Error bars represent SEM.](/cms/asset/59822864-fab2-4db5-8f43-84e952fdaea7/ienz_a_2073442_f0012_c.jpg)
3. Conclusion
AD, accounting for about 70% of all dementia cases, is a chronic, progressive neurodegenerative brain disease in elderly people. There are more than 50 million people living with dementia worldwide and the figure of AD patients will triple by 2050. Accordingly, AD poses a great problem for global health. The current therapeutic agents approved by FDA, including AChE inhibitors (rivastigmine, donepezil, and galantamine) and the NMDA receptor antagonist (menantine), only present modest symptomatic effects and cannot stop, prevent, or reverse the progression of AD. Thus, the development of disease-modifying drugs is a great unmet medical need for AD patients.
Given the complexity of AD, the MTDLs strategy was considered the most promising strategy for the treatment of AD. In this work, based on the MTDLs strategy, a series of novel O-alkyl ferulamide derivatives were designed by introducing propargyl fragments into ferulic acid skeleton. The target compounds were synthesised and evaluated by MAO-A/MAO-B inhibition, antioxidant activity, anti-inflammatory property, anti-Aβ aggregation, and neuroprotective effects. The results in vitro displayed that all the target compounds were highly selective MAO-B inhibitors, and both the secondary amine NR1R2 and alkyl fragment significantly influenced MAO-B inhibitory activity. Among them, compounds 5a, 5d, 5e, 5f, and 5h presented significantly selective MAO-B inhibitory potency with IC50 values of 0.32, 0.56, 0.54, 0.73, and 0.86 μM, respectively. Furthermore, compounds 5a, 5d, 5e, 5f, and 5h indicated potential anti-inflammatory properties by reducing the release of NO and suppressing TNF-α production in LPS-induced BV-2 cells. Compounds 5a, 5d, 5e, 5f, and 5h also showed moderate antioxidants. Moreover, compounds 5a, 5d, 5e, 5f, and 5h remarkably inhibited and disaggregated self-induced Aβ1-42 aggregation, which were supported by TEM images, and displayed a potent neuroprotective effect on Aβ1-42-mediated PC12 cell injury. More importantly, compounds 5a, 5d, 5e, 5f, and 5h presented good blood–brain barrier permeation in vitro and drug-like properties, which was consistent with our design strategy. Further, representative compound 5a was selected to evaluate the BBB permeability in vivo, the PET/CT images analysis with [11C]5f displayed that [11C]5f could penetrate the BBB with a high brain uptake and exhibited good brain clearance kinetic property, suggesting that compound 5f presented good BBB permeability and bioavailability compared with ferulic acid. In general, this work provided an effective strategy to improve the BBB permeability and bioavailability of natural products. Moreover, compound 5f was a potent multi-functional candidate for the treatment of AD and deserved further modification and investigations.
4. Experimental section
4.1. Chemistry
Unless otherwise noted, the reagents required for the chemical synthesis were obtained from Shanghai Titan Scientific Co., Ltd. and were used without purification. All new compounds provided satisfactory 1H NMR and 13 C NMR spectra were recorded on a Varian INOVA spectrometer and used CDCl3 as a solvent, referenced to Tetramethylsilane (TMS). Chemical shifts (δ) are reported in ppm. Splitting patterns are designated as s, single; d, doublet; dd, double-doublet; t, triplet; m, multiplet. The purity of the final synthesised products was evaluated by HPLC analyses which were conducted with a Waters X-Bridge C18 column (4.6 mm × 150 mm, 5 μm) at a flow ratio of 0.8 ml/min. Mobile phase: A: 0.12%TFA in H2O, B: 0.1% TFA in CH3CN. The high-resolution mass spectra were obtained by Waters Xevo G2-XS-Qtof mass spectrometer.
4.1.1. General preparation procedures of 3a–3c
The starting material FA 1 (3 mmol) was dissolved in 20 ml of THF. To this solution, EDCI (4.5 mmol), HOBT (4.5 mmol), and secondary amines 2a–2c (3.5 mmol) were added, respectively, and the reaction mixture was stirred at room temperature overnight. The reaction was monitored through TLC, the solvent was evaporated under reduced pressure after reaction completion. The crude residue was dissolved in 50 ml CH2Cl2, washed with water (2 × 50 ml), saturated NaCl (80 ml), and dried (Na2SO4). The resulting crude was purified by silica gel chromatography (CH2Cl2/acetone = 50:1) to afford the desired 3a–3c, which had been reported in our previous workCitation16.
4.1.1.1. (E)-1-(4-benzylpiperidin-1-yl)-3-(4-hydroxy-3-methoxyphen yl)prop-2-en-1-one (3a)
The starting material FA 1 was treated with secondary amine benzylpiperazine 2a to get intermediate 3a. White solid, yield 51.5%.
4.1.1.2. (E)-1-(3,4-dihydroisoquinolin-2(1H)-yl)-3-(4-hydroxy-3-meth oxyphenyl)prop-2-en-1-one (3b)
The starting material FA 1 was treated with secondary amine 1,2,3,4-tetrahydroisoquinoline 2b to get intermediate 3b. White solid, yield 53.2%.
4.1.1.3. (E)-1-(4-benzylpiperazin-1-yl)-3-(4-hydroxy-3-methoxyphen yl)prop-2-en-1-one (3c)
The starting material FA 1 was treated with secondary amine benzylpiperazine 2d to get intermediate 3c. Light yellow oil, yield 55.9%.
4.1.2. General procedures for the synthesis of target compounds 5a–5i
Alkyl halide 4a–4d (1.3 mmol) was added to a mixture of anhydrous K2CO3 (1.2 mmol), and the key intermediate 3a–3c (1.0 mmol) in 8 ml anhydrous CH3CN. The reaction mixture was heated to 65 °C and stirred for 6–10 h under an argon atmosphere. The reaction was monitored by TLC. On completion of the reaction, the solvent was evaporated under reduced pressure. The crude residue was treated with 30 ml of water and the mixture was extracted with CH2Cl2 (2 × 30 ml). The combined organic phases were washed with saturated NaCl (50 ml), dried over anhydrous Na2SO4, and filtered. The solvent was evaporated under reduced pressure and the residue was purified by silica gel chromatography (petroleum ether/acetone = 50:1) to get compounds 5a–5i.
4.1.2.1. (E)-1-(4-benzylpiperidin-1-yl)-3-(3-methoxy-4-(prop-2-yn-1-yloxy)phenyl)prop-2-en-1-one (5a)
Intermediate 3a was treated with propargyl bromide 4a to get target compound 5a. Light yellow oil, 55.2% yield, 97.5% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 15.3 Hz, 1H, C = CH), 7.29–7.26 (m, 2H, 2 × Ar-H), 7.21 (d, J = 7.2 Hz, 1H, Ar–H), 7.16 − 7.11 (m, 2H, 2 × Ar–H), 7.09 (s, 1H, Ar–H), 7.05 − 6.98 (m, 2H, 2 × Ar–H), 6.77 (d, J = 15.4 Hz, 1H, C = CH), 4.75 (d, J = 1.7 Hz, 2H, OCH2), 4.70–4.67 (m, 1H, 1/2 phCH2), 4.09–4.06 (m, 1H, 1/2 phCH2), 3.88 (s, 3H, OCH3), 3.02 (s, 1H, C≡CH), 2.54–2.52 (m, 3H, NCH2, 1/2NCH2), 2.31–2.29 (m, 1H, 1/2NCH2), 1.81–1.76 (m, 1H, CH), 1.72 (d, J = 13.6 Hz, 2H, CH2), 1.26–1.19 (m, 2H, CH2). 13 C NMR (101 MHz, CDCl3) δ 165.4, 149.7, 148.1, 142.2, 139.9, 129.7, 129.1, 128.3, 126.1, 121.2, 116.0, 115.1, 114.0, 110.6, 78.2, 76.2, 56.7, 56.0, 42.9, 38.3. HR-ESI-MS: Calcd. for C25H27NO3 [M + H]+: 390.2024, found: 390.2066.
4.1.2.2. (E)-1-(4-benzylpiperidin-1-yl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one (5b)
Intermediate 3a was treated with CH3I 4b to get target compound 5b. Light yellow oil, 42.8% yield, 98.1% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 15.6 Hz, 1H, CH = CH), 7.28 (t, J = 7.2 Hz, 2H, 2 × Ar-H), 7.20 (d, J = 7.2 Hz, 1H, Ar-H), 7.14 (d, J = 7.2 Hz, 2H, 2 × Ar-H), 7.09 (d, J = 6.4 Hz, 1H, Ar-H), 7.02 (d, J = 1.6 Hz, 1H, Ar-H), 6.85 (d, J = 8.0 Hz, 1H, Ar-H), 6.75 (d, J = 15.2 Hz, 1H, CH = CH), 4.71–4.69 (m, 1H, 1/2 phCH2), 4.09–4.07 (m, 1H, 1/2 phCH2), 3.91 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.04–3.02 (m, 1H, 1/2 NCH2), 2.63–2.55 (m, 3H, 1/2 NCH2 + NCH2), 1.83–1.77 (m, 1H, CH), 1.74 (d, J = 13.6 Hz, 2H, CH2), 1.28–1.17 (m, 2H, CH2). 13 C NMR (101 MHz, CDCl3) 165.6, 150.4, 149.1, 142.3, 140.0, 129.1, 128.5, 128.3, 126.1, 121.7, 115.4, 111.2, 109.9, 55.9, 46.2, 42.9, 38.4, 32.9, 31.8. HR-ESI-MS: Calcd. for C23H27NO3 [M + H]+: 366.2024, found: 366.2068.
4.1.2.3. (E)-3-(4-(benzyloxy)-3-methoxyphenyl)-1-(4-benzylpiperidin-1-yl)prop-2-en-1-one (5c)
Intermediate 3a was treated with benzyl bromide 4c to get target compound 5c. Light yellow oil, 49.7% yield, 97.4% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 15.2 Hz, 1H, C = CH), 7.41 (d, J = 7.2 Hz, 2H, 2 × Ar–H), 7.36–7.31 (m, 2H, 2 × Ar–H), 7.29–7.23 (m, 4H, 4 × Ar–H), 7.19 (t, J = 7.2 Hz, 1H, Ar–H), 7.13–7.11 (m, 2H, 2 × Ar–H), 7.06–7.01 (m, 2H, 2 × Ar–H), 6.85–6.81 (m, 2H, 2 × Ar–H), 6.74 (d, J = 15.2 Hz, 1H, C = CH), 5.15 (s, 2H, OCH2ph), 4.66–4.65 (m, 1H, 1/2 phCH2), 4.07–4.05 (m, 1H, 1/2 phCH2), 3.89 (s, 3H, OCH3), 3.00–2.99 (m, 1H, 1/2 NCH2), 2.55–2.48 (m, 3H, NCH2 + 1/2 NCH2), 1.81–1.77 (m, 1H, CH), 1.72 (d, J = 13.6 Hz, 2H, CH2), 1.23–1.18 (m, 2H, CH2). 13 C NMR (100 MHz, CDCl3) δ 165.6, 149.7, 149.6, 142.3, 140.0, 139.9, 136.8, 132.7, 129.2, 129.1, 129.0, 128.9, 128.6, 128.4, 128.0, 127.3, 126.1, 121.5, 115.6, 113.7, 110.7, 70.9, 56.1, 46.6, 43.0, 41.3, 38.3, 32.2. HR-ESI-MS: Calcd. for C29H31NO3 [M + H]+: 442.2337, found: 442.2369.
4.1.2.4. (E)-1-(4-benzylpiperidin-1-yl)-3-(4-(cyclohexyloxy)-3-methox yphenyl)prop-2-en-1-one (5d)
Intermediate 3a was treated with cyclohexyl bromide 4d to get target compound 5d. Light yellow oil, 60.2% yield, 97.3% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 15.2 Hz, 1H, C = CH), 7.28 (t, J = 7.2 Hz, 2H, 2 × Ar–H), 7.20 (t, J = 7.2 Hz, 1H, Ar-H), 7.14 (d, J = 7.2 Hz, 2H, 2 × Ar–H), 7.07 (d, J = 6.4 Hz, 1H, Ar-H), 7.02 (s, 1H, Ar-H), 6.83 (d, J = 8.4 Hz, 1H, Ar-H), 6.73 (d, J = 15.2 Hz, 1H, CH = CH), 4.72–4.69 (m, 1H, 1/2 phCH2), 4.10–4.06 (m, 1H, 1/2 phCH2), 3.89 (s, 3H, OCH3), 3.82 (d, J = 6.0 Hz, 2H, OCH2), 3.04–3.03 (m, 1H, 1/2 NCH2), 2.57–2.55 (m, 3H, NCH2 + 1/2 NCH2), 1.91–1.68 (m, 10H, 4 × CH2 + 2 × CH), 1.31–1.18 (m, 4H, 4 × CH2), 1.05–1.02 (m, 2H, CH2). 13 C NMR (101 MHz, CDCl3) δ 165.7, 150.4, 149.6, 142.5, 142.4, 140.0, 129.1, 128.3, 128.2, 126.1, 121.7, 115.1, 114.8, 112.7, 110.8, 74.5, 56.3, 43.0, 38.4, 37.4, 29.9, 26.5, 25.7. HR-ESI-MS: Calcd. for C29H37NO3 [M + H]+: 434.2650, found: 434.2691.
4.1.2.5. (E)-1-(3,4-dihydroisoquinolin-2(1H)-yl)-3-(3-methoxy-4-(pro p-2-yn-1-yloxy)phenyl)prop-2-en-1-one (5e)
Intermediate 3b was treated with propargyl bromide 4a to get target compound 5e. Light yellow oil, 58.2% yield, 98.0% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 15.2 Hz, 1H, C = CH), 7.19–7.12 (m, 5H, 5 × Ar-H), 7.07 (s, 1H, Ar-H), 7.02 (d, J = 8.4 Hz, 1H, Ar-H), 6.84 (d, J = 15.2 Hz, 1H, C = CH), 4.82 (s, 2H, phCH2), 4.78 (d, J = 1.2 Hz, 2H, C≡CHCH2), 3.91 (s, 3H, OCH3), 3.87–3.85 (m, 2H, phCH2), 2.95–3.88 (m, 2H, NCH2), 2.55 (s, 1H, C≡CH). 13 C NMR (100 MHz, CDCl3) 166.0, 149.7, 148.2, 142.6, 129.5, 128.3, 126.9, 126.6, 126.4, 121.3, 116.0, 115.8, 113.9, 110.7, 78.2, 76.2, 56.6, 56.0, 30.9, 29.7. HR-ESI-MS: Calcd. for C22H21NO3 [M + H]+: 348.1555, found: 348.1586.
4.1.2.6. (E)-1-(3,4-dihydroisoquinolin-2(1H)-yl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one (5f)
Intermediate 3b was treated with CH3I 4b to get target compound 5f. Light yellow oil, 76.2% yield, 98.3% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 15.2 Hz, 1H, C = CH), 7.19–7.12 (m, 5H, 5 × Ar-H), 7.06 (s, 1H, Ar-H), 6.86 (d, J = 8.0 Hz, 1H, Ar-H), 6.83 (d, J = 15.6 Hz, 1H, C = CH), 4.83 (s, 2H, phCH2), 3.92 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.89–3.86 (m, 2H, NCH2), 2.95–2.88 (m, 2H, NCH2). 13 C NMR (100 MHz, CDCl3) δ 166.1, 150.6, 149.1, 142.8, 128.3, 126.7, 126.6, 126.6, 121.9, 111.1, 110.0, 56.0, 44.8, 43.6, 29.7. HR-ESI-MS: Calcd. for C20H21NO3 [M + H]+: 324.1555, found: 324.1591.
4.1.2.7. (E)-3-(4-(benzyloxy)-3-methoxyphenyl)-1-(3,4-dihydroisoquinolin-2(1H)-yl)prop-2-en-1-one (5g)
Intermediate 3b was treated with benzyl bromide 4c to get target compound 5g. Light yellow oil, 50.9% yield, 97.8% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 15.2 Hz, 1H, C = CH), 7.34 (d, J = 7.2 Hz, 2H, 2 × Ar-H), 7.28 (t, J = 7.6 Hz, 2H, 2 × Ar-H), 7.22 (d, J = 7.2 Hz, 1H, Ar-H), 7.14–7.00 (m, 4H, 4 × Ar-H), 6.99–6.96 (m, 1H, Ar-H), 6.78 (d, J = 8.8 Hz, 1H, Ar-H), 6.72 (d, J = 15.2 Hz, 1H, Ar-H), 5.09 (s, 2H, phCH2), 4.74 (s, 2H, phCH2), 3.84 (s, 3H, OCH3), 3.79–3.75 (m, 2H, NCH2), 2.86–2.82 (m, 2H, NCH2). HR-ESI-MS: Calcd. for C26H25NO3 [M + H]+: 400.1868, found: 400.1872.
4.1.2.8. (E)-3-(4-(cyclohexyloxy)-3-methoxyphenyl)-1-(3,4-dihydroisoquinolin-2(1H)-yl)prop-2-en-1-one (5h)
Intermediate 3b was treated with cyclohexyl bromide 4d to get target compound 5h. Light yellow oil, 39.2% yield, 97.7% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 15.6 Hz, 1H, C = CH), 7.14–7.10 (m, 4H, 4 × Ar-H), 7.03 (d, J = 8.4 Hz, 1H, Ar-H), 6.99 (d, J = 2.0 Hz, 1H, Ar-H), 6.82 (d, J = 8.0 Hz, 1H, Ar-H), 6.73 (d, J = 15.6 Hz, 1H, C = CH), 4.77 (s, 2H, phCH2), 4.19–4.16 (m, 1H, CH), 3.85–3.79 (m, 5H, OCH3 + phCH2), 2.89–2.86 (m, 2H, NCH2), 1.99–1.95 (m, 2H, CH2), 1.79–1.72 (m, 2H, CH2), 1.55–1.47 (m, 4H, 4 × CH2), 1.27–1.22 (m, 2H, CH2). HR-ESI-MS: Calcd. for C25H29NO3 [M + H]+: 392.2181, found: 392.2216.
4.1.2.9. (E)-1-(4-benzylpiperazin-1-yl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one (5i)
Intermediate 3c was treated with CH3I 4b to get target compound 5i. Light yellow oil, 53.9% yield, 98.3% HPLC purity. 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 15.2 Hz, 1H, CH = CH), 7.33–7.31 (m, 4H, 4 × Ar-H), 7.29–7.26 (m, 1H, Ar-H), 7.10 (dd, J1 = 6.8 Hz, J2 = 1.6 Hz, 1H, Ar-H), 7.02 (d, J = 1.6 Hz, 1H, Ar-H), 6.85 (d, J = 8.4 Hz, 1H, Ar-H), 6.72 (d, J = 15.2 Hz, 1H, CH = CH), 3.91 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.75–3.72 (m, 2H, NCH2), 3.67–3.64 (m, 2H, NCH2), 3.54 (s, 2H, phCH2), 2.49 (t, J = 4.8 Hz, 4H, 2 × NCH2). 13 C NMR (100 MHz, CDCl3) 165.6, 150.5, 149.1, 142.7, 137.6, 129.2, 128.3, 127.3, 121.8, 114.9, 111.1, 109.9, 62.9, 55.9, 52.8. HR-ESI-MS: Calcd. for C22H26N2O3 [M + H]+: 367.1977, found: 367.2006.
4.2. Biological activity experiments
4.2.1. Recombinant human MAO-A and MAO-B inhibition studies
Recombinant human MAO-A and MAO-B were purchased from Sigma-Aldrich and stored at −80 °C. The detailed procedure could reference the previous workCitation18,Citation21. Briefly, tested compounds were prepared in DMSO (2.5 mM) and diluted with potassium phosphate buffer (100 mM, pH 7.40, containing KCl 20.2 mM) to a final volume of 500 μL containing various concentrations of test compounds (0–100 μM) and kynuramine (45 μM for MAO-A and 30 μM for MAO-B). The reactions were initiated by the addition of the enzyme (7.5 μg/mL) and then incubated for 30 min at 37. Then 400 μL NaOH (2 N) and 1000 μL water were added to terminate the enzymatic reactions and the mixtures were centrifuged at 16,000 g for 10 min. The concentrations of the generated 4-hydroxyquinoline were determined by measuring the fluorescence of the supernatant on a Varioskan Flash Multimode Reader (PerkinElmer) with excitation and emission wavelengths at 310 and 400 nm, respectively. IC50 values were calculated from sigmoidal dose-response curves (graphs of the initial rate of kynuramine oxidation versus the logarithm of inhibitor concentration). Each sigmoidal curve was constructed from six different compound concentrations spanning at least three orders of magnitude. Data analyses were carried out with GraphRad Prism 5 employing the one-site competition model. IC50 values were determined in triplicate and expressed as mean ± SD.
4.2.2. Molecular modelling dockingCitation18,Citation21
The crystal structure of human MAO-B in complex with the selective inhibitor 7–(3-chlorobenzyloxy)-4-carboxaldehyde-coumarin (PDB code: 2V60) was obtained from the Protein Data Bank after eliminating the original inhibitors and water molecules. The 3 D Structure of 5a was built and performed geometry optimisation by molecular mechanics. After the addition of Gasteiger charges, removal of hydrogen atoms, the addition of the atomic charges to skeleton atoms, and the assignment of proper atomic types, the further preparation of the inhibitor was accomplished. Docking studies were performed using the AUTODOCK 4.2.6 program. By using Autodock Tools (ADT: version 1.5.6), polar hydrogen atoms were added to amino acid residues, and Gasteiger charges were assigned to all atoms of the enzyme. The resulting enzyme structure was used as an input for the AUTOGRID program. AUTOGRID performed pre-calculated atomic affinity grid maps for each atom type in the ligand. The centre of the grid box was placed with coordinated x = 14.846, y = 128.673, z = 24.971. The dimensions of the active site box were set at 50 × 50 × 50 Å. Flexible ligand docking was performed for the compound. Each docked system was performed by 100 runs of the AUTODOCK search by the Lamarchian genetic algorithm (LGA). A cluster analysis was performed on the docking results using a root mean square (RMS) tolerance of 1.0 and the lowest energy conformation of the highest populated cluster was selected for analysis. Graphic manipulations and visualisations were done by Autodock Tools or Discovery Studio 2.1 software.
4.2.3. Effects on self-mediated Aβ1-42 aggregation
Aβ1-42 was purchased from ChinaPeptides Co., Ltd. The experiments include inhibition experiments and disaggregation experiments. The detailed procedure referenced our previous workCitation25. Briefly, the Aβ1-42 samples were incubated in 50 mM phosphate buffer solution (pH 7.4) at 37 °C for 24 h (final Aβ concentration of 25 μM) with or without the tested compounds (25 μM, final concentration). After incubation, 160 μL of thioflavin T (5 μM in 50 mM glycine-NaOH buffer pH 8.0) was added. Each assay was run in triplicate. Fluorescence was measured with excitation and emission wavelengths at 446 and 490 nm, respectively. The fluorescence intensities were calculated by the expression (1-IFi/IFc) × 100, in which IFi and IFc are the fluorescence intensities obtained for Aβ1-42 in the presence and absence of inhibitors after subtracting the background, respectively.
For the disaggregation of self-induced Aβ fibrils experiment, the Aβ1-42 stock solution was diluted in phosphate buffer solution (pH 7.4). The mixture of the Aβ1-42 (20 μL, 25 μM, final concentration) was incubated at 37 °C for 24 h. The tested compounds (20 μL, 25 μM, final concentration) were then added and incubated at 37 °C for another 24 h. After incubation, 160 μL of 5 μM thioflavin T in 50 mM glycine-NaOH buffer (pH 8.5) was added. Each assay was run in triplicate. The detection method was the same as described above.
4.2.4. Transmission electron microscopy assay
Ten microlitres of the samples, obtained from the ThT assay, were added into a carbon-coated copper/rhodium grid for 2 min. Each grid was incubated with uranyl acetate (1% w/v ddH2O). Upon removal of excess uranyl acetate, the grids were dried for 15 min at room temperature. Images from each sample were taken on a Field Emission Transmission Electron Microscope (JEM-2100F).
4.2.5. Blood − brain barrier permeation assay
The BBB penetration in vitro of compounds was tested through a PAMPA assay. The detailed procedure referenced our previous paperCitation25. Briefly, the commercial drugs were purchased from Sigma and Alfa Aesar. Porcine brain lipid (PBL) was purchased from Avanti Polar Lipids. Both the donor microplate (PVDF membrane, pore size 0.45 mm) and acceptor microplate were obtained from Millipore. The 96-well UV plate (COSTAR) was from Corning Incorporated. The acceptor 96-well microplate was filled with 350 μL of PBS/EtOH (70:30), and the filter membrane was impregnated with 4 μL of PBL in dodecane (20 mg/mL). Compounds were dissolved in DMSO at 5 mg/mL and diluted 50-fold in PBS/EtOH (70:30) to a final concentration of 100 μg/mL. Then 200 μL of the solution was added to the donor wells. The acceptor filter plate was carefully placed on the donor plate to form a sandwich, which was left undisturbed for 18 h at 25 °C. After incubation, the donor plate was carefully removed, and the concentration of compounds in the acceptor wells was determined using the Varioskan Flash Multimode Reader. Every sample was analysed at ten wavelengths in four wells and in at least three independent runs. Pe was calculated using the following expression: Pe = {−VdVa/[(Vd + Va)At]}ln(1 − drugacceptor/drugequilibrium), where Vd is the volume of donor well, Va is the volume in the acceptor well, A is the filter area, t is the permeation time, drug acceptor is the absorbance obtained in the acceptor well, and drug equilibrium is the theoretical equilibrium absorbance. The results are given as the mean ± standard deviation. In the experiment, 11 quality control standards of known BBB permeability were included to validate the analysis set. A plot of the experimental data versus literature values gave a strong linear correlation, Pe(exp) = 0.9163Pe(bibl.) − 0.2247 (R2 = 0.9558). From this equation and the limit established by Di et al. (Pe (bibl.) = 4.0 × 10−6 cm/s) for blood − brain barrier permeation, we concluded that compounds with a permeability >3.44 × 10−6 cm/s could cross the blood − brain barrier.
4.2.6. In vivo PET-CT imaging with [11C]5f in mice
All animal imaging studies were carried out at Massachusetts General Hospital (PHS Assurance of Compliance No. A3596‒01). All mice were socially housed in cages appropriate for the physical and behavioural health of the individual animal and were given unlimited access to food and water, with additional nutritional supplements provided as prescribed by the attending veterinary staff.
4.2.6.1. Radiosynthesis of [11C]5f
The radiosynthesis of [11C]5f referred to our previous work. Briefly, [11C]CH3I was trapped in the solution of precursor 3b (1.0 mg) and NaOH (5.0 mg) in anhydrous DMF (0.3 ml). The mixture was injected into the semi-preparative reversed-phase HPLC (Agilent Eclipse XDB‒C18, 5 μm, 9.4 × 250 mm, eluting with a mobile phase of 38% H2O + 0.1% TFA/62% CH3CN, at the flow rate of 5.0 ml/min). The product-contain collection was reformulated by solid-phase exchange (SPE) C-18 cartridges. The process of [11C]5f preparation was accomplished in 30–40 min after the end of bombardment (EOB), with an overall non-decay corrected radiochemical yields (12–15%, n = 3) at the end of synthesis (EOS) and specific activity of 83.2 GBq/μmol (EOB).
4.2.6.2. Mice PET/CT acquisition and post-processing
Rodent PET/CT imaging studies were conducted in male C57BL/6 mice in groups. Each test group contained 4 mice, anaesthetised with inhalational isoflurane (Patterson Vet Supply, Inc., Greeley, CO, USA) at 2% in a carrier of 2 L/min medial oxygen, and maintained at 1.2% isoflurane for the duration of the imaging scanning. The mice were arranged in a Triumph Trimodality PET/CT/SPECT scanner (Gamma Medica, Northridge, CA, USA). Mice were injected with standard reference or vehicle via a lateral tail vein catheterisation 5-min before the start of PET acquisition for the blocking study. Dynamic PET acquisition lasted for 60 min followed by computed tomography (CT) for anatomic co-registration. Reconstructed PET images were exported from the scanner in DICOM format along with an anatomic CT for analysis. Initial data were imported and analysed using AMIDE softwareCitation31 (an open-source software, Los Angeles, CA, USA) and PMOD (PMOD 4.01, PMOD Technologies Ltd., Zurich, Switzerland).
4.2.6.3. Mice PET‒CT image analysis
Volumes of interest (VOIs) were generated manually in the forms of spheres under the guidance of high‒resolution CT structural images and summed PET data, with a radius no <1 mm to minimise partial volume effects. Time‒activity curves (TACs) were exported as decay‒corrected activity per unit volume. The TACs were expressed as percent injected dose per unit volume for analysis.
Supplemental Material
Download PDF (366.6 KB)Disclosure statement
The authors declare no competing financial interest.
Additional information
Funding
References
- Barnett R. Alzheimer's disease. Lancet 2019;393:1589.
- Citron M. Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov 2010;9:387–98.
- Viña J, Sanz-Ros J. Alzheimer's disease: only prevention makes sense. Eur J Clin Invest 2018;48:e13005.
- Jagust W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci 2018;19:687–700.
- Schedin-Weiss S, Inoue M, Hromadkova L, et al. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Alzheimers Res Ther 2017;9:57.
- Available from: https://www.alzforum.org/therapeutics/rasagiline
- Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement 2016;12:719–32.
- Tamagno E, Guglielmotto M, Vasciaveo V, Tabaton M. Oxidative stress and beta amyloid in Alzheimer's disease. Which comes first: the chicken or the egg? Antioxidants 2021;10:1479.
- de Freitas Silva M, Dias KST, Gontijo VS Jr., et al. Multi-target directed drugs as a modern approach for drug design towards Alzheimer's disease: an update. Curr Med Chem 2018;25:3491–525.
- Cavalli A, Bolognesi ML, Minarini A, et al. Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 2008;51:347–72.
- Zhang P, Xu S, Zhu Z, Xu J. Multi-target design strategies for the improved treatment of Alzheimer's disease. Eur J Med Chem 2019;176:228–47.
- Available from: http://integrity.thomson-pharma.com
- Sgarbossa A, Giacomazza D, di Carlo M. Ferulic acid: a hope for Alzheimer's disease therapy from plants. Nutrients 2015;7:5764–82.
- Nabavi SF, Devi KP, Malar DS, et al. Ferulic acid and Alzheimer's disease: promises and pitfalls. Mini Rev Med Chem 2015;15:776–88.
- Legoabe LJ, Petzer A, Petzer JP. 2-acetylphenol analogs as potent reversible monoamine oxidase inhibitors. Drug Des Devel Ther 2015;9:3635–44.
- Li T, Pan W, Wang K, et al. Novel ferulic acid-donepezil hybrids as multifunctional agents for the treatment of Alzheimer's disease with butyrylcholinesterase, amyloid-β, antioxidant and neuroprotective properties. Lett Drug Des Discov 2017;14:918–29.
- Bai P, Wang K, Zhang P, et al. Development of chalcone-O-alkylamine derivatives as multifunctional agents against Alzheimer's disease. Eur J Med Chem 2019;183:111737.
- Sang Z, Wang K, Zhang P, et al. Design, synthesis, in-silico and biological evaluation of novel chalcone derivatives as multi-function agents for the treatment of Alzheimer's disease. Eur J Med Chem 2019;180:238–52.
- Knez D, Colettis N, Iacovino LG, et al. Stereoselective activity of 1-propargyl-4-styrylpiperidine-like analogues that can discriminate between monoamine oxidase isoforms A and B. J Med Chem 2020;63:1361–87.
- Wang XB, Yin FC, Huang M, et al. Chromone and donepezil hybrids as new multipotent cholinesterase and monoamine oxidase inhibitors for the potential treatment of Alzheimer's disease. RSC Med Chem 2020;11:225–33.
- Tian C, Qiang X, Song Q, et al. Flurbiprofen-chalcone hybrid Mannich base derivatives as balanced multifunctional agents against Alzheimer's disease: design, synthesis and biological evaluation. Bioorg Chem 2020;94:103477.
- Sang Z, Wang K, Shi J, et al. The development of advanced structural framework as multi-target-directed ligands for the treatment of Alzheimer's disease. Eur J Med Chem 2020;192:112180.
- Sang Z, Wang K, Shi J, et al. Apigenin-rivastigmine hybrids as multi-target-directed ligands for the treatment of Alzheimer's disease. Eur J Med Chem 2020;187:111958.
- Cao Z, Yang J, Xu R, et al. Design, synthesis and evaluation of 4′-OH-flurbiprofen-chalcone hybrids as potential multifunctional agents for Alzheimer's disease treatment. Bioorg Med Chem 2018;26:1102–15.
- Sang Z, Wang K, Bai P, et al. Design, synthesis and biological evaluation of novel O-carbamoyl ferulamide derivatives as multi-target-directed ligands for the treatment of Alzheimer's disease. Eur J Med Chem 2020;194:112265.
- Tong Y, Bai L, Gong R, et al. Shikonin protects PC12 cells against β-amyloid peptide-induced cell injury through antioxidant and antiapoptotic activities. Sci Rep 2018;8:26.
- Di L, Kerns EH, Fan K, et al. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem 2003;38:223–32.
- Available from: https://www.molinspiration.com/cgi-bin/properties
- Wang C, Schroeder FA, Hooker JM. Development of new positron emission tomography radiotracer for BET imaging. ACS Chem Neurosci 2017;8:17–21.
- Wang C, Schroeder FA, Wey HY, et al. In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J Med Chem 2014;57:7999–8009.
- Loening AM, Gambhir SS. AMIDE: a free software tool for multimodality medical image analysis. Mol Imaging 2003;2:131–7.