
Abstract
Direct interference with Kelch-like ECH-associated protein 1 (Keap1)-Nrf2 protein-protein interaction (PPI) has recently been introduced as an attractive approach to control life-threatening diseases like myocarditis. The present study aimed to investigate the potential application in myocarditis of a series of novel non-naphthalene derivatives as potential Keap1-Nrf2 PPI inhibitors. Our results indicated that the optimal compound K22 displayed the highest metabolic stability and showed notable Keap1-Nrf2 PPI inhibitory activities in vitro. K22 effectively triggered Nrf2 activation and increased the protein and mRNA expression of Nrf2-regulated genes in H9c2 cells. Moreover, pre-treatment with K22 was shown to mitigate LPS-induced damage to H9c2 cells, causing a marked decrease in the levels of inflammatory factors as well as reactive oxygen species (ROS). Furthermore, K22 was also shown to be non-mutagenic in the Ames test. Overall, our findings suggest that K22 may be a promising drug lead as a Keap1-Nrf2 PPI inhibitor for myocarditis treatment.
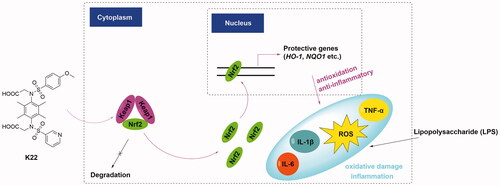
Graphical Abstract
Introduction
The human body is accompanied by reactive oxygen species (ROS) and electrophiles from endogenous or exogenous sources. The in-built cytoprotective mechanisms of our body are capable of up-regulating cytoprotective factors through various detoxification and antioxidant enzymes, thus helping to combat oxidative damage and maintain body homeostasisCitation1. Despite the power of our natural defense systems, persistently high levels of ROS in human cells continue to cause different chronic diseases, such as neurodegenerative and cardiovascular diseases as well as cancer, etcCitation2,Citation3.
The nuclear factor erythroid 2-related factor 2 (Nrf2) has been known to bind to antioxidant response element (ARE) and regulate the transcription of approximately 250 genes and plays an important role in regulating the cellular defense system and helping mitigate oxidative stressCitation4,Citation5. Under basal conditions, the activity of Nrf2 is negatively upregulated by Kelch-like ECH-associated protein 1 (Keap1), which mostly through binding to the Nrf2-ECH homology 2 (Neh2) domain of Nrf2 and ultimately promote its polyubiquitination and proteasomal degradationCitation6. Under stressed conditions, the presence of ROS or electrophiles promotes the covalent modification of Keap1, and accelerates the dissociation of the Cul3-Keap1-Nrf2 complex, leading to the accumulation of Nrf2 and further activation of the Keap1-Nrf2-ARE pathway.Citation6 Thus, the Keap1-Nrf2-ARE pathway is considered an essential pathway to manage counter oxidation and maintain cell homeostasis.
Currently, strategies to upregulate Nrf2 comprise primarily Nrf2 activators and Keap1-Nrf2 protein − protein interaction (PPI) inhibitors. Although approval has been granted by the FDA to Nrf2 activators such as dimethyl fumarate (DMF) for treating patients with relapsing multiple sclerosis (MS), the lack of target sensitivity in most Nrf2 activators has circumscribed their utility in clinical practiceCitation7–9. Alternatively, direct interference with the Keap1-Nrf2 PPI has been advocated as a more appealing alternative for Nrf2 activation. In recent years, a profusion of compounds has been developed as potential Keap1-Nrf2 PPI inhibitors. For example, in 2013, the discovery of compound 1 () by Silvian et al. represents the first Keap1-Nrf2 PPI inhibitor owning a 1,4-diaminonaphthalene core with moderate activity against Keap1 (IC50 = 2.7 μM) via the two-dimensional fluorescence intensity distribution analysis (2 D-FIDA)Citation10. In the same year, compound 2 with tetrahydroisoquinoline core was discovered by Hu et al. via a high-throughput screen (HTS) method using a homogenous fluorescence polarisation assay, and the IC50 of which against Keap1-Nrf2 PPI was 3 μMCitation11. Subsequently, different structural types of small molecules acting as potential Keap1-Nrf2 PPI inhibitors have been gradually disclosed, which include compound 3 with iminocoumarin-benzothiazole coreCitation12, compound 4 with 1,4-isoquinoline coreCitation13, compound 5 with indole coreCitation14, compound 6 with triazole coreCitation15, compound 7 with xylylene coreCitation16, and compound 8 with indoline core ()7, etc. Most of the abovementioned small molecules exhibit notable inhibitory activities against Keap1-Nrf2 PPI and have shown therapeutic potential in in vitro or in vivo models of chronic inflammatory diseases simultaneously. In addition, Kihlberg et al. recently disclosed high-affinity ligands with macrocyclic cores.Citation17
Figure 1. Chemical structures of recently reported small-molecule Keap1-Nrf2 PPI inhibitors.

Notably, among the reported Keap1-Nrf2 PPI inhibitors, the 1,4-bis(arylsulfonamide) benzene core has recently attracted the interest of scientists. Hu et al.Citation18 and Wells et al.Citation19 respectively explored the substituents at the C-2 position of benzene, and both found that compounds with substituents such as O-linked fragments at the C2 position of the benzene exhibited notable inhibitory activity against Keap1. Their findings imply that the 1,4-bis(arylsulfonamido) benzene core represents a promising scaffold for the design of novel Keap1-Nrf2 PPI inhibitors. Owing to the limited structural diversity of currently reported Keap1-Nrf2 PPI inhibitors containing this core, it is of great significance to further explore and enrich the structure types of compounds with this core. In this study, we focussed on the optimisation of the classic 1,4-diaminonaphthalene core and developed a series of benzene scaffold based-derivatives by replacing the defective 1,4-diaminonaphthalene core with benzene or tetramethyl substituted benzene core and using a scaffold fusion strategy to afford the propanoic acid substituted benzene core on the basis of compound 1 (). Subsequently, we evaluated the inhibitory activities against Keap1-Nrf2 PPI of these derivatives, determined the metabolic stabilities of some representative compounds, forecasted the potential binding modes of the optimal compound and explored the potential application of the optimal compound in cardiomyocyte damage induced by lipopolysaccharide (LPS). In addition, a preliminary assessment of the drug-likeness and mutagenic properties of the optimal compound was also performed.
Figure 2. Design of novel non-naphthalene derivatives from compound 1. (A) Crystal structure of compound 1 bound to Keap1 Kelch domain (PDB ID: 4IQK). (B) The design strategy of novel non-naphthalene scaffold-based derivatives.

Results and discussion
Chemistry
Compounds K1–K22 were prepared at first. With p-phenylenediamine (9) as the starting material, compounds K1–K9 could be obtained directly through nucleophilic substitution reaction between 9 and different sulphonyl chloride (2.5 equiv.) with moderate yields (51%–82%) in the presence of pyridine in dichloromethane (Scheme 1). Due to the structural asymmetry of compounds K10–K18, unlike K1–K9, it needs to undergo two nucleophilic substitution reactions to obtain K10–K18 (Scheme 1).
Scheme 1. Synthetic routes of compounds K1–K22. Reagents and conditions: (a). Different sulphonyl chloride (2.5 equiv.), CH2Cl2, pyridine, rt, 6–18 h; (b). Different sulphonyl chloride (1.2 equiv.), CH2Cl2, Et3N, rt, 6–16 h; (c). 4-Methoxybenzenesulfonyl chloride (1.2 equiv.), THF, Et3N, rt, 5–10 h; (d). Ethyl bromoacetate (2.2 equiv.), DMF, K2CO3 (3 equiv.), rt, 16 h; (e). Methanol, 10% NaOH, 85 °C, 5 h; (f). Pyridine-3-sulphonyl chloride (1.2 equiv.), pyridine, THF, rt, 18 h; (g). Ethanol, 10% NaOH, 80 °C, 8 h.

First, nucleophilic substitution reaction between 9 and different sulphonyl chlorides (1.2 equiv.) afforded intermediates 10a–i at the existence of triethylamine. Then the intermediates 10a–i continued to react with 4-methoxybenzenesulfonyl chloride (1.2 equiv.) to obtain the desired products K10–K18 under alkaline conditions (yield 60%–85%). Subsequently, the transformation of K1, K3 or K18 into ethyl ester group substituted intermediates 11a–c could be readily accomplished in high yield in the presence of excessive ethyl bromoacetate. Then, the target products K19–K21 could be easily obtained after hydrolysis of the intermediates 11a–c in 10% NaOH solution. Except that the substrates used were different, the preparation method of compound K22 was roughly similar to that of K19–K21 (Scheme 1).
To obtain the propanoic acid substituted products KP-1 and KP-2, several classic organic chemical reactions were applied. Briefly, 2-bromo-4-nitroaniline (16) was reduced by iron powder afforded 2-bromobenzene-1,4-diamine (17) in a solution of concentrated HCl in ethanol. Next, intermediate 17 underwent a nucleophilic substitution reaction in the presence of excessive 4-methoxybenzenesulfonyl chloride to produce intermediate 18. Heck coupling was then conducted between 18 and benzyl acrylate with the catalysis of palladium (II) acetate and XPhos to get an intermediate 19. The target compound KP-1 could be obtained by the reduction of 19 with hydrogen atmosphere and 10% palladium carbon (yield 72%) (Scheme 2). On the other hand, consecutive nucleophilic substitution reactions of intermediate 17 with 1-methyl-1H-pyrazole-4-sulphonyl chloride and 4-methoxybenzenesulfonyl chloride afforded intermediate 21 successfully. Finally, after heck coupling between 21 and benzyl acrylate and subsequent reduction reaction of intermediate 22 afforded product KP-2 (Scheme 2).
Scheme 2. Synthetic routes of compounds KP-1 and KP-2. Reagents and conditions: (a). Iron powder (4.5 equiv.), HCl (conc.), ethanol, 80 °C, 2 h; (b). 4-Methoxybenzenesulfonyl chloride (2 equiv.), THF, Et3N, rt, overnight; (c). Benzyl acrylate (10 equiv.), palladium (II) acetate (0.1 equiv.), XPhos (0.2 equiv.), N, N-diisopropylethylamine (2 equiv.), DMF, 100 °C, 24 h; (d). 10% Palladium on charcoal, ethanol, H2, rt, 16 h; (e). 1-Methyl-1H-pyrazole-4-sulphonyl chloride (1.1 equiv.), THF, pyridine, rt, 8 h; (f). 4-Methoxybenzenesulfonyl chloride (1.1 equiv.), CH2Cl2, Et3N, rt, 10 h.

Biology
Design strategy
The 1,4-diaminonaphthalene core represents one of the most classical structural cores of non-covalent Keap1-Nrf2 PPI inhibitors. Since the discovery of compound 1 () and the publishing of the crystal structure of Keap1 Kelch-DC bound to compound 1 in 2013Citation10, numerous efforts have been made to explore new Keap1-Nrf2 PPI inhibitors based on this structural core. However, the potential carcinogenic and mutagenic properties of some naphthyl compounds indicated that the 1,4-diamino naphthalene core has poor drug-like properties, which precludes it from qualifying as an optimal core of this series of compoundsCitation20.
Analysis of the crystal structures of compound 1 and Keap1 Kelch-DC suggests several molecular interactions that could account for compound 1’s potency (). The sulphonamides moiety in compound 1 formed hydrogen bonding interactions with the residues Ser508, Ser555 and Ser602 in Kelch-DC, and four pi-pi stacking interactions formed between compound 1 and the residues including Arg415, Tyr334, Tyr525 and Tyr572 (). Since the electron-rich ring of naphthalene is mainly stacked with Arg415, and one of the rings in naphthalene is deeply buried in the polar hole of the central cavity of Kelch-DC, it seems feasible to discard this ring of naphthalene though it may weaken the pi-pi stacking interaction. In view of this, we decided to take the plunge to replace the naphthalene nucleus with a single benzene nucleus while retaining the sulphonamide moiety (). Firstly, we introduced different symmetric substituents at the R1 position by imitating the structural characteristics of compound 1 and designed nine structurally symmetric derivatives (K1–K9). After the best substituent in the R1 position was determined, we next continued to modify the R2 position and designed nine structurally unsymmetric derivatives (K10–K18). After determining the optimal R1 and R2 position, we then went to optimise the R3 position and designed three different derivatives (K19–K21) containing the acetic acid group for improving the inhibitory activity. In order to further optimise the characteristics of the inhibitor, we focussed on modifying the benzene core and finally designed the derivative K22 carrying tetramethyl substituted benzene core (). The reason for introducing a tetramethyl group into the benzene core was to mimic the large conjugation system of the naphthalene ring and retain the hydrophobic interaction with key amino acid residues (e.g. Arg415) on Keap1, although it may reduce the water solubility of the molecule.
Additionally, we also designed two other compounds named KP-1 and KP-2 using a scaffold fusion strategy, both of which were structurally characterised with a propanoic acid group at the C-2 position of the benzene core. In our previous study, we found that Phloretin (PHL) effectively inhibited Keap1-Nrf2 PPI and exhibited activities to suppress high glucose-induced cardiomyocyte oxidation and fibrosis injuryCitation21. In analysing the structural characteristics of PHL, we believed that the integration of the flexible propan-1-one fragment of PHL into compound 1 at the C-2 position of the benzene could compensate for the destroyed stacking interactions with Keap1 after cleaving one benzene of naphthalene in compound 1. Therefore, we introduced a propanoic acid and designed the compound KP-1 (). Based on the in vitro fluorescence polarisation (FP) assay results of KP-1, we finally prepared the compound KP-2 with a hydrophilic N-methyl-pyrazole group instead of one of the 4-methoxyphenyl of KP-1 to further improve the physiochemical properties of the derivatives.
Keap1-Nrf2 PPI inhibitory activity evaluation
The inhibitory potency against Keap1-Nrf2 PPI of the newly designed non-naphthalene derivatives was evaluated by FP assays. According to our design strategy, the nine symmetrical non-naphthalene derivatives K1–K9 were first evaluated. In the initial evaluation, two concentrations (5 and 50 μM) were used to evaluate the potency of these synthesised analogs. The results of inhibitory activities of compounds K1–K9 against Keap1-Nrf2 PPI were shown in . The results showed that compound K3 (R1= 4-methoxyphenyl) exhibited the best inhibitory rate (∼68%) against Keap1-Nrf2 PPI at 50 μM concentration, which was more potent than the other eight derivatives including K1 (R1= 4-phenyl), K2 (R1= 2,4,6-trimethylphenyl), K4 (R1= 4-cyanophenyl), K5 (R1= 4-biphenyl), K6 (R1= benzyl), K7 (R1= pyridine-3-yl), K8 (R1= thiophene-2-yl) and K9 (R1= (5-chlorothiophene)-2-yl), and the IC50 value of K3 was stood at 36.30 μM on Keap1-Nrf2 PPI. Compared to compound 1 (97% inhibition at 50 μM, IC50 = 2.02 μM), the reason for the decrease of activity of the newly designed compounds (e.g. K3) may be caused by replacing of naphthalene ring with the benzene ring destroys the initial stacking interactions with residue Arg415 in Keap1 (). Although the replacement of the naphthalene core of compound 1 with benzene core significantly reduced the inhibitory activity, the experimental results implied that 4-methoxyphenyl substituent in the R1 position seemed important for the maintenance of inhibitory activity of derivatives on Keap1-Nrf2 PPI.
Figure 3. Inhibitory activities of derivatives K1–K18 against Keap1-Nrf PPI by FP assay. (A) Inhibitory rates of K1–K9 towards Keap1-Nrf2 PPI at 5 μM and 50 μM. (B) Binding mode of K3 with Keap1 using molecular docking. (C) Inhibitory rates of K10–K18 towards Keap1-Nrf2 PPI at 5 μM and 50 μM. (D) Binding mode of K18 with Keap1 using molecular docking.

Next, we retained the 4-methoxyphenyl group at the R1 position and introduced different substituents at the R2 position to design and synthesise nine unsymmetrical non-naphthalene derivatives (K10–K18). The inhibitory activities of compounds K10–K18 against Keap1-Nrf2 PPI were synopsised in . The results showed that when the 4-methoxyphenyl in the R2 position was replaced by 4-phenyl (K10), 4-methylphenyl (K11), 2,4,6-trimethylphenyl (K12), 4-trifluoromethylphenyl (K13), 4-biphenyl (K14), benzyl (K15), 2-naphthyl (K16) or quinoline-8-yl (K17), the inhibitory activities of which were markedly decreased compared to those for compound K3 (). But it is still good to see that the pyridin-3-yl substituent at the R2 position (K18) exhibited a slightly higher inhibitory activity against Keap1-Nrf2 PPI than compound K3, with an inhibition rate of 72% at a concentration of 50 μM. We then determined the IC50 value of K18, which showed approximately 31.55 μM on Keap1-Nrf2 PPI. Compared to K3, the better inhibitory activity of K18 may be contributed to the more abundant hydrogen bond interactions formed by the nitrogen atom in pyridin-3-yl and oxygen atom in sulphonyl of K18 with residues Ser555 and Ser508 of Keap1 ().
Inspired by the work of You et al.,Citation22 we hypothesised that the introduction of the acetic acid group at the R3 position may help improve the inhibitory activity of Keap1-Nrf2 PPI of the derivatives. To test this hypothesis, we selected compounds K1, K3 and K18 for the subsequent modification based on the results of K1–K18 in FP assays, and finally designed and synthesised another three non-naphthalene derivatives K19–K21. As expected, when an acetic acid group was introduced at the R3 position, the inhibitory activities of compounds K20 and K21 were significantly improved for compounds K3 and K18, and the IC50 values of K20 and K21 were 1.25 μM and 0.89 μM, respectively (). Compared with compounds K3 and K18, the inhibitory activities of compounds K20 and K21 improved about 29-fold and 35-fold, respectively. Interestingly, the introduction of acetic acid at the R3 position of compound K1 (without 4-methoxy group substitution in phenyl) did not seem to help to increase the inhibitory activity, and the inhibitory rate of K19 remains below 50% at a concentration of 50 μM. These results indicated that acetic acid-substituted compounds with at least one 4-methoxyphenyl group at the R1 position have better inhibitory activity against Keap1-Nrf2 PPI. Compared with K18, the two extra acetic acid groups in K21 form additional hydrogen bonds with key amino acids in Keap1, such as Arg415 and Ser508 (Figure S1A), which may explain the reasons for the increased activity. Meanwhile, compound KP-1 was also prepared and evaluated. The result of the FP assay showed that KP-1 could effectively inhibit Keap1-Nrf2 PPI, and the IC50 value which reached 0.74 μM (), was better than both compound 1 (IC50 = 2.02 μM) and PHL (IC50 = 8.67 μM). The following molecular docking results indicated that the propionic acid group at the C-2 position of the benzene in KP-1 did not form hydrogen bonds with Keap1, but was deeply inserted into the polar hole of the central cavity of kelch DC to form hydrophilic interaction with Arg415 (Figure S1B). Notably, when we further replaced the benzene core of K21 with tetramethyl substituted benzene core (K22), the inhibitory effect of which improved significantly and showed an IC50 of 0.18 μM against Keap1-Nrf2 PPI (), which improved about 4.9-fold than K21. On the other hand, replacing 4-methoxyphenyl group of KP-1 with the hydrophilic N-methyl-pyrazole group obtained KP-2, as shown in , KP-2 (IC50 = 0.11 μM) displayed better inhibitory activity on Keap1-Nrf2 PPI than KP-1, and an approximately 6.7-fold increasing rate. The results of molecular modelling revealed the potential binding mode of KP-2 and Keap1. As shown in Figure S2, unlike KP-1, the propionic acid group of KP-2 formed a sophisticated hydrogen bond interaction with residues Asn414, Arg415, Arg380 and Ser363 in Keap1, which may explain the excellent inhibitory activity of KP-2 against Keap1-Nrf2 PPI. In addition, we also measured the metabolic stability of compound 1, PHL, derivatives K22 and KP-2. As shown in , after 30 min incubation with human liver microsomes, the remaining percentage of K22 was still at a high level, while the values of compound 1, as well as PHL, were 5.6% and 10.3%, respectively, under the same conditions. Surprisingly, although KP-2 exhibited better enzymic inhibitory activity than K22, its lower metabolic stability (61.2% remaining, ) makes it less desirable as a prominent leading compound. With a good balance between enzymic inhibitory activity and metabolic stability, the derivative K22 was selected as the representative compound for further in-depth evaluation. Subsequently, the co-immunoprecipitation assay was performed to prove compound K22 could indeed break the interaction between Keap1-Nrf2. As shown in , when Nrf2 was precipitated in the presence of its primary antibody, Keap1 was also pulled down in the control group. However, Keap1 co-precipitated with Nrf2 in H9c2 cells was markedly decreased upon pre-incubation with compound K22 at 10 μM, suggesting that K22 broke the interaction between Keap1-Nrf2. Based on the results of the aforementioned in vitro assay, the structure-activity relationships of these non-naphthalene derivatives were summarised in .
Figure 4. Dose-response curves, metabolic stability and co-immunoprecipitation analysis of representative compounds. (A) Dose-response curves of compound 1, Phloretin (PHL), K20–K22, KP-1 and KP-2 for inhibition of Keap1-Nrf2 PPI in FP assay; (B) Metabolic stability of compound 1, PHL, K22 and KP-2 in human liver microsomes; (C) Co-immunoprecipitation assay for Nrf2.

Figure 5. Preliminary structure-activity relationships of non-naphthalene scaffold-based derivatives.

Molecular modelling analysis of the optimal non-naphthalene derivative K22 with Keap1 protein
To elucidate the details of the binding mode between the optimal compound and the Keap1 Kelch domain, the dynamic behaviours of K22 to Keap1 were investigated by performing molecular docking, followed by 400 ns molecular dynamics (MD) simulations. As shown in , the root-mean-square deviations (RMSDs) of the backbone atoms (Cα) of Keap1 were quite stable with RMSD fluctuations < 1 Å, while those for K22 were also dynamic stable. These findings suggested the Keap1-K22 complex was dynamic stable and the MD simulation trajectories from 300–400 ns were suitable for further binding mode analysis. The molecular mechanics/generalized Born surface area (MM/GBSA) method based on binding free energy decomposition was employed to highlight the roles of key residues for the binding of K22 to Keap1. The calculation results indicated the key residues for the binding of K22 to Keap1 were Arg415, Ser363, Tyr344, Tyr572, Ser602, Asn414, Tyr525, Arg380, Ala556 and Phe577 (). The hydrogen bonds with residues of Ser363, Asn414, Arg415 and Ser602 showed importance for stabilise the K22 in the binding pocket of Keap1. Compared with the binding mode of K21 and Keap1 (Figure S1A), K22 has more abundant hydrogen bond interactions and hydrophobic effects in Keap1, which may be one of the reasons for its higher inhibitory activity than K21.
Figure 6. Molecular modelling of compound K22 in the binding pocket of Keap1. (A) The root-mean-square deviations (RMSDs) of the backbone atoms (Cα) of Keap1 and heavy atoms of K22. (B) Key residues for the binding of K22 to Keap1. (C) Overview of K22 bound to Keap1. (D) Detailed view of K22 bound to Keap1.

Compound K22 effectively activated NRF2-ARE regulated cytoprotective defense system in vitro
The Keap1-Nrf2-ARE pathway is a key defense mechanism of human cells, and the dysregulation of which is culpable for numerous oxidative stress and inflammatory-related diseases, such as myocarditisCitation23–26. Based on the screening results in vitro, the optimal compound K22 was selected to evaluate whether it has a potential effect on Nrf2 activation or could protect cardiac cells from oxidative stress damage. First, we studied the effect of K22 on the expression of Nrf2-regulated genes in H9c2 cells. As shown in , the results of qRT-PCR indicated that 12 h treatment with 0.5–10 μM K22 increased Nrf2 transcription and Nrf2-regulated genes such as HO-1 and NQO1 in a concentration-dependent manner. Furthermore, the effect of K22 treatment at 10 μM on Nrf2 activation occurred after 4 h and persisted for over 16 h in H9c2 cells
Figure 7. Nrf2 activation effect of K22 on H9c2 cells. (A) Cells were treated with K22 at various concentrations for 12 h. (B) Cells were exposed to K22 (10 μM) at various time points. qRT-PCR assay was applied to analyse the mRNA levels of Nrf2 and Nrf2-targeted gene. GAPDH was used to normalise the expression of these genes with DMSO-treatment group used as the blank control. (C) Cells were treated with K22 at various concentrations for 12 h. (D) Cells were exposed to K22 (10 μM) at various time points. Western blot assay was applied to determine the protein levels of Nrf2 and Nrf2-regulated proteins. (E&F) Statistics of the protein expression levels of Nrf2, HO-1 and NQO1. GAPDH was used as the internal control. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Control group.

, then we measured the expression levels of Nrf2 and Nrf2-downstream proteins by western blot. The results revealed that in H9c2 cells, treatment with 0.5–10 μM K22 concentration-dependently increased protein levels of Nrf2, HO-1 and NQO1 (). Furthermore, the results of the time course study also showed that after 16 h of treatment with 10 μM K22, the expression levels of Nrf2, HO-1 and NQO1 remained high ().
Compound K22 activates Nrf2 downstream genes depends on Nrf2
To further confirm that the Nrf2-regulated enzymes induced by K22 depend on Nrf2, a loss-of-function study on Nrf2 was performed. As shown in , compared with the control group, the mRNA levels of Nrf2 and its regulated genes including HO-1 and NQO1 were both significantly suppressed after Nrf2 siRNA treatment in H9c2 cells, while adding K22 into the Nrf2 siRNA group increased the expression levels of these genes. These results suggest that the activation of Nrf2 downstream genes by K22 is Nrf2-dependent.
Figure 8. The expression levels of Nrf2 and Nrf2-regulated genes after treatment with Nrf2 siRNA and K22. H9c2 cells were incubated with Nrf2 siRNA (50 nM), K22 (10 μM), or Nrf2 siRNA (50 nM) plus K22 (10 μM). qRT-PCR was then used to quantify the expression levels of Nrf2, HO-1 and NQO1 genes. ***p < 0.001.

Compound K22 exhibited cytoprotective effects against LPS-induced injury in H9c2 cells
Since LPS is the main microbial mediator of tissue damage and sepsis caused by Gram-negative bacteria infection, there have been widespread applications of sepsis-induced myocarditis models in screening potential agents for the treatment of myocarditis.Citation27,Citation28 With an eye to evaluating their potential roles, the effects of compound K22 on LPS-induced cardiac injury in cellular models were investigated in this study. First, we evaluated the cytotoxicity of K22 to H9c2 cells by MTS assay. As shown in Figure S3, K22 showed no apparent cytotoxicity till 50 μM concentration, the survival rates of which exceeded 80%. Next, we examined the protective effects of K22 against LPS-induced cell damage. As shown in , treatment with LPS (1 μg/mL) remarkably reduced the viability of H9c2 cells to about 60%, while pre-treatment with K22 increased the viability of H9c2 cells in a concentration-dependent manner. We found that pre-treatment with 10 μM K22 remarkably decreased the secretion of inflammatory factors such as IL-6 (), TNF-α () and IL-1β () induced by LPS in H9c2 cells. In addition, subsequent ROS detection experiments showed that pre-treatment with 10 μM K22 significantly attenuated the increase of LPS-induced ROS levels (), indicating that K22 has a potential protective effect on oxidative stress in H9c2 cells.
Figure 9. Effects of K22 on LPS-induced injury in H9c2 cells. (A) Protective effects of K22 on the LPS-induced cell damage. Cells were pre-treated with K22 (0.5–10 μM) for 12 h and then with LPS (1 μg/mL) for another 12 h. The cell viability was determined by MTS assay. (B–D) Cells were pre-treated with K22 (1–10 μM) for 12 h and then exposed to LPS (1 μg/mL) for another 12 h, and the ratios of IL-6 (B), TNF-α (C) and IL-1β (D) were determined by corresponding ELISA kits, respectively. (E–G) K22 inhibited LPS-induced ROS generation in H9c2 cells. Cells were pre-treated with 10 μM K22 for 12 h and then exposed to LPS (1 μg/mL) for another 12 h. The cells were stained with 10 μM DCFH-DA for 30 min at 37 °C in dark conditions and the fluorescence signals were detected by a flow cytometer. (H) Statistics of ROS levels in H9c2 cells. Data are presented as mean ± SEM. **P < 0.01, ***P < 0.001 vs. Control group.

Drug-likeness and mutagenic properties of K22
In order to analyse the drug-likeness of active compounds K22 and KP-2, the calculations of lipophilicity, physicochemical properties and correlative parameters were performed by SwissADME (http://www.swissadme.ch). As shown in , compared to compound 1, K22 and KP-2 both owing more abundant hydrogen bond receptors, higher topological polar surface area and better aqueous solubility than compound 1. Notably, although compound 4 was originally derived from compound 1 and was reported to possess excellent inhibitory activity against Keap1Citation13, its poor aqueous solubility and exorbitant lipophilicity may not be conducive to its further development (). In addition, K22 was predicted not as a potential P-glycoprotein substrate while KP-2 does, which means K22 may not be susceptible to drug resistance. Furthermore, K22 was not alerted by the PAINS (Pan Assay Interference Structures) screeningCitation29, while compound 1 and KP-2 both showed potential PAINS alert as the existence of the sulphonamide fragment in their structures. Then we also performed the mini-Ames test to determine the mutagenic potential and assess the preliminary safety of compounds K22 and KP-2. The results showed that, in the range of 0–100 μM, both compounds displayed no mutagenic activity towards Salmonella typhimurium TA98 and TA100 with and without S9. In comparison, both compounds 1 and 4 induced >2-fold increases at high dose levels (100 μM) in the presence of S9 compared to the control group (Figure S5). These data demonstrated that benzene and substituted benzene core have better mutagenic properties than naphthalene core. Taken together, these results implied that the structure of K22 is more suitable for further drug development.
Table 1. Drug-likeness properties of compounds 1, 4, K22 and KP-2.
Conclusion
Due to the limited specific therapeutic drugs for myocarditis, it is of great theoretical and practical significance to explore new therapies such as those targeting Keap1-Nrf2 PPI. In this work, we developed a series of non-naphthalene scaffold-based Keap1-Nrf2 PPI inhibitors. Among all these derivatives, the optimal compound K22 not only exhibited excellent inhibitory activity against Keap1-Nrf2 PPI but also owned the highest metabolic stability in human liver microsomes. Further investigations demonstrated that K22 had no apparent cytotoxicity towards H9c2 cells and the cytoprotective effect of which in H9c2 cells mainly via activation of the Nrf2-ARE pathway in vitro. Moreover, under LPS stimulated conditions, the burst of ROS and inflammatory factors (e.g. TNF-α, IL-6 and IL-1β, etc.) could be greatly reversed after incubation with K22 in H9c2 cells. In addition, the results of prediction of the drug-likeness properties and the Ames test also indicated that K22 possesses well druggability. In conclusion, we provided several novel chemotypes carrying the substituted benzene core for the development of direct Keap1–Nrf2 PPI inhibitors, and the optimal compound K22 may be worthy of further development and application in the treatment of myocarditis.
Materials and methods
Chemistry
The spectra of 1H and 13C NMR of target compounds were recorded on a Bruker 400 and 500 MHz spectrometer using tetramethylsilane (TMS) as an internal standard. A Shimadzu LCMS-IT-TOF and Thermo Scientific LTQ-Orbitrap XL in positive or negative ion mode were used to record the HRMS spectra of target compounds. 200–300 mesh silica gel was purchased from Qingdao Haiwan Specialty Chemicals Co., LTD (China). The reactions were monitored by thin-layer chromatography (TLC), and all reagents required for chemical reaction were commercially available reagents without purification unless otherwise specified.
The detailed preparation methods and characterisation of derivatives K1–K22, KP-1 and KP-2 were shown in the Supporting information. Compounds 1 and 4 were prepared following the methods described by Ref.Citation22 and Ref.Citation13, respectively.
Biology
FP assay
The fluorescence polarisation (FP) assays were performed in a similar manner as previously describedCitation30. The experiments were performed briefly on a Synergy H4 microplate reader (BioTek, USA) using the 485 nm excitation and 535 nm emission filters for the FITC. The plates used for the FP measurements were Corning 384-well plates (product #3575). The assay buffer used in this assay consisted of 10 mM HEPES buffer (pH 7.4), 50 mM EDTA, 150 mM NaCl and 0.005% Tween-20. Each well was loaded with a total 40 μL assay solution containing 20 nM FITC-9mer Nrf2 peptide amide (FITC-LDEETGEFL-NH2, 10 μL), 400 nM Keap1 Kelch domain protein (10 μL), and an inhibitor sample at different concentrations (10 μL). Afterwards, the plate was covered and shaken for 30 min at room temperature before FP measurements. Then, the parallel and perpendicular fluorescence intensity (Fǁ and F⊥) relative to the linearly polarised excitation light was measured and the FP was determined. Each experiment was replicated three times. The IC50 values of the tested compound were ascertained from the plot of %inhibition against inhibitor concentration using GraphPad Prism 7.0 (Graphpad Software, Inc., USA).
Metabolic stability
The metabolic stability of representative compounds was evaluated by using human liver microsomes. Briefly, 0.5 mg/mL of human liver microsomes were pre-incubated with 100 mM potassium phosphate buffer (pH 7.4) and 2 mM NADPH at 37 °C for 10 min. The reactions were initiated by adding 10 μM testing compounds. After 0-min, 5-min, 10-min, 15-min, 20-min, 25-min and 30-min, the reactions were terminated by the addition of a mixture of 50 μL MeCN:methanol (1/1, v/v), respectively. After that, the resulting mixtures were centrifuged at 10,000 rpm at 4 °C for 10 min and the supernatant was analysed by LC-MS (AB SCIEX LC-MS system).
Cell culture
H9c2 cells were purchased from the National Collection of Authenticated Cell Cultures (China) and maintained in DMEM (Dulbecco's modified Eagle’s medium) with 10% FBS (fetal bovine serum), 100 U/mL penicillin and 100 mg/mL streptomycin. Cells were cultured at 37 °C under 5% CO2 in a humidified incubator.
Cell cytotoxicity assay
To assess the cell cytotoxicity of the optimal compound K22, an MTS assay was performed. H9c2 cells were cultivated in DMEM medium which contained 10% FBS. The cells were first grown in the logarithmic phase, after which they were incubated in 96-well plates at a 5,000 cells/well density. Each of the groups contained five wells. Subsequently, the compound was dissolved in DMSO and diluted to 50 μM (DMSO < 1%) in concentration. Then, the cells were subjected to treatment with DMSO and K22 (0 to 50 μM) for 24 h at 37 °C, followed by the addition of 10% MTS solution to each well. They were then incubated for another 30 min at 37 °C. To measure the absorbance, a Synergy H4 microplate reader (BioTek, USA) at 490 nm was utilised.
Western blot assay
The primary antibodies including anti-Nrf2 (#33649), anti-HO-1 (#82206), anti-NQO1 (#62262) and anti-GAPDH (#5174) antibodies were purchased from Cell Signalling Technology (USA). Briefly, after treatment with the compounds for a certain time, the protein of the cells was extracted and then quantitated using a BCA protein assay kit (Beyotime, China). After that, the protein samples were separated via SDS-PAGE and transferred to PVDF membranes (Beyotime, China). Subsequently, the membranes were blocked in 5% non-fat milk (Beyotime, China) at room temperature for 1 h, followed by incubation with corresponding primary antibodies at 4 °C overnight. The membranes were first washed three times with 1 × TBST solution, after which they were subjected to a 1 h incubation with the secondary antibodies at room temperature. Finally, the membranes were washed and the signals were detected and analysed by ChemiDoc XRS + systems (Bio-Rad, USA).
Co-immunoprecipitation assay
The interaction between Keap1 and Nrf2 in H9c2 cells was evaluated via co-immunoprecipitation assay using protein A-agarose. Briefly, the treated H9c2 cells were lysed and centrifugated at 4 °C to collect the supernatant, and then reacted with suspended protein A agarose at 4 °C to reduce non-specific binding. After centrifugation for 5 min, the supernatant was incubated with anti-Nrf2 or rabbit IgG for 1 h at 4 °C, followed by the addition of protein A agarose. After shaking overnight, the immunoprecipitates were centrifuged, the supernatant was abandoned, and the pellets were washed three times with lysis buffer. Subsequently, the pellets were suspended in SDS loading buffer and boiled for 10 min. Finally, the precipitated proteins were analysed by western blot with indicated antibodies.
Intracellular ROS determined by flow cytometry
2,7-dichlorodihydrofluorescein diacetate (DCFH-DA, Beyotime, China), a common ROS-sensitive dye, was used to detect ROS production in cells. H9c2 cells were seeded in 6-well plates with a density of 3 × 105 cells per well and incubated with DMEM medium overnight. Then, the cells were pre-treated with DMSO or 10 μM compound K22 for 12 h, before being exposed to 1 μg/mL LPS for another 12 h. After that, cells were collected and stained with 10 μM DCFH-DA in the dark at 37 °C for 30 min in DMEM medium free with FBS. After washing with PBS three times, the fluorescence signal was detected using the FACSCalibur flow cytometer (BD Biosciences, USA).
RNA isolation and qRT-PCR analysis
Total RNA was extracted from H9c2 cells with Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions and in strict compliance with the manufactures’ operating protocols. RNA was transmuted to cDNA by reverse transcriptase (SuperScript VILO™ cDNA Synthesis Kit, Thermo Fisher Scientific, USA). The sequence of primers used for PCR was shown in the Supporting information. To perform quantitative real-time RT-PCR analysis on Nrf2, HO-1 and NQO-1, the Lightcycler 480II real-time quantitative PCR system (Roche, Switzerland) was utilised. These values are presented as multiples of the control. The mRNA expression of all genes was normalised to GAPDH expression.
IL-6, TNF-α and IL-1β production
Levels of IL-6 (IL-6 (m) ELISA kit, P1326, Beyotime, China), TNF-α (TNF-α (m) ELISA kit, PT512, Beyotime, China) and IL-1β (IL-1β (m) ELISA kit, PI301, Beyotime, China) were measured using commercially available kits in compliance with the manufacturer’s operating instructions.
Small interfering RNA (siRNA) transfection
Predesigned siRNA targeting human Nrf2 (EHU093471) and negative control siRNA (SIC002) were purchased from Sigma-Aldrich (Shanghai, China). H9c2 cells were plated in 60 mm dishes at a density of 6 × 105 cells per well. 50 nM siRNA targeting Nrf2 or 50 nM negative control siRNA were transfected into cells by Lipofectamine 3000 (L3000001, Invitrogen, USA). Fresh medium was then added after 24 h incubation and continued to culture for another 48 h. Subsequently, the cells were treated with 10 μM K22 for 6 h and lysed. The expression levels of Nrf2 and Nrf2-regulated genes were then quantified by qRT-PCR.
Molecular modelling
The crystal structure of Keap1 for molecular modelling was obtained from the Protein Data Bank database (PDB entry: 6SP4)Citation31. The processes of molecular docking and MD simulations of K22/KP-2 bound to Keap1 using AutoDock and Amber packages were reported in our previous studyCitation21. Differently, the MD simulation time in this study was set as 400 ns for K22 and 600 ns for KP-2. 1,000 snapshots extracted from the MD simulation trajectories of the last 100 ns for K22 and the last 200 ns for KP-2 respectively were applied to MM/GBSA-based binding free energy decompositions.
Drug-likeness property prediction
The drug-likeness properties of representative compound K22 and KP-2 as well as compound 1 and 4 were predicted by using an available online tool named SwissADME (http://www.swissadme.ch)Citation32.
Water solubility determination
10 mM stock solution in DMSO of test compounds was diluted to different concentrations (1000 μg/mL, 250 μg/mL, 62.5 μg/mL, 15.6 μg/mL, 3.90 μg/mL, 0.97 μg/mL, 0.24 μg/mL). This series of solutions were then injected into an HPLC system for analysis (Agilent 1260 Infinity II LC system, Column: UltimateTM XB-C18 (150 mm × 4.6 mm × 5 μm), underwent a mobile phase (gradient from 10% MeCN/90% H2O to 90% MeCN/10% H2O) and a calibration curve was produced. Subsequently, test compounds were dissolved in phosphate buffer saline (pH 7.4) and this series of mixtures were stirred for 24 h at 25 °C. The samples were then filtered and the filtrates were analysed by the HPLC system (UV absorbance = 256 nm), and the solubility was calculated based on the peak area by quantifying the concentration of test solutions against the calibration curve. Experiments were performed in triplicate.
Mini-Ames test
The mutagenicity of the test compounds was evaluated by an Ames test kit (0211013, IPHASE, Beijing, China), and Salmonella typhimurium strains TA98 and TA100 as well as liver homogenate S9 were applied. Briefly, the bacteria were incubated with a test compound in an exposure medium containing adequate histidine for 90 min at 37 °C. Subsequently, the exposure medium was diluted in an indicator medium without histidine and aliquoted into 48 wells of a 384-well plate. After 72 h, the reversion events to histidine were characterised by the formation of the bacterial colonies, and the colour changes of the medium could be detected by a microplate reader (Synergy H4 microplate reader, BioTek, USA). The experiments were performed in triplicates.
Statistical analysis
Results are reported in the form of means ± SEM. To compare group differences, a one-way analysis of variance (ANOVA) with Tukey’s correction for the post-hoc comparisons was carried out. All the statistical analyses were performed using GraphPad Prism 7.0 software and a statistically significant result was obtained at P < 0.05.
Authors’ contributions
Y.Y. and S.S initiated and supervised the research. Y.S, L.Z, and B.Y designed and performed the research. S.G, Q.L, and M.Z, contributed parts of the research. Y.S, L.Z, and B.Y wrote the manuscript, and all authors read and revised the final manuscript.
Supplemental Material
Download PDF (1.5 MB)Additional information
Funding
References
- Yamamoto M, Kensler TW, Motohashi H. The keap1-nrf2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. 2018;98(3):1169–1203.
- Yang S, Lian G. Ros and diseases: role in metabolism and energy supply. Mol Cell Biochem. 2020;467(1–2):1–12.
- Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol. 2011;7(8):504–511.
- Tonelli C, Chio C II, Tuveson DA. Transcriptional regulation by nrf2. Antioxid Redox Signal. 2018;29(17):1727–1745.
- Sajadimajd S, Khazaei M. Oxidative stress and cancer: the role of nrf2. Curr Cancer Drug Targets. 2018;18(6):538–557.
- Baird L, Yamamoto M. The molecular mechanisms regulating the keap1-nrf2 pathway. Mol Cell Biol. 2020;40(13):e00099–00020.
- Zhou HS, Hu LB, Zhang H, Shan WX, Wang Y, Li X, Liu T, Zhao J, You QD, Jiang ZY. Design, synthesis, and structure-activity relationships of indoline-based kelch-like ech-associated protein 1-nuclear factor (erythroid-derived 2)-like 2 (keap1-nrf2) protein-protein interaction inhibitors. J Med Chem. 2020;63(19):11149–11168.
- Houghton CA, Fassett RG, Coombes JS. Sulforaphane and other nutrigenomic nrf2 activators: can the clinician’s expectation be matched by the reality? Oxid Med Cell Longev. 2016;2016:7857186.
- Fu CY, Chen J, Lu XY, Zheng MZ, Wang LL, Shen YL, Chen YY. Dimethyl fumarate attenuates lipopolysaccharide-induced mitochondrial injury by activating nrf2 pathway in cardiomyocytes. Life Sci. 2019;235:116863.
- Marcotte D, Zeng W, Hus JC, McKenzie A, Hession C, Jin P, Bergeron C, Lugovskoy A, Enyedy I, Cuervo H, et al. Small molecules inhibit the interaction of nrf2 and the keap1 kelch domain through a non-covalent mechanism. Bioorg Med Chem. 2013;21(14):4011–4019.
- Hu L, Magesh S, Chen L, Wang L, Lewis TA, Chen Y, Khodier C, Inoyama D, Beamer LJ, Emge TJ, et al. Discovery of a small-molecule inhibitor and cellular probe of keap1-nrf2 protein-protein interaction. Bioorg Med Chem Lett. 2013;23(10):3039–3043.
- Jiang CS, Zhuang CL, Zhu K, Zhang J, Muehlmann LA, Figueiró Longo JP, Azevedo RB, Zhang W, Meng N, Zhang H. Identification of a novel small-molecule keap1-nrf2 ppi inhibitor with cytoprotective effects on lps-induced cardiomyopathy. J Enzyme Inhib Med Chem. 2018;33(1):833–841.
- Lazzara PR, David BP, Ankireddy A, Richardson BG, Dye K, Ratia KM, Reddy SP, Moore TW. Isoquinoline kelch-like ech-associated protein 1-nuclear factor (erythroid-derived 2)-like 2 (keap1-nrf2) inhibitors with high metabolic stability. J Med Chem. 2020;63(12):6547–6560.
- Cosimelli B, Greco G, Laneri S, Novellino E, Sacchi A, Amendola G, Cosconati S, Bortolozzi R, Viola G. Identification of novel indole derivatives acting as inhibitors of the keap1-nrf2 interaction. J Enzyme Inhib Med Chem. 2019;34(1):1152–1157.
- Bertrand HC, Schaap M, Baird L, Georgakopoulos ND, Fowkes A, Thiollier C, Kachi H, Dinkova-Kostova AT, Wells G. Design, synthesis, and evaluation of triazole derivatives that induce nrf2 dependent gene products and inhibit the keap1-nrf2 protein-protein interaction. J Med Chem. 2015;58(18):7186–7194.
- Abed DA, Lee S, Hu L. Discovery of disubstituted xylylene derivatives as small molecule direct inhibitors of keap1-nrf2 protein-protein interaction. Bioorg Med Chem. 2020;28(6):115343.
- Begnini F, Geschwindner S, Johansson P, Wissler L, Lewis RJ, Danelius E, Luttens A, Matricon P, Carlsson J, Lenders S, et al. Importance of binding site hydration and flexibility revealed when optimizing a macrocyclic inhibitor of the keap1-nrf2 protein-protein interaction. J Med Chem. 2022;65(4):3473–3517.
- Lee S, Abed DA, Nguyen MU, Verzi MP, Hu L. Structure-activity relationships of 1,4-bis(arylsulfonamido)-benzene or naphthalene-n,n’-diacetic acids with varying c2-substituents as inhibitors of keap1-nrf2 protein-protein interaction. Eur J Med Chem. 2022;237:114380.
- Georgakopoulos N, Talapatra S, Dikovskaya D, Dayalan Naidu S, Higgins M, Gatliff J, Ayhan A, Nikoloudaki R, Schaap M, Valko K, et al. Phenyl bis-sulfonamide keap1-nrf2 protein-protein interaction inhibitors with an alternative binding mode. J Med Chem. 2022;65(10):7380–7398.
- Hsu KH, Su BH, Tu YS, Lin OA, Tseng YJ. Mutagenicity in a molecule: Identification of core structural features of mutagenicity using a scaffold analysis. PLOS One. 2016;11(2):e0148900.
- Ying Y, Ye JJ, Sun L, Wang P, Wang H. X. Phloretin prevents diabetic cardiomyopathy by dissociating keap1/nrf2 complex and inhibiting oxidative stress. Front Endocrinol. 2018;9:774.
- Jiang ZY, Lu MC, Xu LL, Yang TT, Xi MY, Xu XL, Guo XK, Zhang XJ, You QD, Sun HP. Discovery of potent keap1-nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J Med Chem. 2014;57(6):2736–2745.
- Hur W, Gray NS. Small molecule modulators of antioxidant response pathway. Curr Opin Chem Biol. 2011;15(1):162–173.
- Magesh S, Chen Y, Hu L. Small molecule modulators of keap1-nrf2-are pathway as potential preventive and therapeutic agents. Med Res Rev. 2012;32(4):687–726.
- Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, Jakel RJ, Johnson JA. Nrf2, a multi-organ protector? Faseb J. 2005;19(9):1061–1066.
- Meng N, Tang H, Zhang H, Jiang C, Su L, Min X, Zhang W, Zhang H, Miao Z, Zhang W, et al. Fragment-growing guided design of keap1-nrf2 protein-protein interaction inhibitors for targeting myocarditis. Free Radic Biol Med. 2018;117:228–237.
- Cheng N, Liang Y, Du X, Ye RD. Serum amyloid a promotes lps clearance and suppresses lps-induced inflammation and tissue injury. EMBO Rep. 2018;19(10):e45517.
- Lakshmikanth CL, Jacob SP, Chaithra VH, de Castro-Faria-Neto HC, Marathe GK. Sepsis: in search of cure. Inflamm Res. 2016;65(8):587–602.
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–2740.
- Inoyama D, Chen Y, Huang X, Beamer LJ, Kong AN, Hu L. Optimization of fluorescently labeled nrf2 peptide probes and the development of a fluorescence polarization assay for the discovery of inhibitors of keap1-nrf2 interaction. J Biomol Screen. 2012;17(4):435–447.
- Ontoria JM, Biancofiore I, Fezzardi P, Ferrigno F, Torrente E, Colarusso S, Bianchi E, Andreini M, Patsilinakos A, Kempf G, et al. Combined peptide and small-molecule approach toward nonacidic thiq inhibitors of the keap1/nrf2 interaction. ACS Med Chem Lett. 2020;11(5):740–746.
- Daina A, Michielin O, Zoete V. Swissadme: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717.