
Abstract
Promising inhibitory activities of the parasite multiplication were obtained upon evaluation of in vivo antimalarial activities of new pyrazolylpyrazoline derivatives against Plasmodium berghei infected mice. Further evaluation of 5b and 6a against chloroquine-resistant strain (RKL9) of P. falciparum showed higher potency than chloroquine. In vitro antileishmanial activity testing against Leishmania aethiopica promastigote and amastigote forms indicated that 5b, 6a and 7b possessed promising activity compared to miltefosine and amphotericin B deoxycholate. Moreover, antileishmanial activity reversal of the active compounds via folic and folinic acids showed comparable results to the positive control trimethoprim, indicating an antifolate mechanism via targeting leishmanial DHFR and PTR1. The compounds were non-toxic at 125, 250 and 500 mg/kg. In addition, docking of the most active compound against putative malarial target Pf-DHFR-TS and leishmanial PTR1 rationalised the observed activities. Molecular dynamics simulations confirmed a stable and high potential binding of 7a against leishmanial PTR1.
1. Introduction
Malaria and leishmaniasis have emerged as thoughtful health problems throughout the history of manhood. According to WHO Report in 2019, although there is a considerable deterioration in the number of malaria cases and deaths, the disease accounts for 228 million cases globally and is one of the top causes of death for children in AfricaCitation1. Despite the efforts in introducing a great number of chemotherapeutic agents to treat malaria, there is still an urgent medical need in the area. The main reason for this is the emergence of resistanceCitation2,Citation3. Resistance has occurred for almost all therapeutic agents approved for the treatment of malaria which represents a major apprehension demanding an instant actionCitation1,Citation3. Therefore, the search for newer and more effective drugs has become a crucial target.
Leishmaniasis is a complex disease that is caused by more than 20 species of Leishmania and is correlated to several clinical manifestations ranging from simple skin lesions around the bite site to fatal visceral formsCitation4,Citation5. More than one billion people are at risk of leishmaniasis in endemic areasCitation6,Citation7. Therefore, there is a continuing necessity to discover new antiprotozoal agents that are effective against multidrug-resistant parasites and inhibitors that target enzymes and proteins macromoleculesCitation8,Citation9. For the folate pathway, dihydrofolate Reductase (DHFR) and Pteridine reductase (PTR1) are validated targets for leishmaniaCitation10. Their main role is to reduce oxidised pteridines like biopterin and folate to active cofactors tetrahydrobiopterin (THB) and tetrahydrofolate (THF), respectively. Nonetheless, utmost leishmania species showed resistance against DHFR-TS inhibitorsCitation11,Citation12, owing to the presence of an alternative salvage pathway regulated by PTR1. Interestingly, the PTR1 enzyme is overexpressed in strains that exhibited antifolate resistance, hence, offering the means to bypass the dihydrofolate reductase-thymidylate synthase (DHFR-TS) pathwayCitation13–15.
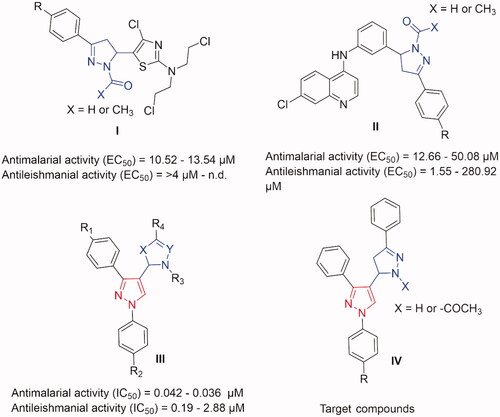
It is well known that pyrazole rings, whether free or conjugated with other heterocyclic rings, demonstrated a wide collection of biological activities, such as antibacterial, antiviral, antitubercular, anti-inflammatory, antioxidant, anticancer, antimalarial and antileishmanialCitation16–29. Also, derivatives containing pyrazoline scaffold were reported to show promising antileishmanial and/or antimalarial effects in a low micromolar range of activities (I and II in )Citation30,Citation31. Interestingly, as we reported earlier, when integrating a pyrazole scaffold in a pyrazoline framework, yielded a promising sub-micromolar range of activities for both antimalarial and antileishmanial effects (III in )Citation32. These interesting biological activity profiles inspired us to synthesise focussed derivatives of pyrazole integrated pyrazoline analogues as potential antimalarial and/or antileishmanial agents (IV).
Figure 1. Some previously reported pyrazoline derivatives (I and II) and pyrazole hybrids with other heterocyclic moieties (III) with dual antimalarial and antileishmanial activity. Compounds (IV) represent our target compounds.
The synthesised compounds were evaluated for their antimalarial activity using Plasmodium berghei infected mice (in vivo approach) where the most active compounds were further evaluated against chloroquine-resistant strain (RKL9) of P. falciparum (in vitro approach). For the antileishmanial activity, the compounds were screened against both promastigote and amastigote forms of Leishmania aethiopica. The reversal of the antileishmanial activity via folic and folinic acid confirmed the antifolate mechanism of the synthesised compounds anticipating PTR1 inhibition. Furthermore, docking experiments on putative malarial Pf-DHFR-TS and leishmanial PTR1 targets rationalised the observed antimalarial and antileishmanial activities.
2. Results and discussion
2.1. Chemistry
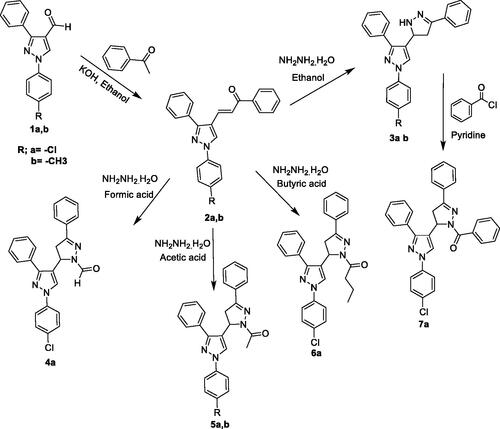
The target compounds were synthesised according to the steps outlined in Scheme 1. Initially, the two intermediate chalcones 2a,b containing α, β-unsaturated ketone group were synthesised by condensation of aldehydes 1a,bCitation33 and acetophenone in the presence of KOH in ethanol. The structures of these compounds were confirmed by IR spectra that showed the absence of characteristic peaks at 2726 and 2669 cm−1 which was attributed to the C-H stretching vibration of the aldehydic groups of 1a,b. Moreover, the 1H-NMR spectrum of these two compounds revealed the disappearance of the singlet at δ 10.1 attributed to the aldehydic peak in addition to the presence of two doublets at δ equals 7.4 and 7.9 corresponding to the methine protons which confirms the formation of the target compounds. The two formed chalcones were cyclized by refluxing with hydrazine hydrate in presence of different solvents like ethanol, formic acid, acetic acid and butyric acid to give the corresponding pyrazolylpyrazoline derivatives 3a, 3b, 4a, 5a, 5b and 6a. The structures of the formed derivatives were proved by the absence of a characteristic intense band around 1665 cm−1 that corresponds to the C = O and the aldehydic characteristic peaks at 2726 and 2669 cm−1 of their IR spectra. In addition, characteristic peaks for the formation of pyrazoline moiety have been observed on their 1H-NMR spectra. These are methylene protons at the C4 of the pyrazoline ring resonated as two doublets of doublet peaks (both integrated for one proton each) at 3.05–3.15 and 3.4–3.55 ppm. The C5 proton peak appeared as a doublet of a doublet at 5.08–5.18 ppm due to vicinal coupling with the two magnetically non-equivalent protons of the methylene group at the C4 position of the pyrazoline ring. The strong deshielding of the C5 protons compared with the C4 protons of the pyrazoline ring was assumed to be due to electron withdrawing neighbours. Finally, the benzoyl derivative of compound 7a was synthesised by refluxing the unsubstituted pyrazolylpyrazoline 3a with benzoyl chloride in presence of pyridine. Generally, the spectral aspects of the pyrazolyl derivatives were confirmed and guided by previous studiesCitation32,Citation34. Representative NMR charts can be found in the Supplementary Material.
Scheme 1. General synthetic route of the target compounds.
2.2. Biological activity
2.2.1. In vivo antimalarial activity testing against Plasmodium berghei
The in vivo biological activities of some of the synthesised pyrazolylpyrazoline derivatives showed promising results against P. berghei ( and ). The in vivo antimalarial activity of the synthesised compounds was evaluated at a dose level of 20 mg/kg (). Those compounds that revealed statistically significant suppression (p < 0.05) were further evaluated at a dose level of 30 mg/kg. Antimalarial testing at the second dose was carried out for three compounds (5b, 6a and 7a) to examine assay variability ().
Table 1. Antiplasmodial activities of the synthesised compounds at 20 mg/kg.
Table 2. Antiplasmodial activities of the synthesised compounds at 30 mg/kg.
By the end of the 4-day suppressive test, compound 6a at a dose level of 20 and 30 mg/kg showed mean parasitaemia of 30.3 and 28.3%, respectively, compared to 52.2 and 59.3% for the negative control ( and ). This specifies a 42.3 and 52.4% suppression for 6a compared to the negative control groups. The mice treated with chloroquine (positive control) were completely free of the parasite on day 4. The growth in percentage suppression with growing the dose of the tested compound is revealing of the presence of a dose-response relationship. Like 6a, 7a displayed significant parasitaemia suppressive effect on day 4 compared to the negative control ( and ). At the dose levels of 20 and 30 mg/kg, the mean parasitaemia for 7a was found to be 33.0 and 41.1%, respectively equivalent to 36.5 and 30.3% suppression of the parasite. However, the decrease in the suppressive effect of increasing the administered dose of 7a implies that the 30 mg/kg dose might have compromised the immune system of the tested mice. Compound 5b showed the most potent suppressive effect among the tested compounds, as seen in and . At the dose levels of 20 and 30 mg/kg the mean parasitaemia was found to be 18.8 and 17.4%, respectively equivalent to 66.7 and 71.2% suppression. Suppressive activity of compound 5b follows a dose-response relationship.
Elucidating some structure-activity relationship (SAR), the methyl substituent on the phenyl group and acetyl substituent on the pyrazoline ring favours the antiplasmodial activity (5b). The presence of bulkier substituents (butanoyl and benzoyl) on the pyrazoline ring appeared to correlate with the activity of compounds 6a and 7a. On the other hand, the absence of substituent on the pyrazoline ring in compound 3b abolished the activity. The presence of smaller substituent (acetyl and formyl) or absence of substituent can correlate to the loss of activity in the case of p-chlorophenyl derivatives 3a, 4a and 5a. Generally, owing to the uniqueness of such compounds as dual-acting antimalarial-antileishmanial hits, we would pursue more studies on them in the near future with a wider array of substituents, in order to extract sharper insights into the structure-activity relationships.
2.2.2. In vitro antimalarial activity testing against chloroquine-resistant (RKL9) strain of Plasmodium falciparum
Compounds 5b and 6a that showed the highest in vivo percent suppression against P. berghei at a dose level of 30 mg/Kg, were further evaluated for their antiplasmodial activities against chloroquine-resistant (RKL9) P. falciparum strain. Results revealed that both compounds showed greater activity than chloroquine phosphate (IC50 = 0.1920 µM) against the chloroquine resistant (RKL9) strain of P. falciparum. Compound 5b was the most potent against RKL9 strains showing around 6-fold higher inhibitory activity than chloroquine (). Comparing 5b to a standard folate inhibitor (pyrimethamine), both were in a similar nano-molar range of inhibitory activity with a slight superiority of pyrimethamine. Interestingly, these results are in coherence with our previous reportCitation32 where the in vitro antimalarial effect of pyrazole-pyrazoline hybrids was in a similar nano-molar range of activity.
Table 3. In vitro anti-plasmodial activity against chloroquine-resistant (RKL9) strain of P. falciparum.
2.2.3. In vitro antileishmanial activity on leishmania aethiopica promastigote and amastigote forms
The assay was used to determine the viability of promastigotes and evaluate the antileishmanial activity of the synthesised compounds. The tested compounds 5b, 6a and 7a showed higher antileishmanial activity than the reference standard miltefosine, whereas compound 5b exhibited comparable activity to the reference standard Amphotericin B deoxycholate (). The result indicated that the presence of a relatively bulky substituent on one of the pyrazolo-N1 rather than its C5 led to a high inhibitory effect on the promastigotes, with IC50 values ranging from 0.05 to 0.89 μM. Furthermore, the presence of an alkyl group rather than hydrogen on the carbonyl of the N1-side chain also improves the antileishmanial activity as indicated by the results observed for compounds 5b, 6a and 7a.
Table 4. Antileishmanial activity is expressed as antipromastigote and antiamastigote activities of the test compounds and reference standards.
Compounds 5b and 7a exhibited good activity against L. aethiopica amastigotes close to the activity determined for miltefosine reference (). Overall, these promising results agree with our previous reportCitation32 where the in vitro antileishmanial activity of pyrazole-pyrazoline hybrids was also in a sub-micromolar range of activity.
2.2.4. Reversal of the antileishmanial activity via folic and folinic acid
Leishmania parasites were exposed to the tested compounds or trimethoprim (the positive control) at concentrations above their IC50 after the addition of either folinic or folic acids. Exposure of the parasite to folinic or folic acid declined the antileishmanial effect of both the tested compounds and trimethoprim. Also, exposure to folic acid together with trimethoprim led to a rise in the parasite survival time up to 100%. This can be clarified by the fact that folic acid (a natural substrate) competed for the active site of PTR1 and leishmanial DHFR enzyme while folinic acid contributed to DNA synthesis without any need to undergo metabolism. Also, folic acid was observed to display greater inhibition of the antileishmanial activity of the tested compounds than folinic acid.
Furthermore, the addition of excess folic acid to parasitic cells after exposure to the tested compounds was performed to investigate its ability to reverse antileishmanial inhibition. All tested compounds and trimethoprim presented reversibility of the antileishmanial inhibition. This designates that the observed antileishmanial activity of the synthesised compounds are mediated via an antifolate mechanism anticipating both leishmanial DHFR and PTR1. Further insights on mechanistic details are elaborated in the modelling section and molecular dynamics simulations for the most active compound 7a.
2.2.5. In vivo acute toxicity test
An acute toxicity study was conducted to assess the acute lethal, physical and behavioural effects of the most active compounds (5b and 6a) after oral administration to mice, as reported earlierCitation35. Oral administration of the two highly active compounds in doses of 125, 250 and 500 mg/kg did not show any significant acute toxic effects on the experimental mice, as shown in . The data indicated that no death was observed during the first 24 h of the experimental period in any of the test groups. The results of the study showed that the median lethal dose (LD50) of synthesised compounds is higher than 500 mg/kg/day for mice through the oral route.
Table 5. Data from the acute toxicity studies.
2.3. Molecular Modelling
2.3.1. Molecular Docking
To elucidate a putative molecular mechanism for the antimalarial activity, we carried out docking experiments of the most active compounds against the quadruple mutant (N51I, C59R, S108N and I164L) Pf DHFR-TS structure. Challenging the most active compounds by such a highly mutant model would provide clues about the in silico binding compared to the reference Pf DHFR-TS binder (pyrimethamine), especially in a resistant variant of malaria.
The docking scores of the most active compounds obviously indicated significant in silico binding towards the mutant Pf DHFR-TS structure, especially when compared to the reference Pf DHFR-TS binder (pyrimethamine) as shown in . Interestingly, their docking scores on the quadruple mutant Pf DHFR-TS (resistant form) are superior to their respective scores against the wild-type Pf DHFR-TS. Particularly, 5b (the most active compound) shows the best score compared to all compounds and is the reference for the quadruple mutant structure. This confirms their inhibitory power against the resistant forms of malaria (see ). This is emphasised by the fact that pyrimethamine (the reference) displayed a worse score against the quadruple mutant compared to the wild-type. Furthermore, 5b and 7a still demonstrate superior scores compared to pyrimethamine against wild-type structure. This specifies a particular binding of the most active compounds towards the mutant Pf DHFR-TS structure as a possible resistance mechanism to malaria.
Table 6. AutoDock Vina docking scores (kcal mol−1) of the most active compounds against Pf DHFR-TS (PDB: 1j3k) and PTR1 (PDB: 2bfm).
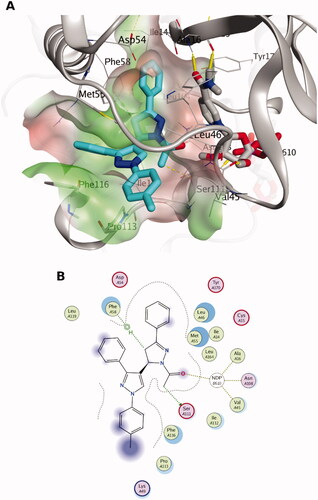
The docking pose of 5b, the most active compound against the chloroquine-resistant (RKL9) strain of P. falciparum (see ), exhibits favourable interactions with the binding site residues of quadruple mutant Pf-DHFR-TS. H-bonding interaction can be observed with the 1-acetyle moiety (carbonyl group as H-bond acceptor) with the cofactor NADPH. Furthermore, the core of the 5b pose appeared to be deeply packed in a hydrophobic region, as shown in . For instance, the 1-acetyl-3-phenyl pyrazoline moiety is packed between the co-factor NADPH and the side chains of Met55, Ile14, Leu46, Ser111 and Phe58. Also, 3-phenyl pyrazole is surrounded by the side chains of Phe116, Met55, Pro113 and Leu119 indicating favourable hydrophobic interactions. Overall, the such postulated binding mode would block the catalytic activity of Pf-DHFR. Based on our analysis of the docking poses of different stereoisomers, we observed some perturbations in their docking scores indicating the importance of stereo-selectivity for Pf-DHFR binding.
Figure 2. The docking pose of the most active compound (5b) as cyan sticks in the binding site of the quadruple mutant Pf-DHFR-TS (PDB code: 1j3k). The hydrophilic and hydrophobic regions are in red and green coloured molecular surfaces, respectively. Non-polar hydrogen atoms were omitted for clarity. The label “NDP-610” represents the NADPH co-factor.
To rationalise the antileishmanial activity and the antifolate mechanism of most active compounds, our attention was devoted primarily to the co-crystal structure of PTR1 as a putative target since the co-crystal structure of the leishmanial DHFR-TS enzyme has not been resolved yet. The most active compounds 5b, 7a and 6a showed a superior docking score compared to trimethoprime (a DHFR and PTR1 inhibitor) and dihydropterine (a natural substrate for PTR1), as seen in . Interestingly, the most active compound 7a against both promastigote and amastigote forms of leishmania displays the best docking score compared to all compounds as well as both reference compounds. This anticipates favourable binding towards PTR1 and hence confirming the antifolate mechanism confirmed by the in vitro experiment (Section Reversal of the antileishmanial activity via folic and folinic acid).
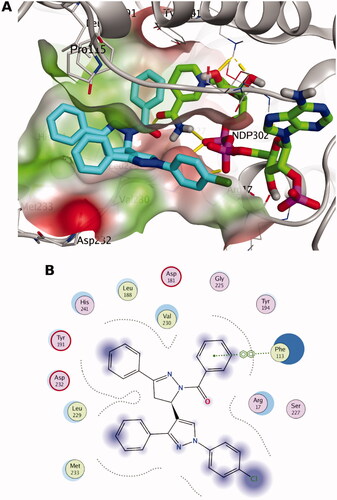
The docking pose of 7a shows mainly hydrophobic interactions with binding site residues blocking the catalytic activity, as seen in . For instance, the 3-phenyl pyrazoline moiety is packed between the co-factor NADPH and the residues Val230, Leu188 and Pro115. The 1-benzoyl pyrazoline moiety demonstrates vdW interaction with Phe113 and hydrophobic interaction with NADPH. Also, 3-phenyl pyrazole group is packed between the residues Val230, Met233 and Pro115 indicating favourable hydrophobic interactions. Overall, the such postulated binding mode would block the catalytic activity of PTR1 and hence inhibiting the leishmanial folate pathway. Again, the poses of different stereoisomers showed differences in the docking scores designating the importance of stereo-selectivity for PTR1 binding.
Figure 3. The docking pose of the most active compound (7a) as cyan sticks in the binding site of PTR1 (PDB code: 7pxx). The hydrophilic and hydrophobic regions are in red and green coloured molecular surfaces, respectively. Non-polar hydrogen atoms were omitted for clarity. The label “NDP-302” represents the NADPH co-factor.
To further confirm the stable binding of 7a pose against leishmanial PTR1 in a time-dependent manner and hence validate PTR1 as a potential target in the folate pathway, we performed molecular dynamics simulation for three systems for 50 ns. The details can be found in the coming Section.
We performed additional docking experiments for the less active compounds 3a and 3b in the binding site of Pf-DHFR and PTR1. We did not find significant score differences between both compounds and the most active ones. The docking scores of 3a and 3b were identical (−11.2 kcal/mol) against Pf-DHFR, which is a comparable score to the range found in the most active compounds (). Likewise, the docking scores of 3a and 3b were −8.9 and −8.5 with PTR1, respectively. Again, this indicates a similar docking range to the most active compounds (). The observed in-vitro anti-leishmanial activity for these compounds (most and least active) lie within the low- and sub-micromolar range of activities. This would be highly challenging to yield significant differences in the output docking scores. Nevertheless, we assume that the lower activity of 3a and 3b can be attributable to their relatively lower lipophilicity compared to the most active compounds 6a and 7a. The calculated lipophilicity (via logP function of Marvin Sketch v17.2.6.0 – ChemAxon – http://www.chemaxon.com) for 3a, 3b, 5b, 6a and 7a were 5.92, 5.83, 5.53, 6.77 and 7.48, respectively. Adequate lipophilicity would enable the ligand to diffuse sufficiently through the lipophilic membranes of the protozoa and reach the macromolecular target for binding events.
2.3.2. Molecular Dynamics simulation
The 7a docking pose in PTR1 was subjected to 50 ns molecular dynamics (MD) simulations for evaluating the stability of its docked pose in a time-dependent manner in the binding site. Furthermore, another run was conducted for the apo PTR1 form and the complexed form with the co-crystal PTR1 structure, to account for its dynamicity as a reference. This results in a total of three MD runs, 50 ns each. Root Mean Square Deviation (RMSD) is a measure of protein backbone stability during the simulation time. RMSD of the apo and complexed with co-crystal ligand forms () reach a converged state at 35 ns with a minor fluctuation in the 0.2 nm range. This reflects the appropriate stability of the protein structure during the two simulation runs. Interestingly, the RMSD profile of the complexed form with 7a exhibited an earlier convergence at 25 ns and a steady state performance until the end of the simulation course. Minor variations of the protein backbone for both complexed forms (co-crystal ligand and 7a) can be observed around 0.2 nm (2 Å) after convergence. This is also in coherence with analysis obtained by the Radius of gyration (Rg) in . Rg is a measure of protein structure compactness during simulation time. There is no great fluctuation in the Rg of the protein complexed with 7a compared to both the apo and co-crystal ligand complex structures since they display an Rg range of 0.05 nm after 25000 frames (25 ns). This gives an indication of the low conformational changes of the protein throughout the simulation, and hence, its stabilityCitation36,Citation37. Per residue root means square fluctuation (RMSF) assesses the conformational changes that occur to each residue of the protein, as shown in . The N-terminal amino acids exhibit the highest RMSF contemplated by the high free movement of their free loops. However, the key binding site amino acids (numbers: Asp232, Val230, Leu188, His241, Tyr191, Leu229, Met233, Phe113) show low RMSF (< 2.5 Å) and comparable fluctuation behaviour to all the three simulated protein forms. This highlights good binding of the complexed ligand (7a) and minimal conformational changes in these residues compared to the apo and co-crystal ligand complex forms. Such good binding is augmented by the analysis of the hydrogen bond count of 7a pose in the binding site during the simulation time, as seen in . It is obvious that at least one H-bonding interaction is formed all over the majority of 50 ns simulations.
Figure 4. MD simulations for the three systems, the apo leishmanial PTR1, 7a-PTR1 complex and co-crystal ligand – PTR1 complex systems. (A) Root mean square deviation (RMSD) of the protein alpha carbon atoms across the 50 ns simulation. (B) The radius of gyration (Rg) for the PTR1 protein across the 50 ns simulation time. The frame number (x-axis) 5000 indicates 50 ns simulation time. (C) Per residue, root means square fluctuation (RMSF). (D) Hydrogen bond counts during the MD simulation for 7a in the binding site during the 50 ns simulation. (E) RMSD of the ligand heavy atoms during the 50 ns simulation of the ligand-complexed systems.
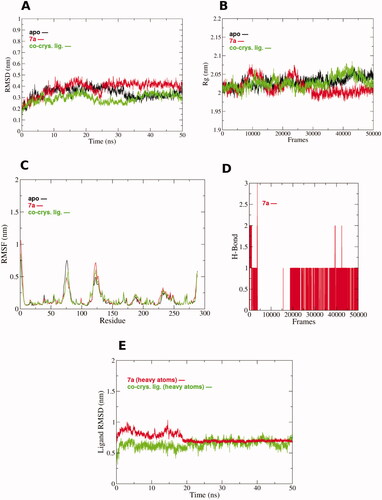
To reveal the ligand positional deviation throughout the simulation, RMSD analysis of the non-hydrogen ligand atoms was performed across the simulation time (). From the beginning of the simulation until 20 ns time, both 7a and co-crystal ligands deviated from their starting pose with some fluctuations around 0.5 nm and 0.75 nm RMSD. Interestingly, after 20 ns both 7a and the co-crystal ligand showed a comparable steady state with superior stability and lower divergence of the 7a pose compared to the co-crystal ligand. This agrees with 7a H-bond count behaviour since a persistent H-bond was formed after 20 ns contributing to it is stable binding in the binding site of PTR1.
We utilised the principal component analysis (PCA) to analyse the conformational sampling of the PTR-1 systems in the simulated subspace via examining their dominant modes of motion. The covariance matrix of atomic fluctuations was diagonalised for predicting the eigenvalues. The first few eigenvectors play a crucial role in the motions of the protein. The first 3 eigenvectors have a larger eigenvalue for the apo structure compared to both 7a and co-crystal complexed forms of PTR1 systems. This reflects greater collective atomic fluctuations of the apo form and implies that the systems complexed with 7a or co-crystal ligand demonstrated reduced motions compared to the apo form.
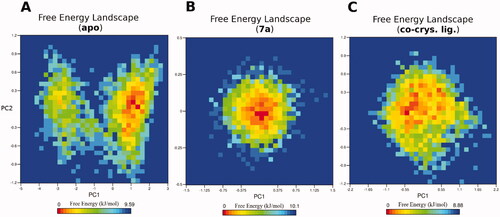
To reveal the ligand influences on the conformational heterogeneity of PTR1, associated free energy landscapes (FEL) were determined as a function of the top two principal components (PC1 and PC2), as illustrated in . FEL can be used to effectively describe conformational redistributions provoked by binding eventsCitation38,Citation39. shows the relative conformational changes of the protein backbone of the three simulated systems. The deeper colour (towards the red colour) in the plot reveals lower-energy conformational metastable states. Interestingly, the simulated apo system of PTR1 visits two separate energy basins, one represents the global minimum of the simulated subspace, while the other is quite narrow and separated by conformations with a low energy barrier from the main basin. This reflects the presence of diverse ensembles of flexible and low-energy conformations during 50 ns simulation. On the other hand, both ligand-complexed systems display comparable FEL profiles implying one main energy basin of the simulated subspace. For instance, the 7a complexed structure is populated by a single and a focussed energy basin indicating a concise range of metastable states during the 50 ns simulation. Likewise, the co-crystal ligand complexed system exhibits similar pattern to the 7a system, however, with a wider energy basin. These results thus clearly highlight that 7a binding to PTR1 can alter the PTR1 conformational subspace towards low energy conformations, and therefore, modulate its function. Overall, the results of the MD simulations confirmed the high potential and stable binding of 7a to PTR1 augmenting the in vitro antileishmanial through the antifolate mechanism.
Figure 5. The free energy landscape (FEL) of the simulated PTR1 systems is based on the principal component analysis. (A) Leishmanial PTR1. (B) 7a-PTR1 complex. (C) Co-crystal ligand – PTR1 complex. The colour bar represents the free energy value in kcal mol − 1. The colour ranges from red to yellow to blue spots indicate the energy minima and energetically favoured protein conformations to more unfavourable high-energy conformations.
3 Material and methods
3.1. Chemistry
Melting points were determined in open glass capillaries using electro thermal BUCHI (B-540) hot storage melting-point apparatus and are uncorrected. Infra-red (IR) spectra were recorded on Shimadzu 8400SP infra-red spectrophotometer using nujol. 1H-NMR spectra were recorded on Bruker Avance DMX400 400 MHz FT-NMR spectrometer using DMSO-d6 as a solvent and the chemical shifts are given in δ (ppm) downfield from tetramethylsilane (TMS) which served as an internal standard. Splitting patterns were designated as follows: s: singlet; d: doublet; m: multiplet. Elemental analyses were performed on Perkin Elmer 2400 elemental analyser and were found within ±0.4% of the theoretical values. Follow-up of the reactions and checking of the purity of the compounds was made by thin layer chromatography (TLC) on silica gel-precoated aluminium sheets (Type 60 GF254, Merck) and the spots were detected by exposure to an iodine chamber for a few minutes.
3.1.1 1-Aryl-3-phenyl-4–(3-phenyl-3-oxopropenyl)-1H-pyrazole (2a,b)
A mixture of the appropriate aldehyde 1a,b (15 mmol)Citation33 and an equimolar amount of acetophenone (1.86 ml) was dissolved in 15 ml of ethanol, and 10 ml of 3% alcoholic KOH was added. The reaction mixture was stirred using a magnetic stirrer at room temperature for 6 h. The resulting solution was then allowed to stand overnight. The formed yellow precipitate was filtered, washed with ethanol, dried and recrystalised from ethanol.
3.1.2 1–(4-Chlorophenyl)-3-phenyl-4–(3-phenyl-3-oxopropenyl)-1H-pyrazole (2a)
Yield, 72.4%; mp, 191–193 °C; IR (Nujol) cm−1: 1665 (C = O); 1610 (C = N); 1456 (C-Cl). 1H-NMR (DMSO-d6) δ(ppm): 7.4 (d, 1H, J = 15.6 Hz, methine-H), 7.4–7.7 (m, 10H, phenyl-H), 7.82 (d, 2H, J = 7.6 Hz p-chlorophenyl-C3,5H), 7.9 (d, 1H, J = 16.0 Hz, methine-H), 8.0 (d, 2H, J = 7.1 Hz p-chlorophenyl-C2,6H), 8.4 (s, 1H, pyrazole-C5H). Analysis calculated for C24H17ClN2O (384.86): C, 74.90; H, 4.45; N, 7.28; Cl, 9.21. Found: C, 75.21; H, 4.71; N, 7.53; Cl, 9.05. Rf [benzene/ethyl acetate (9:1)] = 0.74.
3.1.3 1–(4-Methylphenyl)-3-phenyl-4–(3-phenyl-3-oxopropenyl)-1H-pyrazole (2b)
Yield, 77.7%; mp, 176–1780 C; IR (Nujol) cm−1: 1660 (C = O); 1595 (C = N). 1H-NMR (DMSO-d6) δ(ppm): 7.4 (d, 1H, J = 15.6 Hz, methine-H), 7.38–7.65 (m, 10H, phenyl-H), 7.82 (d, 2H, J = 7.6 Hz, p-chlorophenyl-C3,5H), 7.9 (d, 1H, J = 15.9 Hz, methine-H), 7.98 (d, 2H, J = 7.1 Hz, p-chlorophenyl- C2,6H), 8.4 (s, 1H, pyrazole-C5H). Analysis calculated for C25H20N2O (364.44): C, 82.39; H, 5.53; N,7.70. Found: C, 82.70; H, 5.24; N, 7.91. Rf [benzene/ethyl acetate (9:1)] = 0.80.
3.1.4 1-Aryl-3-phenyl-4–(3-phenyl-2-pyrazolin-5-yl)-1H-pyrazole (3a,b)
A mixture of 2a,b (2.6 mmol) and an equimolar amount of hydrazine hydrate (0.13 ml) was dissolved in 15 ml of ethanol. The mixture was heated under reflux for 30 min with continuous stirring using a magnetic stirrer. The reaction mixture was then cooled and the formed white precipitate was filtered, washed with ethanol, dried and recrystalised from ethanol.
3.1.5 1–(4-Chlorophenyl)-3-phenyl-4–(3-phenyl-2-pyrazolin-5-yl)-1H-pyrazole (3a)
Yield, 78.6%; mp, 207–209 °C; IR (Nujol)cm−1: 3314 (N-H); 1600 (C = N); 1455 (C-Cl). 1H-NMR (DMSO-d6) δ(ppm): 3.05–3.15 (dd, 1H, J = 4.6, 17.6 Hz, pyrazoline-C4H), 3.4–3.55 (dd, 1H, J = 11.6, 17.6 Hz, pyrazoline-C4H), 5.08–5.18 (dd,1H, J = 4.6, 11.6 Hz, pyrazoline-C5H), 7.25–7.75 (m, 15H, p-chlorophenyl-H, phenyl-H, pyrazoline- NH), 8.05 (s, 1H, pyrazole-C5H). Analysis calculated for C24H19ClN4 (398.89): C, 72.27; H, 4.80; N,14.05; Cl, 8.89. Found: C, 72.56; H, 5.01; N, 13.91; Cl, 8.70. Rf [benzene/ethyl acetate (9:1)] = 0.57.
3.1.6 1–(4-Methylphenyl)-3-phenyl-4–(3-phenyl-2-pyrazolin-5-yl)-1H-pyrazole (3b)
Yield, 75.76%; mp, 211–213 °C; IR (cm−1): 3322 (N-H); 1600 (C = N). 1H-NMR (DMSO-d6) δ(ppm): 2.3 (s, 3H, CH3), 3.1–3.2 (dd, 1H, J = 4.5, 17.5 Hz, pyrazoline-C4H), 3.6–3.7 (dd, 1H, J = 11.5, 17.5 Hz, pyrazoline-C4H), 5.8–5.9 (dd, 1H, J = 4.5, 11.6 Hz, pyrazoline-C5H), 7.18–7.68 (m, 15H, p-methylphenyl-H, phenyl-H, pyrazoline-NH), 7.8 (s, 1H, pyrazole-C5H). Analysis calculated for C25H22N4 (378.47): C, 79.34; H, 5.86; N, 14.80. Found: C, 79.62; H, 6.08; N, 15.15. Rf [benzene/ethyl acetate (9:1)] = 0.61
3.1.7 1–(4-Chlorophenyl)-3-phenyl-4–(1-formyl-3-phenyl-2-pyrazolin-5-yl)-1H-pyrazole (4a)
A mixture of 2a (1.3 mmol, 0.50 gm) and equimolar amount of hydrazine hydrate (0.07 ml) was dissolved in 20 ml of formic acid. The mixture was heated under reflux for 6 h with continuous stirring using a magnetic stirrer. The reaction mixture was then cooled and the formed white precipitate was filtered, washed, dried and recrystalised from ethanol. Yield, 72.7%; mp, 221–223 °C; IR (Nujol) cm−1: 1667 (C = O); 1600 (C = N); 1446 (C-Cl). 1H-NMR (DMSO-d6) δ(ppm): 3.1–3.2 (dd, 1H, J = 4.6, 17.6 Hz, pyrazoline-C4H), 3.7–3.8 (dd, 1H, J = 11.6, 17.6 Hz, pyrazoline-C4H), 5.7–5.8 (dd, 1H, J = 4.6, 11.6 Hz, pyrazoline-C5H), 7.25 − 7.7 (m, 14H, p-chlorophenyl-H, phenyl-H), 7.9 (s, 1H, pyrazole-C5H), 8.9 (s, 1H, CHO). Analysis calculated for C25H19ClN4O (426.90): C, 70.34; H, 4.49; N,13.12; Cl, 8.30. Found: C, 70.51; H, 4.31; N, 12.87; Cl, 8.51. Rf [benzene/ethyl acetate (9:1)] = 0.55.
3.1.8 1–(4-Chlorophenyl)-3-phenyl-4–(1-acetyl-3-phenyl-2-pyrazolin-5-yl)-1H-pyrazole (5a,b)
A mixture of 2a,b (1.3 mmol) and an equimolar amount of hydrazine hydrate (0.07 ml) was dissolved in 15 ml of glacial acetic acid. The mixture was heated under reflux for 4 h with continuous stirring using a magnetic stirrer. The reaction mixture was then cooled and the formed white precipitate was filtered, washed, dried and recrystalised from ethanol.
3.1.9 1–(4-Chlorophenyl)-3-phenyl-4–(1-acetyl-3-phenyl-2-pyrazolin-5-yl)-1H-pyrazole (5a)
Yield, 75.4%; mp, 233–236 °C; IR (Nujol) cm−1: 1665 (C = O); 1595 (C = N); 1455 (C-Cl). 1H-NMR (DMSO-d6) δ(ppm): 2.5 (s, 3H, CH3), 3.1–3.2 (dd, 1H, J = 4.4, 17.4 Hz, pyrazoline-C4H), 3.6–3.7 (dd, 1H, J = 11.8, 17.4 Hz, pyrazoline-C4H), 5.8–5.9 (dd, 1H, J = 4.3, 11.7 Hz, pyrazoline-C5H), 7.25 − 7.78 (m, 14H, p-chlorophenyl-H, phenyl-H), 7.81 (s, 1H, pyrazole-C5H). Analysis calculated for C26H21ClN4O (440.92): C, 70.82; H, 4.80; N,12.71; Cl, 8.04. Found: C, 70.68; H, 4.72; N, 12.38; Cl, 8.19. Rf [benzene/ethyl acetate (9:1)] = 0.59.
3.1.10 1–(4-Methylphenyl)-3-phenyl-4–(1-acetyl-3-phenyl-2-pyrazolin-5-yl)-1H-pyrazole (5b)
Yield, 70.40%; mp, 217–219 °C; IR (Nujol) cm−1: 1670 (C = O); 1615(C = N). 1H-NMR (DMSO-d6) δ(ppm): 2.3 (s, 3H, p-methylphenyl-H), 2.4 (s, 3H, acetyl-H), 3.1–3.2 (dd, 1H, J = 4.4, 17.5 Hz, pyrazoline-C4H), 3.6–3.7 (dd, 1H, J = 11.6, 17.5 Hz, pyrazoline-C4H), 5.8–5.9 (dd, 1H, J = 4.4, 11.6 Hz, pyrazoline-C5H), 7.18–7.68 (m, 14H, p-chlorophenyl-H, phenyl-H), 7.8 (s, 1H, pyrazole-C5H). Analysis calculated for C27H24N4O (420.51): C, 77.12; H, 5.75; N, 13.32. Found: C, 76.86; H, 5.47; N, 13.55. Rf [benzene/ethyl acetate (9:1)] = 0.65.
3.1.11 1–(4-Chlorophenyl)-3-phenyl-4–(1-butanoyl-3-phenyl-2-pyrazolin-5-yl)-1H-pyrazole (6a)
A mixture of 2a (1.3 mmol, 0.5 gm) and an equimolar amount of hydrazine hydrate (0.07 ml) was dissolved in 15 ml of butyric acid. The mixture was heated under reflux for 6 h with continuous stirring using a magnetic stirrer. The reaction mixture was then concentrated, cooled and poured onto crushed ice (30 g). Finally, the white precipitate formed was filtered, washed with water, dried and recrystalised from ethanol: water (6:1) mixture. Yield, 78.65%; mp, 235–237 °C; IR (Nujol) cm−1: 1665(C = O); 1600 (C = N); 1450 (C-Cl). 1H-NMR (DMSO-d6) δ(ppm): 1.0–1.1 (t, 3H, CH3), 1.7–1.9 (m, 2H, CH2CH3), 2.7–2.9 (m, 2H, CH2CH2), 3.1–3.2 (dd, 1H, J = 4.5, 17.4 Hz, pyrazoline-C4H), 3.6–3.7 (dd, 1H, J = 11.7, 17.4 Hz, pyrazoline-C4H), 5.8–5.9 (dd, 1H, J = 4.5, 11.6 Hz, pyrazoline-C5H), 7.25–7.78 (m, 14H, p-chlorophenyl-H, phenyl-H), 7.8 (s, 1H, pyrazole-C5H). Analysis calculated for C28H25ClN4O (468.98): C, 71.71; H, 5.37; N, 11.95; Cl, 7.56. Found: C, 71.94; H, 5.61; N, 11.84; Cl, 7.38. Rf [benzene/ethyl acetate (9:1)] = 0.62.
3.1.12 1–(4-Chlorophenyl)-3-phenyl-4–(1-benzoyl-3-phenyl-2-pyrazolin-5-yl)-1H-pyrazole (7a)
To a solution of 3a (1.2 mmol, 0.48 gm) in dry pyridine (5 ml) an equivalent amount of benzoyl chloride (0.20 ml) was added. The reaction mixture was then heated in a boiling water bath for 20 min, cooled and poured onto crushed ice (30 g). The white precipitate formed was separated by filtration, washed with water, dried and recrystalised from ethanol. Yield, 75.4%; mp, 237–239 °C; IR (cm−1): 1650 (C = O); 1605 (C = N); 1455 (C-Cl). 1H-NMR (DMSO-d6) δ(ppm): 3.2–3.3 (dd, 1H, J = 4.8, 17.4 Hz, pyrazoline-C4H), 3.8–3.9 (dd, 1H, J = 11.8, 17.8 Hz, pyrazoline-C5H), 6.0–6.1 (dd, 1H, J = 4.6, 12.0 Hz, pyrazoline-C5H), 7.25– 7.95 (m, 19H, p-chlorophenyl-H, phenyl-H), 8.2 (s, 1H, pyrazole-C5H). Analysis calculated for C31H23ClN4O (502.99): C, 74.02; H, 4.61; N, 11.14; Cl, 7.05. Found: C, 73.79; H, 4.83; N, 10.88; Cl, 7.21. Rf [benzene/ethyl acetate (9:1)] = 0.70.
3.2. Biological evaluation
3.2.1. In vivo antimalarial activity testing against Plasmodium berghei
The in vivo antimalarial activities of the synthesised compounds were performed by using the standard 4-day suppressive test as described by Fidock et al.Citation40. The practical procedures were performed as described earlierCitation32. Briefly, Swiss albino mice of both sexes, weighing 24–38 g and 4–6 weeks of age, were acclimatised for a period of 7 days at room temperature (23–25 °C) and relative humidity of 60–65%. On day 0, the test mice were injected intravenously with 0.2 ml of 2 × 107 parasitised erythrocytes infected with P. berghei ANKA strain. After 2 h of injection, the infected mice were weighed and arbitrarily divided into 37 groups of five mice per cage. Groups 1–35 (35 cages) received the compounds orally, each at 20 mg/kg dose levels and served as treatment groups. Group 36 received the drug vehicle (7% Tween, 3% ethanol in distilled water) and served as a negative control, while Group 37 received chloroquine phosphate at the dose level of 20 mg/kg and served as a positive control. On days 1–3, the treatment groups were treated with the same single dose of the synthesised compound at 24 h intervals. On day 4 (i.e. 24 h after the last dose), a blood smear from all test animals was prepared using Giemsa stain. The level of parasitemia was determined microscopically by counting 5 fields of approximately 100 erythrocytes per field. The difference between the mean value for the negative control group (taken as 100%) and those of the experimental groups was calculated and expressed as percent suppression or activity. Chloroquine-treated mice were completely cured of the parasite. The in vivo antimalarial activity testing procedure was repeated for three compounds (5b, 6a and 7a) at a dose level of 30 mg/Kg since they showed better percent suppression than the other compounds.
3.2.2. In vitro antimalarial activity testing against chloroquine-resistant (RKL9) strain of Plasmodium falciparum
The P. falciparum strain RKL9 was maintained in continuous culture using the standard method described by Trager and JensenCitation41. Compounds (5b and 6a), the two most active compounds at a dose level of 30 mg/Kg, were further examined for their antiplasmodial activities against chloroquine resistant (RKL9) P. falciparum strain as reported earlierCitation32.
3.2.3. In vitro antileishmanial activity on L. aethiopica promastigotes and amastigote forms
All the tested compounds were evaluated for their antileishmanial activity on both the promastigote and amastigote forms as reported earlierCitation32.
3.2.4. Reversal of the antileishmanial activity via folic and folinic acid
This test was carried out on the in vitro growth assay for promastigotes and based on the previously published methodologyCitation42. All experimental steps were performed as reported earlierCitation43.
3.2.5. In vivo acute toxicity test
Compounds that have shown good activity (5b and 6a) were tested for their oral acute toxicity in mice according to previously reported proceduresCitation35. Four groups of animals each group consisting of six male mice were used for testing acute toxicity. Mice in groups one and two were given 125 and 250 mg/kg/day of the synthesised compounds respectively and the third group was given the highest dose (500 mg/kg/day) and the fourth group was treated with the vehicle (control group) at a maximum dose volume of 1 ml/100 g of body weight by oral route. After the substance has been administered, food was withheld for a further 2 h period. The mice were observed closely as reported earlierCitation44,Citation45.
3.3. Molecular Modelling
3.3.1. Molecular docking
The prepared structure of the quadruple mutant (N51I, C59R, S108N and I164L) Pf DHFR-TS (PDB code:1j3k) was adapted from previous reportsCitation26,Citation27. The leishmanial PTR1 (PDB: 7pxx) was retrieved from the Protein Data Bank (PDB) and prepared using “Quickprep” and “Structure Preparation" modules of MOE. The PDB files were converted to PDBQT files by employing a python script (prepare_receptor4.py) provided by the MGLTools package (version 1.5.4)Citation46 for AutoDock Vina (version 1.1.2)Citation47 docking experiments.
The most active compounds (5b, 6a and 7a) and their stereoisomers were built and prepared by MOE as reported earlierCitation26. The resulting conformers were saved as an SD files for the docking experiments. The SD files were converted and split into PDB files by MOE, which was further converted into PDBQT files by an MGLTools (version 1.5.4) python script (prepare_ligand4.py) for AutoDock Vina docking experiments.
AutoDock Vina (version 1.1.2) was used for docking experiments of the most active compounds against both quadruple mutants (N51I, C59R, S108N and I164L) Pf DHFR-TS and leishmanial PTR1 structures. We employed default docking parameters and the size of the docking grid was 22 Å × 22 Å × 22 Å, with a grid spacing of 1 Å. The centre of the grid box was adjusted on the centre of mass of the co-crystalized ligands. By default, the docking was terminated when the maximum energy difference between the best-scored pose and the worst one was 3 kcal/mol. This docking setup was validated by a successful pose-retrieval docking experiment for the co-crystal ligand on both PDB crystal structures.
3.3.2. Molecular Dynamics
The molecular dynamics simulations were carried out as reported earlier in some proceduresCitation48,Citation49. Molecular dynamics simulations and systems build-up were carried out using GROMACS 2020.3Citation50. The protein-ligand complex was solvated in a triclinic box of SPC216 with explicit water modelCitation51. The system was then neutralised by NaCl molecules at 0.1 M concentration. A steepest descent minimisation algorithm was applied for system energy minimisation setting 10 kJ/mol and 50,000 steps as convergence criteria. NVT followed by NPT equilibration was completed for 500 ps each at 300 K temperature and 1 atm pressure. Then, a production run was carried out for 50 ns at the NPT ensemble. The coordinates of the trajectory were saved each 10 ps time interval resulting in 5000 frames for the whole 50 ns simulation time. The V-rescale modified Berendsen thermostatCitation52 was used for temperature coupling for each equilibration run, while Berendsen couplingCitation53 was used for pressure coupling with a 2 ps time constant for equilibration and production runs. However, Parrinello-Rahman pressure coupling schemeCitation54 was employed for pressure coupling for the production runs. A Verlet cut-off scheme was used for searching neighbouring atoms and Van Der Waals calculations with cut-off and switch list distances of 1.2 and 1.0 nm, respectively. Particle Mesh Ewald methodCitation55 was used for the calculations of long-range electrostatics within 1.2 nm. Bond lengths were constrained using the LINear Constraint Solver (LINCS) algorithmCitation56. CHARMM36 all-atom force fieldCitation57 was used for topology and parameter generation of the protein molecules, and SwissParam serverCitation58 was used for ligand parameterisation. For all simulations, a leap-frog integrator was used with a steps size of 2 fs. Different analysis metrics, such as root mean squared deviation (RMSD), the radius of gyration (Rg), root mean squared fluctuation (RMSF), hydrogen-bond (H-bond) count, PCA (principal component analysis), and free energy landscape (FEL) were calculated via GROMCS and MDtraj toolsCitation59 and some were plotted using XMGRACECitation60.
4 Conclusion
Nine target pyrazoline compounds were synthesised in acceptable yields (70–78%). The in vivo antimalarial activity of the synthesised compounds was evaluated against P. berghei. Compounds 5b, 6a and 7a showed inhibition of the parasite multiplication by 66.7, 42.3 and 36.5%, respectively, at a dose level of 20 mg/kg; and 71.2, 52.4 and 30.3%, respectively, at a dose level of 30 mg/kg. The two most active compounds (5b and 6a) were evaluated in vitro for their antimalarial activity against chloroquine-resistant strain (RKL9) of P. falciparum and were both found to be more potent than the standard chloroquine with IC50 values of 0.0368 and 0.0946 µM, respectively.
The compounds were evaluated for their in vitro antileishmanial activity against Leishmania aethiopica promastigote and amastigote forms. Interestingly, the results showed that compounds 5b, 6a and 7b were more potent than the standard miltefosine with IC50 values of 0.05, 0.89 and 0.08 μM for the promastigote form, respectively. Furthermore, the reversal of antileishmanial activity of the active compounds via folic and folinic acids showed analogous results to the positive control Trimethoprim. This designates the antifolate mechanism for the antileishmanial activity of these compounds anticipating both leishmanial DHFR-TS and PTR1 enzymes as putative targets.
The in vivo acute toxicity test exhibited that 5b and 6a compounds were non-toxic at 125, 250 and 500 mg/kg. Molecular docking of the most active compounds against putative malarial Pf-DHFR-TS and leishmanial PTR1 targets justified the observed activities. The docking poses of 5b and 7a displayed superior performances compared to pyrimethamine against both wild-type and quadruple mutant (resistant) Pf-DHFR-TS, with superior scores towards the mutant form, implying high potential binding to the resistant Pf-DHFR-TS. Moreover, the most active compounds demonstrated a superior scores against the leishmanial PTR1 compared to the references, dihydropterine and trimethoprim. Interestingly, molecular dynamics simulations of three leishmanial PTR1 systems for 50 ns each, for the apo, the complexed with the most active 7a, and the co-crystal complex systems, highlighted the stable and privileged binding of 7a towards PTR1 in a time-dependent manner. These findings highlight that 7a exerts its antileishmanial activity via inhibiting leishmanial PTR1.
Author contributions
All authors have given approval to the final version of the manuscript. All authors agree to be accountable for the content of the work.
Ethics statements
The animal study was reviewed and approved by the protocols used in this study followed the guidelines set in ‘The Guide for the Care and Use of Laboratory Animals’, and obtained approval by Animal Care & Use Committee (ACUC), Faculty of Pharmacy, Alexandria University, No. ACUC17/18 at 29/4/2017.
Supplemental Material
Download PDF (234.2 KB)Acknowledgement
TMI would like to acknowledge Bibliotheca Alexandrina High-Performance Computing (BA-HPC) for granting access to perform the molecular dynamics simulations. The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant Code: (22UQU4290565DSR64).
Disclosure statement
The authors have no other relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- W.H.O. World malaria report. https://www.who.int/publications/i/item/9789241565721; 2019.
- Cheviet T, Wein S, Bourchenin G, et al. beta-hydroxy- and beta-aminophosphonate acyclonucleosides as potent inhibitors of Plasmodium falciparum growth. J Med Chem 2020;63:8069–87.
- Madhav H, Hoda N. An insight into the recent development of the clinical candidates for the treatment of malaria and their target proteins. Eur J Med Chem 2021;210:112955.
- Goncalves GA, Spillere AR, das Neves GM, et al. Natural and synthetic coumarins as antileishmanial agents: a review. Eur J Med Chem 2020;203:112514.
- Bekhit AA, El-Agroudy E, Helmy A, et al. Leishmania treatment and prevention: natural and synthesized drugs. Eur J Med Chem 2018;160:229–44.
- Pigott DM, Bhatt S, Golding N, et al. Global distribution maps of the leishmaniases. eLife 2014;3:e02851 1–21.
- W.H.O., Leishmaniasis. https://www.who.int/health-topics/leishmaniasis#tab=tab_1; 2020.
- Ogutu B. Artemether and lumefantrine for the treatment of uncomplicated Plasmodium falciparum malaria in sub-Saharan Africa. Expert Opin Pharmacother 2013;14:643–54.
- Nwaka S, Hudson A. Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov 2006;5:941–55.
- Rajasekaran R, Chen YP. Potential therapeutic targets and the role of technology in developing novel antileishmanial drugs. Drug Discov Today 2015;20:958–68.
- Vickers TJ, Beverley SM. Folate metabolic pathways in Leishmania. Essays Biochem 2011;51:63–80.
- Nare B, Hardy LW, Beverley SM. The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J Biol Chem 1997;272:13883–91.
- Corona P, Gibellini F, Cavalli A, et al. Structure-based selectivity optimization of piperidine–pteridine derivatives as potent Leishmania pteridine reductase inhibitors. J Med Chem 2012;55:8318–29.
- Dube D, Periwal V, Kumar M, et al. 3D-QSAR based pharmacophore modeling and virtual screening for identification of novel pteridine reductase inhibitors. J Mol Model 2012;18:1701–11.
- de Souza Moreira D, Ferreira RF, Murta SMF. Molecular characterization and functional analysis of pteridine reductase in wild-type and antimony-resistant Leishmania lines. Experiment Parasitol 2016;160:60–6.
- Nayak N, Ramprasad J, Dalimba U. New INH–pyrazole analogs: design, synthesis and evaluation of antitubercular and antibacterial activity. Bioorg Med Chem Lett 2015;25:5540–5.
- Li Y-R, Li C, Liu J-C, et al. Synthesis and biological evaluation of 1,3-diaryl pyrazole derivatives as potential antibacterial and anti-inflammatory agents. Bioorg Med Chem Lett 2015;25:5052–7.
- Meng FJ, Sun T, Dong WZ, et al. Discovery of novel pyrazole derivatives as potent neuraminidase inhibitors against influenza H1N1 virus. Archiv Der Pharmazie 2016;349:168–74.
- Chuang H, Huang L-CS, Kapoor M, et al. Design and synthesis of pyridine-pyrazole-sulfonate derivatives as potential anti-HBV agents. MedChemComm 2016;7:832–6.
- Hafez HN, El-Gazzar A-RBA, Al-Hussain SA. Novel pyrazole derivatives with oxa/thiadiazolyl, pyrazolyl moieties and pyrazolo[4,3-d]-pyrimidine derivatives as potential antimicrobial and anticancer agents. Bioorg Med Chem Lett 2016;26:2428–33.
- Shi JB, Tang WJ, qi XB, et al. Novel pyrazole-5-carboxamide and pyrazole–pyrimidine derivatives: synthesis and anticancer activity. Eur J Med Chem 2015;90:889–96.
- Özdemir A, Altıntop MD, Kaplancıklı ZA, et al. Synthesis and evaluation of new 1, 5-Diaryl-3-[4-(methyl-sulfonyl) phenyl]-4, 5-dihydro-1H-pyrazole derivatives as potential antidepressant agents. Molecules 2015;20:2668–84.
- Viveka S, Dinesha D, Shama P, et al. Design, synthesis, anticonvulsant and analgesic studies of new pyrazole analogues: a Knoevenagel reaction approach. RSC Advances 2015;5:94786–95.
- Mabkhot YN, Kaal NA, Alterary S, et al. Synthesis, in-vitro antibacterial, antifungal, and molecular modeling of potent anti-microbial agents with a combined pyrazole and thiophene pharmacophore. Molecules 2015;20:8712–29.
- Faidallah HM, Al-Mohammadi MM, Alamry KA, Khan KA. Synthesis and biological evaluation of fluoropyrazolesulfonylurea and thiourea derivatives as possible antidiabetic agents. J Enzyme Inhib Med Chem 2016;31(Suppl 1):157–163.
- Bekhit AA, Saudi MN, Hassan AMM, et al. Synthesis, in silico experiments and biological evaluation of 1,3,4-trisubstituted pyrazole derivatives as antimalarial agents. Eur J Med Chem 2019;163:353–66.
- Bekhit AA, Saudi MN, Hassan AM, et al. Synthesis, molecular modeling and biological screening of some pyrazole derivatives as antileishmanial agents. Future Med Chem 2018;10:2325–44.
- Tageldin GN, Fahmy SM, Ashour HM, et al. Design, synthesis and evaluation of some pyrazolo[3,4-d]pyrimidines as anti-inflammatory agents. Bioorg Chem 2018;78:358–71.
- Atta KFM, Ibrahim TM, Farahat OOM, et al. Synthesis, modeling and biological evaluation of hybrids from pyrazolo[1,5c]pyrimidine as antileishmanial agents. Future Med Chem 2017;9:1913–29.
- Ramirez-Prada J, Robledo SM, Velez ID, et al. Synthesis of novel quinoline-based 4,5-dihydro-1H-pyrazoles as potential anticancer, antifungal, antibacterial and antiprotozoal agents. Eur J Med Chem 2017;131:237–54.
- Insuasty B, Ramirez J, Becerra D, et al. An efficient synthesis of new caffeine-based chalcones, pyrazolines and pyrazolo[3,4-b][1,4]diazepines as potential antimalarial, antitrypanosomal and antileishmanial agents. Eur J Med Chem 2015;93:401–13.
- Bekhit AA, Hassan AM, El Razik HAA, et al. New heterocyclic hybrids of pyrazole and its bioisosteres: design, synthesis and biological evaluation as dual acting antimalarial-antileishmanial agents. Eur J Med Chem 2015;94:30–44.
- Bekhit AA, Ashour HMA, Bekhit AE-DA, et al. Synthesis of some pyrazolyl benzenesulfonamide derivatives as dual anti-inflammatory antimicrobial agents. J Enzyme Inhib Med Chem 2009;24:296–309.
- Faour WH, Mroueh M, Daher CF, et al. Synthesis of some new amide-linked bipyrazoles and their evaluation as anti-inflammatory and analgesic agents. J Enzyme Inhib Med Chem 2016;31:1079–94.
- Bekhit AA, Baraka AM. Novel milrinone analogs of pyridine-3-carbonitrile derivatives as promising cardiotonic agents. Eur J Med Chem 2005;40:1405–13.
- Arba M, Wahyudi ST, Brunt DJ, et al. Mechanistic insight on the remdesivir binding to RNA-Dependent RNA polymerase (RdRp) of SARS-cov-2. Comp Biol Med 2021;129:104156.
- Ismail MI, Ragab HM, Bekhit AA, Ibrahim TM. Targeting multiple conformations of SARS-CoV2 papain-like protease for drug repositioning: an in-silico study. Comp Biol Med 2021;131:104295.
- Pandey P, Rane JS, Chatterjee A, et al. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: an in silico study for drug development. J Biomol Struct Dyn 2021;39:6306–16.
- Pandey P, Prasad K, Prakash A, Kumar V. Insights into the biased activity of dextromethorphan and haloperidol towards SARS-CoV-2 NSP6: in silico binding mechanistic analysis. J Mol Med 2020;98:1659–73.
- Fidock DA, Rosenthal PJ, Croft SL, et al. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov 2004;3:509–20.
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science 1976;193:673–5.
- Mendoza-Martinez C, Galindo-Sevilla N, Correa-Basurto J, et al. Antileishmanial activity of quinazoline derivatives: synthesis, docking screens, molecular dynamic simulations and electrochemical studies. Eur. J. Med. Chem 2015;92:314–31.
- Temraz MG, Elzahhar PA, El-Din ABA, et al. Anti-leishmanial click modifiable thiosemicarbazones: design, synthesis, biological evaluation and in silico studies. Eur J Med Chem 2018;151:585–600.
- Bekhit AA, Hymete A, Damtew A, et al. Synthesis and biological screening of some pyridine derivatives as anti-malarial agents. J Enzyme Inhib Med Chem 2012;27:69–77.
- Bekhit AA, Fahmy HTY. Design and synthesis of some substituted 1H‐Pyrazolyl‐oxazolidines or 1H‐Pyrazolyl‐thiazolidines as anti‐inflammatory‐antimicrobial agents. Archiv Der Pharmazie 2003;336:111–8.
- Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model 1999;17:57–61.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61.
- Elghoneimy LK, Ismail MI, Boeckler FM, et al. Facilitating SARS CoV-2 RNA-dependent RNA polymerase (RdRp) drug discovery by the aid of HCV NS5B palm subdomain binders: in silico approaches and benchmarking. Comp Biol Med 2021;134:104468.
- Bekhit AA, Nasralla SN, El-Agroudy EJ, et al. Investigation of the anti-inflammatory and analgesic activities of promising pyrazole derivative. Eur J Pharm Sci 2022;168:106080.
- Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015;1–2:19–25.
- Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A 2001;105:9954–60.
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys 2007;126:014101.
- Berendsen HJC, Postma JPM, Gunsteren WFv, et al. Molecular dynamics with coupling to an external bath. J Chem Phys 1984;81:3684–90.
- Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys 1981;52:7182–90.
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J Chem Phys 1993;98:10089–92.
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comp Chem 1997;18:1463–72.
- Huang J, MacKerell AD Jr.CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comp Chem 2013;34:2135–45.
- Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comp Chem 2011;32:2359–68.
- McGibbon RT, Beauchamp KA, Harrigan MP, et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys J 2015;109:1528–32.
- Turner PX. Version 5.1. 19. Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR; 2005.