
Abstract
The DNA methyltransferases (DNMTs) were found in mammals to maintain DNA methylation. Among them, DNMT1 was the first identified, and it is an attractive target for tumour chemotherapy. DC_05 and DC_517 have been reported in our previous work, which is non-nucleoside DNMT1 inhibitor with low micromolar IC50 values and significant selectivity towards other S-adenosyl-L-methionine (SAM)-dependent protein methyltransferases. In this study, through a process of similarity-based analog searching, a series of DNMT1 inhibitors were designed, synthesized, and evaluated as anticancer agents. SAR studies were conducted based on enzymatic assays. And most of the compounds showed strong inhibitory activity on human DNMT1, especially WK-23 displayed a good inhibitory effect on human DNMT1 with an IC50 value of 5.0 µM. Importantly, the pharmacokinetic (PK) profile of WK-23 was obtained with quite satisfying oral bioavailability and elimination half-life. Taken together, WK-23 is worth developing as DNMT1-selective therapy for the treatment of malignant tumour.
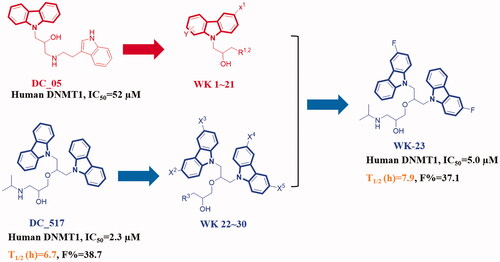
Graphical Abstract
1. Introduction
Epigenetic modification, like DNA methylation, plays a major role in the expression of genetic information. Methylation of DNA at C-5 of cytosine is one of the most studied modifications of the mammalian genomeCitation1. DNMTs (DNA methyltransferases), which include DNMT1, DNMT3A, and DNMT3B, have been identified in humansCitation2. DNMT1 is the most abundant among the three and is responsible for the maintenance of CpG methylation patterns in mammals with hemimethylated CpG dinucleotides serving as preferred substratesCitation3. DNMT1 plays a significant role in the structural modification of chromosomes and the regulation of gene expressionCitation1. The methylation of the 5-carbon on cytosine residues (5mC) in CpG dinucleotides was the first described covalent modification of DNA and is one of the most extensively characterised modifications of chromatin, and thus, DNMT inhibitors have become useful tools for treating cancersCitation4.
So far, two types of DNMT1 inhibitors have been thus found, namely nucleoside analogs and non-nucleoside analogs. Nucleoside analogs, such as the U.S. Food and Drug Administration (FDA) approved 5-azacytidine and 5-aza-2′-deoxycytidineCitation5, or 2-pyrimidone-1-β-D–riboside (zebularine)Citation6, or the dinucleotide derivative SGI-110Citation7, exert their effects by incorporation into DNA inducing substantial DNA methylation inhibition and reactivation of hypermethylated genes. However, these drugs are unstable, show low specificity, and have obvious toxic side effectsCitation8. Therefore, specific concern has been given about non-nucleosides. As shown in , various non-nucleoside analogs have been reported, including the followed natural compounds, such as genisteinCitation9, (-)-epigallocatechin 3-O-gallate (EGCG)Citation10, and curcuminCitation11; repurposed drugs, such as the antihypertensive drug hydralazineCitation12, procainamideCitation13, and 7bCitation14; novel inhibitors, such as phthalimido-L-tryptophan (RG108)Citation15, the quinolone derivative SGI-1027Citation16, and the recently reported selective DNMT1 inhibitor GSK3482364Citation17, and three small molecules identify from a DNMT focussed library including CSC027480404, CSC026286840, and CSC027694519Citation18. Another recent research identified two 3-bromo-3-nitroflavanones 3b and 4a as potent non-nucleoside DNMT inhibitors with good activity and stability targeting DNA methylationCitation19. Compared with nucleoside analogs, non-nucleoside analogs are less likely to be incorporated into DNA and hence provide a relatively safe method to target DNA methylation.
Figure 1. The approved or clinically investigated DNMT inhibitors.

There are two non-nucleoside analogs DC_05 and DC_517 have been reported as specific and highly potent inhibitors of DNMT1 via biochemical and cellular assaysCitation20. With an aim to improve PK properties, and decreased the side effect and toxicity, we designed and synthesised a series of derivatives based on the structures of DC_05 and DC_517. According to the DNMT1 enzyme inhibition assays, we obtained WK-23 as a specific DNMT1 inhibitor with excellent inhibitory activity (IC50 = 5.0 µM). In the further in vivo pharmacokinetic (PK) study, WK-23 displayed a good plasma exposure and an acceptable oral bioavailability of F% = 37.1.
2. Materials and methods
2.1. Chemistry
All anhydrous reactions were performed under a nitrogen atmosphere. The reaction progress was monitored by thin layer-chromatography (TLC) using silica gel F254 plates. Melting points (uncorrected) were determined on an XRC-1 micro melting point apparatus. Infra-red spectra (IR) were recorded on a PerkinElmer Spectrum Two FT-IR instrument. High-resolution mass spectra (HRMS) were taken on a Thermo-Fisher LTQ Orbitrap XL instrument. The 1H and 13 C NMR (nuclear magnetic resonance spectra) experiments were performed by Bruker AM-600 spectrometer using TMS (tetramethylsilane) as the internal standard. Column chromatography was run on 200–300 mesh silica gel from Qingdao Ocean Chemicals (Qingdao, Shandong, China). Unless otherwise indicated, all materials were obtained from commercially available sources and used without further purification.
2.2. General procedures
2.2.1. 2-Chloro-N-(4-fluorophenyl) aniline (a2)
To a mixture of NaOtBu (24.00 g, 250.0 mmol), [HPtBu3][BF4] (1.02 g, 3.5 mmol) and Pd(OAc)2 (560.0 mg, 2.5 mmol) in toluene (200 ml) and stirred to disperse it well. Then added 2-chloraniline (6.40 g, 50.0 mmol) (a1) and 4-bromofluorobenzene (8.75 g, 50.0 mmol). After the addition was completed, the temperature was raised to 110 °C and refluxed for 4 h under a nitrogen atmosphere. After completion of the reaction as indicated by TLC (PE/EA = 20:1) (petroleum ether/ethyl acetate), cool to room temperature, quenched by water (100 ml), extracted with EtOAc (ethyl acetate) (200 ml × 2). The organic layers were combined and washed with brine, dried with anhydrous Na2SO4, and evaporated in vacuo to obtain the crude products of 12.60 g. The crude products were further purified by column chromatography with an eluting system of petroleum ether (100%) to give a2 as a light-yellow liquid (10.30 g) in a 93% yield.
Liquid. HRMS-ESI calcd. for C12H10FNCl [M + H]+ 222.0486, found 222.0533.
1H NMR (600 MHz, DMSO-d6) δ 7.64 (s, 1H), 7.40 (dd, J = 8.0, 1.4 Hz, 1H), 7.17 (td, J = 7.7, 7.2, 1.5 Hz, 1H), 7.15–7.08 (m, 5H), 6.86 (ddd, J = 8.8, 6.3, 1.6 Hz, 1H).
13C NMR (151 MHz, DMSO-d6) δ 157.62 (d, J = 237.3 Hz), 141.33, 139.45 (d, J = 2.2 Hz), 130.43, 128.27, 122.63, 121.54, 121.37 (d, J = 7.8 Hz), 117.85, 116.12 (d, J = 22.3 Hz).
2.2.2. 2-Chloro-4-fluoro-N-(4-fluorophenyl)aniline (b2)
Liquid. HRMS-ESI calcd. for C12H9F2NCl [M + H]+ 240.0392, found 240.0440.
1H NMR (600 MHz, DMSO-d6) δ 7.63 (s, 1H), 7.42 (dd, J = 8.6, 3.0 Hz, 1H), 7.18 (dd, J = 9.0, 5.5 Hz, 1H), 7.13–7.03 (m, 3H), 7.01–6.94 (m, 2H).
13C NMR (151 MHz, DMSO-d6) δ 157.21 (d, J = 236.3 Hz), 156.71 (d, J = 240.6 Hz), 140.35 (d, J = 2.1 Hz), 137.91 (d, J = 2.7 Hz), 124.61 (d, J = 10.6 Hz), 120.79 (d, J = 8.4 Hz), 119.86 (d, J = 7.8 Hz), 117.45 (d, J = 25.6 Hz), 116.11 (d, J = 22.3 Hz), 115.29 (d, J = 21.8 Hz).
2.2.3. General procedure for intermediates a3 and b3
2.2.3.1. 3-Fluoro-9H-carbazole (a3)
To a mixture of NaOtBu (21.87 g, 227.8 mmol), [HPtBu3][BF4] (925.0 mg, 3.2 mmol) and Pd(OAc)2 (510.0 mg, 2.3 mmol) in 1,4-dioxane (250 ml) and stirred to disperse it well. Then added a2 (10.07 g, 45.5 mmol). After the addition was completed, the temperature was raised to 110 °C and refluxed for 4 h under a nitrogen atmosphere. After completion of the reaction as indicated by TLC (PE/EA = 20:1), cool to room temperature and quenched by water (100 ml), extracted with EtOAc (200 ml × 2). The organic layers were combined and washed with brine, dried with anhydrous Na2SO4, and evaporated in vacuo to give the grey black crude products 10.20 g. The crude products were further purified by column chromatography with an eluting system of PE/EA (100:1–20:1). The residue after concentration in vacuo was triturated with petroleum ether, the mixture was filtered to obtain a white solid a3 3.80 g in 45% yield.
m.p. 134–136 °C. HRMS-ESI calcd. for C12H9FN [M + H]+ 186.0719, found 186.0767.
1H NMR (600 MHz, DMSO-d6) δ 11.30 (s, 1H), 8.12 (d, J = 7.8 Hz, 1H), 7.95 (dd, J = 9.4, 2.6 Hz, 1H), 7.53–7.45 (m, 2H), 7.40 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 7.23 (td, J = 9.1, 2.6 Hz, 1H), 7.19–7.09 (m, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.83 (d, J = 232.2 Hz), 141.2, 136.63, 126.57, 123.29 (d, J = 9.8 Hz), 122.62 (d, J = 4.2 Hz), 121.10, 118.89, 113.60 (d, J = 25.3 Hz), 112.22 (d, J = 9.3 Hz), 111.66, 106.17 (d, J = 23.6 Hz).
2.2.3.2. 3,6-Difluoro-9H-carbazole (b3)
m.p. 149–151 °C. HRMS-ESI calcd. for C12H8F2N [M + H]+ 204.0625, found 204.0672.
1H NMR (600 MHz, DMSO-d6) δ 11.34 (s, 1H), 7.97 (dd, J = 9.4, 2.6 Hz, 2H), 7.49 (dd, J = 8.8, 4.4 Hz, 2H), 7.26 (td, J = 9.1, 2.6 Hz, 2H).
13C NMR (151 MHz, DMSO-d6) δ 156.63 (d, J = 232.4 Hz), 137.69, 122.96 (dd, J = 10.0, 4.3 Hz), 114.39 (d, J = 25.5 Hz), 112.62 (d, J = 9.2 Hz), 106.53 (d, J = 23.8 Hz).
2.2.4. General procedure for intermediates a4 and b4
2.2.4.1. 3-Fluoro-9-(oxiran-2-ylmethyl)-9H-carbazole (a4)
At ambient temperature, a3 (2.40 g, 13.0 mmol) was dissolved in DMF (50 ml), followed by the addition of KOH (0.80 g, 14.3 mmol). After 5 min stirring under 0 °C, epichlorohydrin (2.40 g, 26.0 mmol) was added dropwise. Then the mixture was warmed to room temperature and stirred for another 3 h. After a3 was completely consumed, the solution was poured into 20 ml water, and extracted with EtOAc (50 ml × 2). The organic layers were combined and washed with brine, dried with anhydrous Na2SO4, and evaporated in vacuo to give 3.80 g of black liquid crude product. The crude products were further purified by column chromatography with an eluting system of PE/EA (80:1–20:1) to give off brown solid a4 1.90 g in 60% yield.
m.p. 39–41 °C. HRMS-ESI calcd. for C15H13FNO [M + H]+ 242.0981, found 242.1030.
1H NMR (600 MHz, DMSO-d6) δ 8.16 (d, J = 7.8 Hz, 1H), 8.01 (dd, J = 9.2, 2.6 Hz, 1H), 7.70–7.63 (m, 2H), 7.51–7.44 (m, 1H), 7.31 (td, J = 9.2, 2.6 Hz, 1H), 7.24–7.16 (m, 1H), 4.80 (dd, J = 15.8, 3.1 Hz, 1H), 4.42 (dd, J = 15.9, 5.8 Hz, 1H), 3.31 (dq, J = 5.8, 3.1 Hz, 1H), 2.79–2.73 (m, 1H), 2.57 (dd, J = 5.1, 2.6 Hz, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.71 (d, J = 232.9 Hz), 141.22, 136.86, 126.34, 122.60 (d, J = 9.9 Hz), 121.74 (d, J = 4.1 Hz), 120.69, 118.97, 113.26 (d, J = 25.4 Hz), 110.68 (d, J = 9.2 Hz), 109.92, 105.86 (d, J = 23.8 Hz), 50.31, 44.64, 44.35.
2.2.4.2. 3,6-Difluoro-9-(oxiran-2-ylmethyl)-9H-carbazole (b4)
m.p. 121–123 °C. HRMS-ESI calcd. for C15H12F2NO [M + H]+ 260.0887, found 260.0932.
13C NMR (151 MHz, DMSO-d6) δ 156.98 (d, J = 233.3 Hz), 138.22, 122.58 (dd, J = 10.1, 4.1 Hz), 114.53 (d, J = 25.5 Hz), 111.55 (d, J = 9.1 Hz), 106.70 (d, J = 24.0 Hz), 50.79, 45.08, 45.01.
2.2.5. General procedure for intermediates d1, d2, d3
2.2.5.1. 1,3-Bis(3-fluoro-9H-carbazol-9-yl)propan-2-ol (d1)
To solution of a4 (0.90 g, 3.7 mmol) and a3 (0.69 g, 3.7 mmol) in acetone 8 ml was added KOH (0.42 g, 7.5 mmol) and anhydrous Na2SO4 (0.53 g, 3.7 mmol). The resultant mixture was stirred at room temperature overnight. After TLC indicated the total conversion, water was added to quench the reaction and the mixture was washed with EtOAc (20 ml × 2). The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. Flash column chromatography utilising PE/EA (10:1) as the eluting afforded d1.
Intermediates d2 and d3 were prepared like that described for d1.
m.p. 153–155 °C. HRMS-ESI calcd. for C27H21F2N2O [M + H]+ 427.1622, found 427.1670.
1H NMR (600 MHz, DMSO-d6) δ 8.15 (d, J = 7.7 Hz, 2H), 7.99 (dd, J = 9.2, 2.5 Hz, 2H), 7.63 (dd, J = 8.9, 4.3 Hz, 2H), 7.58 (d, J = 8.3 Hz, 2H), 7.43 (t, J = 7.6 Hz, 2H), 7.28 (td, J = 9.2, 2.5 Hz, 2H), 7.18 (t, J = 7.4 Hz, 2H), 5.27 (s, 1H), 4.60–4.46 (m, 4H), 4.40 (s, 1H).
13C NMR (151 MHz, DMSO-d6) δ 157.04 (d, J = 232.9 Hz), 141.86, 137.60, 126.62, 123.01 (d, J = 9.6 Hz), 122.21 (d, J = 4.0 Hz), 121.13, 119.16, 113.56 (d, J = 25.2 Hz), 111.21 (d, J = 9.1 Hz), 110.43, 106.25 (d, J = 23.7 Hz), 69.23, 47.62.
2.2.5.2. 1,3-Bis(3,6-difluoro-9H-carbazol-9-yl) propan-2-ol (d2)
m.p. 207–209 °C. HRMS-ESI calcd. for C27H19F4N2O [M + H]+ 463.1434, found 463.1480.
1H NMR (600 MHz, DMSO-d6) δ 8.01 (dd, J = 9.2, 2.5 Hz, 4H), 7.67 (dd, J = 9.0, 4.3 Hz, 4H), 7.32 (td, J = 9.2, 2.6 Hz, 4H), 5.22 (s, 1H), 4.59 (dd, J = 14.9, 3.3 Hz, 2H), 4.50 (dd, J = 14.9, 8.7 Hz, 2H), 4.35 (s, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.84 (d, J = 232.9 Hz), 138.51, 122.52 (dd, J = 10.0, 4.3 Hz), 114.34 (d, J = 25.4 Hz), 111.65 (d, J = 9.0 Hz), 106.60 (d, J = 23.9 Hz), 69.26, 47.74.
2.2.5.3. 1-(3,6-Difluoro-9H-carbazol-9-yl)-3-(3-fluoro-9H-carbazol-9-yl) propan-2-ol (d3)
m.p. 207–209 °C. HRMS-ESI calcd. for C27H19F4N2O [M + H]+ 463.1434, found 463.1480.
1H NMR (600 MHz, DMSO-d6) δ 8.01 (dd, J = 9.2, 2.5 Hz, 4H), 7.67 (dd, J = 9.0, 4.3 Hz, 4H), 7.32 (td, J = 9.2, 2.6 Hz, 4H), 5.22 (s, 1H), 4.59 (dd, J = 14.9, 3.3 Hz, 2H), 4.50 (dd, J = 14.9, 8.7 Hz, 2H), 4.35 (s, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.84 (d, J = 232.9 Hz), 138.51, 122.52 (dd, J = 10.0, 4.3 Hz), 114.34 (d, J = 25.4 Hz), 111.65 (d, J = 9.0 Hz), 106.60 (d, J = 23.9 Hz), 69.26, 47.74.
2.2.6. 9,9′-(2-(Oxiran-2-ylmethoxy) propane-1,3-diyl) bis (3-fluoro-9H-carbazole) (d4)
Powder KOH (208.0 mg, 3.7 mmol) and anhydrous Na2SO4 (484.0 mg, 3.4 mmol) were added to a d1 solution (1.45 g, 3.4 mmol) in acetone (20 ml) and stirred for 5 min at 0 °C. Epichlorohydrin (3.15 g, 34 mmol) was added dropwise and the resultant mixture was stirred at room temperature for 6 h. Upon completion, water (20 ml) was added to quench the reaction, and the mixture was partitioned between EtOAc and H2O. The aqueous layer was extracted by EtOAc, and the combined organics were washed with saturated aqueous NaCl, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography using PE/EA (50:1–10:1) as the eluent. White solid, yield 73%.
Intermediates d5 and d6 were prepared like that described for d4.
2.2.7. General procedure for preparation of the target compounds (WK-1–WK-11)
2.2.7.1. 1-((2-(1H-indol-3-yl)ethyl)amino)-3-(3-fluoro-9H-carbazol-9-yl)propan-2-ol (WK-1)
A mixture of intermediate compound a4 (120.0 mg, 0.5 mmol, 1 equiv.) and aliphatic amines (318.7 mg, 2.0 mmol, 4 equiv.) dissolved in 5 ml EtOH, was introduced into a 10 ml sealed tube. The mixture was stirred at 60 °C and monitored by TLC until compound a4 was completely consumed. The mixture was extracted with EtOAc and water. The organic layer was combined, washed with saturated NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. Final flash column chromatography utilising PE/EA (1:1) as the eluent afforded 1-((2-(1H-indol-3-yl)ethyl)amino)-3-(3-fluoro-9H-carbazol-9-yl)propan-2-ol (WK-1) as a white solid, 64.0 mg (yield 32.10%). m.p. 137–139 °C. HRMS-ESI calcd. for C25H25FN3O [M + H]+ 402.1982, found 402.1962. Intermediates WK-2–WK-9 were prepared in a procedure similar to that described for WK-1.
IR (KBr), ν, cm−1: 3273, 2930, 1487, 1459, 1439, 1351, 1281, 1170, 889, 795, 727.
m.p. 137–139 °C. HRMS-ESI calcd. for C25H25FN3O [M + H]+ 402.1982, found 402.1962.
1H NMR (600 MHz, DMSO-d6) δ 10.79 (s, 1H), 8.14 (d, J = 7.6 Hz, 1H), 7.97 (d, J = 8.9 Hz, 1H), 7.67–7.55 (m, 2H), 7.52 (d, J = 7.9 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.23 (t, J = 8.1 Hz, 1H), 7.20–7.11 (m, 2H), 7.06 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 5.02 (s, 1H), 4.46 (dd, J = 14.9, 5.0 Hz, 1H), 4.27 (dd, J = 14.8, 6.8 Hz, 1H), 3.98 (s, 1H), 2.83 (dd, J = 22.5, 6.5 Hz, 4H), 2.61 (ddd, J = 44.5, 11.8, 5.5 Hz, 2H), 1.99 (brs, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.48 (d, J = 232.7 Hz), 141.45, 137.15, 136.24, 127.29, 126.08, 122.58, 122.38 (d, J = 9.9 Hz), 121.61 (d, J = 4.4 Hz), 120.79, 120.53, 118.52, 118.30, 118.10, 112.99 (d, J = 25.4 Hz), 112.59, 111.31, 110.72 (d, J = 8.9 Hz), 110.01, 105.62 (d, J = 23.8 Hz), 68.87, 52.98, 50.35, 47.19, 25.57.
2.2.7.2. 1-(3-Fluoro-9H-carbazol-9-yl)-3-((4-fluorophenethyl)amino)propan-2-ol (WK-2)
White solid, 94.0 mg (yield 49%). IR (KBr), ν, cm−1: 3059, 2822, 1603, 1511, 1486, 1465, 1279, 1224, 1168, 890, 743.
m.p. 109–111 °C. HRMS-ESI calcd. for C23H23F2N2O [M + H]+ 381.1778, found 381.1762.
1H NMR (600 MHz, DMSO-d6) δ 8.14 (d, J = 7.7 Hz, 1H), 7.98 (dd, J = 9.3, 2.6 Hz, 1H), 7.59–7.56 (m, 2H), 7.43 (t, J = 8.2 Hz, 1H), 7.29–7.23 (m, 3H), 7.17 (t, J = 7.4 Hz, 1H), 7.09 (t, J = 8.9 Hz, 2H), 5.04 (s, 1H), 4.43 (dd, J = 14.8, 5.0 Hz, 1H), 4.25 (dd, J = 14.8, 6.8 Hz, 1H), 3.95 (t, J = 5.9 Hz, 1H), 2.77–2.67 (m, 4H), 2.57 (ddd, J = 38.6, 5.6 Hz, 2H), 1.99 (brs, 1H).
13C NMR (151 MHz, DMSO-d6) δ 160.67 (d, J = 240.8 Hz), 156.50 (d, J = 232.6 Hz), 141.45, 137.15, 136.64 (d, J = 3.2 Hz), 130.37 (d, J = 8.0 Hz), 126.10, 122.40 (d, J = 9.9 Hz), 121.62 (d, J = 4.1 Hz), 120.57, 118.56, 114.84 (d, J = 20.8 Hz), 113.02 (d, J = 25.4 Hz), 110.72 (d, J = 9.2 Hz), 110.01, 105.67 (d, J = 23.7 Hz), 68.84, 52.93, 51.19, 47.17, 35.08.
2.2.7.3. 4-(2-((3-(3-Fluoro-9H-carbazol-9-yl)-2-hydroxypropyl)amino)ethyl)phenol (WK-3)
White solid, 119.0 mg (yield 48%). IR (KBr), ν, cm−1: 2932, 1597, 1518, 1488, 1465, 1465, 1271, 1245, 1169, 888, 739.
m.p. 140–142 °C. HRMS-ESI calcd. for C23H24FN2O2 [M + H]+ 379.1822, found 379.1802.
1H NMR (600 MHz, DMSO-d6) δ 9.14 (brs, 1H), 8.14 (d, J = 7.7 Hz, 1H), 7.97 (dd, J = 9.2, 2.6 Hz, 1H), 7.60–7.58 (m, 2H), 7.44 (t, J = 7.7 Hz, 1H), 7.26 (td, J = 9.1, 2.7 Hz, 1H), 7.17 (t, J = 7.4 Hz, 1H), 7.00 (d, J = 8.3 Hz, 2H), 6.67 (d, J = 8.4 Hz, 2H), 4.98 (brs, 1H), 4.44 (dd, J = 14.8, 5.0 Hz, 1H), 4.25 (dd, J = 14.8, 6.8 Hz, 1H), 3.96 (p, J = 5.6 Hz, 1H), 2.72–2.63 (m, 2H), 2.63–2.57 (m, 2H), 2.53 (dd, J = 11.9, 6.1 Hz, 1H), 1.88 (s, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.96 (d, J = 232.7 Hz), 155.90, 141.91, 137.61, 130.83, 129.91, 126.57, 122.86 (d, J = 9.9 Hz), 122.08 (d, J = 4.0 Hz), 121.02, 119.01, 115.47, 113.49 (d, J = 25.1 Hz), 111.20 (d, J = 8.9 Hz), 110.49, 106.12 (d, J = 23.7 Hz), 69.25, 53.38, 51.97, 47.64, 35.54.
2.2.7.4. 1-(3-fluoro-9H-carbazol-9-yl)-3-((2-(2-methoxyphenoxy) ethyl)amino) propan-2-ol (WK-4)
White solid, 69.0 mg (yield 41%). IR (KBr), ν, cm−1: 3352, 1591, 1507, 1490, 1467, 1255, 1221, 1124, 1017, 745.
m.p. 55–57 °C. HRMS-ESI calcd. for C24H26FN2O3 [M + H]+ 409.1927, found 409.1901.
1H NMR (600 MHz, DMSO-d6) δ 8.15 (d, J = 7.7 Hz, 1H), 7.98 (dd, J = 9.2, 2.7 Hz, 1H), 7.67–7.61 (m, 2H), 7.44 (t, J = 8.1 Hz, 1H), 7.27 (td, J = 9.1, 2.7 Hz, 1H), 7.18 (t, J = 7.4 Hz, 1H), 6.99–6.93 (m, 2H), 6.92–6.84 (m, 2H), 5.13 (s, 1H), 4.47 (dd, J = 14.8, 5.1 Hz, 1H), 4.29 (dd, J = 14.8, 6.9 Hz, 1H), 4.02 (hept, J = 5.5, 5.0 Hz, 3H), 3.74 (s, 3H), 3.43–3.34 (m, 1H), 2.88 (t, J = 5.6 Hz, 2H), 2.68 (dd, J = 11.8, 4.7 Hz, 1H), 2.60 (dd, J = 11.8, 6.3 Hz, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.52 (d, J = 232.4 Hz), 149.17, 148.06, 141.46, 137.16, 126.14, 122.42 (d, J = 9.9 Hz), 121.65 (d, J = 4.1 Hz), 121.05, 120.72, 120.58, 118.57, 113.61, 113.06 (d, J = 25.4 Hz), 112.20, 110.76 (d, J = 9.2 Hz), 110.02, 105.67 (d, J = 23.7 Hz), 68.82, 68.22, 55.45, 52.90, 48.45, 47.14.
2.2.7.5. 1-((3-Butoxypropyl)amino)-3-(3-fluoro-9H-carbazol-9-yl)propan-2-ol (WK-5)
White solid, 91.0 mg (yield 59%). IR (KBr), ν, cm−1: 3257, 2931, 2862, 1575, 1487, 1466, 1281, 1167, 1111, 887, 738.
m.p. 71–73 °C. HRMS-ESI calcd. for C22H30FN2O2 [M + H]+ 373.2291, found 373.2270.
1H NMR (600 MHz, DMSO-d6) δ 8.15 (d, J = 7.7 Hz, 1H), 7.98 (dd, J = 9.2, 2.6 Hz, 1H), 7.66–7.60 (m, 2H), 7.45 (t, J = 8.3 Hz, 1H), 7.28 (td, J = 9.2, 2.6 Hz, 1H), 7.18 (t, J = 7.4 Hz, 1H), 5.04 (s, 1H), 4.45 (dd, J = 14.8, 4.9 Hz, 1H), 4.27 (dd, J = 14.9, 6.9 Hz, 1H), 3.96 (p, J = 5.6 Hz, 1H), 3.40 (t, J = 6.4 Hz, 2H), 3.33 (t, J = 6.5 Hz, 2H), 2.59–2.51 (m, 4H), 1.86 (s, 1H), 1.64 (p, J = 6.6 Hz, 2H), 1.50–1.41 (m, 2H), 1.30 (h, J = 7.4 Hz, 2H), 0.86 (s, 3H).
13C NMR (151 MHz, DMSO-d6) δ 156.51 (d, J = 232.7 Hz), 141.48, 137.18, 126.09, 122.40 (d, J = 9.9 Hz), 121.64 (d, J = 3.9 Hz), 120.57, 118.55, 113.01 (d, J = 25.3 Hz), 110.75 (d, J = 9.1 Hz), 110.02, 105.67 (d, J = 23.7 Hz), 69.67, 68.83, 68.41, 53.25, 47.26, 46.72, 31.37, 29.79, 18.91, 13.79.
2.2.7.6. 1-(3-Fluoro-9H-carbazol-9-yl)-3-((2-hydroxypropyl)amino)propan-2-ol (WK-6)
White solid, 74.0 mg (yield 56%). IR (KBr), ν, cm−1: 3305, 1586, 1488, 1463, 1170, 950, 791, 743.
m.p. 118–120 °C. HRMS-ESI calcd. for C18H22FN2O2 [M + H]+ 317.1665, found 317.1649.
1H NMR (600 MHz, DMSO-d6) δ 8.16 (d, J = 7.8 Hz, 1H), 7.99 (dd, J = 9.2, 2.7 Hz, 1H), 7.70–7.63 (m, 2H), 7.47 (t, J = 7.5 Hz, 1H), 7.30 (td, J = 9.2, 2.7 Hz, 1H), 7.19 (t, J = 7.4 Hz, 1H), 5.39 (s, 1H), 4.82 (s, 1H), 4.46 (dd, J = 14.9, 5.1 Hz, 1H), 4.34 (dd, J = 14.8, 6.9 Hz, 1H), 4.13 (q, J = 4.6 Hz, 1H), 3.85–3.77 (m, 1H), 2.79 (dd, J = 12.1, 4.0 Hz, 1H), 2.71 (dd, J = 12.1, 7.3 Hz, 1H), 2.62 (dd, J = 12.0, 3.9 Hz, 1H), 2.57–2.51 (m, 1H), 1.06 (d, J = 6.2 Hz, 3H).
13C NMR (151 MHz, DMSO-d6) δ 157.02 (d, J = 232.7 Hz), 141.89, 137.58, 126.62, 122.94 (d, J = 9.8 Hz), 122.15 (d, J = 4.1 Hz), 121.07, 119.12, 113.54 (d, J = 25.4 Hz), 111.25 (d, J = 9.1 Hz), 110.51, 106.18 (d, J = 23.6 Hz), 68.08, 64.54, 56.59, 52.61, 47.50, 21.86.
2.2.7.7. 2-((3-(3-Fluoro-9H-carbazol-9-yl)-2-hydroxypropyl)amino)propan-1-ol (WK-7)
White solid, 100.0 mg (yield 76%). IR (KBr), ν, cm−1: 3415, 2976, 1487, 1468, 1283, 1168, 1052, 885, 856, 742.
m.p. 119–121 °C. HRMS-ESI calcd. for C18H22FN2O2 [M + H]+ 317.1665, found 317.1650.
1H NMR (600 MHz, DMSO-d6) δ 8.15 (d, J = 7.5 Hz, 1H), 7.98 (dd, J = 9.3, 2.6 Hz, 1H), 7.67–7.61 (m, 2H), 7.45 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.29 (td, J = 9.1, 2.6 Hz, 1H), 7.18 (t, J = 7.4 Hz, 1H), 5.07 (d, J = 5.1 Hz, 1H), 4.57 (t, J = 5.4 Hz, 1H), 4.44 (dd, J = 14.9, 4.8 Hz, 1H), 4.28 (dd, J = 14.9, 7.0 Hz, 1H), 3.99–3.89 (m, 1H), 3.30 (dt, J = 9.6, 4.5 Hz, 1H), 3.25–3.18 (m, 1H), 2.67 (dd, J = 11.4, 4.7 Hz, 1H), 2.56 (dt, J = 11.5, 6.3 Hz, 1H), 2.48 (d, J = 7.1 Hz, 1H), 1.91 (s, 1H), 0.87 (d, J = 6.3 Hz, 3H).
13C NMR (151 MHz, DMSO-d6) δ 156.96 (d, J = 232.3 Hz), 141.94, 137.64, 126.55, 122.86 (d, J = 9.6 Hz), 122.09 (d, J = 4.0 Hz), 121.02, 119.01, 113.47 (d, J = 25.1 Hz), 111.29 (d, J = 9.2 Hz), 110.56, 106.12 (d, J = 23.8 Hz), 69.89, 65.95, 55.37, 51.39, 47.95, 17.74.
2.2.7.8. 1-(3-Fluoro-9H-carbazol-9-yl)-3-(((R)-2-phenylpropyl)amino)propan-2-ol (WK-8)
White solid, 83.0 mg (yield 53%). IR (KBr), ν, cm−1: 2960, 1589, 1486, 1462, 1279, 1162, 1099, 884, 793, 700.
m.p. 100–102 °C. HRMS-ESI calcd. for C24H26FN2O [M + H]+ 377.2029, found 377.2006.
1H NMR (600 MHz, DMSO-d6) δ 8.14 (d, J = 7.6 Hz, 1H), 7.97 (dd, J = 9.3, 2.6 Hz, 1H), 7.61–7.53 (m, 2H), 7.47–7.41 (m, 1H), 7.31–7.21 (m, 5H), 7.20–7.15 (m, 2H), 5.01 (s, 1H), 4.42 (dd, J = 14.8, 5.1 Hz, 1H), 4.23 (dd, J = 14.8, 6.7 Hz, 1H), 3.93 (p, J = 5.7 Hz, 1H), 2.87 (h, J = 7.0 Hz, 1H), 2.68 (dd, J = 11.5, 7.1 Hz, 1H), 2.62 (dd, J = 11.5, 7.1 Hz, 1H), 2.56 (dd, J = 11.9, 5.1 Hz, 1H), 2.51 (d, J = 6.7 Hz, 1H), 1.82 (s, 1H), 1.22 (d, J = 7.0 Hz, 3H).
13C NMR (151 MHz, DMSO-d6) δ 156.96 (d, J = 232.7 Hz), 146.26, 141.90, 137.59, 128.73, 127.55, 126.57, 126.45, 122.85 (d, J = 9.5 Hz), 122.07 (d, J = 4.0 Hz), 121.02, 119.01, 113.48 (d, J = 25.4 Hz), 111.17 (d, J = 9.1 Hz), 110.46, 106.12 (d, J = 23.7 Hz), 69.18, 57.33, 53.43, 47.55, 39.73, 20.25.
2.2.7.9. 1-(3-Fluoro-9H-carbazol-9-yl)-3-((4-fluorobenzyl)amino)propan-2-ol (WK-9)
White solid, 84.0 mg (yield 55%). IR (KBr), ν, cm−1: 2924, 1606, 1587, 1516, 1487, 1223, 1171, 889, 790, 740.
m.p. 130–132 °C. HRMS-ESI calcd. for C22H21F2N2O [M + H]+ 367.1622, found 367.1608.
1H NMR (600 MHz, DMSO-d6) δ 8.14 (d, J = 7.8 Hz, 1H), 7.98 (dd, J = 9.3, 2.6 Hz, 1H), 7.61 (dd, J = 8.7, 4.3 Hz, 2H), 7.48–7.41 (m, 1H), 7.36 (dd, J = 8.5, 5.8 Hz, 2H), 7.28 (td, J = 9.1, 2.7 Hz, 1H), 7.18 (t, J = 7.4 Hz, 1H), 7.12 (t, J = 8.9 Hz, 2H), 5.10–4.96 (m, 1H), 4.47 (dd, J = 14.9, 4.6 Hz, 1H), 4.28 (dd, J = 14.9, 7.1 Hz, 1H), 3.99 (s, 1H), 3.74–3.61 (m, 2H), 2.60–2.52 (m, 2H), 2.38 (brs, 1H).
13C NMR (151 MHz, DMSO-d6) δ 161.53 (d, J = 241.6 Hz), 156.96 (d, J = 232.7 Hz), 141.93, 137.63, 137.41 (d, J = 2.8 Hz), 130.29 (d, J = 7.9 Hz), 126.55, 122.86 (d, J = 9.9 Hz), 122.10 (d, J = 4.1 Hz), 121.02, 119.01, 115.26 (d, J = 21.0 Hz), 113.47 (d, J = 25.2 Hz), 111.23 (d, J = 9.1 Hz), 110.51, 106.12 (d, J = 23.7 Hz), 69.37, 53.08, 52.82, 47.81.
2.2.7.10. (2S)-tert-butyl 4-(3-(3-fluoro-9H-carbazol-9-yl)-2-hydroxypropyl)-2-(hydroxymethyl)piperazine-1-carboxylate (WK-10)
A mixture of intermediate compound a4 (200.0 mg, 0.8 mmol, 1 equiv.) and e1 (367 mg, 1.7 mmol, 2.1 equiv.) dissolved in 5 ml ethanol (EtOH), was introduced into a 10 ml sealed tube. The mixture was stirred at 60 °C and monitored by TLC until compound a4 was completely consumed. The mixture was treated with EtOAc and water. The organic layer was washed with saturated NaCl and it was dried over anhydrous Na2SO4 and concentrated in vacuo. Final flash column chromatography utilising dichloromethane/methanol (DCM/MeOH) (20:1) as the eluent afforded (2S)-tert-butyl 4-(3-(3-fluoro-9H-carbazol-9-yl)-2-hydroxypropyl)-2-(hydroxymethyl)piperazine-1-carboxylate (WK-10). White solid, 111.0 mg (yield 29%).
IR (KBr), ν, cm−1: 3387, 1667, 1487, 1465, 1416, 1281, 1166, 1129, 859, 799, 721.
m.p. 71–73 °C. HRMS-ESI calcd. for C25H33FN3O4 [M + H]+ 458.2455, found 458.2461.
1H NMR (600 MHz, DMSO-d6) δ 8.14 (t, J = 6.8 Hz, 1H), 7.98 (ddd, J = 9.2, 6.6, 2.6 Hz, 1H), 7.70–7.58 (m, 2H), 7.45 (dt, J = 16.1, 7.7 Hz, 1H), 7.28 (dtd, J = 20.9, 9.2, 2.6 Hz, 1H), 7.18 (td, J = 7.4, 4.8 Hz, 1H), 5.13–4.60 (m, 2H), 4.49 (dd, J = 14.9, 3.0 Hz, 1H), 4.34–4.23 (m, 1H), 3.99 (dd, J = 77.4, 27.9 Hz, 2H), 3.84–3.67 (m, 2H), 3.47 (s, 1H), 3.23–2.93 (m, 2H), 2.78 (dd, J = 72.6, 7.9 Hz, 1H), 2.47–2.29 (m, 2H), 2.09–1.90 (m, 2H), 1.40 (s, 9H).
13C NMR (151 MHz, DMSO-d6) δ 156.52 (d, J = 232.3 Hz), 141.71, 137.40, 126.09, 122.45 (d, J = 9.7 Hz), 121.68 (d, J = 4.2 Hz), 120.51, 118.53, 112.99 (d, J = 24.7 Hz), 111.06 (d, J = 9.1 Hz), 110.34, 105.60 (d, J = 23.7 Hz), 78.75, 67.63, 66.69, 62.10, 53.88, 53.25, 53.04, 52.18, 47.69, 28.10.
2.2.7.11. 1-(3-Fluoro-9H-carbazol-9-yl)-3-((S)-3-(hydroxymethyl)piperazin-1-yl)propan-2-ol (WK-11)
A mixture of WK-10 (201.0 mg, 0.4 mmol, 1 equiv.) and 2 N hydrogen chloride-1, 4-Dioxane solution 5 ml was introduced into a 10 ml sealed tube. The mixture was stirred at room temperature for 2 h. After WK-10 was completely consumed, the mixture was treated with EtOAc and water. The organic layer was combined, washed with saturated NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. Final flash column chromatography utilising DCM/MeOH (20:1) as the eluent afforded 1-(3-fluoro-9H-carbazol-9-yl)-3-((S)-3-(hydroxymethyl)piperazin-1-yl)propan-2-ol (WK-11). White solid, 120.0 mg (yield 77%).
IR (KBr), ν, cm−1: 3311, 2925, 1629, 1585, 1486, 1463, 1322, 1282, 886, 799, 721.
m.p. 35–37 °C. HRMS-ESI calcd. For C20H25FN3O2 [M + H]+ 358.1931, found 358.1950.
1H NMR (600 MHz, DMSO-d6) δ 8.14 (d, J = 7.7 Hz, 1H), 7.98 (dd, J = 9.2, 2.5 Hz, 1H), 7.69–7.57 (m, 2H), 7.45 (t, J = 7.7 Hz, 1H), 7.29 (td, J = 9.2, 2.6 Hz, 1H), 7.18 (t, J = 7.4 Hz, 1H), 5.31 (s, 1H), 5.12 (s, 1H), 4.45 (dt, J = 14.8, 4.5 Hz, 1H), 4.31 (dd, J = 14.9, 6.7 Hz, 1H), 4.07 (s, 1H), 3.53 (s, 2H), 3.47 (dt, J = 18.3, 5.6 Hz, 1H), 3.13–3.02 (m, 2H), 2.95–2.77 (m, 3H), 2.47–2.34 (m, 2H), 2.30 (q, J = 11.0, 10.0 Hz, 1H), 2.16 (q, J = 12.5, 12.1 Hz, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.54 (d, J = 232.6 Hz), 141.53, 137.23, 126.13, 122.46 (d, J = 9.9 Hz), 121.69 (d, J = 4.1 Hz), 120.60, 118.62, 113.06 (d, J = 25.3 Hz), 110.98 (d, J = 9.0 Hz), 110.22, 105.70 (d, J = 23.7 Hz), 67.16, 61.36, 59.81, 56.09, 54.99, 50.29, 47.53, 42.99.
2.2.7.12. 1-((2-(1H-indol-3-yl)ethyl)amino)-3-(3,6-difluoro-9H-carbazol-9-yl)propan-2-ol (WK-12)
A mixture of intermediate compound b4 (100.0 mg, 0.4 mmol, 1 equiv.) and tryptamine (247.0 mg, 1.5 mmol, 3.75 equiv.) dissolved in 5 ml EtOH, was introduced into a 10 ml sealed tube. The mixture was stirred at 60 °C and monitored by TLC until compound b4 was completely consumed. The mixture was treated with EtOAc and water. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. Final flash column chromatography utilising PE/EA (1:2) as the eluent afforded 1-((2-(1H-indol-3-yl)ethyl)amino)-3-(3,6-difluoro-9H-carbazol-9-yl)propan-2-ol (WK-12). Off-white solid, 63.0 mg (yield 39%). WK-13–WK-19 were prepared in a procedure similar to that described for WK-12.
IR (KBr), ν, cm−1: 3285, 1735, 1492, 1470, 1146, 1124, 854, 797, 787, 732.
m.p. 130–132 °C. HRMS-ESI calcd. for C25H24F2N3O [M + H]+ 420.1887, found 420.1867.
1H NMR (600 MHz, DMSO-d6) δ 10.78 (s, 1H), 7.99 (dd, J = 9.2, 2.7 Hz, 2H), 7.59 (dd, J = 9.0, 4.3 Hz, 2H), 7.52 (d, J = 7.9 Hz, 1H), 7.33 (d, J = 8.1 Hz, 1H), 7.29–7.22 (m, 2H), 7.15 (s, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 5.01 (s, 1H), 4.45 (dd, J = 14.9, 4.8 Hz, 1H), 4.27 (dd, J = 14.9, 6.9 Hz, 1H), 3.95 (s, 1H), 2.90–2.75 (m, 4H), 2.60 (ddd, J = 46.7, 11.8, 5.6 Hz, 2H), 1.88 (brs, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.27 (d, J = 232.7 Hz), 138.04, 136.24, 127.29, 122.60, 121.89 (dd, J = 10.0, 4.2 Hz), 120.79, 118.31, 118.10, 113.78 (d, J = 25.4 Hz), 112.62, 111.32, 111.13 (d, J = 9.0 Hz), 105.99 (d, J = 23.8 Hz), 68.94, 52.92, 50.36, 47.35, 25.58.
2.2.7.13. 4-(2-((3-(3,6-Difluoro-9H-carbazol-9-yl)-2-hydroxypropyl) amino) ethyl) phenol (WK-13)
White solid, 51.0 mg (yield 33%). IR (KBr), ν, cm−1: 2934, 1586, 1494, 1471, 1269, 1247, 1189, 1148, 849, 788.
m.p. 143–145 °C. HRMS-ESI calcd. for C23H23F2N2O2 [M + H]+ 397.1728, found 397.1709.
1H NMR (600 MHz, DMSO-d6) δ 9.14 (s, 1H), 7.99 (dd, J = 9.2, 2.7 Hz, 2H), 7.59 (dd, J = 9.0, 4.3 Hz, 2H), 7.29 (td, J = 9.1, 2.7 Hz, 2H), 7.00 (d, J = 7.9 Hz, 2H), 6.67 (d, J = 7.9 Hz, 2H), 5.01 (s, 1H), 4.43 (dd, J = 14.9, 4.8 Hz, 1H), 4.25 (dd, J = 14.8, 6.9 Hz, 1H), 3.93 (t, J = 5.8 Hz, 1H), 2.72–2.62 (m, 2H), 2.63–2.56 (m, 3H), 2.53 (d, J = 6.2 Hz, 1H), 1.96 (s, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.28 (d, J = 232.9 Hz), 155.41, 138.03, 130.44, 129.44, 121.90 (dd, J = 9.8, 4.2 Hz), 114.99, 113.80 (d, J = 25.5 Hz), 111.14 (d, J = 9.0 Hz), 106.00 (d, J = 23.8 Hz), 68.89, 52.89, 51.54, 47.35, 35.14.
2.2.7.14. 1-(3,6-Difluoro-9H-carbazol-9-yl)-3-((4-fluorophenethyl)amino)propan-2-ol (WK-14)
White solid, 36.0 mg (yield 23%). IR (KBr), ν, cm−1: 2819, 1511, 1491, 1470, 1145, 938, 825, 788.
m.p. 114–116 °C. HRMS-ESI calcd. for C23H22F3N2O [M + H]+ 399.1684, found 399.1658.
1H NMR (600 MHz, DMSO-d6) δ 8.00 (dd, J = 9.2, 2.6 Hz, 2H), 7.58 (dd, J = 9.0, 4.3 Hz, 2H), 7.32–7.24 (m, 4H), 7.10 (t, J = 8.9 Hz, 2H), 5.03 (s, 1H), 4.43 (dd, J = 14.9, 4.8 Hz, 1H), 4.25 (dd, J = 14.9, 6.9 Hz, 1H), 3.93 (t, J = 5.9 Hz, 1H), 2.76–2.68 (m, 4H), 2.60 (dd, J = 11.8, 5.2 Hz, 1H), 2.55–2.52 (m, 1H), 1.92 (s, 1H).
13C NMR (151 MHz, DMSO-d6) δ 161.10 (d, J = 241.1 Hz), 156.72 (d, J = 232.8 Hz), 138.46, 137.08 (d, J = 2.9 Hz), 130.80 (d, J = 7.8 Hz), 122.34 (dd, J = 10.0, 4.3 Hz), 115.26 (d, J = 21.0 Hz), 114.23 (d, J = 25.4 Hz), 111.55 (d, J = 9.0 Hz), 106.46 (d, J = 23.8 Hz), 69.33, 53.30, 51.62, 47.78, 35.52.
2.2.7.15. 1-(3,6-Difluoro-9H-carbazol-9-yl)-3-(phenethylamino)propan-2-ol (WK-15)
White solid, 75.0 mg (yield 51%). IR (KBr), ν, cm−1: 2846, 1585, 1491, 1478, 1298, 1181, 1142, 936, 855, 784.
m.p. 91–93 °C. HRMS-ESI calcd. for C23H23F2N2O [M + H]+ 381.1778, found 381.1757.
1H NMR (600 MHz, DMSO-d6) δ 8.00 (dd, J = 9.2, 2.6 Hz, 2H), 7.59 (dd, J = 9.0, 4.3 Hz, 2H), 7.33–7.26 (m, 4H), 7.25–7.20 (m, 2H), 7.18 (t, J = 7.3 Hz, 1H), 5.03 (s, 1H), 4.43 (dd, J = 14.9, 4.8 Hz, 1H), 4.26 (dd, J = 14.9, 6.9 Hz, 1H), 3.94 (s, 1H), 2.78–2.67 (m, 4H), 2.60 (dd, J = 11.8, 5.2 Hz, 1H), 2.53 (dd, J = 11.8, 6.0 Hz, 1H), 1.87 (s, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.27 (d, J = 232.8 Hz), 140.48, 138.02, 128.41 (d, J = 65.3 Hz), 125.80, 121.89 (dd, J = 10.0, 4.3 Hz), 113.79 (d, J = 25.4 Hz), 111.12 (d, J = 9.1 Hz), 106.01 (d, J = 23.9 Hz), 68.89, 52.87, 51.22, 47.33, 36.02.
2.2.7.16. 1-(3,6-Difluoro-9H-carbazol-9-yl)-3-((2-(2-methoxyphenoxy) ethyl) amino) propan-2-ol (WK-16)
White solid, 77.0 mg (yield 47%). IR (KBr), ν, cm−1: 3375, 1586, 1508, 1493, 1471, 1255, 1221, 1126, 865, 747.
m.p. 93–95 °C. HRMS-ESI calcd. for C24H25F2N2O3 [M + H]+ 427.1833, found 427.1815.
1H NMR (600 MHz, DMSO-d6) δ 8.00 (dd, J = 9.2, 2.6 Hz, 2H), 7.65 (dd, J = 9.0, 4.3 Hz, 2H), 7.30 (td, J = 9.2, 2.6 Hz, 2H), 6.96 (td, J = 7.4, 1.9 Hz, 2H), 6.88 (dtd, J = 17.0, 7.4, 1.8 Hz, 2H), 5.10 (s, 1H), 4.46 (dd, J = 14.9, 4.9 Hz, 1H), 4.29 (dd, J = 14.9, 6.9 Hz, 1H), 4.06–3.96 (m, 3H), 3.73 (s, 3H), 2.87 (t, J = 5.4 Hz, 2H), 2.66 (dd, J = 11.8, 4.9 Hz, 1H), 2.58 (dd, J = 11.8, 6.2 Hz, 1H), 2.23 (s, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.77 (d, J = 232.7 Hz), 149.61, 148.54, 138.51, 122.39 (dd, J = 10.0, 4.3 Hz), 121.48, 121.17, 114.30 (d, J = 25.4 Hz), 114.02, 112.65, 111.63 (d, J = 9.0 Hz), 106.50 (d, J = 23.8 Hz), 69.38, 68.73, 55.90, 53.33, 48.95, 47.76.
2.2.7.17. 1-((3-butoxypropyl)amino)-3-(3,6-difluoro-9H-carbazol-9-yl)propan-2-ol (WK-17)
White solid, 52.0 mg (yield 34%). IR (KBr), ν, cm−1: 3261, 2931, 2863, 2820, 1586, 1492, 1475, 1146, 939, 844, 789.
m.p. 94–96 °C. HRMS-ESI calcd. for C22H29F2N2O2 [M + H]+ 391.2197, found 391.2179.
1H NMR (600 MHz, DMSO-d6) δ 8.00 (dd, J = 9.2, 2.6 Hz, 2H), 7.64 (dd, J = 9.0, 4.3 Hz, 2H), 7.31 (td, J = 9.1, 2.6 Hz, 2H), 5.02 (s, 1H), 4.45 (dd, J = 14.9, 4.7 Hz, 1H), 4.27 (dd, J = 14.9, 6.9 Hz, 1H), 3.94 (p, J = 5.7 Hz, 1H), 3.40 (t, J = 6.4 Hz, 2H), 3.33 (t, J = 6.5 Hz, 3H), 2.59–2.51 (m, 3H), 1.93 (s, 1H), 1.64 (p, J = 6.6 Hz, 2H), 1.51–1.41 (m, 2H), 1.30 (h, J = 7.4 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H).
13C NMR (151 MHz, DMSO-d6) δ 156.76 (d, J = 232.8 Hz), 138.53, 122.38 (dd, J = 10.0, 4.3 Hz), 114.26 (d, J = 25.4 Hz), 111.61 (d, J = 9.0 Hz), 106.50 (d, J = 23.9 Hz), 70.13, 69.39, 68.87, 53.68, 47.89, 47.19, 31.83, 30.29, 19.37, 14.25.
2.2.7.18. 1-(3,6-Difluoro-9H-carbazol-9-yl)-3-((2-hydroxypropyl)amino)propan-2-ol (WK-18)
White solid, 80.0 mg (yield 62%). IR (KBr), ν, cm−1: 3300, 2968, 1585, 1494, 1478, 1147, 1099, 938, 865, 788.
m.p. 130–132 °C. HRMS-ESI calcd. for C18H21F2N2O2 [M + H]+ 335.1571, found 335.1554.
1H NMR (600 MHz, DMSO-d6) δ 8.00 (dd, J = 9.2, 2.7 Hz, 2H), 7.64 (dd, J = 9.0, 4.3 Hz, 2H), 7.32 (td, J = 9.1, 2.7 Hz, 2H), 5.05 (s, 1H), 4.52 (s, 1H), 4.45 (dd, J = 14.9, 4.9 Hz, 1H), 4.28 (dd, J = 14.8, 6.9 Hz, 1H), 3.95 (p, J = 5.5 Hz, 1H), 3.71 (h, J = 6.1 Hz, 1H), 2.57 (dd, J = 11.8, 4.9 Hz, 1H), 2.54–2.51 (m, 1H), 2.41 (d, J = 5.8 Hz, 2H), 2.05 (s, 1H), 1.05 (d, J = 6.2 Hz, 3H).
13C NMR (151 MHz, DMSO-d6) δ 156.76 (d, J = 232.8 Hz), 138.50, 122.38 (dd, J = 10.0, 4.3 Hz), 114.29 (d, J = 25.5 Hz), 111.61 (d, J = 8.9 Hz), 106.51 (d, J = 23.9 Hz), 69.29, 65.70, 57.89, 53.50, 47.76, 22.04.
2.2.7.19. 2-((3-(3,6-Difluoro-9H-carbazol-9-yl)-2-hydroxypropyl)amino)propan-1-ol (WK-19)
White solid, 97.0 mg (yield 75%). IR (KBr), ν, cm−1: 2977, 1490, 1473, 1300, 1177, 1149, 1049, 846, 793, 786.
m.p. 136–138 °C. HRMS-ESI calcd. for C18H21F2N2O2 [M + H]+ 335.1571, found 335.1558.
1H NMR (600 MHz, DMSO-d6) δ 8.00 (dd, J = 9.2, 2.6 Hz, 2H), 7.65 (dd, J = 9.0, 4.3 Hz, 2H), 7.32 (td, J = 9.1, 2.7 Hz, 2H), 5.07 (s, 1H), 4.58 (s, 1H), 4.44 (dd, J = 14.9, 4.5 Hz, 1H), 4.28 (dd, J = 14.9, 7.1 Hz, 1H), 3.92 (s, 1H), 3.30 (dd, J = 10.7, 4.8 Hz, 1H), 3.22 (d, J = 7.4 Hz, 1H), 2.67 (dd, J = 11.4, 4.8 Hz, 1H), 2.55 (h, J = 6.3 Hz, 1H), 2.49–2.43 (m, 1H), 1.97 (s, 1H), 0.88 (d, J = 6.3 Hz, 3H).
13C NMR (151 MHz, DMSO-d6) δ 156.76 (d, J = 232.8 Hz), 138.52, 122.38 (dd, J = 9.9, 4.1 Hz), 114.26 (d, J = 25.4 Hz), 111.70 (d, J = 9.0 Hz), 106.49 (d, J = 23.8 Hz), 69.92, 65.91, 55.37, 51.29, 48.10, 17.71.
2.2.7.20. General procedure for compound WK-20 and WK-21. 1-((2-(1H-indol-3-yl)ethyl)amino)-3-(9H-pyrido[3,4-b]indol-9-yl)propan-2-ol (WK-20)
Powder KOH was added to a 9H-β-carboline solution in N, N-dimethylformamide (DMF) at ambient temperature and stirred for 30 min until dissolved. Epichlorohydrin was added via syringe, and the mixture was stirred at room temperature. Upon completion, the solution was partitioned between EtOAc and H2O. The crude products were used for the next step without further purification. A mixture of crude products (200.0 mg) and tryptamine (aromatic amines) (284.0 mg, 1.8 mmol) dissolved in 5 ml EtOH, was introduced into a 10 ml sealed tube. The mixture was stirred at 60 °C. Upon completion, the mixture was treated with EtOAc and water. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. Finally purified by preparative thin layer chromatography to afford 1-((2-(1H-indol-3-yl)ethyl)amino)-3-(9H-pyrido[3,4-b]indol-9-yl)propan-2-ol (WK-20). Off-white solid, 44.0 mg (yield 27%).
IR (KBr), ν, cm−1: 3051, 2921, 1625, 1454, 1329, 1217, 1033, 742, 730.
m.p. 85–87 °C. HRMS-ESI calcd. for C24H25N4O [M + H]+ 385.2028, found 385.2025.
1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 9.07 (s, 1H), 8.36 (d, J = 5.2 Hz, 1H), 8.26 (d, J = 7.8 Hz, 1H), 8.12 (d, J = 5.2 Hz, 1H), 7.71 (d, J = 8.3 Hz, 1H), 7.62–7.47 (m, 2H), 7.33 (d, J = 8.1 Hz, 1H), 7.27 (t, J = 7.4 Hz, 1H), 7.15 (d, J = 2.2 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 5.14 (s, 1H), 4.57 (dd, J = 14.8, 4.4 Hz, 1H), 4.41 (dd, J = 14.8, 7.1 Hz, 1H), 4.05 (t, J = 5.9 Hz, 1H), 2.87 (s, 4H), 2.78–2.62 (m, 2H).
13C NMR (151 MHz, DMSO) δ 138.71, 133.84, 128.52, 123.15, 122.19, 121.35, 119.82, 118.77, 118.66, 114.82, 111.84, 111.10, 68.98, 52.95, 50.38, 47.77, 26.81, 25.42.
2.2.7.21. 1-((2-(1H-indol-3-yl)ethyl)amino)-3-(9H-pyrido[2,3-b]indol-9-yl)propan-2-ol (WK-21)
Off-white solid, 16.0 mg (yield 17%). IR (KBr), ν, cm−1: 3359, 3293, 2920, 1592, 1573, 1464, 1415, 1213, 772, 732.
m.p. 107–109 °C. HRMS-ESI calcd. for C24H25N4O [M + H]+ 385.2028, found 385.2020.
1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 8.56 (d, J = 7.6 Hz, 1H), 8.45 (d, J = 4.5 Hz, 1H), 8.21 (d, J = 7.7 Hz, 1H), 7.75 (d, J = 8.3 Hz, 1H), 7.52 (t, J = 8.5 Hz, 2H), 7.34 (d, J = 8.1 Hz, 1H), 7.31–7.21 (m, 2H), 7.16 (d, J = 2.2 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 5.62 (s, 1H), 4.50 (d, J = 5.7 Hz, 2H), 4.33 (s, 1H), 2.95 (ddt, J = 27.8, 21.8, 9.8 Hz, 6H).
13C NMR (151 MHz, DMSO) δ 138.71, 133.84, 128.52, 123.15, 122.19, 121.35, 119.82, 118.77, 118.66, 114.82, 111.84, 111.10, 68.98, 52.95, 50.38, 47.77, 26.81, 25.42.
2.2.7.22. 1-((1,3-Bis(3,6-difluoro-9H-carbazol-9-yl)propan-2-yl)oxy)-3-(isopropylamino)propan-2-ol (WK-22)
A mixture of intermediate compound d5 (330.0 mg, 0.6 mmol, 1 equiv.) and tryptamine (aromatic amines) (375.0 mg, 6.4 mmol, 10.7 equiv.) dissolved in 5 ml isopropyl alcohol, was introduced into a 10 ml sealed tube. The mixture was stirred at 60 °C and monitored by TLC until compound d5 was completely consumed. The mixture was treated with EtOAc and H2O. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. Final flash column chromatography utilising EA/MeOH (20:1) as the eluent afforded 1-((1, 3-bis(3,6-difluoro-9H-carbazol-9-yl)propan-2-yl)oxy)-3-(isopropylamino)propan-2-ol (WK-22). White solid, 78.0 mg (yield 21%).
IR (KBr), ν, cm−1: 2982, 1622, 1547, 1489, 1298, 1181, 1169, 1145, 850, 790.
m.p. 89–91 °C. HRMS-ESI calcd. for C33H32F4N3O2 [M + H]+ 578.2431, found 578.2413.
1H NMR (600 MHz, DMSO-d6) δ 8.11–7.96 (m, 4H), 7.71 (ddd, J = 19.5, 9.0, 4.2 Hz, 4H), 7.40–7.27 (m, 4H), 4.68–4.58 (m, 4H), 4.20 (td, J = 7.6, 4.0 Hz, 1H), 2.71 (dp, J = 8.0, 3.8 Hz, 1H), 2.39 (dd, J = 9.4, 4.5 Hz, 1H), 2.24 (q, J = 7.4, 5.9 Hz, 2H), 1.85 (s, 2H), 1.71 (dd, J = 12.0, 3.4 Hz, 1H), 1.35 (dd, J = 11.9, 8.2 Hz, 1H), 0.73 (dd, J = 27.3, 6.3 Hz, 6H).
13C NMR (151 MHz, DMSO-d6) δ 172.71, 157.22, 155.68, 137.82 (d, J = 6.1 Hz), 122.12 (dd, J = 6.5, 3.5 Hz), 114.00 (d, J = 25.4 Hz), 111.12 (d, J = 9.0 Hz), 106.27 (d, J = 23.5 Hz), 77.75, 73.83, 67.16, 48.84, 48.08, 45.39, 45.35, 21.83, 21.74, 21.42.
2.2.7.23. General methods for preparation of target compounds (WK-23–WK-25). 1-((1,3-bis(3-fluoro-9H-carbazol-9-yl)propan-2-yl)oxy)-3-(isopropylamino)propan-2-ol (WK-23)
A mixture of intermediate compound d4 (460.0 mg, 1.0 mmol, 1 equiv.) and tryptamine (590.0 mg, 10.0 mmol, 10 equiv.) dissolved in 5 ml isopropyl alcohol, was introduced into a 10 ml sealed tube. The mixture was stirred at 60 °C and monitored by TLC until compound d4 was completely consumed. The mixture was treated with EtOAc and H2O. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. Final flash column chromatography utilising EA/MeOH (20:1) as the eluent afforded 1-((1,3-bis(3-fluoro-9H-carbazol-9-yl)propan-2-yl)oxy)-3-(isopropylamino)propan-2-ol (WK-23). White solid, 86.0 mg (yield 17%).
White solid, 86.0 mg (yield 17%). IR (KBr), ν, cm−1: 3325, 2978, 1588, 1490, 1469, 1286, 1169, 1052, 879, 741.
m.p. 195–196 °C. HRMS-ESI calcd. for C33H34F2N3O2 [M + H]+ 541.2619, found 542.2587.
1H NMR (600 MHz, DMSO-d6) δ 8.18 (d, J = 7.7 Hz, 2H), 8.11 (brs, 1H), 8.02 (dt, J = 9.2, 2.8 Hz, 2H), 7.74–7.64 (m, 4H), 7.48 (qd, J = 7.2, 1.2 Hz, 2H), 7.36–7.29 (m, 2H), 7.21 (td, J = 7.4, 4.2 Hz, 2H), 5.00 (brs, 1H), 4.77–4.58 (m, 4H), 4.27 (tt, J = 8.2, 4.2 Hz, 1H), 3.18 (tt, J = 10.1, 7.4, 3.3 Hz, 1H), 2.68 (p, J = 6.5 Hz, 1H), 2.61 (dd, J = 9.4, 4.4 Hz, 1H), 2.36 (dd, J = 9.4, 7.7 Hz, 1H), 2.03–1.98 (m, 1H), 1.61 (dd, J = 12.6, 9.8 Hz, 1H), 1.01 (d, J = 6.5 Hz, 3H), 0.92 (d, J = 6.5 Hz, 3H).
13C NMR (151 MHz, DMSO-d6) δ 156.67 (d, J = 233.3 Hz), 141.20 (d, J = 3.9 Hz), 136.90 (d, J = 9.9 Hz), 126.39, 122.61 (dd, J = 9.7, 4.6 Hz), 121.76 (dd, J = 6.1, 3.8 Hz), 120.81 (d, J = 3.6 Hz), 118.97, 113.36 (d, J = 25.4 Hz), 110.76 (d, J = 8.9 Hz), 109.92 (d, J = 2.9 Hz), 105.98 (dd, J = 24.0, 5.4 Hz), 77.98, 73.17, 64.81, 49.52, 46.79, 45.16, 45.04, 18.67, 17.92.
2.2.7.24. 1-((1,3-Bis(3-fluoro-9H-carbazol-9-yl)propan-2-yl)oxy)-3-(cyclopropylamino)propan-2-ol (WK-24)
White solid, 69.0 mg (yield 51%). IR (KBr), ν, cm−1: 2929, 1629, 1585, 1486, 1463, 1323, 1282, 1167, 797, 743, 720.
m.p. 57–59 °C. HRMS-ESI calcd. for C33H32F2N3O2 [M + H]+ 540.2463, found 540.2451.
1H NMR (600 MHz, DMSO-d6) δ 8.16 (dd, J = 7.8, 2.9 Hz, 2H), 8.00 (dt, J = 9.1, 2.8 Hz, 2H), 7.68 (ddd, J = 19.9, 9.9, 6.1 Hz, 3H), 7.62 (d, J = 8.3 Hz, 1H), 7.48–7.42 (m, 2H), 7.30 (td, J = 9.0, 2.0 Hz, 2H), 7.19 (td, J = 7.4, 4.6 Hz, 2H), 4.71–4.58 (m, 4H), 4.20 (tt, J = 8.3, 4.1 Hz, 1H), 3.99 (s, 1H), 2.74 (dt, J = 7.6, 3.8 Hz, 1H), 2.46 (dd, J = 9.5, 4.8 Hz, 1H), 2.32 (dd, J = 9.5, 7.3 Hz, 1H), 1.84 (dd, J = 12.1, 3.8 Hz, 1H), 1.58 (tt, J = 6.8, 3.6 Hz, 1H), 1.52 (dd, J = 12.2, 7.8 Hz, 1H), 0.17 (dtt, J = 14.1, 10.7, 5.0 Hz, 2H), −0.03–-0.13 (m, 2H).
13C NMR (151 MHz, DMSO-d6) δ 156.68 (d, J = 232.9 Hz), 141.20 (d, J = 11.5 Hz), 136.98 (d, J = 5.6 Hz), 126.31, 122.62 (d, J = 9.8 Hz), 121.80 (d, J = 4.3 Hz), 120.81, 118.92, 113.26 (d, J = 25.2 Hz), 110.78 (dd, J = 8.9, 2.7 Hz), 109.94 (d, J = 4.8 Hz), 105.95 (d, J = 23.7 Hz), 77.98, 73.99, 67.50, 51.66, 45.26, 29.84, 5.92, 5.66.
2.2.7.25. 1-((1,3-Bis(3-fluoro-9H-carbazol-9-yl)propan-2-yl)oxy)-3-(tert-butylamino)propan-2-ol (WK-25)
White solid, 26.0 mg (yield 19%). IR (KBr), ν, cm−1: 3353, 2779, 1584, 1488, 1323, 1283, 1167, 857, 797, 743, 721.
m.p. 183–185 °C. HRMS-ESI calcd. for C34H36F2N3O2 [M + H]+ 556.2776, found 556.2768.
1H NMR (600 MHz, DMSO-d6) δ 8.18 (d, J = 7.8 Hz, 2H), 8.02 (dt, J = 9.2, 2.2 Hz, 2H), 7.74–7.63 (m, 4H), 7.46 (q, J = 8.1 Hz, 2H), 7.32 (qd, J = 9.0, 2.6 Hz, 2H), 7.20 (q, J = 7.4 Hz, 2H), 5.04 (brs, 1H), 4.66 (qd, J = 15.3, 6.1 Hz, 4H), 4.29 (tt, J = 8.1, 4.1 Hz, 1H), 3.18 (s, 1H), 2.64 (dd, J = 9.3, 4.2 Hz, 1H), 2.46–2.41 (m, 1H), 2.15 (d, J = 11.4 Hz, 1H), 1.62 (t, J = 10.8 Hz, 1H), 1.21 (s, 1H), 0.98 (s, 9H).
13C NMR (151 MHz, DMSO-d6) δ 156.70 (d, J = 232.7 Hz), 141.22 (d, J = 7.2 Hz), 136.97 (d, J = 11.1 Hz), 126.46, 122.65 (d, J = 9.9 Hz), 121.82 (d, J = 2.2 Hz), 120.90 (d, J = 8.9 Hz), 119.02, 113.44 (dd, J = 25.2, 3.2 Hz), 110.79 (t, J = 9.8 Hz), 109.93 (d, J = 5.1 Hz), 106.05 (dd, J = 23.7, 10.4 Hz), 78.01, 73.19, 65.50, 45.21, 45.16, 43.89, 25.06, 0.15.
2.2.7.26. General methods for preparation of target compounds (WK-26, WK-2 8–30). (2R)-tert-butyl 4-(3-((1-(3,6-difluoro-9H-carbazol-9-yl)-3-(3-fluoro-9H-carbazol-9-yl)propan-2-yl)oxy)-2-hydroxypropyl)-2-(hydroxymethyl)piperazine-1-carboxylate (WK-26)
A mixture of intermediate compound d6 (100.0 mg, 0.2 mmol, 1 equiv.) and e1 (86.0 mg, 0.4 mmol, 2 equiv.) dissolved in 5 ml isopropyl alcohol, was introduced into a 10 ml sealed tube. The mixture was stirred at 60 °C and monitored by TLC until compound d4 was completely consumed. The mixture was treated with EtOAc and H2O. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. Final flash column chromatography utilising PE/EA (1:1) as the eluent afforded (2 R)-tert-butyl 4-(3-((1-(3,6-difluoro-9H-carbazol-9-yl)-3-(3-fluoro-9H-carbazol-9-yl)propan-2-yl)oxy)-2-hydroxypropyl)-2-(hydroxymethyl)piperazine-1-carboxylate (WK-26). White solid, 42.0 mg (yield 29%).
IR (KBr), ν, cm−1: 3403, 2932, 1676, 1488, 1465, 1167, 1147, 1120, 857, 793, 745.
m.p. 74–76 °C. HRMS-ESI calcd. for C40H44F3N4O5 [M + H]+ 717.3264, found 717.3263.
1H NMR (600 MHz, DMSO-d6) δ 8.16 (t, J = 7.3 Hz, 1H), 8.06–7.97 (m, 3H), 7.75–7.60 (m, 4H), 7.46 (dt, J = 15.9, 7.7 Hz, 1H), 7.32 (dtd, J = 26.0, 9.2, 2.6 Hz, 3H), 7.19 (q, J = 7.1 Hz, 1H), 4.76–4.39 (m, 5H), 4.23 (dt, J = 8.0, 4.1 Hz, 1H), 3.84–3.59 (m, 2H), 3.42 (d, J = 8.1 Hz, 1H), 3.34–3.24 (m, 1H), 3.21 (s, 1H), 2.79 (s, 1H), 2.59–2.51 (m, 1H), 2.43–2.31 (m, 2H), 2.00–1.87 (m, 1H), 1.67–1.44 (m, 1H), 1.43–1.31 (m, 11H), 1.29–1.18 (m, 2H).
13C NMR (151 MHz, DMSO-d6) δ 156.69 (d, J = 232.8 Hz), 156.52 (d, J = 233.2 Hz), 141.27, 137.83, 137.01, 126.35 (d, J = 6.0 Hz), 122.64 (d, J = 10.0 Hz), 122.19 (dd, J = 10.1, 4.2 Hz), 121.82, 120.79, 118.91, 114.12 (d, J = 25.5 Hz), 113.27 (d, J = 25.0 Hz), 111.23 (d, J = 9.4 Hz), 110.75 (d, J = 9.2 Hz), 109.97 (d, J = 11.3 Hz), 106.35 (d, J = 23.8 Hz), 105.93 (d, J = 23.8 Hz), 78.54, 77.89, 73.97, 73.65, 66.09, 60.26, 58.68, 54.97, 52.87, 51.95, 45.30, 28.08.
2.2.7.27. 1-((1-(3,6-Difluoro-9H-carbazol-9-yl)-3-(3-fluoro-9H-carbazol-9-yl) propan-2-yl)oxy)-3-((R)-3-(hydroxymethyl)piperazin-1-yl)propan-2-ol (WK-27)
A mixture of WK-26 (207.0 mg, 0.3 mmol, 1 equiv.) and 2 N hydrogen chloride-1, 4-Dioxane solution 5 ml was introduced into a 10 ml sealed tube. The mixture was stirred at room temperature and monitored by TLC until WK-26 was completely consumed. The mixture was extracted with EtOAc and H2O, the organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. Final flash column chromatography utilising DCM/MeOH (15:1) as the eluent afforded 1-((1-(3,6-difluoro-9H-carbazol-9-yl)-3-(3-fluoro-9H-carbazol-9-yl)propan-2-yl)oxy)-3-((R)-3-(hydroxymethyl)piperazin-1-yl)propan-2-ol (WK-27). White solid, 123.0 mg (yield 69%).
IR (KBr), ν, cm−1: 3343, 2933, 1585, 1488, 1465, 1167, 1146, 1120, 858, 792, 746.
m.p. 150–152 °C. HRMS-ESI calcd. for C35H36F3N4O3 [M + H]+ 617.2740, found 617.2728.
1H NMR (600 MHz, DMSO-d6) δ 9.18 (brs, 1H), 8.71 (brs, 1H), 8.17 (d, J = 7.7 Hz, 1H), 8.02 (ddd, J = 18.9, 9.1, 2.3 Hz, 3H), 7.78–7.63 (m, 4H), 7.52–7.40 (m, 1H), 7.39–7.27 (m, 3H), 7.19 (t, J = 7.5 Hz, 1H), 5.38 (dt, J = 8.9, 4.6 Hz, 1H), 4.64 (dt, J = 11.1, 5.6 Hz, 4H), 4.22 (h, J = 5.8 Hz, 1H), 3.95 (t, J = 5.2 Hz, 1H), 3.51–3.45 (m, 1H), 2.91 (d, J = 11.7 Hz, 1H), 2.81 (s, 1H), 2.70 (s, 1H), 2.61–2.52 (m, 1H), 2.48–2.37 (m, 1H), 2.34–2.09 (m, 3H), 1.94–1.62 (m, 2H), 1.50–1.36 (m, 1H), 1.22 (d, J = 12.6 Hz, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.67 (d, J = 233.0 Hz), 156.48 (d, J = 233.5 Hz), 141.33, 137.84, 137.04, 126.37, 122.64 (d, J = 9.8 Hz), 122.13 (dd, J = 10.0, 4.3 Hz), 121.80 (d, J = 4.1 Hz), 120.86, 118.95, 114.12 (d, J = 25.6 Hz), 113.30 (d, J = 24.9 Hz), 111.28 (d, J = 9.0 Hz), 110.86 (d, J = 8.4 Hz), 110.05, 106.39 (d, J = 23.8 Hz), 106.01 (d, J = 23.8 Hz), 77.86, 73.41, 66.57, 59.62, 59.19, 55.84, 51.56, 49.34, 45.44, 45.28, 42.52.
2.2.7.28. 1-((2-(1H-indol-3-yl)ethyl)amino)-3-((1-(3,6-difluoro-9H-carbazol-9-yl)-3-(3-fluoro-9H-carbazol-9-yl)propan-2-yl)oxy)propan-2-ol (WK-28)
White solid, 72.0 mg (yield 55%). IR (KBr), ν, cm−1: 2928, 1585, 1488, 1463, 1281, 1168, 1146, 1120, 857, 792, 744.
m.p. 113–115 °C. HRMS-ESI calcd. for C40H36F3N4O2 [M + H]+ 661.2790, found 661.2784.
1H NMR (600 MHz, DMSO-d6) δ 11.03 (s, 1H), 8.34 (s, 1H), 8.11 (d, J = 7.8 Hz, 1H), 8.05–7.92 (m, 3H), 7.79–7.62 (m, 4H), 7.53 (d, J = 7.9 Hz, 1H), 7.48–7.41 (m, 1H), 7.42–7.29 (m, 4H), 7.18 (d, J = 2.0 Hz, 1H), 7.13 (dt, J = 14.2, 7.3 Hz, 2H), 7.06–7.00 (m, 1H), 4.99 (s, 1H), 4.77–4.59 (m, 4H), 4.22 (p, J = 7.1 Hz, 1H), 3.11 (s, 1H), 2.85 (dd, J = 11.6, 6.0 Hz, 2H), 2.61 (ddq, J = 18.8, 11.6, 6.8 Hz, 2H), 2.52 (d, J = 4.4 Hz, 1H), 2.33–2.27 (m, 1H), 2.05 (d, J = 10.8 Hz, 1H), 1.75–1.65 (m, 1H).
13C NMR (151 MHz, DMSO-d6) δ 156.69 (d, J = 233.1 Hz), 156.49 (d, J = 233.4 Hz), 141.26, 137.80, 136.93, 136.28, 126.79, 126.40, 122.99, 122.62 (d, J = 9.7 Hz), 122.15 (dd, J = 10.0, 4.0 Hz), 121.76 (d, J = 4.0 Hz), 121.22, 120.82, 118.96, 118.46, 118.18, 114.21 (d, J = 25.7 Hz), 113.40 (d, J = 25.5 Hz), 111.57, 111.26 (d, J = 9.0 Hz), 110.83 (d, J = 8.8 Hz), 110.00, 106.42 (d, J = 23.7 Hz), 106.01 (d, J = 23.8 Hz), 78.24, 73.29, 64.63, 54.97, 49.66, 47.51, 45.29, 45.20, 21.76.
2.2.7.29. 1-((3-Butoxypropyl)amino)-3-((1-(3,6-difluoro-9H-carbazol-9-yl)-3-(3-fluoro-9H-carbazol-9-yl)propan-2-yl)oxy)propan-2-ol (WK-29)
White solid, 92.0 mg (yield 73%). IR (KBr), ν, cm−1: 3323, 2932, 2867, 1585, 1488, 1467, 1296, 1109, 855, 794, 745.
m.p. 151–153 °C. HRMS-ESI calcd. for C37H41F3N3O3 [M + H]+ 632.3100, found 632.3088.
1H NMR (600 MHz, DMSO-d6) δ 8.24 (brs, 1H), 8.17 (d, J = 7.8 Hz, 1H), 8.03 (ddd, J = 20.0, 9.1, 2.6 Hz, 3H), 7.80–7.67 (m, 4H), 7.49 (t, J = 7.3 Hz, 1H), 7.42–7.29 (m, 3H), 7.20 (t, J = 7.5 Hz, 1H), 4.98 (brs, 1H), 4.74–4.60 (m, 4H), 4.22 (p, J = 6.2 Hz, 1H), 3.42–3.31 (m, 5H), 3.07 (s, 1H), 2.44–2.31 (m, 2H), 2.29–2.23 (m, 1H), 1.99–1.89 (m, 1H), 1.69–1.53 (m, 3H), 1.48 (p, J = 6.6 Hz, 2H), 1.31 (h, J = 7.4 Hz, 2H), 0.88 (td, J = 7.4, 2.6 Hz, 3H).
13C NMR (151 MHz, DMSO-d6) δ 156.70 (d, J = 233.1 Hz), 156.50 (d, J = 233.5 Hz), 141.27, 137.82, 136.97, 126.42, 122.63 (d, J = 9.8 Hz), 122.14 (dd, J = 9.9, 4.0 Hz), 121.79 (d, J = 4.4 Hz), 120.85, 118.99, 114.21 (d, J = 25.4 Hz), 113.41 (d, J = 25.4 Hz), 111.29 (d, J = 9.2 Hz), 110.87 (d, J = 8.7 Hz), 110.04, 106.43 (d, J = 24.0 Hz), 106.02 (d, J = 23.8 Hz), 78.18, 73.21, 69.87, 67.15, 64.38, 49.58, 45.26, 45.18, 45.00, 31.27, 25.67, 18.91, 13.86.
2.2.7.30. 1-((1-(3,6-Difluoro-9H-carbazol-9-yl)-3-(3-fluoro-9H-carbazol-9-yl) propan-2-yl)oxy)-3-isopropoxypropan-2-ol (WK-30)
Pale yellow liquid, 43.0 mg (yield 36%). IR (KBr), ν, cm−1: 3323, 2932, 2867, 1585, 1488, 1467, 1296, 1109, 855, 794, 745.
Liquid. HRMS-ESI calcd. for C33H32F3N2O3 [M + H]+ 561.2365, found 561.2338.
1H NMR (600 MHz, DMSO-d6) δ 8.16 (d, J = 7.7 Hz, 1H), 8.02 (ddd, J = 14.3, 9.2, 2.5 Hz, 3H), 7.69 (ddd, J = 16.8, 8.8, 4.1 Hz, 4H), 7.46 (t, J = 8.1 Hz, 1H), 7.37–7.28 (m, 3H), 7.19 (td, J = 7.4, 3.6 Hz, 1H), 4.64 (d, J = 5.9 Hz, 4H), 4.20 (p, J = 6.9, 6.3 Hz, 1H), 4.01 (d, J = 5.2 Hz, 1H), 2.74 (p, J = 6.1 Hz, 1H), 2.69–2.61 (m, 1H), 2.43–2.31 (m, 3H), 2.13 (dd, J = 9.7, 6.4 Hz, 1H), 0.71 (dd, J = 15.1, 6.1 Hz, 6H).
13C NMR (151 MHz, DMSO-d6) δ 156.69 (d, J = 233.0 Hz), 156.49 (d, J = 233.1 Hz), 141.27, 137.82, 137.01, 126.31, 122.65 (d, J = 9.8 Hz), 122.16 (dd, J = 9.9, 4.2 Hz), 121.81 (d, J = 4.1 Hz), 120.82, 118.92, 114.04 (d, J = 25.7 Hz), 113.24 (d, J = 25.3 Hz), 111.16 (d, J = 8.9 Hz), 110.78 (d, J = 9.6 Hz), 109.96, 106.32 (d, J = 24.0 Hz), 105.97 (d, J = 23.8 Hz), 77.91, 72.30, 70.63, 68.83, 68.19, 45.44, 45.25, 21.87, 21.60.
2.3. Biological section
2.3.1. DNMT1 inhibition assays
2.3.1.1. ELISA DNMT1 activity assay
All the compounds were first screened using an ELISA EpiQuik DNA methyltransferase (DNMT) activity/inhibitor assay kit (Epigentek). To measure the effects of the compounds on human DNMT1 activity, 200 nM purified DNMT1 was incubated with 50 µM and 100 µM of the different compounds and S-adenosylmethionine in the DNMT assay buffer in the assay plate at 37 °C for 2 hCitation36. Next, every sample was incubated with the capture and detection antibody, followed by incubation with developer solution for 10 min at room temperature. The absorbance was measured at 450 nm using a POLARstar Omega microplate reader (BMG). S-Adenosylhomocysteine (AdoHcy) was used as a positive control. Methylation inhibition assays for EZH2, LSD, and G9a were performed in modified Tris, pH 9.0, buffer using AlphaLisa technology. An amount of 10 µL of the reaction system contained a corresponding concentration of SAM (Sigma) (EZH2 50 µM, LSD 50 µM, and G9a 50 µM), which was the Km value in each enzymatic reaction, plus 100 nM biotinylated peptide H3 (1–21) (synthesis by GLChina) and the relevant enzyme concentration (0.03 nM EZH2, 0.03 nM LSD, and 0.03 nM G9a). The proteins were preincubated with various compound concentrations for 15 min at room temperature before the substrate and SAM were added. After 60 min of incubation at room temperature, acceptor and donor AlphaLisa beads were added according to the manufacturer’s recommendations. The signals were read in Alpha mode with an EnSpire multimode plate reader (PerkinElmer). IC50 values were derived by fitting the data for the inhibition percentage to a dose-response curve by non-linear regression in GraphPad Prism 5.0Citation20.
2.3.1.2. Radioactive methylation assay
The DNMT1 radioactive methylation inhibition assays were performed in 30 µL reactions containing 0.1 µM adenosyl-L-methionine S-[methyl-3H] (3H-SAM, 15 Ci/mmol, PerkinElmer), 0.25 µg/mL poly (dI-dC)·poly (dI-dC) (Sigma), 40 nM DNMT1 in 50 mM Tris-HCl, pH 8.0, 1 mM DTT, 5% glycerol, and 100 µg/mL BSA. The proteins were preincubated with a range of compound concentrations for 15 min at room temperature before adding the substrate and [3H] SAM. After 60 min of incubation at 37 °C, the reaction systems were transferred to a MultiScreen HTS filter plate (Millipore), and the plate was washed 3 times with doubly distilled H2O via a vacuum. The radioactivity was determined by liquid scintillation counting (MicroBeta, PerkinElmer). IC50 values were derived by fitting the data for the inhibition percentage to a dose-response curve by non-linear regression in GraphPad Prism 5.0.
2.3.2. Cell lines and culture conditions
The human lung cancer cells (A549) and human colon cancer cells (HCT116) were cultured in an RPMI-1640 medium containing 10% FBS, 100 U/mL streptomycin, and 100 U/mL penicillin at 37 °C in a humidified atmosphere with 5% CO2. All compounds were dissolved in DMSO and stock solutions were stored at −20 °C. Reagents were freshly diluted to the marked concentrations with a culture medium before use. DMSO concentration in experimental conditions never exceeded 0.1% (v/v). All cell lines were provided by the cell laboratory, School of pharmaceutical sciences, Guangzhou Medical University.
2.3.3. MTT assay
Cell viability was detected using an MTT assay kit. Cells were seeded into 96-well plates at a density of ∼1.0 × 104/well. 24 h later, sextuplicate wells were treated with media and new compounds at a fixed concentration (50 µM). After 24, 48, and 72 h, the drug-containing medium was replaced by a 100 µL fresh medium with 5 mg/mL MTT solution. After 4 h of incubation, the medium with MTT was removed, and 100 µL of DMSO was added to each well. The plates were gently agitated until the purple formazan crystals were dissolved, and the OD490 was determined using a microscope (Olympus BX53, Japan). The data were calculated and plotted as the percent viability compared to the control.
2.4. PK study
In vivo pharmacokinetic properties of WK-22, WK-23, WK-27, and DC_517 were performed by Medicilon Company, Shanghai, China. SPF-grade SD male rats (8 groups, n = 3 rats per group) with a body weight of 230–260 g were purchased from Shanghai SIPPR-BK LAB Animal Ltd., Shanghai, China, and used for the pharmacokinetic analysis of the tested compounds. All animals were deprived of food overnight after the cannulation surgery. Subsequently, the tested compounds WK-23 and DC_517 were dissolved/suspended in 5% DMSO, 10% Solutol, and 85% water, and the tested compounds WK-22 and WK-27 were dissolved/suspended in 5% DMSO, 40% PEG400, and 55% saline for intravenous administration (i.v.) and oral administration (p.o.), respectively. A final dosage of 2.0 and 10.0 mg/kg rat of the formulated compounds was administered for i.v. and p.o. purposes, respectively, and the blood samples were taken at various time points during a 24 h period. At different time points, blood samples were collected from the femoral vein. The plasma samples were obtained after centrifugation (6800 g, 6 min, 2–8 °C) and stored at −80 °C until the assay. The AUC (area under concentration-time curve) was calculated through the trapezoidal rule with extrapolation to time infinity. The concentration of the compounds in the blood was analysed by LC-MS/MS (Shimadzu liquid chromatographic system and SCIEX Triple Quad 5500+ mass spectrometer, Applied Biosystems, Ontario, Canada). The Tmax, T1/2, and Cmax value was obtained through visual inspection of the plasma concentration-time curve. The Vss value was generated from DAS 3.2.8 software. The F value of WK-23 was calculated with the formula: F = (AUC p.o. × Dose i.v.)/(AUC i.v. × Dose p.o.) × 100%.
2.5. Molecular docking
Molecular docking was performed by the Glide program packed in Maestro (Maestro, Schrödinger, LLC, New York, NY, 2020.). The crystal structure of human DNMT1 was used as a template (PDB code: 4WXX) and removed the water moleculesCitation20. The docking procedure was initiated by the optimisation of protein structure using the Protein preparation Wizard module. Then the inhibitor was optimised by the Ligand Preparation module to generate stereoisomers and protonation states. Extra precision (XP) mode was used to perform the molecular docking, and the final result was selected through the Glide score function.
3. Results and discussion
3.1. Chemistry
The preparation routes of all target compounds are outlined in Scheme 1. First, we synthesised the key intermediates, and the general synthetic routes are illustrated in Scheme 1. Briefly, the commercially available 2-chloroaniline or 2-chloro-4-fluoroaniline reacted with bromofluorobenzene by the Buchwald-Hartwig coupling reaction and then the transition metal-catalyzed C-C coupling ring was constructed for 3 or 3, 6 substitution carbazoleCitation21. Fluorine-substituted carbazoles reacted with epichlorohydrin to get epoxy intermediatesCitation22. The general synthetic route of piperazine sidechain is illustrated in Scheme S1. Finally, the epoxy intermediates interacted with different amine compounds under basic conditions by nucleophilic substitution reaction to achieve 30 target compoundsCitation23. The structure of these compounds was confirmed by 1H NMR, 13 C NMR, high-resolution mass spectra, and infra-red spectra.
Scheme 1. Reagents and conditions: (a) NaOtBu, Pd(OAc)2, [HPtBu3][BF4], 4-bromofluorobenzene, toluene, reflux, 4 h; (b) Pd(OAc)2, [HPtBu3][BF4], NaOtBu, 1,4-dioxane, reflux, 18 h; (c) Epichlorohydrin, KOH, DMF, r.t. (d) Amines, EtOH, 60 °C; (e) 3-fluoro-9H-carbazole (a3) or 3,6-difluoro-9H-carbazole (b3), KOH, Na2SO4, acetone, r.t.; (f) Epichlorohydrin, KOH, Na2SO4, acetone, r.t.; (g) Amines, isopropanol, 60 °C.
![Scheme 1. Reagents and conditions: (a) NaOtBu, Pd(OAc)2, [HPtBu3][BF4], 4-bromofluorobenzene, toluene, reflux, 4 h; (b) Pd(OAc)2, [HPtBu3][BF4], NaOtBu, 1,4-dioxane, reflux, 18 h; (c) Epichlorohydrin, KOH, DMF, r.t. (d) Amines, EtOH, 60 °C; (e) 3-fluoro-9H-carbazole (a3) or 3,6-difluoro-9H-carbazole (b3), KOH, Na2SO4, acetone, r.t.; (f) Epichlorohydrin, KOH, Na2SO4, acetone, r.t.; (g) Amines, isopropanol, 60 °C.](/cms/asset/459c3bc7-c62b-480e-bfd8-6e595945a245/ienz_a_2079640_sch0001_b.jpg)
3.2. DNMT1 inhibition assays
DNMT1 inhibition test was performed on 30 candidates to verify their biochemical activities. The EpiQuik DNA methyl-transferase (DNMT) activity/inhibitor assay kit (Epigentek) was used to identify the activity of synthetic compounds. It was found that 11 compounds could inhibit DNMT1 activity by >80% (, ), and these compounds had similar potency to that of DC_517 against DNMT1 at a concentration of 50 µM and 100 µM.
Figure 2. Schematic showing design for binding DNMT1 pharmacophore.

Table 1. Biochemical assay results for DC_05 and DC_517 analogues against DNMT1 catalytic activity. 
3.3. Radioactive methylation assay against DNMT1
To quantitatively analyse DNMT1 inhibitory effect, the H3-labelled radioactive methylation assay was conducted to measure the methyltransferase activity of DNMT1 at a range of concentrations for these compounds. As shown in , WK-22 (IC50 = 4.9 µM) and WK-23 (IC50 = 5.0 µM) showed similar inhibitory activity as DC_517 (IC50 = 2.3 µM), whereas the other compounds were less potent. This result helped us to uncover sufficient information for the study of hit optimisation and structure-activity relationships (SARs).
3.4. Structure–activity relationships investigation
All the carbazole derivatives summarised in Scheme 1, including twenty-one derivatives of DC_05 and nine derivatives of DC_517 were prepared and biologically evaluated for the DNMT1 inhibitory activity. The experimental data of compounds bearing monocarbazole or dicarbazole as DNMT1 inhibitors are displayed in , respectively. The structure-activity relationships of DC_05, DC_517, and its analogs were determined and investigated by comparing the DNMT1 inhibitory activity.
Accordingly, as shown in , the catalytic site of DNMT1 is thereby includes two parts, namely, the cofactor binding site SAM pocket and the substrate binding site cytosine pocket. In our previous study, we conducted the molecular docking study and found that the compounds occupied the SAM pocket and cytosine pocket of DNMT1 (PDB code 4DA4)Citation20.
As shown in , WK-1 and WK-12 with fluorine substitute at C-3 or C-3,6 position, showed IC50 values of 28 and 17 µM, respectively, stronger than the lead compound DC_05 (IC50 = 52 µM). And when comparing the activity between WK-13, WK-14, WK-17, WK-18, and WK-19 from WK-3, WK-2, WK-5, WK-6, and WK-7, respectively, we found that difluoro-carbazole derivatives exhibited a better DNMT1 inhibition activity than the derivatives only replaced single fluorine on C-3 position.
WK-20 and WK-21 with a nitrogen heterocyclic replaced at region β, which showed lower inhibitory activity than DC_05 at 100 µM. It could be that the lone pair on the nitrogen is not tolerated in the pocket either because the environment is non-polar or because there is actually already an atom with a lone pair that leads to a repulsive effect.
The result of enzyme inhibitory activities showed that different amines at region γ will lead to a different activity. When it comes to monocarbazole, the decreasing order of influence of amines in inhibition activity was tryptamine, phenethylamines, and aliphatic amines. It could be corresponding to the space structure of the cytosine pocket since the structures linked to the amines are too small to occupy the cytosine pocket completely. But WK-6, WK-7, WK-18, and WK-19 still showed a relatively potent activity, possibly because the derivatives form extra hydrogen bonds via its hydroxyl to bind to the amino acid residue of the cytosine pocket. Similarly, phenethylamines derivative WK-13 (IC50 = 19 µM), which has a hydrogen donor on the aromatic ring exhibited a better activity. The inhibition of WK-2 and WK-9 are inferiors to DC_05, which suggests a long linking chain is optimal for the distance of binding targets. This derivation is also consistent with the activity data of dicarbazole molecules. In addition, WK-26 lost its activity probably because the Boc-protecting group resulted in steric hindrance while WK-24 was also inactive perhaps because the cyclopropyl group reduces hydrophobic and weakens the binding with the cytosine pocket.
3.5. Methyltransferase enzymatic selectivity profiling
Based on the DNMT1 Inhibition Assays data, some derivatives showed a considerable DNMT1 inhibition activity. Monocarbazole molecules that exhibited better inhibition on DNMT1 than DC_05, or dicarbazole molecules that exhibited better inhibition than DC_517 at 50 µM and 100 µM, were chosen to promote the next step. In addition to DNMT1, there are many other methyltransferases that can bind with S-adenosyl-L-methionine (SAM) to facilitate transmethylation reactionsCitation24,Citation25. To investigate the selectivity of these potent compounds for DNMT1, we then evaluated the inhibitory activities against DNMT1 and other important methyltransferases at the concentration of 50 µM, including DNMT3A/3L, DNMT3B/3L, and other SAM-dependent enzymesCitation1,Citation2,Citation26, such as EZH2Citation27, LSD1Citation28, G9a (histone H3 lysine 9 methyltransferase)Citation29.
As shown in , the enzymatic selectivity and inhibitory activity were measured at the concentration of 50 µM. Compounds WK-1, WK-12, and WK-13 displayed a strong activity on DNMT1 while showing hardly any inhibitory activities against DNMT3B/3L, EZH2, and G9a. Compounds WK-22, WK-23, and WK-27 displayed a similar activity targeting DNMT1 to that on DNMT3A/3L, DNMT3B/3L, and LSD1, while showed nearly no inhibitory activities against EZH2 and G9a. As shown in Table S1, WK-23 exhibited good selectivity on EZH2 and G9a (Relative selectivity index = 0.85 and 0.89), while had a similar effect on DNMT 3 A/3L, DNMT 3B/3L, and LSD1 to that on DNMT1 (Relative selectivity index = 0.06, 0.02 and 0.2). Here, the selectivity of non-nucleoside DNMT1 inhibitors is somewhat predictable because other methyltransferases mentioned above catalyse different substrates and share very low homology with DNMT1, even in their catalytic domainCitation30–32. Subsequently, the screened compounds were evaluated in a dose-dependent assay against HCT116 (human colon cancer cell line) and A549 (human lung carcinoma cell line).
Figure 3. Enzymatic selectivity of preliminarily screened compounds (50 μM).

3.6. Pharmacokinetics (PKs)
By virtue of the favourable DNMT1 inhibitory activity and specificity, WK-22, WK-23, WK-27 were selected for further PK evaluation in Sprague-Dawley (SD) Rats with DC_517 used as the referenceCitation33. As shown in , WK-22 gave an AUC of 867.2 ± 15.9 h ng/mL and oral bioavailability of 27.0%, whereas WK-27 gave an AUC of 427.2 ± 60.0 h ng/mL and oral bioavailability of 1.8%. This is perhaps because the introduction of piperazine leads to the reduction of permeability and absorption into the blood. WK-23 displayed a favourable plasma exposure (AUC0-t = 1064.9 ± 121.2 h ng/mL) and an acceptable oral bioavailability (F% = 37.1 ± 1.7), which is equivalent to DC_517 (AUC0-t = 1022.7 ± 60.9 h ng/mL, F% = 38.7 ± 2.9). Most strikingly, the elimination half-life of WK-23 (T1/2 = 7.9 h) has an advantage over DC_517 (T1/2 = 6.7 h).
Table 2. The PK properties of selected compound WK-22, WK-27, WK-23, and DC_517
3.7. Evaluation of antitumor activity in cells
Collectively, compounds WK-1, WK-12, WK-13, WK-19, WK-22, and WK-23 showed significant DNMT1 inhibiting activity, and we concomitantly explored the anti-proliferative effect of these most interesting compounds on cancer cell lines. As shown in , we tested these molecules in HCT116Citation34 and A549Citation35. Notably, WK-22 and WK-23 displayed the highest anti-proliferative effects in these two types of cancer cell lines, which is consistent with those of the DNMT1 inhibition assays. The results also demonstrated that WK-22 and WK-23 led to obvious dose-dependence and time-dependence anti-proliferation in HCT116 cancer cells (). Moreover, in comparison with A549, generally, these compounds are more sensitive to HCT116 cells. Regarding the Pharmacokinetics study, WK-23 exhibited an advantage in elimination half-life (T1/2 = 7.9 h) and oral bioavailability (F% = 37.1 ± 1.7) over other compounds. Consequently, we further explored the binding mode of WK-23 to DNMT1.
Figure 4. Effect of WK-23 and DC_517 on the viability of human colon cell lines and human lung cell lines. WK-23 has stronger concentration dependence and time dependence than DC_517, which inhibited the viability of HCT116 and A549 cells in the cell viability assay. Significance between groups was analysed by one-way analysis of variance (ANOVA) using IBM SPSS software. *p < 0.05.

Table 3. Derivatives’ Inh % at 50 μM on HCT116 and A549 cell lines
3.8. Molecular docking
Molecular docking was performed to explore the binding mode of WK-23 to DNMT1. The docking procedure was validated by reproducing the SAH binding mode with a root-mean-square deviation (RMSD) of 0.965 Å (Figure S1). According to our previous study, there is no obvious difference between the enantiomers of DC_517, which implies the racemic of WK-23 is applicable for molecular docking simulations with DNMT1 (PDB code 4WXX). As shown in , the binding pattern of WK-23 with DNMT1 is similar to that of DC_517Citation20. One carbazolyl of WK-23 occupied the SAM pocket, and the other carbazolyl forms cation − π interactions with R1574 and W1170. Besides, the amino group of WK-23 interacts with the main chain of F1145, and the hydroxyl group forms hydrogen bonds with E1168. These polar interactions further stabilise the binding of the inhibitor to DNMT1.
Figure 5. Putative binding mode of WK-23 with DNMT1 (PDB code 4WXX). The protein (white) is shown as a cartoon. The compound (deep salmon) and the side chain of F1145, E1168, W1170, and R1574 were shown in sticks (blue).

4. Conclusion
In summary, a series of novel carbazole-based derivatives were designed, synthesised, and evaluated for their biological activity. The structure-activity relationship of their anti-proliferative activity was explored. Among these compounds, WK-22 and WK-23 displayed appreciable human DNMT1 inhibitory activity in the micromolar range (IC50 = 4.9 µM and 5.0 µM). Simultaneously, both WK-22 and WK-23 has promising anti-proliferative effect on A549 and HCT116 cell lines. In further in vivo pharmacokinetic study, WK-23 displayed a better plasma exposure and prolonged elimination half-life (T1/2 = 7.9 h), especially the more acceptable oral bioavailability of (F% = 37.1) than WK-22 (F% = 27.0). Concomitantly, the molecule docking showed the binding pattern of WK-23 with DNMT1 is similar to that of DC_517, forming stable binding to DNMT1.
In conclusion, due to its favourable biological performance, compound WK-23 warrants further assessment as a potential therapeutic agent for the treatment of human cancers.
Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.
Supplemental Material
Download PDF (8.3 MB)Disclosure statement
The authors declare that they have no financial, conflict of interest, or personal relationships that may appear to hinder or affect this work. No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med 2018;378:1537–34.
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007;128:683–92.
- Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet 2000;9:2395–402.
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012;150:12–27.
- Chuang JC, Yoo CB, Kwan JM, et al. Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2′-deoxycytidine. Mol Cancer Ther 2005;4:1515–20.
- Takemura Y, Satoh M, Hatanaka K, et al. Zebularine exerts its antiproliferative activity through s phase delay and cell death in human malignant mesothelioma cells. Biosci Biotechnol Biochem 2018;82:1159–64.
- Kuang YT, El-Khoueiry A, Taverna P, et al. Guadecitabine (SGI-110) priming sensitizes hepatocellular carcinoma cells to oxaliplatin. Mol Oncol 2015;9:1799–814.
- Seidel C, Florean C, Schnekenburger M, et al. Chromatin-modifying agents in anti-cancer therapy. Biochimie 2012;94:2264–79.
- Fang MZ, Chen DP, Sun Y, et al. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res 2005;11:7033–42.
- Fang MZ, Wang YM, Ai N, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res 2003;63:7563–70.
- Dikmen M. Comparison of the effects of curcumin and RG108 on NGF-induced PC-12 Adh cell differentiation and neurite outgrowth. J Med Food 2017;20:376–85.
- Zambrano P, Segura-Pachero B, Perez-Cardenas E, et al. A phase I study of hydralazine to demethylate and reactivate the expression of tumor suppressor genes. BMC Cancer 2005;5:44.
- Cornacchia E, Golbus J, Maybaum J, et al. Hydralazine and procainamide inhibit T-cell DNA methylation and induce autoreactivity. J Immunol 1988;140:2197–200.
- Castellano S, Kuck D, Viviano M, et al. Synthesis and biochemical evaluation of δ(2)-isoxazoline derivatives as DNA methyltransferase 1 inhibitors. J Med Chem 2011;54:7663–77.
- Asgatay S, Champion C, Marloie G, et al. Synthesis and evaluation of analogues of N-phthaloyl-L-tryptophan (RG108) as inhibitors of DNA methyltransferase 1. J Med Chem 2014;57:421–34.
- Datta J, Ghoshal K, Denny WA, et al. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res 2009;69:4277–85.
- Gilmartin AG, Groy A, Core ER, et al. In vitro and in vivo induction of fetal hemoglobin with a reversible and selective DNMT1 inhibitor. Haematologica 2020;106:1979–87.
- Juarez-Mercado KE, Prieto-Martinez FD, Sanchez-Cruz N, et al. Expanding the structural diversity of DNA methyltransferase inhibitors. Pharmaceuticals 2020;14:17.
- Pechalrieu D, Dauzonne D, Arimondo PB, et al. Synthesis of novel 3-halo-3-nitroflavanones and their activities as DNA methyltransferase inhibitors in cancer cells. Eur J Med Chem 2020;186:111829.
- Chen SJ, Wang YL, Zhou W, et al. Identifying novel selective non-nucleoside DNA methyltransferase 1 inhibitors through docking-based virtual screening. J Med Chem 2014;57:9028–41.
- Bedford RB, Betham M. N-H carbazole synthesis from 2-chloroanilines via consecutive amination and C-H activation. J Org Chem 2006;71:8.
- Molette J, Routier J, Abla N, et al. Identification and optimization of an aminoalcohol-carbazole series with antimalarial properties. ACS Med Chem Lett 2013;4:1037–41.
- Bombrun A, Gerber P, Casi G, et al. 3,6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J Med Chem 2003;46:4.
- Svedruzic ZM. Mammalian cytosine DNA methyltransferase DNMT1: enzymatic mechanism, novel mechanism-based inhibitors, and RNA-directed DNA methylation. Curr Med Chem 2008;15:92–106.
- Song JK, Teplova M, Ishibe-Murakami S, et al. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 2012;335:709–12.
- Egger G, Liang GN, Aparicio A, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004;429:457–63.
- Huang X, Yan J, Zhang M, et al. Targeting epigenetic crosstalk as a for EZH2-aberrant solid tumors. Cell 2018;175:186–34.
- Meier K, Brehm A. Chromatin regulation: how complex does it get? Epigenetics 2014;9:1485–95.
- Milner CM, Campbell RD. The G9a gene in the human major histocompatibility complex encodes a novel protein containing ankyrin-like repeats. Biochem J 1993; 290:811–8.
- Esteller M. Molecular origins of cancer: epigenetics in cancer. N Engl J Med 2008;358:1148–59.
- Rivera CM, Ren B. Mapping human epigenomes. Cell 2013;155:39–55.
- Yoo J, Medina-Franco JL. Inhibitors of DNA methyltransferases: insights from computational studies. Curr Med Chem 2012;19:3475–87.
- Wei WL, An YL, Li ZW, et al. Simultaneous determination of resibufogenin and its eight metabolites in rat plasma by LC-MS/MS for metabolic profiles and pharmacokinetic study. Phytomedicine 2019;60:152971.
- Estève PO, Chin HG, Smallwood A, et al. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 2006;20:3089–103.
- Fu JL, Zhou HX, Chen J, et al. Low expression of PRKCDBP promoted cisplatin resistance in lung adenocarcinoma by DNMT1 and TNF-α. Oncol Rep 2020;44:1616–26.
- Xiao DL, Dasgupta C, Chen M, et al. Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovasc Res 2014;101:373–82.