
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
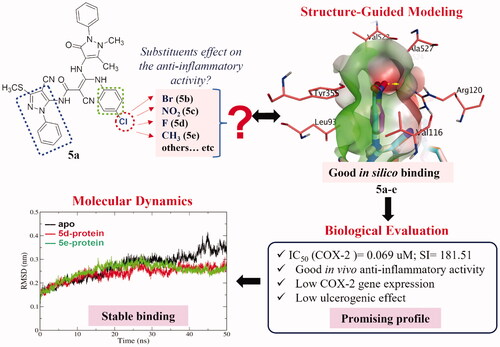
A structure-guided modelling approach using COX-2 as a template was used to investigate the effect of replacing the chloro atom located at the chlorophenyl ring of amide-linked bipyrazole moieties, aiming at attaining better anti-inflammatory effect with a good safety profile. Bromo, fluoro, nitro, and methyl groups were revealed to be ideal candidates. Consequently, new bipyrazole derivatives were synthesised. The in vitro inhibitory COX-1/COX-2 activity of the synthesised compounds exhibited promising selectivity. The fluoro and methyl derivatives were the most active candidates. The in vivo formalin-induced paw edoema model confirmed the anti-inflammatory activity of the synthesised compounds. All the tested derivatives had a good ulcerogenic safety profile except for the methyl substituted compound. In silico molecular dynamics simulations of the fluoro and methyl poses complexed with COX-2 for 50 ns indicated stable binding to COX-2. Generally, our approach delivers a fruitful matrix for the development of further amide-linked bipyrazole anti-inflammatory candidates.
Graphical Abstract
1. Introduction
Inflammation is a protective reaction performed by body tissues in response to harmful stimuli, resulting in pain, heat, redness, and/or swelling. This process, if performed correctly, usually leads to recovery and healingCitation1. However, if it is not properly controlled, inflammation might result in persistent tissue damageCitation2.
Arachidonic acid is an important substrate in the biosynthesis of mediators called eicosanoids, usually produced during inflammatory processes. Examples of which are leukotrienes as the products of 5-lipoxygenase (5-LOX) in addition to prostaglandins (PGs) and thromboxanes (TXs) as the products of cyclooxygenases (COX)Citation3–5. Other mediators and other signalling molecules that are usually secreted by the immune system during inflammation include histamine, oxygen- and nitrogen-derived free radicals, and serotoninCitation6. In regular tissues, the production of prostaglandin, the lipid autacoid generated during the metabolism of arachidonic acid by COX enzymes, is usually very low. However, a dramatic increase in its production is observed in inflamed tissuesCitation7. Accordingly, COXCitation8,Citation9 is the target of most anti-inflammatory agents, namely nonsteroidal anti-inflammatory drugs (NSAIDs)Citation10. Nevertheless, drugs treating inflammation by targeting other mechanisms are availableCitation11.
COX enzymes exist in several distinct isoforms; the most important of which in inflammation are COX-1 and COX-2Citation10,Citation12. The constitutive COX-1 is responsible for cytoprotection of the gastrointestinal (GI) tract together with controlling renal function, while the inducible COX-2 is upregulated by pro-inflammatory mediators such as endotoxins, mitogens, or cytokines, and is thus considered a major cause of inflammatory conditionsCitation13. Despite the important effects of cytokines in the activity of many cells, they are highly implicated in the development of inflammation as a result of bacterial infection and/or exposure to lipopolysaccharides (LPS)Citation14–17. The production of proinflammatory cytokines, not only results in upregulation of COX-2, but also leads to the release of interleukin (IL)-1β, IL-8, tumour necrosis factor-alpha (TNF-α), IL-6, and IL-12 which are highly implicated in inflammation.
The co-existence of these two distinct isoforms of COX enzymes presents an argument regarding the benefit/risk ratio of using selective versus non-selective COX inhibitors. Although long-term use of non-selective NSAIDs is known to result in gastrointestinal disorders such as ulceration, the long-term use of COX-2 selective inhibitors with high selectivity index results in cardiovascular (CV) disordersCitation18. Accordingly, a balance between the inhibitory effect of anti-inflammatory drugs on COX-2 versus COX-1 is highly recommended to avoid the aforementioned side effectsCitation19. Consequently, the development of effective anti-inflammatory agents with minimal side effects still represents a major challenge to all research aimed at relieving inflammatory conditions.
The pyrazole nucleus is a key structural motif in heterocyclic and medicinal chemistry due to its ability to display a wide range of bioactivities including antimicrobial, anticancer, and anti-inflammatory. In fact, numerous pyrazole compounds have already been used in clinical settings as nonsteroidal anti-inflammatory medications, namely phenylbutazone, sulfinpyrazone, celecoxib, and others. Later on, a number of pyrazole nucleus alterations were carried out to create new compounds with safer profiles than the ones already present. As an example, Tewari et al., using nimesulide as the reference medication, synthesised a novel series of pyrazole derivatives and tested them in vivo for their anti-inflammatory effect in a carrageenan-induced rat paw edoema model. According to molecular modelling studies, pyrazole analogs interact with the COX-2 active site by forming hydrogen bonds, π–π interaction, and cation–π interaction. These interactions prolong the residence time of the ligand in the active site, increasing the anti-inflammatory activity of the compoundsCitation20. Keche et al. also synthesised a series of pyrazole derivatives that showed promising anti-inflammatory activity by inhibiting significantly TNF-α and interleukin-6 relative to dexamethasoneCitation21. Moreover, pyrazole nucleus is the core structure of SC-558 ()Citation20. Structure-activity relationship studies on celecoxib and SC-558 revealed some essential features for binding with the COX-2 enzyme active site, which include the necessity of the presence of N1-, C3- and C5-substituents. In addition, the presence of a substituted phenyl ring at the 5-position appeared to be crucial for the complete binding of these compounds with the active site of the COX-2 enzyme ()Citation22. As a part of our continuous search for potent anti-inflammatory agents with minimal gastrointestinal side effects, compounds bearing amide-linked bipyrazole moieties have proven to be highly beneficial in reducing inflammation without causing the expected gastrointestinal ulcerationCitation2. Furthermore, the presence of a chlorophenyl ring on the side chain present at the C5-position proved to increase the protective effect against induced paw edoema by 1.5-fold higher than the chloro-free derivative.
Figure 1. Logical flow and the rational of the study.

In this study, we aim to explore the possible reason for the pronounced effect of the chloro atom at the para-position of the phenyl ring on the side chain present at the C5-position. We started with structure-guided modelling using COX-2 as a template for fine-tuning the possible substitution pattern via the aid of a docking protocol. The employed substituents were selected to be of various topologies (such as size and weight) and electronic features (electron withdrawing and donating). Then, we executed a synthetic protocol to afford the development of the recommended substituents from the modelling approach. Subsequently, these compounds were biologically evaluated for their in vitro anti-inflammatory activities against COX-1 and COX-2. Moreover, in vivo formalin-induced paw edoema test, acute ulcerogenic testing, and the COX/LOX gene expression profile quantified via real-time PCR were performed. Finally, the most active compounds were subjected to molecular dynamics (MD) simulations to confirm their stable binding to COX-2. A brief description of the study logical flow is shown in .
2. Methods and materials
2.1. Chemistry
All reagents and solvents were purchased from local suppliers and were dried and purified when necessary, by standard techniques. Melting points were determined in open glass capillaries using Thomas–Hoover melting point apparatus. Infra-red spectra (IR) were recorded in cm−1, using KBr discs, by a Perkin-Elmer 1430 Infra-red spectrophotometer. Nuclear magnetic resonance (1H-NMR and 13 C-NMR) were determined using a Bruker 300 ultrashield spectrophotometer and dimethyl sulfoxide d6 (DMSO-d6) as a solvent. The data were reported as chemical shifts or δ values (ppm) relative to tetramethylsilane (TMS) as internal standard. Elemental microanalyses were performed at the regional centre for Mycology and Biotechnology, Al-Azhar University, and the values were within ± 0.4% of the theoretical values. Reaction progress was monitored by thin-layer chromatography (TLC) on silica gel sheets (60 GF254, Merck). The spots were visualised by exposure to iodine vapour or UV-lamp at λ 254 nm for few seconds.
2.1.1. 5-Amino-3-methylsulfanyl-1H-pyrazole-4-carbonitrile (2)Citation2
A solution of 2-bis(methylthio)methylene)malononitrile (17 g, 100 mmol) and phenyl hydrazine (10.8 g, 100 mmol) in methanol (200 ml) was heated under reflux for 3 h. The reaction mixture was concentrated under reduced pressure and the separated solid product was filtered and recrystallised from methanol to give colourless needles (22 g, 96%), mp; 140–142 °C; reported mp: 141–142 °CCitation2.
2.1.2. 2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)acetamide (3)Citation2
The aminopyrazole 2 (4.6 g, 20 mmol) was added to a mixture of cyanoacetic acid (2.55 g, 30 mmol) and acetic anhydride (30 ml) and heated at 50 °C for 5 h. The mixture was allowed to cool then poured into ice-cold water. The precipitate formed was filtered, washed with water, dried, and crystallised from methanol. Colourless needles (4.4 g, 74%), m.p: 186–187 °C. IR (KBr, cm−1): 3253 (NH), 3048, 2950, 2907 (CH), 2224 (CN), 1704 (C = O), 1594 (C = N), 1563, 1524, 1459 (C = C), 1319, 1075 (C-S-C). 1H-NMR (300 MHz, DMSO-d6, δ ppm): 2.59 (s, 3H, SCH3), 3.99 (s, 2H, CH2), 7.46–7.60 (m, 5H, phenyl-H), 11.17 (s, 1H, NH, D2O exchangeable). 13 C-NMR (75 MHz, δ ppm): 14.09 (CH3), 26.46 (CH2), 89.10 (pyrazole C4), 112.24, 115.58 (CN), 124.54 (phenyl C2,6), 129.35 (phenyl C4), 130.03 (phenyl C3,5), 137.41 (phenyl C1), 141.21 (pyrazole C3), 150.77 (pyrazole C5), 162.65 (C = O). Anal. Calcd for C14H11N5OS (297.34): C, 56.55; H, 3.73; N, 23.55. Found: C, 56.68; H, 3.72; N, 23.74.
2.1.3. General procedure for the synthesis of 2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3-(methylsulfanyl)-3- (substituted amino) acrylamides (4a-e)
The cyanoacetamide derivative 3 (1.19 g; 4 mmol) was added to a well-stirred ice-cold suspension of finely divided KOH (0.22 g; 4 mmol) in dry DMF (8 ml) followed by the appropriate aryl isothiocyanate (4 mmol). The mixture was stirred at 0–5 °C for 2 h, allowed to reach room temperature then stirring was continued for an overnight. Dimethyl sulphate (0.5 g, 0.38 ml, 4 mmol) was then added and stirring was maintained for 24 h. The reaction mixture was poured onto ice-cold water and the separated product was extracted with chloroform three times. The combined chloroform extract was dried over anhydrous Na2SO4. The chloroform was evaporated under reduced pressure leaving an oily compound that was used into the next step without further purification.
2.1.4. General procedure for the synthesis of 2-Cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3–(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-ylamino)-3-(substituted amino)acrylamides (5a-e)
A mixture of the acrylamide 4 (5 mmol) and 4-aminoantipyrine (1 g; 5 mmol) was fused at 170–180 °C in oil bath for 2 h. Ethanol (20 ml) was added and the reaction mixture was refluxed for additional 2 h then concentrated and left to stand for an overnight. The separated solid product was filtered, washed with ethanol, dried, and crystallised from ethanol–DMF (4:1). IR (KBr, cm1): 3376, 3312, 3187 (NH), 2228–2224 (CN), 1662, 1631 (C = O), 1590 (C = N), 1308, 1093 (C-S-C).
2.1.5. 3-(Chlorophenylamino)-2-cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3–(2,3-dihydro-1,5-dimethyl-3-oxo-2- phenyl-1H-pyrazol-4-ylamino)acrylamide (5a)
Light brown crystals (2.35 g, 74%), mp: 294–295 °CCitation2.
2.1.6. 3-(Bromophenylamino)-2-cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3–(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-ylamino)acrylamide (5 b)
Light brown crystals (2.89 g, 85%), mp: 284–285 °C, 1H-NMR (300 MHz, DMSO-d6, ppm): 2.41 (s, 3H, CCH3), 2.68 (s, 3H, S-CH3), 3.16 (s, 3H, N-CH3), 7.42–7.54 (m, 14 H, aromatic-H), 8.160, 10.09, 12.75 (3 s, each 1H, 3NH, D2O exchangeable). 13 C-NMR (75 MHz, ppm): 14.05 (C-CH3), 16.08 (SCH3), 35.82 (N-CH3), 99.84 (pyrazole C4), 107.39 (acrylamide C2), 114.13 (pyrazolinone C4), 116.26, 116.53 (CN), 120.38 (pyrazolinone phenyl C2,6), 121.99 (chlorophenylamino C2,6), 125.64 (pyrazolinone phenyl C4), 126.56 (phenyl C4), 127.92 (phenyl C2,6), 128.39 (Chlorophenylamino C4), 129.50 (pyrazolinone C5), 129.75 (pyrazolinone phenyl C3,5), 129.98 (phenyl C3,5), 131.96 (phenylamino C3,5), 138.87 (pyrazolinonephenyl C1), 141.73 (phenyl C1), 143.39 (phenylamino C1), 152.79 (pyrazole C3), 153.91 (pyrazole C5), 155.04, 157.62 (C = O), 170.80 (acrylamide C3). Anal. Calculated for C32H26BrN9O2S (680.58): C, 56.47; H, 3.85; N, 18.52. Found: C, 56.29; H, 3.63; N, 18.59.
2.1.7. 3-(Nitrophenylamino)-2-cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3–(2,3-dihydro-1,5-dimethyl-3-oxo-2- phenyl-1H-pyrazol-4-ylamino)acrylamide (5c)
Yellowish white crystals (2.39 g, 74%), mp: 279–281 °C, 1H-NMR (300 MHz, DMSO-d6, ppm): 2.70(s, 3H, CCH3), 2.74 (s, 3H, S-CH3), 3.08 (s, 3H, N-CH3), 7.34–8.19 (m, 14 H, aromatic-H), 8.45, 10.55, 12.85 (3 s, each 1H, 3NH, D2O exchangeable). 13 C-NMR (75 MHz, ppm): 12.61 (C-CH3), 14.08 (SCH3), 36.49 (N-CH3), 108.08 (pyrazole C4), 112.85 (acrylamide C2), 114.85 (pyrazolinone C4), 118.88, 119.48 (CN), 121.25 (pyrazolinone phenyl C2,6), 123.35 (chlorophenylamino C2,6), 124.66 (pyrazolinone phenyl C4), 125.77 (phenyl C4), 126.52 (phenyl C2,6), 128.38 (Chlorophenylamino C4), 129.23 (pyrazolinone C5), 129.40 (pyrazolinone phenyl C3,5), 129.75 (phenyl C3,5), 129.97 (phenylamino C3,5), 137.76 (pyrazolinonephenyl C1), 138.74 (phenyl C1), 141.27 (phenylamino C1), 149.39 (pyrazole C3), 152.80 (pyrazole C5), 156.18, 157.03 (C = O), 162.81(acrylamide C3). Anal. Calculated for C32H26N10O4S (646.68): C, 59.43; H, 4.05; N, 21.66. Found: C, 59.19; H, 4.33; N, 21.59.
2.1.8. 3-(Fluorophenylamino)-2-cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3–(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-ylamino)acrylamide (5d)
Light brown crystals (2.14 g, 69%), mp: 291–292 °C, 1H-NMR (300 MHz, DMSO-d6, ppm): 2.52 (s, 3H, CCH3), 2.72 (s, 3H, S-CH3), 3.17 (s, 3H, N-CH3), 6.8–7.55 (m, 14 H, aromatic-H), 8.15, 8.55, 12.79 (3 s, each 1H, 3NH, D2O exchangeable). 13 C-NMR (75 MHz, ppm): 13.67 (C-CH3), 16.294 (SCH3), 35.80 (N-CH3), 97.48 (pyrazole C4), 99.51 (acrylamide C2), 115.33 (pyrazolinone C4), 116.36, 117.12 (CN), 120.95(pyrazolinone phenyl C2,6), 123.73 (chlorophenylamino C2,6), 124.65 (pyrazolinone phenyl C4), 125.09 (phenyl C4), 125.73 (phenyl C2,6), 126.70 (Chlorophenylamino C4), 127.25 (pyrazolinone C5), 128.40 (pyrazolinone phenyl C3,5), 129.83 (phenyl C3,5), 129.98 (phenylamino C3,5), 139.02 (pyrazolinonephenyl C1), 141.77 (phenyl C1), 142.28 (phenylamino C1), 150.18 (pyrazole C3), 155.07 (pyrazole C5), 157.45, 160.65 (C¼O), 170.78 (acrylamide C3). Anal. Calculated for C32H26FN9O2S (619.67): C, 62.02; H, 4.23; N, 20.34. Found: C, 62.17; H, 4.39; N, 19.99.
2.1.9. 3-(Tolylamino)-2-cyano-N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl)-3–(2,3-dihydro-1,5-dimethyl-3-oxo-2-phenyl-1H-pyrazol-4-ylamino)acrylamide (5e)
Yellowish crystals (2.19 g, 71%), mp: 287–289 °C, 1H-NMR (300 MHz, DMSO-d6, ppm): 2.44 (s, 3H, pyrazolo-CH3), 2.70 (s, 3H, S-CH3), 2.9 (s, 3H, phenyl-CH3), 3.17 (s, 3H, N-CH3), 7.43–7.55 (m, 14 H, aromatic-H), 7.97, 8.16, 12.8 (3 s, each 1H, 3NH, D2O exchangeable). 13 C-NMR (75 MHz, ppm): 10.39, 13.76 (phenyl-C-CH3and pyrazolo-C-CH3), 14.92 (SCH3), 35.84 (N-CH3), 73.77 (pyrazole C4), 99.25 (acrylamide C2), 101.3 (pyrazolinone C4), 115.2, 116.80 (CN), 120.21 (pyrazolinone phenyl C2,6), 121.03 (chlorophenylamino C2,6), 123.73 (pyrazolinone phenyl C4), 125.72 (phenyl C4), 126.61 (phenyl C2,6), 128.38 (Chlorophenylamino C4), 129.23 (pyrazolinone C5), 129.46 (pyrazolinone phenyl C3,5), 129.67 (phenyl C3,5),130.53 (phenylamino C3,5), 137.764 (pyrazolinonephenyl C1), 138.66 (phenyl C1), 149.40 (phenylamino C1), 151.22 (pyrazole C3), 152.80 (pyrazole C5), 157.01, 160.51 (C = O), 162.81 (acrylamide C3). Anal. Calculated for C33H29N9O2S (615.71): C, 64.37; H, 4.75; N, 20.47. Found: C, 64.19; H, 4.53; N, 20.59.
2.2. Molecular Modelling
2.2.1. Molecular docking
Coordinates for COX-1 and COX-2 crystal structures were retrieved from the Protein Data Bank (PDB codes: 1EQG and 6BL4, respectively) and handled consequently with Molecular Operating Environment program (MOE)Citation23. Unneeded chains, ions, water, and other molecules were removed. Then preparation was performed via “QuickPrep” module at default settings.
The molecules were built and prepared as reported earlierCitation24 using MOE. The molecules were further converted into PDBQT files by an MGLTools (version 1.5.4) python script (prepare_ligand4.py)Citation25. The 3D and 2D depictions of ligand-protein interactions were produced using MOE.
The docking experiments against COX-1 and COX-2 were performed using VinaXB and rescored in-place using AutoDock Vina (version 1.1.2). The size of the docking grid was 22 Å × 22 Å × 22 Å, with a grid spacing of 1 Å for both proteins. To enhance exhaustive sampling of the generated poses, the exhaustiveness parameter was set at 27 (three times default sampling). The rest of the settings were kept at default.
2.2.2. Molecular Dynamics
The molecular dynamics simulations were carried out as reported earlierCitation26,Citation27. Molecular dynamics simulations and systems build up were carried out using GROMACS 2020.3Citation28,Citation29. CHARMM36 all-atom force field was applied for topology and parameter generation of the protein molecules, and SwissParam server was utilised for ligands 5d and 5e parameterisation. Analysis metrics, such as, RMSD, Rg, RMSF and H-bond count were calculated via GROMCS. All analysis charts were constructed using XMGRACECitation30,Citation31.
2.3. Biological screening
2.3.1. Animal
Male mice weighing between 20–25 g were obtained from the animal house of the Faculty of Pharmacy, Beirut Arab University, Lebanon. Animals were housed in standard polypropylene cages, four per cage, under a 12:12-h light/dark cycle with free access to food and water.
2.3.2. Ethical consideration
All experiments were performed at Beirut Arab University laboratories after obtaining approval from the Investigation Review Board under the number 2022-H-0077-P-R-0467. Animal care and handling for the research were performed in accordance with the regulations and guidelines stipulated by the Institutional Animal Care and Use Guidelines (IACUG) at Beirut Arab University, Lebanon, certified by the Ministry of Public Health: (1/141).
2.3.3. In vitro COX study
The inhibitory COX activity of the tested compounds and the reference was quantified via enzyme immunoassay using Cayman colorimetric COX (ovine) inhibitor screening assay kit (Catalog No. 560131, Cayman Chemicals, Ann Arbour, MI). The reaction buffer (0.1 N TrisHCl, pH 8.0, containing 5 mM EDTA and 2 mM phenol), haem, and inactive COX-1 or inactive COX-2 was placed in the designated tubes. The tubes were incubated for 10 min at 37 °C. The reaction was then initiated by adding arachidonic acid to all the test tubes, which were vortexed and incubated at 37 °C for another 2 min. To stop the enzyme catalysis, 1 M of HCL was added to each tube. Reagents were pipetted into the 96-well plate to perform the enzyme immunoassay. The plate was developed then by Ellman’s Reagent. The absorbance was measured at a wavelength between 405 and 420 nm using a 96-well Tecan Safire plate reader. Indomethacin and celecoxib were used as reference standards in the study. The assays were performed in triplicate and the IC50 values were calculated from the concentration curves using the GraphPad software PRISM.
2.3.4. Formalin-induced paw edoema assay
The anti-inflammatory effect of the studied compounds was assessed using formalin-induced paw edoema. The dose of the Chloro derivative (5a) was applied, according to our previous reportsCitation32, while the doses of the other derivatives were calculated as equimolar to 5a. This experiment consist of seven groups (n = 5 mice in each group). Compounds were injected intraperitoneally to mice, 30 min before subplantar injection of formalin. The negative control group received only the vehicle. Groups of animals pre-treated with indomethacin (5 mg/kg) or celecoxib (20 mg/kg) were considered as positive controls. Mice received a subplantar injection of 200 μl of a 5% (w/v) of formalin at the right hind paw. The volume of the paw was measured by plethysmometer (Ugo Basile, Italy) immediately after and then 4 h after the formalin injection to determine the difference in paw volume. The percentages of paw edoema inhibition were calculated using the following equations:
After the procedure, the animals were sacrificed and the inflamed paw tissues were cut. They were frozen in liquid nitrogen and stored at −80 °C until they were used for RT-PCR.
2.3.5. Real-time PCR
RT-PCR was performed for the detection of gene expression of COX-2, COX-1, and LOX. Total RNA was isolated from homogenised paw tissues using TRIzol reagent. The concentration and quality of RNA preparations were determined spectrophotometrically (ND-1000 Spectrophotometer, NanoDrop Technologies, Wilmington, Delaware, USA) at 260 and 260/280 nm. Standardised amounts of RNA were reverse-transcribed to cDNA using the QuantiTect Reverse Transcription kit.
The primers sequences for COX-2, COX-1, and LOX as well as the housekeeping gene were designed from the sequence list of the GeneBank database (National Centre for Biotechnology Information, NCBI) using Beacon designer 8 software, and then blasted against the GeneBank database sequences.
Real-Time PCR was performed using 5x FIREPol® EvaGreen® qPCR Mix detected by the Rotor-Gene Q thermocycler (Qiagen, USA). The Master Mix, in each reaction tube, includes cDNA, H2O, SYBR Green, as well as the forward and reverse primer of the genes of interest.
The cycling conditions were as follows: initial denaturation at 95 °C for 15 min and 45 cycles of amplification (denaturation at 95 °C for 15 s, annealing at 56 °C for 30 s, and extension at 72 °C for 45 s). Mouse COX-2, COX-1, and LOX, mRNA relative expression levels were determined by using the 2−ΔΔCt method.
2.3.6. Acute ulcerogenic side effect of the compounds
After sacrificing the animals, their stomach were dissected along the greater curvature, rinsed with saline, pinned on a dissecting tray, and examined for the presence of ulcers. An ulcer index was calculated using the Abouzeit–Har scale where normal stomach scored 0, petechial haemorrhage 1, ulcers <1 mm long 3, from 1 to <2 mm long 4, from 2 to <3 mm long 5, from 3 to 4 mm long 6, and very large ulcers received 7 pointsCitation33. The ulcer index for each stomach was obtained by the summation of the total ulcer points.
2.3.7. Statistical analysis
The data are presented as means ± SD. The differences between control and treatment groups were tested by one-way analysis of variance (ANOVA) followed by the Tukey post hoc test. Differences were considered significant when p < 0.05. Statistical analysis was performed using the SPSS 23 software.
3. Results and discussion
3.1. Rational and modelling
We have previously reported the synthesis and anti-inflammatory activity of some new amide-linked bipyrazoles, amongst which compounds I and its chloro congener 5a () showed interesting biological results. Remarkably, 5a exhibited a significantly superior anti-inflammatory profile compared with its parent compound ICitation2. This observation posed an important question about the importance of the substitution pattern at this specific position. Investigating this in the current study, we modelled different substitution patterns. We replaced the chloro atom with fluoro and bromo atoms (representing other halogens), nitro group (as an electron withdrawing group), methyl group (simple alkyl group), isopropyl moiety (branched alkyl group), and different ring systems, e.g. phenyl and 5-membered system. Amusingly, substitution patterns with the fluoro, bromo, nitro and methyl groups produced acceptable docking scores and were able to occupy the narrow cleft formed by Val116, Arg120, Tyr355, and Ala527 in the COX-2 enzyme (more details are in the Molecular Docking Section). However, larger substituents, including isopropyl, and 5- and 6-membered ring system, demonstrated a visible steric clash with the residues of narrow cleft, and hence exhibited modest scores, as shown in .
Figure 2. (A) The docking poses of 5a-e for chloro, bromo, nitro, fluoro and methyl substituents, as cyan, pale rose, orange and purple sticks, respectively, in the binding site of COX-2. (B) Focussed view at the substituents of 5a-e. (C), (D) and (E), Focussed view at substituents isopropyl, 5-membered dummy ring and 6-membered phenyl groups, respectively. Polar and non-polar regions of the binding site were presented by red and green coloured molecular surface, respectively. Dashed lines indicate favourable interactions, while red arrows represent steric clashes and unfavourable interactions. Non-polar hydrogen atoms were omitted for clarity.

Guided by these findings, a group of compounds reflecting chloro, fluoro, nitro, and methyl groups (5a-e; and Citation2(A)) were synthesised in this study and subjected to thorough biological testing to evaluate their anti-inflammatory and safety profiles.
3.2. Chemistry
The synthesis of the intermediate and target compounds is illustrated in the following scheme (). The starting compounds, 5-amino-3-methylsulfanyl-1Hpyrazole-4-carbonitrile (2), and the cyanoacetamide derivative (3) were prepared according to the procedure reported in the literatureCitation2,Citation34. The ketene N,S-acetals (4a-e) were prepared through a two-step procedure to prevent hydrolysis of the CN group into the corresponding CONH2. In the first step, the appropriate aryl isothiocyanate was added to an ice-cold mixture of 3 and KOH in DMF at 0–5 °C, cooling was maintained for a further 2 h, then the reaction mixture was allowed to reach room temperature and left for overnight to attain complete reaction with the aryl isothiocyanates. The second step in the afore-mentioned procedure involves the addition of dimethyl sulphate to the reaction to accomplish the final required S-methylation of the products. The obtained products were separated as an oily material that was used as crude without purification to prepare the target compounds 5a-e through their fusion with 4-aminoantipyrine at 170 °C. IR spectra of the final products 5a-e indicated the presence of three NH bands, a signal for the cyano group and two carbonyl signals. 1H-NMR spectra of the synthesised target compounds revealed the presence of three or four singlets equivalent to the three or four aliphatic methyl protons in compounds 5b-d and 5e, respectively. Fourteen aromatic protons appeared as a multiplet and finally, the three D2O exchangeable protons of the three NH groups appeared at their expected downfield shifts (refer to ).
Figure 3. Synthesis of the target compounds 5a-e. Reagents and reaction conditions: i: C6H5NH2, EtOH, Reflux; ii: cyanoacetic acid, acetic anhydride, 50 °C; iii: 4-R-C6H4NCS, KOH, DMF, 0 °C; iv: 4-aminoantipyrine, oil bath at 170 °C.

3.3. Molecular Modelling
3.3.1. Molecular Docking
This section delivers in silico insights on recommending 5a-e compounds for in vitro COX-2/COX-1 inhibitory activity.
3.3.1.1. Selection of the docking tool and rescoring
AutoDock VinaCitation35 as a docking tool was able to show an enhanced screening performance using the DEKOIS 2.0Citation36,Citation37 benchmark set for COX-1 and COX-2 enzymes. This indicates an efficient recognition of the active ligands in a pool of their false positive decoysCitation36,Citation37. In addition, performing docking employing AutoDock Vina produced successful pose-retrieval for the X-ray co-crystallised ligand for both enzymes. These findings encouraged us to use AutoDock Vina as a predictive tool for 5a-e compounds. Exploring the halogen-bond (XB) effect, as a possible interaction component for 5a (with chloro group), 5 b (bromo group) and 5d (fluoro group), directed us to utilise the modified version of AutoDock Vina that includes an XB scoring term, referred to as AutoDock VinaXB.Citation38,Citation39 Therefore, we employed AutoDock VinaXB as a guiding docking tool for our pose and scoring prediction. In addition, to pinpoint whether XB has an impact on the binding event of these compounds or not, we re-scored in-place the produced poses by VinaXB with the original AutoDock Vina. If identical scores were reproduced by AutoDock Vina and VinaXB, this would suggest the absence of the XB term contribution to docked poses. shows the identical scores for both VinaXB and AutoDock Vina for the 5a-e indicating that there is no contribution of the XB term in VinaXB. This can be verified via visual inspection ( and Citation4) since the backbone carbonyl groups (XB-acceptor) of the narrow cleft are in the far proximity to the halogen groups of 5a,b,d (XB-donor).
Table 1. The docking score of the active compounds in the binding site of COX-2.
3.3.1.2. Selection of an X-ray crystal structure and description of the docking poses
For the COX-1 enzyme, the X-ray co-crystal structure of PDB code: 1EQG was selected for docking simulations. Exploring the docking score distribution of the synthesised compounds against COX-1 revealed low in silico binding (data not shown). On the other hand, for COX-2, we utilised the X-ray co-crystal structure of PDB code: 6BL4 for our docking runs. The docking scores of 5a-5e showed a narrow range distribution (-7.8 to −8.1) indicating comparable in silico affinity of these compounds. Checking the docking scores revealed that celecoxib had the best score compared with all docked compounds, while indomethacin lied within the score range of 5a-5e.
Predicting the selectivity of 5a-5e towards COX-2 over COX-1, we found that these compounds cannot afford a specific binding to the binding site of the COX-1. This finding is likely ascribable to the fact that the general size and topology of the COX-1 binding site is smaller than that of COX-2Citation40–42. Besides, the average molecular weight of 5a-e is 645.7 ± 25 g/mol, which is obviously above the average range of the diverse and representative COX-1 and COX-2 ligands reported in DEKOIS 2.0 benchmark sets (the average molecular weights of COX-1 and COX-2 in DEKOIS 2.0 benchmark set are 333.8 ± 62.97 and 373.6 ± 50.65, respectively).
All predicted compounds 5a-e showed comparable in silico binding poses and interaction pattern in COX-2, as shown in . The postulated binding poses of 5a, 5 b, 5d and 5e show H-bonding interactions with Arg120 and Tyr115, and several hydrophobic interactions with the hydrophobic side chains of the binding site of COX-2, as seen in . For instance, the chlorophenylamino group (5a) exhibits hydrophobic interactions with the hydrophobic residues of the narrow cleft, e.g. Val116, Tyr355 and Ala527. Furthermore, the N-(4-cyano-3-(methylsulfanyl)-1-phenyl-1H-pyrazol-5-yl) moiety displayed favourable interactions with Lys83, Arg120 and Tyr115, respectively, as well as hydrophobic interaction with Leu123. Interestingly, the other oxo-phenyl-pyrazolyl moiety shows favourable hydrophobic interactions with ILE112 and Val89. Upon elucidating the substituents of interest, all of them were packed in the narrow cleft as elaborated in the “Rational Section” forming mainly favourable hydrophobic interactions with the surrounding residues. In addition, the nitro group of 5c appeared to possess an H-bonding interaction with the side chain of Arg120. Overall, these predictions endorse 5a-e compounds for further biological testing and in vitro COX-2/COX-1 inhibition assay.
Figure 4. (A) Overlay of the docking pose of 5a (cyan), 5b (pale rose), 5d (orange) and 5e (purple) in the binding site of COX-2 (PDB: 6BL4). Non-polar hydrogen atoms were omitted for clarity. (B) Interactions of 5a docking pose with COX-2 protein in a 2 D depiction.

3.3.2. Molecular Dynamics simulations
After performing the in vitro anti-inflammatory assay against COX-1 and COX-2, compounds 5d and 5e demonstrated the best activities (more details are in the Biological Evaluation Section). We performed 50 ns molecular dynamics (MD) simulations for 5d and 5e poses to assess their stability while binding to COX-2 in a time-dependent manner. In addition, another MD run was conducted for the apo COX-2 form as a reference for comparison purposes. This results in a total of three 50 ns MD simulation runs.
An analysis of root mean square deviation (RMSD) is displayed in . RMSD is a measure of protein stability during the simulation time and measures the alpha carbon atoms of the protein backbone. RMSD of the apo and complexed (5d and 5e) forms () are comparable to each other for the first 25 ns of simulation, with a RMSD range of 0.15–0.3 nm. This reflects an acceptable low range of 0.15 nm RMSD for them all. However, further simulation time from 25 to 50 ns reflects more steady behaviour for both complex forms compared with higher fluctuations for the apo form. The dynamics of these two complexed forms converged after 25 ns of simulation, highlighting the idea that the structural changes present in the complex converged to a more stable structure compared with the apo form. This indicates that the binding poses of both 5d and 5e are capable to stabilise the protein in a time-dependent manner.
Figure 5. (A) Root mean square deviation (RMSD) of the protein alpha carbon atoms during the simulation for the apo and complexed forms with 5d and 5e. (B) Radius of gyration (Rg) for the COX-2 protein in the apo and complexed forms with 5d and 5e during the simulation time. The 50000 ps simulation time (x- axis) implies 50 ns time. (C) Per residue root mean square fluctuation (RMSF) for the apo and complexed forms with 5d and 5e. (D) and (E) Hydrogen bond count during the MD simulation for both 5d and 5e in the binding site, respectively.

This is also in coherence with the analysis obtained by the Radius of gyration (Rg) in . Rg is a measure of protein structure compactness during the simulation time. There is no great fluctuation in the Rg of the protein complex with 5d or 5e compared with the apo structure. The only exception can be observed after 40 ns to 50 ns where the apo form showed higher fluctuations in the Rg compared with both complexed forms. This reflects that the protein undergoes lower conformational changes in the complexed form with either 5d or 5e throughout the simulation, and hence, better stability compared with the apo formCitation43,Citation44. The residue root mean square fluctuation (RMSF) assesses the conformational changes that occur to each residue of the protein, as shown in . Generally, the apo and complexed forms exhibit comparable per residue fluctuations profiles. The protein structural loops showed higher fluctuations especially for the apo form. For instance, at the N-terminal residues Ala33-Glu73 displayed high RMSF values reached to 0.5 nm for the apo form. Besides, loop residues His207-Leu230 exhibited values up to 0.4 nm for the apo form and lower values (< 0.3 nm) for the complexed forms. These values contemplated by the high flexibility of such structural loops. However, the key binding site amino acids (numbers: Ly83, Val 89, Arg120, Tyr255 and Glu524) showed lower RMSF values (< 0.2 nm) indicating good binding of the complexed poses 5d and 5e and minimal conformational changes in these regions.
The analysis of the H-bond count of 5d and 5e poses confirms their acceptable binding in the binding site during the simulation time, as seen in , respectively. It is clear that at least one H-bonding interaction is formed through the 50 ns simulation for both 5d and 5e poses, however with higher occurrence for 5e in most of the trajectory frames. Overall, the results of the MD simulations confirmed the stable and acceptable binding of both 5d and 5e poses to COX-2 suggested by the in vitro and docking experiment.
3.4. Biological evaluation
3.4.1. In vitro COX study
At the site of inflammation, eicosanoids are produced from the cellular membrane lipid pool by the activation of the phospholipase A2 family, which mobilises fatty acids, particularly arachidonic acid. The latter is metabolised by two major routes, the cyclooxygenase (COX) and lipoxygenase (LOX) pathwaysCitation45,Citation46. Distinct COX isoforms exist, mainly COX-1 and COX-2. COX-1 is expressed constitutively in most cells to form the housekeeper prostanoids responsible for gastric epithelial cytoprotection and homeostasis. COX-2 is the important source of prostanoids formation in inflammationCitation47. The enzyme immunoassay performed on the tested compounds revealed a good selectivity towards COX-2 relative to COX-1 (). Consequently, the tested compounds may have good anti-inflammatory activity by inhibiting COX-2 while conserving the gastric epithelial integrity by maintaining a relatively high IC50 towards the COX-1 receptor. In fact, celecoxib displayed the highest IC50 and indomethacin the lowest activity. All tested compounds’ results lay between celecoxib and indomethacin. Consequently, the lower selectivity of the tested compounds relative to celecoxib can lead to a better cardiovascular side effect profileCitation18. Examination of the selectivity ratios of the synthesised compounds against COX-2/COX-1 receptors revealed that all compounds possess a promising selectivity ranging from 55.6 to 181.2. In addition, the COX-2 inhibitory activities lied in a narrow range of IC50 values for all compounds from 0.060 to 0.135 µM. These observations are in coherence with the in silico predictions since the 5a-e exhibited comparable scores in a narrow range, and did not show specific binding to COX-1 reflecting their promising selectivity towards COX-2 enzyme.
Table 2. COX-1 and COX-2 inhibition of the tested compounds.
3.4.2. In vivo studies
3.4.2.1. Formalin-induced paw edoema assay
Acute inflammation is rapid in onset and of a few hours’ duration. It is characterised by four cardinal signs: hotness, redness, swelling, and pain. Those develop experimentally by the subcutaneous administration of Formalin. Immediately, edoema, hyperalgesia, and erythema develop due to the migration of neutrophils to the site and the generation of pro-inflammatory agents such as bradykinin, histamine, tachykinins, reactive oxygen, and nitrogen species. Thus, edoema is quantified by measuring the increase in paw size, which is maximal around 4 h post formalin injectionCitation48. Accordingly, in the current study, edoema inhibition was recorded after 30 min and 4 h of edoema induction. Results () showed higher percentages of inhibition after 30 min for compound 5a as compared with the positive control indomethacin, and a similar inhibition for 5c and 5e to the positive control celecoxib. At 4 h, compounds 5a and 5c showed a higher inhibition than both indomethacin and celecoxib. Overall, these results reflect promising in vivo anti-inflammatory performance for most of these compounds.
Figure 6. Biological results for the formalin-induced paw edoema assay.

3.4.2.2. Real-time PCR
All tested compounds showed a significantly lower COX-2 gene expression as compared with the vehicle group and an almost equivalent effect to indomethacin, reflecting the results shown in the paw edoema test. Consequently, we can conclude that these agents exert their activity mainly by inhibiting the COX-2 enzyme expressed in the inflammation process. COX-1 was mainly maintained in compounds 5a and 5c which reflect the gastro protection effect with the use of those two compounds. On the other hand, LOX was not affected by any tested compound nor by indomethacin as reported by previous reports such as Ku et al studyCitation49 ().
Table 3. Real-time PCR results.
3.4.2.3. Acute ulcerogenic side effect of the compounds
NSAIDs are known for their GI side effects that are manifested endoscopically as lesions ranging from sub-epithelial haemorrhages, erosions, to ulcers. The GI side effects are due to topical injury of the mucosa and a systemic effect due to prostaglandin depletion derived from inhibition of COX-1Citation50,Citation51. Accordingly, in the current study, histological examination of the gastric structure revealed an intact to almost intact mucosa for compounds 5a-e comparable to the negative control group (). Moderate to large ulcers were detected in the groups which received 5e and indomethacin, respectively. The results of 5e and indomethacin can be linked to the inhibition of COX-1, while 5a, 5 b, 5c, and celecoxib preserved the integrity of the mucosa since COX-1 protective effect was maintained. The effect of 5e on the mucosa can be attributed to its electron donating ability since it is the only electron donating group usedCitation50,Citation51.
Table 4. Acute ulcerogenic side effect of the tested compounds.
4. Conclusion
The aim of the current study is to investigate the effect of various substituents replacing the chloro atom at the p-position of the phenyl ring present in the previously synthesised amide-linked bipyrazole derivative (5a) on the anti-inflammatory activity. To achieve that, we employed a structure-guided approach, where we modelled different groups representing halogens (e.g. chloro, bromo,and fluoro atoms), alkyl (e.g. methyl and isopropyl) groups, electron withdrawing group (e.g. nitro) 4- and 5-memebered systems. Guided by docking scheme using VinaXB and rescoring using AutoDock Vina, only derivatives with chloro (5a), bromo (5 b), nitro (5c), fluoro (5d) and methyl (5e) groups were recommended for potential binding in the narrow cleft of COX-2 formed by Val116, Arg120, Tyr355 and Ala527. Consequently, these new bipyrazole derivatives (5a-e) were synthesised and characterised with the desired substituents. Various biological evaluations of these derivatives were performed to characterise their anti-inflammatory and safety profiles, whether in vitro or in vivo. The in vitro inhibitory COX-1/COX-2 activity results revealed a nano-molar range of activity of the synthesised compounds against COX-2, in addition to promising selectivity indices towards COX-2 over COX-1 compared with Celecoxib and Indomethacin as references which may suggest a better cardiovascular safety profile.
A closer look at the results indicated that the fluoro (5d) and methyl (5e) derivatives showed the best in vitro anti-inflammatory activity with good selectivity indices for COX-2 vs. COX-1. Furthermore, most of the tested compounds showed promising in vivo formalin-induced paw edoema anti-inflammatory activity after 30 min and 4 h. Interestingly, nitro and chloro substitution led to a significantly higher inhibition of paw edoema and higher COX-1 gene expression relative to indomethacin, reflecting a low incidence of gastrointestinal ulcerative potential. Furthermore, except for the methyl-substituted compound, all the other tested derivatives showed a good ulcerogenic safety profile. Lastly, in silico molecular dynamics (MD) simulations for the most active compounds in vitro (5d and 5e) were conducted. The fluoro (5d) and methyl (5e) docked poses complexed with COX-2 were subjected for 50 ns MD simulation and exhibited good and stable binding to COX-2 compared with the apo COX-2 form via different metrics, such as RMSD, Rg, RMSF and H-bond count. Overall, this study provides a successful ground for further development of other amide-linked bipyrazole candidates having potent anti-inflammatory activity and good safety profile as well as a baseline for further pharmacodynamic and pharmacokinetic studies of the current compounds.
Supplemental Material
Download PDF (1.7 MB)Acknowledgements
TMI acknowledges the cluster of Bibliotheca Alexandrina High-Performance Computing for granting access to perform the molecular dynamics simulations. The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant Code: [22UQU4290565DSR47].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- MA Ragab H, Bekhit AA, AF Rostom S, Bekhit E-DA. Compounds containing azole scaffolds as cyclooxygenase inhibitors: a review. Curr Topics Med Chem 2016;16:3569–81.
- Faour WH, Mroueh M, Daher CF, et al. Synthesis of some new amide-linked bipyrazoles and their evaluation as anti-inflammatory and analgesic agents. J Enzyme Inhib Med Chem 2016;31:1079–94.
- Mak TW, Saunders ME, Jett, BD. Primer to the immune response. Boston: Newnes; 2013.
- Piomelli D. Arachidonic acid in cell signaling. Curr Opin Cell Biol 1993;5:274–80.
- Marks AD, Lieberman M, Peet A. The molecular biology of cancer. In: Marks basic medical biochemistry a clinical approach. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2018:759–812.
- Anwikar S, Bhitre M. Study of the synergistic anti-inflammatory activity of Solanum xanthocarpum Schrad and Wendl and Cassia fistula Linn. Int J Ayurveda Res 2010;1:167.
- Sharma PK, Kumar S, Kumar P, et al. Synthesis and biological evaluation of some pyrazolylpyrazolines as anti-inflammatory–antimicrobial agents. Eur J Med Chem 2010;45:2650–5.
- Zou H, Yuan C, Dong L, et al. Human cyclooxygenase-1 activity and its responses to COX inhibitors are allosterically regulated by nonsubstrate fatty acids. J Lipid Res 2012;53:1336–47.
- AlFadly ED, Elzahhar PA, Tramarin A, et al. Tackling neuroinflammation and cholinergic deficit in Alzheimer's disease: multi-target inhibitors of cholinesterases, cyclooxygenase-2 and 15-lipoxygenase. Eur J Medi Chem 2019;167:161–86.
- Qiu K-M, Yan R, Xing M, et al. Synthesis, biological evaluation and molecular modeling of dihydro-pyrazolyl-thiazolinone derivatives as potential COX-2 inhibitors. Bioorg Med Chem 2012;20:6648–54.
- Yuan C, Sidhu RS, Kuklev DV, et al. Cyclooxygenase allosterism, fatty acid-mediated cross-talk between monomers of cyclooxygenase homodimers. J Biol Chem 2009;284:10046–55.
- Hajjar ER, Cafiero AC, Hanlon JT. Polypharmacy in elderly patients. Am J Geriatric Pharmacother 2007;5:345–51.
- Ragab FA, Gawad NMA, Georgey HH, Said MF. Synthesis of novel 1, 3, 4-trisubstituted pyrazoles as anti-inflammatory and analgesic agents. Eur J Med Chem 2013;63:645–54.
- Guo B, Lager KM, Henningson JN, et al. Experimental infection of United States swine with a Chinese highly pathogenic strain of porcine reproductive and respiratory syndrome virus. Virology 2013;435:372–84.
- Marza AD, Abdullah FFJ, Ahmed IM, et al. Involvement of nervous system in cattle and buffaloes due to Pasteurella multocida B: 2 infection: a review of clinicopathological and pathophysiological changes. J Adv Veter Anim Res 2015;2:252–62.
- Moreland LW Rheumatology and immunology therapy: A to Z essentials. USA: Springer Science & Business Media, 2004.
- Rubin E HM, Reisner Essentials of Rubin's pathology. USA: Lippincott Williams & Wilkins, 2009.
- Mohy El‐Din MM, Senbel AM, Bistawroos AA, et al. A novel COX‐2 inhibitor pyrazole derivative proven effective as an anti‐inflammatory and analgesic drug. Basic Clin Pharmacol Toxicol 2011;108:263–73.
- Abd El Razik HA, Mroueh M, Faour WH, et al. Synthesis of new pyrazolo [3, 4‐d] pyrimidine derivatives and evaluation of their anti‐inflammatory and anticancer activities. Chem Biol Drug Des 2017;90:83–96.
- Khloya P, Kumar S, Kaushik P, et al. Synthesis and biological evaluation of pyrazolylthiazole carboxylic acids as potent anti-inflammatory–antimicrobial agents. Bioorg Med Chem Lett 2015;25:1177–81.
- Keche AP, Hatnapure GD, Tale RH, et al. Synthesis, anti-inflammatory and antimicrobial evaluation of novel 1-acetyl-3,5-diaryl-4,5-dihydro (1H) pyrazole derivatives bearing urea, thiourea and sulfonamide moieties. Bioorg Med Chem Lett 2012;22:6611–5.
- Fioravanti R, Bolasco A, Manna F, et al. Synthesis and biological evaluation of N-substituted-3, 5-diphenyl-2-pyrazoline derivatives as cyclooxygenase (COX-2) inhibitors. Eur J Med Chem 2010;45:6135–8.
- Molecualr Operating Environment (MOE 2018), Chemical Computing Group Inc.: Montreal, http://www.chemcomp/com.
- Shaaban MM, Ragab HM, Akaji K, et al. Design, synthesis, biological evaluation and in silico studies of certain aryl sulfonyl hydrazones conjugated with 1,3-diaryl pyrazoles as potent metallo-β-lactamase inhibitors. Bioorg Chem 2020;105:104386.
- Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model 1999;17:57–61.
- Elghoneimy LK, Ismail MI, Boeckler FM, et al. Facilitating SARS CoV-2 RNA-dependent RNA polymerase (RdRp) drug discovery by the aid of HCV NS5B palm subdomain binders: in silico approaches and benchmarking. Comput Biol Med 2021;134:104468.
- Bekhit AA, Nasralla SN, El-Agroudy EJ, et al. Investigation of the anti-inflammatory and analgesic activities of promising pyrazole derivative. Eur J Pharm Sci 2022;168:106080.
- Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015;1-2:19–25.
- Huang J, MacKerell AD. Jr, CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 2013;34:2135–45.
- Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 2011;32:2359–68.
- P, Turner (2005) XMGRACE, Version 5.1. 19. Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR.
- Domiati S, Mehanna M, Ragab H, et al. Investigation of chronic efficacy and safety profile of two potential anti-inflammatory bipyrazole-based compounds in experimental animals. J Inflamm Res 2018;11:143–53.
- Abouzeit-Har MS, Verimer T, Long JP. Effect of long term estrogen and lithium treatment on restraint induced gastric erosion in intact and ovariectomized rats. Die Pharmazie. 1982;37:593–5.
- Tominaga Y, Honkawa Y, Hara M, Hosomi A. Synthesis of pyrazolo [3, 4‐d] pyrimidine derivatives using ketene dithioacetals. J Heterocyclic Chem 1990;27:775–83.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61.
- Ibrahim TM, Bauer MR, Boeckler FM. Applying DEKOIS 2.0 in structure-based virtual screening to probe the impact of preparation procedures and score normalization. J Cheminformatics 2015;7:1–16.
- Bauer MR, Ibrahim TM, Vogel SM, Boeckler FM. Evaluation and optimization of virtual screening workflows with DEKOIS 2.0–a public library of challenging docking benchmark sets. J Chem Inform Model 2013;53:1447–62.
- Wilcken R, Zimmermann MO, Lange A, et al. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem 2013;56:1363–88.
- Koebel MR, Schmadeke G, Posner RG, Sirimulla S. AutoDock VinaXB: implementation of XBSF, new empirical halogen bond scoring function, into AutoDock Vina. J Cheminform 2016;8:27.
- Charlier C, Michaux C. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a new strategy to provide safer non-steroidal anti-inflammatory drugs. Eur J Med Chem 2003;38:645–59.
- Dannhardt G, Kiefer W. Cyclooxygenase inhibitors–current status and future prospects. Eur J Med Chem 2001;36:109–26.
- Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996;384:644–8.
- Arba M, Wahyudi ST, Brunt DJ, et al. Mechanistic insight on the remdesivir binding to RNA-Dependent RNA polymerase (RdRp) of SARS-cov-2. Comput Biol Med 2021;129:104156.
- Ismail MI, Ragab HM, Bekhit AA, Ibrahim TM. Targeting multiple conformations of SARS-CoV2 papain-like protease for drug repositioning: an in-silico study. Comput Biol Med 2021;131:104295.
- Morteau O. Prostaglandins and inflammation: the cyclooxygenase controversy. Arch Immunol Ther Exps 2000;48:473–80.
- Robbins S Robbins and Cotran pathologic basis of disease. Philadelphia, PA: Saunders/Elsevier, 2010.
- Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 2011;31:986–1000.
- Pitsillides AA (2004) Inflammation Protocols. Methods In Molecular Biology, Volume 225. Edited by P. G. Winyard and D. A. Willoughby. $99.50. Humana Press, Totowa, NJ, 2003. 380 pages. ISBN 0-86903-970-6. Rheumatology 43 814–814.
- Ku EC, Lee W, Kothari HV, Scholer DW. Effect of diclofenac sodium on the arachidonic acid cascade. Am J Med 1986;80:18–23.
- Sostres C, Gargallo CJ, Arroyo MT, Lanas A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best practice & research. Clin Gastroenterol 2010;24:121–32.
- Laine L. The gastrointestinal effects of nonselective NSAIDs and COX-2-selective inhibitors. Sem Arthr Rheum 2002;32:25–32.