
Abstract
A new series of co-drugs was designed based on hybridising the dihydropteroate synthase (DHPS) inhibitor sulphonamide scaffold with the COX-2 inhibitor salicylamide pharmacophore through biodegradable linkage to achieve compounds with synergistic dual inhibition of COX-2/PGE2 axis and DHPS enzyme to enhance antibacterial activity for treatment of septicaemia. Compounds 5 b, 5j, 5n and 5o demonstrated potent in vitro COX-2 inhibitory activity comparable to celecoxib. 5j and 5o exhibited ED50 lower than celecoxib in carrageenan-induced paw edoema test with % PGE2 inhibition higher than celecoxib. Furthermore, 5 b, 5j and 5n showed gastric safety profile like celecoxib. Moreover, in vivo antibacterial screening revealed that, 5j showed activity against S.aureus and E.coli higher than sulfasalazine. While, 5o revealed activity against E.coli higher than sulfasalazine and against S.aureus comparable to sulfasalazine. Compound 5j achieved the target goal as potent inhibitor of COX-2/PGE2 axis and in vivo broad-spectrum antibacterial activity against induced septicaemia in mice.
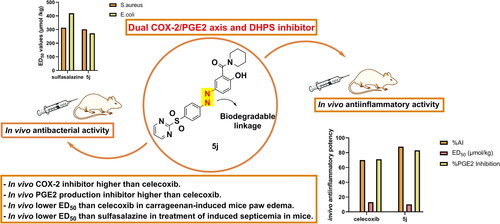
Graphical Abstract
1. Introduction
Prostaglandin E2 (PGE2) is an oxygenated arachidonic acid metabolite produced mainly by the action of Cyclooxygenase enzymes (COX)Citation1. Being a pro-inflammatory mediator, PGE2 activates macrophages, neutrophils, and mast cells at early stages of inflammationCitation2,Citation3. Additionally, it had been demonstrated as a powerful immunosuppressant inhibiting pathogen-killing via alveolar macrophages and phagocytosis. As a result, it is considered a potent risk factor during inflammationCitation4,Citation5. Moreover, COX-2 isozyme, a key enzyme in the process of PGE2 biosynthesis, had been reported to be induced by infections of the pathogenic Streptococcus pyogenes. In addition, several bacterial toxins including streptococcal cytolysin S (SLS) and streptococcal cytolysin O (SLO) produced by S. pyogenes together with pneumolysin and Clostridium difficile toxin A have been reported as potent inducers of COX-2 expressionCitation6. besides, it had been confirmed that COX-2/PGE2 production were up-regulated and promoted biofilm formation by Staphylococcus aureus thereby enhances its adherence to the human fibronectin and finally motivates its growthCitation7. Hence, blocking COX-2/PGE2 pathway introduces a new, an effective and an unexpected manner to overcome bacterial infections. Accordingly, celecoxib, the selective COX-2 inhibitor, had been reported to increase sensitivity of Staphylococcus aureus to antibioticsCitation8. Interestingly, celecoxib increased the utilisation of ampicillin by bacteria by inhibiting antibiotic resistance genes as well as increasing membrane permeabilityCitation9,Citation10. Furthermore, it had been reported that the commonly available antibiotics chloramphenicol, cefuroxime and Oxytetracycline exerted a synergistic effect with celecoxib, against MRSA strains with a 4-fold reduction in the cefuroxime and chloramphenicol MICs and 2-fold decrease in the Oxytetracycline MICCitation11,Citation12,Citation13. On the other hand, sulphonamides were the first antibacterial agents to be discovered by Gerhard Domagk, since then they have been applied for different clinical indicationsCitation14. Moreover, they exert their action by competitive inhibition of dihydropteroate synthase enzyme (DHPS), a significant anti-folate targetCitation15. Additionally, the sulphonamide moiety constitutes the prime pharmacophore of most selective COX-2 inhibitors as celecoxib and its derivativesCitation16. In fact, sulphonamides comprise an interesting class of drugs with a wide scope of pharmacological activities such as antibacterialCitation17,Citation18, hypoglycemicCitation19, diureticCitation20, carbonic anhydrase enzyme inhibitory propertiesCitation21–24, antithyroidCitation25 and anti-inflammatory agentsCitation26. This may be attributed to sulphonamides' phenylamino and sulphonyl amino groups essential for various biological activities as well as metal coordinationCitation27. Furthermore, Sulfasalazine (I), is a sulphonamide prodrug degraded by colonic bacteria via azo reductase enzyme to sulfapyridine, an antibiotic, and 5-aminosalicylic acid, an anti-inflammatory agent, compounds with different modes of actionCitation28,Citation29. It has been demonstrated that, sulfasalazine exerted its anti-inflammatory action via inhibiting a number of immunological processes, including lymphocyte proliferation, interleukin-2 synthesis in addition to interleukin-1 production by monocytes, but was not classified as an NSAIDCitation30. On the other hand, salicylamides had been previously reported as inhibitors of the two-component regulatory system (TCS) in bacteriaCitation31,Citation32. Besides, "5-substituted salicylamides" mainly designed to act as topical antibiotics, were serendipitously discovered to have innate anti-inflammatory activity. Therefore, those 5-substituted salicylamides as were referred to as lipophilic, antibacterial, anti-inflammatory drugs (LAADs). Salifluor (II), , the first-generation lead drug candidate of LAADs, had an excellent efficacy against a wide variety of Gram-positive and Gram-negative oral organisms. Trifluorosal (III), , a second-generation LAAD-type salicylamide, underwent early-stage pharmaceutical development for gingivities treatment. Among the third generation, naphthafluor (IV), , is currently in development as a treatment for acne as well as gingivitisCitation33. Besides, salicylamide derivatives V, exhibited significant COX-2 inhibition with significant COX-2 selectivity comparable to celecoxibCitation34. As well, salicylamide derivatives have been regarded as one of the promising candidates owing to their interesting biological activities, including antimycobacterialCitation35,Citation36, antifungalCitation37, antibacterialCitation31,Citation38, antineoplasticCitation39,Citation40, antianthelminthicCitation41, antiparasiticCitation42 as well as anti-inflammatory activitiesCitation43,Citation44. In view of the aforesaid facts, designing new candidates targeting COX-2/DHPS enzymes together with PGE2 production would be more effective in combating inflammatory bacterial infections. Thus, in the current study, we utilised the DHPS inhibiting properties of sulphonamides and the COX-2 inhibiting properties of salicylamides as well as their beneficial antibacterial effects to provide a monotherapy approach targeting COX-2 and DHPS enzymes being effective against bacterial infections together with their sequential inflammatory actions with lower evidence of resistance. Accordingly, herein we designed some new biodegradable molecules combining sulphonamide moiety linked to N-Substituted-2-hydroxybenzamides through azo linkage. The newly synthesised compounds were evaluated in vitro for their COX-1/COX-2 inhibitory activities and their antibacterial activities against human pathogenic bacteria including a number of Gram-positive and Gram-negative bacteria and the most active compounds were further evaluated in vivo for their anti-inflammatory and antibacterial efficacies. In addition, their effect on PGE2 production and ulcerogenic effect were also evaluated. Moreover, molecular docking studies were performed to profile the binding pattern of the potential dual COX-2/DHPS inhibitors with the active site of the targeted COX-2 and dihydropteroate synthase (DHPS) enzymes. Furthermore, in silico predictions of physicochemical parameters, drug likeness score and acquiescence to the Lipinski’s rule of five (RO5) were carriedout to the biologically active compounds to estimate their suitability to act as possible orally active dug candidates.
Figure 1. Rational design of dual COX-2/PGE2 axis and DHPS inhibitors.

2. Experimental
2.1. Chemistry
All chemicals were bought from commercial sources and used directly without further purification. Open-glass capillaries were used in measuring melting points on Stuart Scientific melting point apparatus (SMP10) and are uncorrected. The reactions' rates were followed up by thin layer chromatography (TLC) on silica gel pre-coated Merck aluminium GF254 plates, and the spots were visualised at λ 254 nm using UV-lamp for few seconds' exposure to iodine vapours. Thermo Scientific Nicolet iS20 Fourier Transform Infra-red Spectrometer was used for recording Infra-red spectra (IR), using KBr discs, Faculty of Pharmacy, Mansoura University. Proton nuclear magnetic resonance spectra (1H-NMR) were scanned on JNM-ECA II 500 MHz JEOL Spectrometer at the NMR unit, Faculty of Science, Mansoura University and on Bruker Avance III 400 MHz Spectrometer at Centre for Drug Discovery Research and Development-Faculty of Pharmacy, Ein Shams University using deuterated dimethylsulphoxide (DMSO-d6) as a solvent. Data were interpreted as chemical shifts expressed in δ values (ppm) relative to tetramethylsilane (TMS) as internal standard. Signal splitting type was indicated by one of the following letters: s = singlet, d = doublet, t = triplet, q = quartette, dd = doublet of doublet and m = multiplet. 13 C-NMR spectra were scanned on JNM-ECA II 125 MHz JEOL Spectrometer at the NMR unit, Faculty of Science, Mansoura University. Electron impact mass spectra (EI-MS) were scanned on Direct Inlet part mass analyser in Thermo Scientific GCMS model ISQ at the Regional Centre for Mycology and Biotechnology, Al-Azhar University, Cairo. Elemental analyses (C, H, N and S) was the tool used for checking purity of the new compounds using FLASH 2000 CHNS/O analyser, Thermo Scientific at the Regional Centre for Mycology and Biotechnology (RCMB), Al-Azhar University. The results were within ±0.4% of the calculated values for the proposed formulae. ChemDraw Professional 16.0.1.4. was used for naming the new compounds according to the naming algorithm developed by CambridgeSoft Corporation compounds 2aCitation45 and 2 b-2dCitation46,Citation47,Citation48 were prepared as previously reported.
2.1.1. General procedure for the synthesis of compounds 5a-o
The appropriate sulphonamide derivative (2 mmol) was suspended in hydrochloric acid (17%, 2 ml) then ethanol was added until a clear solution was obtained the solution was cooled from 0 °C to −5 °C and a solution of NaNO2 (2 mmol, 0.138 gm) in water (1 ml) was added dropwise. After stirring at 0 °C for 15–30 min, a solution of the appropriate N-substituted-2-hydroxybenzamide (2 mmol) in NaOH (10%, 2 ml) was added to the diazonium salt dropwise and the reaction mixture was stirred overnight at room temperature. The formed precipitate was filtered off, washed with water and crystallised from ethanol.
2.1.1.1. N-Butyl-2-hydroxy-5-((4-sulfamoylphenyl) diazenyl) benzamide (5a)
Pale brown crystals; yield 95%, m.p. 227–229 °C. IR (KBr, cm−1): 3529 (OH), 3377, 3269 (NH2, NH), 1646 (C = O), 1496 (N = N), 1305, 1149 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 0.92 (t, J = 7.4 Hz, 3H, NH-CH2-CH2-CH2-CH3), 1.31–1.42 (m, 2H, NH-CH2-CH2-CH2-CH3), 1.52–1.61 (m, 2H, NH-CH2-CH2-CH2-CH3), 3.36–3.39 (m, 2H, NH-CH2-CH2-CH2-CH3), 7.08 (d, J = 8.6 Hz, 1H, salicyl-C3-H),7.51 (s, 2H, sulfamoylphenyl NH2, D2O exchangeable), 7.78–7.82 (m, 4H, 1-sulfamoylphenyl-C2,6,3,5-H), 7.97 (dd, J = 8.6, 2.4 Hz, 1H, salicyl-C4-H), 8.57 (d, J = 2.4 Hz, 1H, salicyl-C6-H), 9.14 (t, J = 5.7 Hz, 1H, CH2-NH, D2O exchangeable), 13.48 (s, 1H, OH, D2O exchangeable) 0.13 C-NMR (125 MHz, DMSO-D6) δ 13.72 (NH-CH2-CH2-CH2-CH3), 19.67 (NH-CH2-CH2-CH2-CH3), 30.82 (NH-CH2-CH2-CH2-CH3), 46.47 (NH-CH2-CH2-CH2-CH3), 115.31(salicyl-C3), 118.68 (salicyl-C1), 121.79 (sulfamoylphenyl-C2,6), 125.44 (salicyl-C4), 125.83(salicyl-C6), 126.77 (sulfamoylphenyl-C3,5), 144.49 (sulfamoylphenyl-C4), 150.34 (salicyl-C5), 151.68 (sulfamoylphenyl-C1), 163.42 (salicyl-C2), 168.58 (C = O). Anal. Calcd.for C17H20N4O4S (376.43): C, 54.24; H, 5.36; N, 14.88; S, 8.52. Found: C, 54.37; H, 5.48; N, 14.66; S, 8.63.
2.1.1.2. N-Butyl-2-hydroxy-5-((4-(N-(thiazol-2-yl) sulfamoyl) phenyl) diazenyl) benzamide (5 b)
Shiny orang crystals; yield 97%, m.p. 239–241 °C. IR (KBr, cm−1): 3528 (OH), 3395, 3373 (NH), 1637 (C = O), 1491 (N = N), 1298, 1143 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 0.91 (t, J = 7.5 Hz, 3H, NH-CH2-CH2-CH2-CH3), 1.30–1.40 (m, 2H, NH-CH2-CH2-CH2-CH3), 1.52–1.60 (m, 2H, NH-CH2-CH2-CH2-CH3), 3.41–3.47 (m, 2H, NH-CH2-CH2-CH2-CH3), 6.88 (d, J = 4.7 Hz, 1H,thiazolyl-C5-H), 7.10 (d, J = 8.9 Hz, 1H, salicyl-C3-H), 7.29 (d, J = 4.7 Hz, 1H,thiazolyl-C4-H), 7.94 − 8.01 (m, 5H, 1-sulfamoyl phenyl-C2,3,5,6-H and salicyl-C4-H), 8.59 (s, 1H, salicyl-C6-H), 9.12 (t, J = 5.7 Hz, 1H, CH2-NH, D2O exchangeable), 12.84 (s, 1H, sulfamoyl NH, D2O exchangeable), 13.52 (s, 1H, OH, D2O exchangeable). Anal. Calcd.for C20H21N5O4S2 (459.54): C, 52.27; H, 4.61; N, 15.24; S, 13.95. Found: C, 52.39; H, 4.72; N, 15.13; S, 14.07.
2.1.1.3. N-Butyl-2-hydroxy-5-((4-(N-(pyrimidin-2-yl) sulfamoyl) phenyl) diazenyl) benzamide (5c)
Orang crystals; yield 95%, m.p. 228–230 °C. IR (KBr, cm−1): 3653 (OH), 3465, 3425 (NH), 1646 (C = O), 1494 (N = N), 1344,1146 (SO2).1H-NMR (400 MHz, DMSO-d6) δ 0.91 (t, J = 7.5 Hz, 3H, NH-CH2-CH2-CH2-CH3), 1.31–1.40 (m, 2H, NH-CH2-CH2-CH2-CH3), 1.52 − 1.60 (m, 2H, NH-CH2-CH2-CH2-CH3), 3.41–3.48 (m, 2H, NH-CH2-CH2-CH2-CH3), 7.06 (t, J = 4.6 Hz, 1H, pyrimidinyl-C5-H), 7.11 (d, J = 8.8 Hz, 1H, salicyl-C3-H), 7.96 − 8.00 (m, 3H, 1-sulfamoyl phenyl-C2,6-H and salicyl-C4-H), 8.17 (d, J = 8.6 Hz, 2H, sulfamoyl phenyl-C3,5-H), 8.52 (d, J = 4.6 Hz, 2H, pyrimidinyl-C4,6-H), 8.59 (d, J = 2.4 Hz, 1H, salicyl-C6-H), 9.12 (t, J = 5.7 Hz, 1H, CH2-NH, D2O exchangeable), 12.04 (s, 1H, 1-sulfamoyl NH, D2O exchangeable), 13.52 (s, 1H, OH, D2O exchangeable). 13 C-NMR (125 MHz, DMSO-D6) δ 13.73 (NH-CH2-CH2-CH2-CH3), 19.69 (NH-CH2-CH2-CH2-CH3), 30.85 (NH-CH2-CH2-CH2-CH3), 46.51 (NH-CH2-CH2-CH2-CH3), 115.70 (pyrimidinyl-C5), 118.83 (salicyl-C3), 122.47 (salicyl-C1), 124.07 (sulfamoylphenyl-C2,6), 126.15 (salicyl-C4), 129.11 (salicyl-C6), 137.11(sulfamoylphenyl-C3,5) 144.53(sulfamoylphenyl-C4), 152.10 (salicyl-C5), 154.10 (sulphamoylphenyl-C1), 156.79 (pyrimidinyl-C4,6), 158.70 (pyrimidinyl-C2), 163.97 (salicyl-C2), 168.27 (C = O). Anal. Calcd.for C21H22N6O4S (454.51): C, 55.50; H, 4.88; N, 18.49; S, 7.05. Found: C, 55.62; H, 4.98; N, 18.37; S, 7.13. EIMS m/z (% relative abundance): 457.96 (2.96) (M+•+3), 454.49 (10.42) (M+•), 432.78 (18.08), 396.36 (43.52), 327.07 (28.63), 115.05 (38.74), 57.04 (48.32), 44.92 (82.56) 43.03 (100) (base peak).
2.1.1.4. N-Butyl-2-hydroxy-5-((4-(N-(4-methylpyrimidin-2yl) sulfamoyl) phenyl) diazenyl) benzamide (5d)
Deep orang crystals; yield 97%, m.p. 235–238 °C. IR (KBr, cm−1): 3676 (OH), 3525, 3394 (NH), 1643 (C = O), 1495 (N = N), 1347, 1149 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 0.91 (t, J = 7.3 Hz, 3H, NH-CH2-CH2-CH2-CH3), 1.30 − 1.39 (m, 2H, NH-CH2-CH2-CH2-CH3), 1.52 − 1.60 (m, 2H, NH-CH2-CH2-CH2-CH3), 2.32 (s, 3H,pyrimidinyl-4-CH3), 3.30–3.36 (m, 2H, NH-CH2-CH2-CH2-CH3), 6.91 (d, J = 5.2 Hz, 1H, pyrimidinyl-C5-H), 7.11 (d, J = 8.9 Hz, 1H, salicyl-C3-H), 7.95–7.99 (m, 3H, 1-sulfamoyl phenyl-C2,6-H, salicyl-C4-H), 8.17 (d, J = 8.6 Hz, 2H, 1-sulfamoyl phenyl-C3,5-H), 8.32 (d, J = 5.2 Hz, 1H, pyrimidinyl-C6-H), 8.60 (d, J = 2.4 Hz, 1H, salicyl-C6-H), 9.13 (t, J = 5.6 Hz, 1H, CH2-NH, D2O exchangeable), 11.96 (s, 1H, sulfamoyl NH, D2O exchangeable), 13.53 (s, 1H, OH, D2O exchangeable). Anal. Calcd.for C22H24N6O4S (468.53): C, 56.40; H, 5.16; N, 17.94; S, 6.84. Found: C, 56.52; H, 5.17; N, 17.83; S, 6.94.
2.1.1.5. N-Benzyl-2-hydroxy-5-((4-sulfamoylphenyl) diazenyl) benzamide (5e)
Orang crystals; yield 95%, m.p. 250–252 °C. IR (KBr, cm−1): 3452 (OH), 3400, 3361 (NH2), 3268 (NH), 1640 (C = O), 1494 (N = N), 1337, 1150 (SO2). 1H- NMR (400 MHz, DMSO-d6) δ 4.56 (s, 2H, -benzyl-CH2), 7.12 (d, J = 9.0 Hz, 1H, salicyl-C3-H), 7.28 (s, 2H, benzyl-C2,6-H), 7.33–7.37 (m, 3H, benzyl-C3,4,5-H),7.53 (s, 2H, sulfamoylphenylNH2, D2O exchangeable), 7.97–8.03 (m, 5H, sulfamoylphenyl-C2,3,5,6-H and salicyl-C4-H), 8.64 (s, 1H, salicyl-C6-H), 9.71 (s, 1H, CH2-NH, D2O exchangeable), 13.30 (br.s, 1H, OH, D2O exchangeable) 0.13 C-NMR (125 MHz, DMSO-d6) δ 42.65 (benzyl-CH2), 116.13 (salicyl-C3), 118.95 (salicyl-C1), 122.66 (sulfamoylphenyl-C2,6), 126.06 (salicyl-C4), 126.60 (salicyl-C6), 127.09 (benzyl-C2,6), 127.49 (benzyl-C4), 128.49 (benzyl-C3,5), 138.82 (sulfamoylphenyl-C3,5), 144.46 (benzyl-C1), 145.40 (sulfamoylphenyl-C4), 152.00 (salicyl-C5), 153.68 (sulfamoylphenyl-C1), 163.91(salicyl-C2), 168.13 (C = O). Anal. Calcd.for C20H18N4O4S (410.45): C, 58.53; H, 4.42; N, 13.65; S, 7.81. Found: C, 58.79; H, 4.55; N, 13.42; S, 7.89. EIMS m/z (% relative abundance): 410.42 (4.8) (M+•), 393.47 (10.02), 314.40 (13.69), 167.46 (27.82), 96.14 (32.86), 69.04 (100) (base peak), 53.96 (37.56), 43.98 (43.98).
2.1.1.6. N-Benzyl-2-hydroxy-5-((4-(N-(thiazol-2-yl) sulfamoyl) phenyl) diazenyl) benzamide (5f)
Deep brown crystals; yield 90%, m.p. 281–283 °C. IR (KBr, cm−1): 3566 (OH), 3450, 3371 (NH), 1642 (C = O), 1493 (N = N), 1297, 1139 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 4.55 (d, J = 5.4 Hz, 2H, benzyl-CH2), 6.86 (d, J = 4.5 Hz, 1H, thiazolyl-C5-H), 7.08 (d, J = 9.2 Hz, 1H, salicyl-C3-H), 7.27 (d, J = 4.5 Hz, 2H, benzyl-C2,6-H), 7.32 − 7.38 (m, 4H, benzyl-C3,4,5-H and thiazolyl-C4-H), 7.90 − 8.01 (m, 5H, sulfamoyl phenyl-C2,3,5,6-H and salicyl-C4-H), 8.61 (d, J = 2.5 Hz, 1H, salicyl-C6-H), 9.86 (t, J = 5.4 Hz,1H, CH2-NH, D2O exchangeable),12.06 (s, 1H, sulfamoyl NH, D2O exchangeable), 13.33 (s, 1H, OH, D2O exchangeable). Anal. Calcd.for C23H19N5O4S2 (493.56): C, 55.97; H, 3.88; N, 14.19; S, 12.99. Found: C, 56.09; H, 3.99; N, 14.08; S, 13.11.
2.1.1.7. N-Benzyl-2-hydroxy-5-((4-(N-(pyrimidin-2-yl) sulfamoyl) phenyl) diazenyl) benzamide (5 g)
Deep yellow crystals; yield 93%, m.p. 275–277 °C. IR (KBr, cm−1): 3568 (OH), 3393, 3356 (NH), 1648 (C = O), 1493 (N = N), 1337,1145 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 4.55 (d, J = 5.4 Hz, 2H, benzyl-CH2), 7.06 (t, J = 4.8 Hz, 1H, pyrimidinyl-C5-H), 7.13 (d, J = 8.9 Hz, 1H, salicyl-C3-H), 7.24–7.29 (m, 2H, benzyl-C2,6-H), 7.33 − 7.39 (m, 3H, benzyl-C3,4,5-H), 7.98 (d, J = 7.8 Hz, 2H, sulfamoyl phenyl-C2,6-H), 8.02 (dd, J = 8.9, 2.6, 1H, salicyl-C4-H), 8.17 (d, J = 7.8 Hz, 2H, sulfamoyl phenyl-C3,5-H), 8.52 (d, J = 4.8 Hz, 2H, pyrimidinyl-C4,6-H), 8.63 (d, J = 2.6 Hz, 1H, salicyl-C6-H), 9.65 (t, J = 5.4 Hz, 1H, CH2-NH, D2O exchangeable) 12.06 (s, 1H, sulfamoyl NH, D2O exchangeable), 13.30 (s, 1H, OH, D2O exchangeable). 13 C-NMR (126 MHz, DMSO-D6) δ 42.62 (benzyl-CH2), 115.94 (pyrimidinyl-C5), 118.77(salicyl-C3), 122.45(salicyl-C1), 126.04 (sulfamoylphenyl-C2,6), 126.57 (salicyl-C4), 127.05 (salicyl-C6), 127.44 (benzyl-C2,6), 128.44 (benzyl-C4), 129.06 (benzyl-C3,5), 132.94 (sulfamoylphenyl-C3,5), 138.74 (sulfamoylphenyl-C4), 141.74 (benzyl-C1), 144.59 (salicyl-C5), 154.05 (sulfamoylphenyl-C1), 156.76 (pyrimidinyl-C4,6), 158.50 (pyrimidinyl-C2), 163.62 (salicyl-C2), 168.09 (C = O). Anal. Calcd. for C24H20N6O4S (488.52): C, 59.01; H, 4.13; N, 17.20; S, 6.56. Found: C, 59.13; H, 4.24; N, 17.09; S, 6.64.
2.1.1.8. 4-((4-Hydroxy-3-(piperidine-1-carbonyl) phenyl) diazenyl) benzenesulfonamide (5 h)
Yellow crystals; yield 95%, m.p. 220–222 °C. IR (KBr, cm−1): 3568 (OH), 3384, 3346 (NH2), 1681 (C = O), 1482 (N = N), 1349, 1148 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 1.41–1.61 (m, 6H, piperidinyl-C3,4,5-H), 3.14–3.25, 3.51–3.65 (2 m, 4H, piperidinyl-C2,6-H), 7.08 (d, J = 8.7 Hz, 1H, salicyl-C3-H), 7.52 (s, 2H, sulfamoyl NH2, D2O exchangeable), 7.71 (d, J = 2.5 Hz, 1H, salicyl-C6-H), 7.89 (dd, J = 8.7, 2.5 Hz, 1H, salicyl-C3-H), 7.95 − 8.01 (m, 4H, sulfamoyl phenyl-C2,3,5,6-H), 10.87 (s, 1H, OH, D2O exchangeable). Anal. Calcd. for C18H20N4O4S (388.44): C, 55.66; H, 5.19; N, 14.42; S, 8.25. Found: C, 55.78; H, 5.29; N, 14.30; S, 8.36.
2.1.1.9. 4-((4-Hydroxy-3-(piperidine-1-carbonyl) phenyl) diazenyl)-N-(thiazol-2-yl) benzenesulfonamide (5i)
Orang crystals; yield 90%, m.p. 164–166 °C. IR (KBr, cm−1): 3651 (OH), 3451 (NH), 1574 (C = O), 1481 (N = N), 1328, 1138 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 1.42–1.61 (m, 6H, piperidinyl-C3,4,5-H), 3.14–3.25, 3.51–3.65 (2 m, 4H, piperidinyl-C2,6-H), 6.87 (d, J = 4.5 Hz, 1H, thiazolyl-C5-H), 7.07 (d, J = 8.7 Hz, 1H, salicyl-C3-H), 7.28 (d, J = 4.5 Hz, 1H, thiazolyl-C4-H), 7.69 (d, J = 2.4 Hz, 1H, salicyl-C6-H), 7.87 (dd, J = 8.7, 2.4 Hz, 1H, salicyl-C4-H), 7.91–7.99 (m, 4H, sulfamoyl phenyl-C2,3,5,6-H), 10.87 (s, 1H, sulfamoyl NH, D2O exchangeable), 12.86 (s, 1H, OH, D2O exchangeable) 0.13 C-NMR(126 MHz, DMSO-D6) δ 24.05 (piperidinyl-C4), 25.63 (piperidinyl-C3,5), 47.34 (piperidinyl-C2,6), 108.60 (thiazolyl-C5), 116.41(salicyl-C3), 119.31(salicyl-C1), 122.53(sulfamoylphenyl-C2,6), 124.63(salicyl-C4), 125.66(salicyl-C6), 126.17 (thiazolyl-C4), 127.19 (sulfamoylphenyl-C3,5), 143.51(sulfamoylphenyl-C4), 144.93 (salicyl-C5), 153.69 (sulfamoylphenyl-C1), 157.39 (salicyl-C2),165.61(C = O),169.07(thiazolyl-C2). Anal. Calcd.for C21H21N5O4S2 (471.55): C, 53.49; H, 4.49; N, 14.85; S, 13.60. Found: C, 53.61; H, 4.59; N, 14.73; S, 13.68.
2.1.1.10. 4-((4-Hydroxy-3-(piperidine-1-carbonyl) phenyl) diazenyl)-N-(pyrimidin-2-yl) benzenesulfonamide (5j)
Orang crystals; yield 93%, m.p. 267–269 °C. IR (KBr, cm−1): 3651 (OH), 3448 (NH), 1581 (C = O), 1498 (N = N), 1349, 1166 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 1.41–1.60 (m, 6H, piperidinyl-C3,4,5-H), 3.14–3.24, 3.52–3.66 (2 m, 4H, piperidinyl-C2,6-H), 7.04–7.08 (m, 2H, pyrimidinyl-C5-H, salicyl-C3-H), 7.70 (d, J = 2.2 Hz, 1H, salicyl-C6-H), 7.87 (dd, J = 8.5, 2.2 Hz, 1H, salicyl-C4-H), 7.95 (d, J = 8.5 Hz, 2H, sulfamoyl phenyl-C2,6-H), 8.14 (d, J = 8.5 Hz, 2H, sulfamoyl phenyl-C3,5-H), 8.51 (d, J = 4.8 Hz, 2H, pyrimidinyl-C4,6-H), 10.90 (s, 1H, sulfamoyl NH, D2O exchangeable), 12.02 (br.s, 1H, OH, D2O exchangeable). Anal. Calcd.for C22H22N6O4S (466.52): C, 56.64; H, 4.75; N, 18.01; S, 6.87. Found: C, 56.68; H, 4.87; N, 17.89; S, 6.88.
2.1.1.11. 4-((4-Hydroxy-3-(piperidine-1-carbonyl) phenyl) diazenyl)-N-(4-methylpyrimidin-2-yl) benzenesulfonamide (5k)
Orang crystals; yield 97%, m.p. 208–210 °C. IR (KBr, cm−1): 3652 (OH), 3567, 3448 (NH), 1612 (C = O), 1472 (N = N), 1337, 1165 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 1.41–1.62 (m, 6H, piperidinyl-C3,4,5-H), 2.32 (s, 3H, pyrimidinyl-4-CH3), 3.15–3.25, 3.52–3.66 (2 m, 4H, piperidinyl-C2,6-H), 6.90 (d, J = 5.3 Hz, 1H, pyrimidinyl-C5-H), 7.19 (d, J = 8.8 Hz, 1H, salicyl-C3-H), 7.74 (d, J = 2.4 Hz, 1H, salicyl-C6-H), 7.88 (dd, J = 8.8, 2.4 Hz, 1H, salicyl-C4-H), 7.94 (d, J = 8.6 Hz, 2H, sulfamoyl phenyl-C2,6-H), 8.14 (d, J = 8.6 Hz, 2H, sulfamoyl phenyl-C3,5-H), 8.31 (d, J = 5.3 Hz, 1H, pyrimidinyl-C6-H), 10.89 (s, 1H, sulfamoyl NH, D2O exchangeable), 11.99 (br.s, 1H, OH, D2O exchangeable) 0.13 C-NMR (125 MHz, DMSO-D6) δ 23.22 (pyrimidinyl-4-CH3-C), 24.07 (piperidinyl-C4), 25.69 (piperidinyl-C3,5), 47.25 (piperidinyl-C2,6) 112.45 (pyrimidinyl-C5), 116.44 (salicyl-C3), 119.36 (salicyl-C1), 122.31(sulfamoylphenyl-C2,6), 125.62 (salicyl-C4), 126.24 (salicyl-C6), 129.25 (sulfamoylphenyl-C3,5), 137.59 (sulfamoylphenyl-C4), 145.00 (salicyl-C5), 153.93(sulfamoylphenyl-C1), 156.42 (pyrimidinyl-C6), 157.51(pyrimidinyl-C2), 159.49 (salicyl-C2), 165.62 (pyrimidinyl-C4), 168.54 (C = O). Anal. Calcd.for C22H22N6O5S (482.52): C, 54.76; H, 4.60; N, 17.42; S, 6.64. Found: C, 54.84; H, 4.64; N, 17.34; S, 6.72. EIMS m/z (% relative abundance): 482.49 (11.78) (M+•), 458.70 (48.60), 395.30 (34.33), 360.25 (41.93), 349.02 (50.06), 308.18 (51.43), 282.03 (100) (base peak), 193.02 (35.65).
2.1.1.12. 4-((4-Hydroxy-3-(morpholine-4-carbonyl) phenyl) diazenyl) benzenesulfonamide (5 l)
Pale brown crystals; yield 93%, m.p. 272–274 °C. IR (KBr, cm−1): 3450 (OH), 3340 (NH2), 1610 (C = O), 1489 (N = N), 1345, 1154 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 3.53–3.69 (m, 8H, morpholinyl-H), 7.11 (d, J = 8.8 Hz, 1H, salicyl-C3-H), 7.50 (s, 2H, sulfamoyl NH2, D2O exchangeable), 7.77 (d, J = 2.4 Hz, 1H, salicyl-C6-H), 7.91 (dd, J = 8.8, 2.4 Hz, 1H, salicyl-C4-H), 7.96 − 8.01 (m, 4H, sulfamoyl phenyl-C2,3,5,6-H), 11.00 (s, 1H, OH, D2O exchangeable). Anal. Calcd.for C17H18N4O5S (390.41): C, 52.30; H, 4.65; N, 14.35; S, 8.21. Found: C, 52.41; H, 4.77; N, 14.25; S, 8.29.
2.1.1.13. 4-((4-Hydroxy-3-(morpholine-4-carbonyl)phenyl)diazenyl)-N-(thiazol-2-yl)benzenesulfonamide (5 m)
Brown crystals; yield 95%, m.p. 235–237 °C. IR (KBr, cm−1): 3653 (OH), 3454 (NH), 1603 (C = O), 1483 (N = N), 1411, 1141 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 3.52–3.69 (m, 8H, morpholinyl-H), 6.86 (d, J = 4.6 Hz, 1H, thiazolyl-C5-H), 7.14 (d, J = 8.9 Hz, 1H, salicyl-C3-H), 7.27 (d, J = 4.6 Hz, 1H, thiazolyl-C4-H), 7.74 (d, J = 2.5 Hz, 1H, salicyl-C6-H), 7.88 (dd, J = 8.9, 2.5 Hz, 1H, salicyl-C4-H), 7.92–7.98 (m, 4H, sulfamoyl phenyl-C2,3,5,6-H), 11.06 (s, 1H, sulfamoyl NH, D2O exchangeable), 12.87 (br.s, 1H, OH, D2O exchangeable) 0.13 C-NMR (125 MHz, DMSO-D6) δ 46.62 (morpholinyl-C3,5), 66.50 (morpholinyl-C2,6), 108.62 (thiazolyl-C5), 116.52 (salicyl-C3), 119.32 (salicyl-C1), 120.87 (sulfamoylphenyl-C2,6), 124.91(salicyl-C4), 125.16 (salicyl-C6), 125.55 (thiazolyl-C4), 129.02(sulfamoylphenyl-C3,5), 137.16 (sulfamoylphenyl-C4), 145.96 (salicyl-C5), 153.08 (sulfamoylphenyl-C1), 159.79 (salicyl-C2), 168.64 (C = O), 169.21(thiazolyl-C2). Anal. Calcd.for C20H19N5O5S2 (473.52): C, 50.73; H, 4.04; N, 14.79; S, 13.54. Found: C, 50.85; H, 4.12; N, 14.68; S, 13.58. EIMS m/z (% relative abundance): 476.32 (3.62) (M+•+3), 473.46 (13.41) (M+•), 401.27 (19.48), 393.28 (38.19), 371.15 (17.19), 157.01 (15.13), 125.34 (20.48), 84.94 (24.86), 70.99 (34.08), 44.41 (100) (base peak).
2.1.1.14. 4-((4-Hydroxy-3-(morpholine-4-carbonyl) phenyl) diazenyl)-N-(pyrimidin-2-yl) benzenesulfonamide (5n)
Shiny brown crystals; yield 95%, m.p. 241–243 °C. IR (KBr, cm−1): 3651 (OH), 3450 (NH), 1583 (C = O), 1500 (N = N), 1335, 1162 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 3.51–3.67 (m, 8H, morpholinyl-H), 7.05 (t, J = 4.9 Hz, 1H, pyrimidinyl-C5-H), 7.11 (d, J = 8.8 Hz, 1H, salicyl-C3-H), 7.75 (d, J = 2.5 Hz, 1H, salicyl-C6-H), 7.89 (dd, J = 8.8, 2.5 Hz, 1H, salicyl-C4-H), 7.95 (d, J = 8.6 Hz, 2H, sulfamoyl phenyl-C2,6-H), 8.15 (d, J = 8.6 Hz, 2H, sulfamoyl phenyl-C3,5-H), 8.51 (d, J = 4.9 Hz, 2H, pyrimidinyl-C4,6-H), 11.03 (s, 1H, sulfamoyl NH, D2O exchangeable), 11.97 (s, 1H, OH, D2O exchangeable). 13 C-NMR (125 MHz, DMSO-D6) δ 46.91(morpholinyl-C3,5), 66.35 (morpholinyl-C2, 6), 116.57(pyrimidinyl-C5), 119.22(salicyl-C3), 122.49(salicyl-C1), 123.19(sulfamoylphenyl-C2,6), 124.78(salicyl-C4), 126.50(salicyl-C6), 129.05(sulfamoylphenyl-C3,5), 132.15(sulfamoylphenyl-C4), 145.02(salicyl-C5), 154.13(sulfamoylphenyl-C1), 156.80(pyrimidinyl-C4,6), 157.54(pyrimidinyl-C2), 158.39 (salicyl-C2), 165.98 (C = O). Anal. Calcd.for C21H20N6O5S (468.49): C, 53.84; H, 4.30; N, 17.94; S, 6.84. Found: C, 53.92; H, 4.34; N, 17.83; S, 6.92.
2.1.1.15. 4-((4-Hydroxy-3-(morpholine-4-carbonyl) phenyl) diazenyl)-N-(4-methylpyrimidin-2-yl) benzenesulfonamide (5o)
Shiny brown crystals; yield 95%, m.p. 205–207 °C. IR (KBr, cm−1): 3652 (OH), 3450 (NH), 1612 (C = O), 1472 (N = N), 1337, 1165 (SO2). 1H-NMR (400 MHz, DMSO-d6) δ 2.32 (s, 3H, pyrimidinyl-4-CH3), 3.51–3.66 (m, 8H, morpholinyl-H), 6.90 (d, J = 5.3 Hz, 1H, pyrimidinyl-C5-H), 7.19 (d, J = 8.8 Hz, 1H, salicyl-C3-H), 7.74 (d, J = 2.4 Hz, 1H, salicyl-C6-H), 7.88 (dd, J = 8.8, 2.4 Hz, 1H, salicyl-C4-H), 7.94 (d, J = 8.6 Hz, 2H, sulfamoyl phenyl-C2,6-H), 8.14 (d, J = 8.6 Hz, 2H, sulfamoyl phenyl-C3,5-H), 8.31 (d, J = 5.3 Hz, 1H, pyrimidinyl-C6-H), 11.21 (s, 1H, sulfamoyl NH, D2O exchangeable), 11.85 (s, 1H, OH, D2O exchangeable). Anal. Calcd.for C22H22N6O5S (482.52): C, 54.76; H, 4.60; N, 17.42; S, 6.64. Found: C, 54.84; H, 4.71; N, 17.32; S, 6.68.
2.2. Biological evaluation
2.2.1. In vitro COX-1 and COX-2 enzymatic assay
All the synthesised target compounds were screened for their COX-1 and COX-2 inhibitory activities according to the previously reported methodsCitation49,Citation50 (page S34, supplementary file).
2.2.2. Carrageenan-induced paw edoema in mice
According to the reported procedures for carrageenan-induced paw edoema test in miceCitation51, this test was carried out for compounds 5 b, 5j, 5n and 5o. (Approved by HU-IACUC)Citation52 (Pages S34 and S35, supplementary file).
2.2.3. Determination of ED50
ED50 of compounds 5 b, 5j, 5n and 5o was calculated as reportedCitation53. (Page S35, supplementary file).
2.2.4. Estimation of rat plasma PGE2
Plasma PGE2 concentration of compounds 5 b, 5j, 5n and 5o was also measured as previously reportedCitation54. (Approved by HU-IACUC)Citation52 (Page S35, supplementary file).
2.2.5. Gastric ulcerogenic activity
The acute gastric ulcerogenic effect of compounds 5 b, 5j, 5n and 5o in adult male Wistar rats was evaluated as previously reportedCitation55. (Approved by HU-IACUC)Citation52 (Pages S35 and S36, supplementary file).
2.2.6. Antibacterial screening
2.2.6.1. Inhibition-zone measurements
Inhibition zones of the target compounds was calculated as previously reportedCitation56. (Page S36, supplementary file).
2.2.6.2. Minimal inhibitory concentration (MIC) measurement
MIC of test compounds was measured according to the reported methodsCitation57. (Page S36, supplementary file).
2.2.6.3. Minimal bactericidal concentration (MBC) measurement
MBC tests were also carried out as reportedCitation58. (Page S37, supplementary file).
2.2.7. In vivo antibacterial screening in mice (bacteremic infection)
The in vivo antibacterial activity of compounds 5 b, 5j, 5n and 5o against E.coli and Staphylococcus aureus using sulfasalazine as a reference drugCitation59 was performed as reportedCitation60. (Approved by HU-IACUC)Citation52 (Pages S37 and S38, supplementary file).
2.3. Docking studies
Molecular Operating Environment (MOE 2018.0802) softwareCitation61,Citation62 https://www.chemcomp.com/Research-Citing_MOE.htm, Chemical Computing Group (Chemical Computing Group, Quebec, Canada, Montreal, Canada) was utilised in performing Molecular docking studies. Protein preparation was performed as previously reportedCitation63, page S38, supplementary file.
2.4. In silico prediction of physicochemical properties, drug likeness score, pharmacokinetics, toxicity profile and ligand efficiency metrics
In the present study, prediction of the physicochemical properties was performed using Molinspiration chemoinformatic server, pharmacokinetics by Pre-ADMET calculator, drug likeness score and toxicological effects by Osiris property explorer, page S38, supplementary file.
3. Results and discussion
3. 1. Chemistry
The synthetic routes for the preparation of the intermediates and target compounds are outlined in Scheme 1. Aminolysis reaction of methyl salicylate (1) with the selected primary or secondary amines was employed to obtain N-Substituted-2-hydroxybenzamides (2a-d) in good yields and with pure regioselectivity as reported previouslyCitation45,Citation64. The target azo compounds (5a-o) were attained by diazotisation of the appropriate sulphonamide with NaNO2 and HCl at 0 °C followed by addition of the appropriate benzamide (2a-d). Moreover, integrity of the structures of the newly synthesised compounds was justified by microanalysis, IR, 1H-NMR, 13 C-NMR and MS data (see chemistry experimental section and supporting information). The IR spectra of compounds 5a-o were characterised by the absorption bands specific for OH, NH, C = O, N = N and SO2 functional groups. 1H-NMR spectra of compounds 5a-d were characterised by appearance of one D2O exchangeable triplet signal around δ 9.12 ppm stands for n-butyl NH proton in addition to two D2O exchangeable singlets stands for sulfamoyl NH proton in the range δ 11.96 − 12.84 ppm for 5 b, 5c and 5d and sulfamoyl NH2 protons at δ 7.51 ppm for 5a together with OH proton around δ 13.52 ppm. Moreover, compounds 5e-g were distinguished by appearance of one signal around δ 4.55 ppm corresponding to benzyl CH2 protons, three D2O exchangeable signals; in the range δ 9.65 − 9.9.86 ppm corresponding to n-benzyl NH proton, around δ 12.06 ppm corresponding to sulfamoyl NH proton for 5f and 5 g, around δ 7.53 ppm corresponding to sulfamoyl NH2 protons for 5e and around δ 13.30 ppm corresponding to OH proton. Besides, compounds 5 h-k were differentiated by three multiplets in the ranges δ 1.41–1.62, 3.14–3.25 and 3.51–3.66 ppm corresponding to the ten piperidinyl protons, in addition to one D2O exchangeable singlet around δ 10.90 ppm corresponding to sulfamoyl NH proton for 5i, 5j and 5k and around δ 7.52 ppm corresponding to sulfamoyl NH2 protons for 5 h together with one D2O exchangeable singlet in the range δ 10.87–12.86 ppm corresponding to OH proton. Furthermore, compounds 5 l-o were distinguished by appearance of multiplet signal in the range δ 3.51–3.69 ppm stands for the eight morpholinyl protons together with two D2O exchangeable singlets; around δ 7.50 ppm corresponding to sulfamoyl NH2 protons for 5 l, in the range δ 11.03–11.21 ppm corresponding to sulfamoyl NH proton for 5 m, 5n and 5o and in the range δ 11.00–12.87 ppm corresponding to OH proton. 13 C-NMR spectra proved the appearance of C = O signal at its expected region (δ165.61–168.64 ppm). It also showed the presence of signals assigned to n-butyl moiety around δ 13.72, 19.67, 30.82 and 46.47 ppm for compounds 5a and 5c, signal assigned to benzyl-CH2 around δ 42.65 ppm for compounds 5e and 5 g, three signals around δ 24.05, 25.63 and 47.34 ppm corresponding to piperidinyl carbons for compounds 5i and 5k, in addition to two signal around δ 46.62 and 66.50 ppm assigned to morpholinyl carbons for compounds 5 m and 5n. Finally, MS spectra showed the molecular ion peak (M+•) at m/z 454.49 for 5c, at 410.42 for 5e, at 482.49 for 5k and at 473.46 for 5 m.
Scheme 1. Synthesis of the target co-drugs (5a-o).

3.2. Biological evaluation
3.2.1. In vitro COX-1 and COX-2 enzymatic assay
Compounds 5a-o were screened for their in vitro COX-1/COX-2 inhibitory activities using an ovine COX-1/human recombinant COX-2 assay kit (Catalog no. 560131; Cayman Chemicals Inc. Ann Arbour, MI, USA). Celecoxib and Rofecoxib were used as reference selective COX-2 inhibitors and indomethacin and Diclofenac sodium were used as reference non-selective COX inhibitors. The data recorded in revealed that all compounds were more potent COX-2 inhibitors than Diclofenac sodium. Compared to indomethacin, compounds 5 b, 5j, 5n and 5o were more potent COX-2 inhibitors while compounds 5c, 5i and 5 l were equipotent. Moreover, compounds 5d, 5 g, 5k and 5 m were nearly comparable to indomethacin. All compounds exhibited weak inhibition of COX-1 compared to indomethacin. Considering Celecoxib, compound 5j displayed equipotent COX-2 inhibitory activity while compounds 5 b, 5c, 5i, 5 l, 5n and 5o were nearly comparable to Celecoxib. Interestingly, all compounds showed balanced weak COX-1 and more potent COX-2 inhibition with selectivity indices (SI) ranging from 58 to 239. The resulted SI values exceeded those for the non-selective COX inhibitors while being inferior to selective COX-2 inhibitors. This could also be considered advantageous by avoiding the cardiovascular side effects of the highly selective COX-2 inhibitorsCitation65. Careful inspection of the structures of the tested compounds revealed that compounds 5 b, 5j, 5n and 5o were the most potent COX-2 inhibitors in this series. The highest activity was observed with the piperidin-1-yl and sulfadiazine derivative 5j. Replacement of 4-sulfamoylphenyl moiety in compound 5a with thiazol-2-yl (5 b), pyrimidin-2-yl (5c) and 4-methylpyrimidin-2-yl (5d) respectively seemed to greatly enhance COX-2 inhibitory activity with higher selectivity indices. As observed with compounds 5e-g, introduction of benzyl moiety decreased COX-2 inhibitory activity. Moreover, the presence of 4-sulfamoylphenyl moiety in compounds 5e and 5 h did not enhance COX-2 inhibitory activity making them the least active compounds in this series. Concerning compounds 5i-5k, 5 l, 5n and 5o, introduction of piperidin-1-yl and morpholin-1-yl moieties to methyl salicylate portion of the compound significantly enhanced COX-2 inhibitory activity comparable to that of Celecoxib.
Table 1. In vitro COX-1 and COX-2 inhibitory IC50 values and COX SI values of the target compounds (5a-o).
3.2.2. Carrageenan-induced paw oedema in mice
Although most of the newly synthesised compounds exhibited a promising in vitro anti-inflammatory activity, it is important to assess their efficacy inside the biological system this can be attributed to their azo co-drug nature. As a result, they must be subjected to azo reductase enzyme, a metabolising enzyme in the colon, liberating two active metabolites; the sulphonamide portion and the 5-amino salicylamide portion being ready for systemic absorption. Consequently, the synthesised compounds, 5 b, 5j, 5n and 5o showing the most potent and selective COX-2 inhibitory activity were subjected to in vivo carrageenan-induced paw oedema bioassay in mice using celecoxib and Diclofenac as reference drugs. The results illustrated in and revealed that, after 2 h, both 5j and 5o exhibited similar pharmacokinetic profiles to celecoxib as revealed from their rapid onset of action. Interestingly, after 2 h, compound 5j showed more potent inhibition of paw edoema than both celecoxib and Diclofenac. To conclude the anti-inflammatory activity of the tested compounds, the anti-inflammatory activity after 8 h was taken as a point for comparison which showed that, compound 5j showed the best % inhibition of paw oedema (88.50%) being more than that was displayed by both celecoxib (70.87%) and Diclofenac (69.13%). Furthermore, compound 5o displayed comparable inhibition of paw oedema (72.17%) to that of celecoxib and Diclofenac. Compounds 5 b and 5n also showed moderate anti-inflammatory activities (56.09% and 60.87%, respectively). Furthermore, the ED50 values () showed that all compounds were nearly equipotent (10.94–18.69 µmol/kg). However, compound 5j was found to be the most potent among this series with ED50 = 10.94 µmol/kg exceeding those for celecoxib (13.07 µmol/kg) and Diclofenac (11.46 µmol/kg). In addition, the ED50 of compound 5o (13.27 µmol/kg) was the same as those for celecoxib.
Figure 2. Effects of the target compounds 5 b, 5j, 5n and 5o on the thickness of carrageenan-induced paw edoema in mice along interval of 0 – 8 h after injecting carrageenan (mm) together with the reference drugs celecoxib and Diclofenac in a dose of 10 µmol/kg.

Table 2. Effects of the target compounds 5 b, 5j, 5n and 5o on carrageenan-induced paw edoema in mice (mm), their percentage anti-inflammatory activity and their ED50 values (µmol/kg) (95% confidence level).
3.2.3. Estimation of rat plasma PGE2
Estimating serum levels of PGE2 is a critical parameter to assess the in vivo anti-inflammatory efficacies of COX-2 inhibitors. Consequently, compounds 5 b, 5j, 5n and 5o were subjected to estimation of PGE2 in rat serum and results were summarised in (Table 3, page S4 supplementary file). The results showed that, the most active compound was 5j recording plasma PGE2% inhibition of 83.25% higher than reference celecoxib (% inhibition = 71.18) and Diclofenac (% inhibition = 79.10). Moreover, compound 5o (72.92%) showed PGE2-diminishing activity comparable to that of celecoxib but slightly lower than that of Diclofenac. Also, 5 b and 5n were acceptable PGE2-lowering agents with % PGE2-inhibition of 65.70% and 55.18%, respectively. Finally, the results showed good relevance between selective COX-2 inhibitory activity and decreasing production of plasma PGE2 as one of the prime mediators released through COX-2 enzyme pathway.
Figure 3. % inhibition of rat serum PGE2 production after 8 h of injecting 10 µmol/kg b.wt of the tested compounds as well as the reference drugs celecoxib and Diclofenac.

3.2.4. Gastric ulcerogenic activity
Inclusive monitoring of the isolated fasted-rat stomachs revealed a normal stomach texture for compounds 5 b, 5j and 5n as well as the reference celecoxib whereas compound 5o expressed signs of gastric ulcers and hyperaemia as the reference Diclofenac. Furthermore, the degree of inflammatory reactions of the tested compounds in the gastric layers was confirmed by histopathological examination (). The results disclosed the gastrointestinal safety profile of compounds 5 b, 5j and 5n as well as celecoxib. Nevertheless, the tested compound 5o exhibited erosion in the gastric layers as the reference Diclofenac.
Figure 4. The ulcerogenic effect of the tested compounds 5 b, 5j, 5n and 5o as well as celecoxib and Diclofenac as reference drugs on gastric layers.

3.2.5. In vitro antibacterial screening
All the newly synthesised compounds were evaluated for their in vitro antibacterial activities against the human pathogens: Staphylococcus aureus (RCMB 0100183), Staphylococcus epidermidis (RCMB 0100183), Streptococcus mutans (RCMB 0100172), and Bacillus subtilis (RCMB 0100162) as examples of Gram-positive bacteria and Pseudomonas aeruginosa (RCMB 0100243), Escherichia coli (RCMB 010052), Salmonella typhi (RCMB 0100104), Shigella dysenteriae (RCMB 0100542) and Proteus vulgaris (RCMB 010085) as examples of Gram-negative bacteria using Ampicillin and levofloxacin as standard Gram-positive and Gram-negative antibacterial agents respectively[6] (Table 5, page S5 supplementary file) see experimental section. The results showed that, most of the tested compounds did not exhibit significant in vitro antibacterial activity, whereas compounds (5 b, 5i, 5j, 5k, 5 m, 5n, and 5o) displayed weak in vitro antibacterial activity this could be assigned to the fact that the investigated compounds were azo co-drugs. Hence, metabolic biotransformation by azo-reductase enzyme into their active metabolites is a requirement for expressing their activities.
3.2.6. In vivo antibacterial screening in mice
Compounds 5 b, 5j, 5n and 5o as well as sulfasalazine as a reference drug were evaluated for their in vivo antibacterial efficacy against experimental bacteremic infections caused by S. aureus as gram-positive and E.coli as gram-negative bacteria in mice and their ED50 values were determined, summarised and listed in . Concerning infection caused by S. aureus, all tested compounds improved the survival rate of model mice compared to positive control group which died. Compound 5j displayed the most potent activity with ED50 value of 302.1 µmol/kg being slightly potent S.aureus inhibitor than sulfasalazine (313.7 µmol/kg). Moreover, compounds 5 b, 5n and 5o showed good antibacterial activity with ED50 values ranging from 352.7 to 413.1 µmol/kg. By investigating the inhibitory activity against E.coli, all the tested compounds displayed antibacterial activity higher than sulfasalazine activity (ED50 = 272.7 − 415.7 µmol/kg vs 419.2 µmol/kg). Compound 5j was the most potent one against E.coli (ED50 = 272.7 µmol/kg). Furthermore, compound 5 b was found to have equal inhibitory effects against both S.aureus and E.coli (ED50 = 413.1 and 415.7 µmol/kg, respectively).
Table 3. The protective effects of promising compounds and the reference drug, sulfasalazine against bacteremic infections in mice ED50 values in µmol/kg/day (95% confidence limit).
3.3. Docking studies of the potential dual COX-2/DHPS inhibitors
We docked the target azo-dye prodrugs (5 b), (5n), (5o), and (5j) into COX-2 active site (pdb entry 3LN1)Citation66 which showed potential anti-inflammatory in both the in vitro () and in vivo ( and ) testing. We also docked the expected metabolite of (5j) into DHPS active site (pdb entry 3TZF)Citation67 after exposure to the azo-reductaseCitation68into the biological system which showed the most potential anti-bacterial activity in the in vivo testing ().
3.3.1. Docking of the compounds (5 b), (5n), (5o), and (5j) as COX-2 inhibitors:
The docking solutions of the compounds (5 b), (5n), (5o), and (5j) () verified the potential activities against COX-2 as anti-inflammatory agents because the drug-receptor interactions of the best poses are very comparable to that of the co-crystallised ligand inhibitor, celecoxib (). All the docked compounds interacted with the Leu 338, Arg499, and Ser 339 amino acids which are redundantly reportedCitation69 as a part of the polar side pocket of COX-2 active site and interaction with it is required for the selective inhibition of the enzyme. On the other side, we docked compound (5f) as one of the compounds of the least potencies against COX-2 () to realise the reasons stand behind such low potency. Docking solution of (5f) showed failure of the compound to relax freely into the active site in a way to interact with the polar side pocket amino acids except for Ser339. And, it lacks the carboxylic acid group which might have enabled it interact with Arg120 as an alternative path to inhibit the enzymeCitation69.
Figure 5. 2 D-style of docking solutions of the potential COX-2 inhibitors, 5 b; 5n; 5o; 5j, and Celecoxib as the co-crystallised ligand inhibitor in the active site of COX-2 (PBD code 3LN1). Besides, the docking solution of compound (5f) as the least potent inhibitor to visualise its distinct binding pattern with the same active site. Amino acid residues presented by the protein sequence code of three letters and numbers; interaction forces presented by green dotted lines which are categorised according to the description scheme associated with the figure.

3.3.2. Docking of compound (5j-metabolite) as a potential DHPS inhibitor:
Docking solution of (5j-metabolite) revealed that the compound interacted with the receptor active site with acceptable conformity to the co-crystallised ligand inhibitor, Sulfamethoxazole (). The metabolite interacted with Pro64, and Lys221but failed to interact with Phe28 and Ser222 amino acids. It is worthy to mention that the metabolite failed to sit into the active site aligned with the sulfamethoxazole but preferred to invert on its horizontal axis in the opposite direction to relax freely. This might be a reason for the failure of the compound to interact with both Phe28, and Ser222 amino acids as shown in ().
Figure 6. In the left side: 2 D-style of the docking solutions of the co-crystallised ligand inhibitor, sulfamethoxazole, and 5j-metabolite in the active site of DHPS (PBD code 3TZF) to visualise the interaction forces with the active site. In the right side: there are the corresponding 3 D-style docking solutions of sulfamethoxazole, and 5j-metabolite to visualise the orientation of the docked compound in the active site. Amino acid residues presented by the protein sequence code of three letters and numbers, atoms assigned by colours, (blue nitrogen; red oxygen; yellow sulphur; grey carbon). Interaction forces in the 2 D-style are categorised according to the description scheme associated with the figure while in the 3 D-style, it appeared as brown dotted lines for the hydrogen bonding and green dotted lines for the hydrophobic interactions.

3.4. In silico prediction of the physicochemical properties, drug likeness score, pharmacokinetics, toxicity profile and ligand efficiency metrics
Early prediction of the physicochemical and pharmacokinetic properties of new drug candidates is an infrastructure for lead optimisation as well as drug development processCitation70. Accordingly, the in silico physicochemical characters, drug likeness, pharmacokinetic parameters, toxicity profile and ligand efficiency metrics of the most active compounds 5 b, 5j, 5n and 5o were predicted by MolinspirationCitation71. Pre-ADMETCitation72 and Osiris property explorerCitation73 online soft wares (Table 6, pages S7 and 8 supplementary file).
The calculated values predicted that all the tested compounds comply with Lipinski’s rule of 5 and satisfied Veber’s criteria. Moreover, the results predicted good oral bioavailability and an acceptable molecular flexibility. Additionally, their percentages of absorption (%ABS; calculated as %ABS = 109–0.345 × TPSA)Citation74, as well as their solubility satisfied the solubility requirement (>0.0001 mg/L)Citation75,Citation76. Furthermore, all the compounds expected to have low cell permeability in both Caco-2 and MDCK cell models but with low CNS penetration. Meanwhile, all of them were predicted to be excellently absorbed and strongly bound to plasma proteins (>90%)Citation76,Citation77. In another context, the values of LE and LLE for the most active compounds regarding their COX-2 inhibitory activity were calculated by the reported equationsCitation78, then compared to celecoxib as a reference drug. The results expected the expediency of these compounds as lead-like candidates (for more details see pages S6, 7 and 8 supplementary file).
4. Conclusion
Seeking for damping bacterial resistance together with broadening spectrum of the biological activities there by dominating inflammatory bacterial infections, our study employed pharmacophoric hybridisation strategy by combining the antibacterial features of sulphonamides with the anti-inflammatory and antibacterial features of salicylamides through azo-linkage to design a novel series of biodegradable compounds that may be powerful tools to combat resistant bacteria as well as their consecutive inflammatory diseases. The newly synthesised compounds were challenged in vitro for their expected antibacterial and COX-inhibitory activities. Compounds 5 b, 5j, 5n and 5o had been identified as the most potent COX-2 inhibitors among the series with IC50 values ranging from 0.05 µm to 0.07 µm and SI values from 153.6 to 239.2. Moreover, all compounds displayed moderate in vitro antibacterial activities with MIC values ranging from 25 − 200 µg/ml which may be attributed to their prodrug nature and they become active only inside the biological system after being exposed to azo reductase enzyme. Consequently, the most active COX-2 inhibitors 5 b, 5j, 5n and 5o were evaluated for their in vivo anti-inflammatory and antibacterial activities. Surprisingly, compounds 5j and 5o were found to be the most potent inhibitors of carrageenan induced paw oedema in mice with % inhibition 88.50% and 72.17%, respectively and ED50 values of 10.94 µmol/kg and 13.27 µmol/kg, respectively. In addition, investigating rat serum concentration of PGE2 revealed that all of the four compounds are potent PGE2 inhibitors with % inhibition in the range of 55.18% − 83.25% which confirmed their COX-2 inhibitory properties. Moreover, in vivo antibacterial activity against S.aureus and E.coli infections confirmed their potent antibacterial action with ED50 values in the range of 302.1 − 413.1 µmol/kg against S.aureus and 272.7 − 419.2 µmol/kg against E.coli. Furthermore, docking studies of the same four compounds into COX-2 active site emphasised their anti-inflammatory potential in addition to being harmonised to a great extent with their IC50 values and selectivity indices obtained from the in vitro assay. Besides, docking of 5j metabolite into DHPS active site illustrated the inhibitory activity of the new candidates against bacteria. Finally, in silico prediction of the pharmacokinetic properties and toxicity profile as well as ligand efficiency metrics furnished extra support for the lead-like character of the target compounds. Compound 5j achieved the target goal as potent in vivo dual COX-2/DHPS inhibitor. In general, the antibacterial characters together with anti-inflammatory properties of the newly synthesised compounds confirmed their ability for further optimisation process as potential antibacterial agents.
| Abbreviations | ||
| COX-2 | = | cyclooxygenase-2 isozyme |
| PGE2 | = | prostaglandin E2 |
| DHPS | = | dihydropteroate synthase enzyme |
| SLS | = | streptococcal cytolysin S |
| SLO | = | streptococcal cytolysin O |
| NSAIDs | = | non-steroidal antiinflammatory drugs |
Supplemental Material
Download PDF (4.8 MB)Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Funding
References
- Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol 2006;119:1737–40.
- Nakayama T, Mutsuga N, Yao L, Tosato G. Prostaglandin E2 promotes degranulation-independent release of MCP-1 from mast cells. J Leukoc Biol 2006;79:95–104.
- Kalinski P. Regulation of Immune Responses by Prostaglandin E2. J Immunol 2012;188:21–8.
- Serezani CH, Chung J, Ballinger MN, et al. Prostaglandin E 2 Suppresses Bacterial Killing in Alveolar Macrophages by Inhibiting NADPH Oxidase. Am J Respir Cell Mol Biol 2007;37:562–70.
- Obermajer N, Wong JL, Edwards RP, et al. PGE(2)-driven induction and maintenance of cancer-associated myeloid-derived suppressor cells. Immunol Invest 2012;41:635–57.
- Blaschke U, Beineke A, Klemens J, et al. Induction of Cyclooxygenase 2 by Streptococcus pyogenes Is Mediated by Cytolysins. J Innate Immun 2017;9:587–97.
- Wang Y, Ren B, Zhou X, et al. Growth and adherence of Staphylococcus aureus were enhanced through the PGE2 produced by the activated COX-2/PGE2 pathway of infected oral epithelial cells. PLoS One 2017;12:e0177166–21.
- Annamanedi M, Kalle AM. Celecoxib sensitizes Staphylococcus aureus to antibiotics in macrophages by modulating SIRT1. PLoS One 2014;9:e99285–9.
- Kalle AM, Rizvi A. Inhibition of bacterial multidrug resistance by celecoxib, a cyclooxygenase-2 inhibitor. Antimicrob Agents Chemother 2011;55:439–42.
- Varma GYN, Kummari G, Paik P, Kalle AM. Celecoxib potentiates antibiotic uptake by altering membrane potential and permeability in Staphylococcus aureus. J Antimicrob Chemother 2019;74:3462–72.
- Ahmed EF, El-baky RMA, Ahmed ABF, et al. Antibacterial activity of some non-steroidal anti-inflammatory drugs against bacteria causing urinary tract infection. Am J Infect Dis Microbiol 2017;5:66–73.
- Elaine WLC, Ling W, Yee ZY, et al. Synergistic effect of non - steroidal anti - inflammatory drugs (NSAIDs) on antibacterial activity of cefuroxime and chloramphenicol against methicillin - resistant Staphylococcus aureus. Integr Med Res 2010;10: 70–4.
- Altaf M, Ijaz M, Ghaffar A, et al. Antibiotic susceptibility profile and synergistic effect of non-steroidal anti-inflammatory drugs on antibacterial activity of resistant antibiotics (Oxytetracycline and Gentamicin) against methicillin resistant Staphylococcus aureus (MRSA). Microb Pathog 2019;137:103755.
- Jeśman C, Młudzik A, Cybulska M. [History of antibiotics and sulphonamides discoveries]. Pol Merkur Lekarski 2011;30:320–2.
- El-attar MAZ, Shaaban OG, Elbayaa RY, et al. Design, synthesis, antibacterial evaluation and molecular docking studies of some new quinoxaline derivatives targeting dihyropteroate synthase enzyme. Bioorg Chem 2018;76:437–48.
- Bano S, Javed K, Ahmad S, et al. Synthesis and biological evaluation of some new 2-pyrazolines bearing benzene sulfonamide moiety as potential anti-inflammatory and anti-cancer agents. Eur J Med Chem 2011;46:5763–8.
- Zhang H, He S, Peng Y, et al. European Journal of Medicinal Chemistry Design, synthesis and antimicrobial evaluation of novel benzimidazole-incorporated sulfonamide analogues. Eur J Med Chem 2017;136:165–83.
- Davison EK, Mcgowan JE, Li FF, et al. Bioorganic & Medicinal Chemistry C-2 derivatized 8-sulfonamidoquinolines as antibacterial compounds. Bioorg Med Chem 2021;29:115837.
- Taha M, Irshad M, Imran S, et al. Synthesis of piperazine sulfonamide analogs as diabetic-II inhibitors and their molecular docking study. Eur J Med Chem 2017;141:530–7.
- Sethi KK, Verma SM, Tanç M, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of the cytosolic mammalian carbonic anhydrase isoforms I, II and VII with benzene sulfonamides incorporating 4,5,6,7-tetrachlorophthalimide moiety. Bioorg Med Chem 2013;21:5168–74.
- Küçükbay FZ, Küçükbay H, Tanc M, Supuran CT. Synthesis and carbonic anhydrase I, II, IV and XII inhibitory properties of N-protected amino acid - sulfonamide conjugates. J Enzyme Inhib Med Chem 2016;31:1476–83.
- Eldehna WM, Al-Ansary GH, Bua S, et al. Novel indolin-2-one-based sulfonamides as carbonic anhydrase inhibitors: synthesis, in vitro biological evaluation against carbonic anhydrases isoforms I, II, IV and VII and molecular docking studies. Eur J Med Chem 2017;127:521–30.
- Buğday N, Küçükbay FZ, Küçükbay H, et al. Synthesis of novel dipeptide sulfonamide conjugates with effective carbonic anhydrase I, II, IX, and XII inhibitory properties. Bioorg Chem 2018;81:311–8.
- Küçükbay H, Buğday N, Küçükbay FZ, et al. Synthesis and carbonic anhydrase inhibitory properties of novel 4-(2-aminoethyl)benzenesulfonamide-dipeptide conjugates. Bioorg Chem 2019;83:414–23.
- Ghorab MM, Ragab FA, Heiba HI, et al. In vitro anticancer screening and radiosensitizing evaluation of some new quinolines and pyrimido[4,5-b]quinolines bearing a sulfonamide moiety. Eur J Med Chem 2010;45:3677–84.
- Al-said MS, Ghorab MM, Al-qasoumi SI, et al. European Journal of Medicinal Chemistry Synthesis and in vitro anticancer screening of some novel benzenesulfonamides. Eur J Med Chem 2010;45:3011–8.
- Liu Y, Chen Y, Zhang HY. Handbook of macrocyclic supramolecular assembly. Singapore: Springer; 2020. doi:10.1007/978-981-13-1744-6
- PEPPERCORN MA, Boston M. Sulfasalazine. Pharmacology, clinical use, toxicity, and related new drug development 1984;101:377–86.
- Hoult RS. Section 1 mode of action pharmacological and biochemical actions of sulphasalazine. Drugs 1986;32:18–26.
- Bach MK, Brashler JR, Johnson MA. Inhibition by sulfasalazine of ltc synthetase and of rat liver glutathione s-transferases. Biochem Pharmacol 1985;34:2695–704.
- Stephenson K, Yamaguchi Y, Hoch JA. The mechanism of action of inhibitors of bacterial two-component signal transduction systems. J Biol Chem 2000;275:38900–4.
- Hilliard JJ, Goldschmidt RM, Licata L, et al. Multiple mechanisms of action for inhibitors of histidine protein kinases from bacterial two-component systems. Antimicrob Agents Chemother 1999;43:1693–9.
- Dunn JA, Coburn RA, Evans RT, et al. Novel topically active antimicrobial and anti-inflammatory compounds for acne. Dermatologic, Cosmeceutic, Cosmet Dev Ther Nov Approaches 2007;243–9.
- El-Nagar MKS, Abdu-Allah HHM, Salem OIA, et al. Novel N-substituted 5-aminosalicylamides as dual inhibitors of cyclooxygenase and 5-lipoxygenase enzymes: synthesis, biological evaluation and docking study. Bioorg Chem 2018;78:80–93.
- Férriz JM, Vávrová K, Kunc F, et al. Salicylanilide carbamates: antitubercular agents active against multidrug-resistant Mycobacterium tuberculosis strains. Bioorg Med Chem 2010;18:1054–61.
- Imramovsky A, Vinšová J, Férriz JM, et al. New antituberculotics originated from salicylanilides with promising in vitro activity against atypical mycobacterial strains. Bioorganic & Medicinal Chemistry 2009;17:3572–9.
- Imramovsky A, Vinšová J, Férriz JM, et al. Salicylanilide esters of N-protected amino acids as novel antimicrobial agents. Bioorganic & Medicinal Chemistry Letters 2009;19:348–51.
- Imramovsky A, Pesko M, Kralova K, et al. Investigating spectrum of biological activity of 4- and 5-chloro-2-hydroxy-n-[2-(arylamino)-1-alkyl-2-oxoethyl]benzamides. Molecules 2011;16:2414–30.
- Deng W, Guo Z, Guo Y, et al. Acryloylamino-salicylanilides as EGFR PTK inhibitors. Bioorg Med Chem Lett 2006;16:469–72.
- Benesova L, Minarik M, Jancarikova D, et al. Multiplicity of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors. Anticancer Res 2010;1672:1667–71.
- Bartram DJ, Leathwick DM, Taylor MA, et al. Veterinary Parasitology The role of combination anthelmintic formulations in the sustainable control of sheep nematodes. Vet Parasitol 2012;186:151–8.
- Bak A, Kos J, Michnova H, et al. Consensus-based pharmacophore mapping for new set of N - (disubstituted-phenyl)-3. Int J Mol Sci 2020;21:6583.
- Brown ME, Fitzner JN, Stevens T, et al. Salicylanilides: selective inhibitors of interleukin-12p40 production. Bioorg Med Chem 2008;16:8760–4.
- Angom RS, Zhu J, Wu AT, et al. LCC-09, a novel salicylanilide derivative, exerts anti-inflammatory effect in vascular endothelial cells. J Inflamm Res 2021;14:4551–65.
- Lima RN, Silva VR, De Santos L, et al. Fast synthesis of amides from ethyl salicylate under microwave radiation in a solvent-free system. RSC Adv 2017;7:56566–74.
- Fahmy HHI, Soliman GA. Synthesis of New Salicylamide Derivatives with Evaluation of Their Antiinflammatory. Analgesic and Antipyretic Activities 2001;24:180–9.
- Gnanaprakasam B, Milstein D. Synthesis of amides from esters and amines with liberation of H 2 under neutral conditions. J Am Chem Soc 2011;133:1682–5.
- Modak A, Dutta U, Kancherla R, Maity S, Bhadra M, Mobin SM, Maiti D. Predictably selective (sp3)C-O bond formation through copper catalyzed dehydrogenative coupling: Facile synthesis of dihydro-oxazinone derivatives. Org Lett 2014;16:2602–5.
- AlFadly ED, Elzahhar PA, Tramarin A, et al. Tackling neuroinflammation and cholinergic deficit in Alzheimer's disease: multi-target inhibitors of cholinesterases, cyclooxygenase-2 and 15-lipoxygenase. Eur J Med Chem 2019;167:161–86.
- Rashad AY, Kassab SE, Daabees HG, et al. Febuxostat-based amides and some derived heterocycles targeting xanthine oxidase and COX inhibition. Synthesis, in vitro and in vivo biological evaluation, molecular modeling and in silico ADMET studies. Bioorg Chem 2021;113:104948.
- Alfayomy AM, Abdel-Aziz SA, Marzouk AA, Shaykoon MSA, et al. Design and synthesis of pyrimidine-5-carbonitrile hybrids as COX-2 inhibitors: anti-inflammatory activity, ulcerogenic liability, histopathological and docking studies. Bioorg Chem 2021;108:104555.
- El-Dershaby NH, El-Hawash SA, Kassab SE, et al. 2021. Design, Synthesis and Biological Evaluation of Some Novel Sulfonamide Derivatives as Dual Antiinflammatory and Antibacterial Agents, Institutional Animal Care and Use Committee (HU-IACUC), Faculty of Science, Helwan University, Egypt.: Approval No. HU2021/Z/AE0721-01.
- El-Hawash SAM, Badawey ESAM, El-Ashmawey IM. Nonsteroidal antiinflammatory agents-part 2 antiinflammatory, analgesic and antipyretic activity of some substituted 3-pyrazolin-5-ones and 1,2,4,5,6,7-3H-hexahydroindazol-3-ones. Eur J Med Chem 2006;41:155–65.
- Shabaan MA, Kamal AM, Faggal SI, et al. Synthesis and biological evaluation of pyrazolone analogues as potential anti-inflammatory agents targeting cyclooxygenases and 5-lipoxygenase. Arch Pharm (Weinheim) 2020;353:1900308.
- Srivastava SK, Nath C, Gupta MB, Vrat S, et al. Protection against gastric ulcer by verapamil. Pharmacol Res 1991;23:81–6.
- Al-Omary FAM, Abou-Zeid LA, Nagi MN, et al. Non-classical antifolates. Part 2: synthesis, biological evaluation, and molecular modeling study of some new 2,6-substituted-quinazolin-4-ones. Bioorg Med Chem 2010;18:2849–63.
- CLSI CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. Clin Lab Standars Inst 2018;32:18.
- Weinstein MP, Patel JB, Burnhman C-A, ZImmer BL. MO7: methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically standard, approval CDM-A. Wayne, PA: Clinical and Laboratory Standards Institute; 2018.
- Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 2001;15:1633–40.
- Yamaguchi K, Domon H, Miyazaki S, et al. In vitro and in vivo antibacterial activities of CS-834, a new oral carbapenem. Antimicrob Agents Chemother 1998;42:555–63.
- Bussiere DE, Xie L, Srinivas H, et al. Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nat Chem Biol 2020;16:15–23.
- Abdellatif KRA, Abdelall EKA, Lamie PF, et al. New pyrazole derivatives possessing amino/methanesulphonyl pharmacophore with good gastric safety profile: design, synthesis, cyclooxygenase inhibition, anti-inflammatory activity and histopathological studies. Bioorg Chem 2020;95:103540.
- Wang M, Lu J, Wang M, et al. Discovery of SHP2-D26 as a First, Potent, and Effective PROTAC Degrader of SHP2 Protein. J Med Chem 2020;63:7510–28.
- Basha A, Lipton M, Weinreb SM. T&r&e&-on Letters No. $3, pp 4171 - 4174, 1977. Tetrahedron Lett 1977;18:4171–4.
- Patrono C. Cardiovascular effects of nonsteroidal anti-inflammatory drugs. Curr Cardiol Rep 2016;18:25–8.
- Wang JL, Limburg D, Graneto MJ, et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: the second clinical candidate having a shorter and favorable human half-life. Bioorg Med Chem Lett 2010;20:7159–63.
- Yun M-K, Wu Y, Li Z, et al. Catalysis and sulfa drug resistance in dihydropteroate synthase. Science 2012;335:1110–4.
- Misal SA, Gawai KR. Azoreductase: a key player of xenobiotic metabolism. Bioresour Bioprocess 2018;5:17.
- Kassab SE, Khedr MA, Ali HI, Abdalla MM. Discovery of new indomethacin-based analogs with potentially selective cyclooxygenase-2 inhibition and observed diminishing to PGE2 activities. Eur J Med Chem 2017;141:306–21.
- Can NÖ, Osmaniye D, Levent S, et al. Design, synthesis and biological assessment of new thiazolylhydrazine derivatives as selective and reversible hMAO-A inhibitors. Eur J Med Chem 2018;144:68–81.
- Molinspiation. Available from: https://www.molinspiration.com/ [last accessed 4 Feb 2022].
- Pre-ADMET. Available from: https://preadmet.bmdrc.kr/ [last accessed 14 Feb 2022].
- Osiris Property Explorer. Available from: https://www.organic-chemistry.org/prog/peo/ [last accessed 16 Feb 2022].
- Zhao YH, Abraham MH, Le J, et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res 2002;19:1446–57.
- Ahsan MJ, Govindasamy J, Khalilullah H, et al. POMA analyses as new efficient bioinformatics’ platform to predict and optimise bioactivity of synthesized 3a,4-dihydro-3H-indeno[1,2-c]pyrazole-2-carboxamide/carbothioamide analogues. Bioorg Med Chem Lett 2012;22:7029–35.
- Hassan NW, Saudi MN, Abdel-Ghany YS, et al. Novel pyrazine based anti-tubercular agents: design, synthesis, biological evaluation and in silico studies. Bioorg Chem 2020;96:103610.
- Elzahhar PA, Abd El Wahab SM, Elagawany M, et al. Expanding the anticancer potential of 1,2,3-triazoles via simultaneously targeting Cyclooxygenase-2, 15-lipoxygenase and tumor-associated carbonic anhydrases. Eur J Med Chem 2020;200:112439.
- Hopkins AL, Keserü GM, Leeson PD, et al. The role of ligand efficiency metrics in drug discovery. Nat Rev Drug Discov 2014;13:105–21.