
Abstract
The present study aimed to investigate the antitumor effect of simultaneous inhibition of dihydrofolate reductase (DHFR) enzyme. We designed some novel pyrazolo[3,4-d]pyrimidines bearing different amino acid conjugates as efficient antifolate agents attributable to their structural similarity with methotrexate (MTX) and MTX-related antifolates. All compounds were tested to screen their enzymatic inhibition against DHFR compared with the reference drug MTX and for their in vitro antitumor cytotoxicity against six MTX-resistant cancer cell lines. The flow cytometry indicated that the most potent compound 7f arrested MCF-7 cells in the S-phase and induced apoptosis. Western blot for visualisation proved the ability of compound 7f to induce the expression of proapoptotic caspases and Bax proteins in MCF-7 breast cancer cell line beside its ability to diminish the expression of antiapoptotic Bcl-2 protein. Molecular modelling studies concluded that compound 7f displayed better binding energy than that of the normal ligand MTX.
New pyrazolo[3,4-d]pyrimidine derivatives 7a–m which are structurally similar to the classical methotrexate (MTX) and MTX-related antifolates were synthesised as antitumor agents.
Novel N-acyl amino acid compound 7f exhibited marked DHFR inhibition activity that are parralel to both the molecular docking results and cytotoxic activity.
Compound 7f could induce the expression of proapoptotic caspases and Bax proteins in MCF-7 breast cancer cell line beside its ability to diminish the expression of antiapoptotic Bcl-2 protein.
All prepared compounds obey Lipinski rule of five except compound 7f.
HIGHLIGHTS
GRAPHICAL ABSTRACT
Introduction
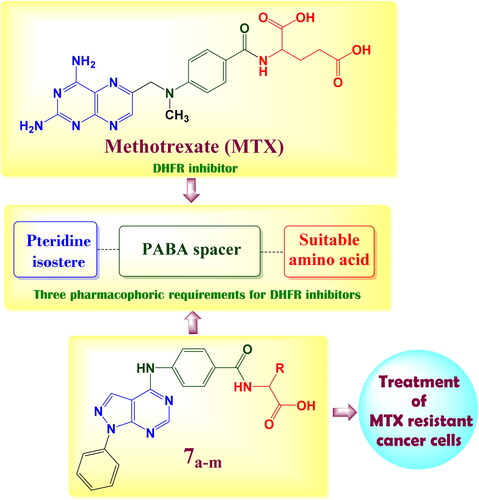
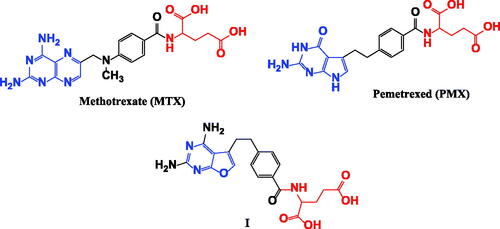
Dihydrofolate reductase (DHFR) is a critical enzyme in folic acid metabolism; it promotes catalysis for conversion of dihydrofolate (DHF) to tetrahydrofolate (THF), which is essential for purine de novo synthesis and thymidylate synthesis in cell proliferationCitation1, therefore inhibition of DHFR enzyme exhibits great antitumor activity as it suppresses de novo nucleotide biosynthesis leading to an imbalance of purine and pyrimidine precursors and rendering cells incapable of undergoing a proper DNA replicationCitation2. The most common dihydrofolate reductase inhibitors (DHFRIs) are the classical antifolates such as methotrexate (MTX), aminopterine, pralatrexate and pemetrexed (PMX)Citation3. The chemical structure of MTX prototype is composed of 3 main important pharmacophores: pteridine nucleus, 4-amino benzoic acid and glutamic acidCitation4 (). With mismatch, pemetrexed (PMX) elucidates ascendant DHFR inhibition effect although it accommodates pyrrolo[2,3-d]pyrimidine nucleus instead of pteridineCitation5 as well the furo[2,3-d]pyrimidine in analogue I was also reported as potential DHFR inhibitorCitation6 as shown in .
Figure 1. Structural requirements for DHFR inhibition activity.
The previous findings encouraged us to synthesise novel pyrazolo[3,4-d]pyrimidine derivatives incorporate a different series of amino acid conjugates 7a–m as shown in Scheme 2. Our aim is based on the isosteric replacement of the pteridine nucleus of MTX with pyrazolo[3,4-d]pyrimidine scaffold with the retention of 4-aminobenzoyl spacer and either glutamic acid or different series of amino acids to achieve a significant inhibitory level of DHFR enzyme as in and therefore, achieve considerable cytotoxic activities against different MTX-resistant cell lines. We selected different series of amino acid conjugates depending on their different hydrophilic or hydrophobic behaviours beside their different aliphatic or aromatic nature hoping to achieve superior DHFR inhibition activity that exceed classical antifolates and to treat the classical antifolates resistant cancer cell lines.
Figure 2. The structural approach between the synthesised novel pyrazolo[3,4-d]pyrimidines 7a–m with MTX reference drug.
![Figure 2. The structural approach between the synthesised novel pyrazolo[3,4-d]pyrimidines 7a–m with MTX reference drug.](/cms/asset/b14cebe3-3f00-40a6-aeaa-218563a3f9a1/ienz_a_2142786_f0002_c.jpg)
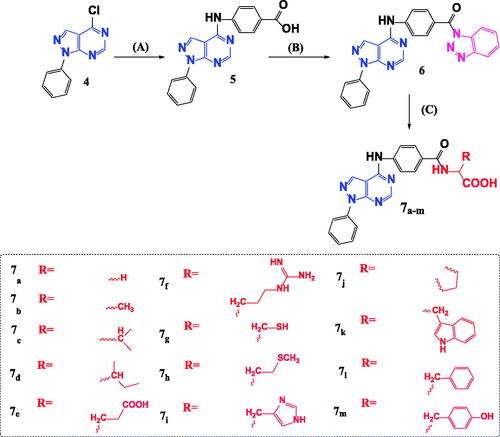
Scheme 2. Synthetic pathway for preparation of novel N-acyl amino acid conjugates 7a–m. Reagents and conditions: A = 4-aminobenzoic acid, isopropanol, heat, 16–18 h. B = 1,2,3 Benzotriazole, SOCI2, DCM, room temp. C = Amino acid, TEA, Acetonitrile, heat 70° C.
Results and discussion
Chemistry
Our starting synthetic material ethyl (ethoxymethylene)cyanoacetate 1 was cyclized with phenyl hydrazine to obtain ethyl 5-amino-1-phenyl-1H-pyrazole-4-carboxylate 2Citation7 which underwent further cyclisation with formamide to afford the pyrazolo[3,4-d]pyrimidinone compound 3Citation8 as shown in Scheme 1.
Scheme 1. Synthesis of 4-chloro-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine 4. Reagents and conditions: A = Phenyl hydrazine, ethanol, 80° C, 4 h. B = HCONH2, 190° C, 8 h. C = POCI3, 106° C, 6 h.
![Scheme 1. Synthesis of 4-chloro-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine 4. Reagents and conditions: A = Phenyl hydrazine, ethanol, 80° C, 4 h. B = HCONH2, 190° C, 8 h. C = POCI3, 106° C, 6 h.](/cms/asset/bf7e0ca6-7489-48e4-b578-f7eccd42a238/ienz_a_2142786_sch0001_c.jpg)
Compound 3 was chlorinated using phosphorous oxychloride to obtain 4-chloro-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine 4Citation9 (Scheme 1) which reacted with 4-amino benzoic acid to give the key intermediate 4-{(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino}benzoic acid (5)Citation10 as shown in Scheme 2.
Reaction of our key intermediate 5 with 1H-benzotriazole afforded the novel N-acyl benzotriazole compound 6 that was condensed with a series of different amino acids to obtain N-acyl amino acids compounds (7a–m) (Scheme 2).
The novel N-acyl benzotriazole derivative 6 was characterised using melting point and TLC techniques in different solvent systems. The structure of compound 6 was confirmed using 1HNMR and 13CNMR whereas its 1HNMR revealed the absence of any signals in aliphatic region however; it comprised numerous signals in aromatic region integrated into 15 aromatic protons which are divided into; 2 doublet signals at 7.69 and 8.62 ppm integrated for 8 protons, 2 triplet signals at 7.39 and 7.59 ppm integrated for 5 protons. In addition, the appearance of two characteristic singlet signals at 8.23 ppm confirm the presence of 3-H and 6-H position of pyrazolo[3,4-d]pyrimidine nucleus. 13CNMR data of compound 6 elucidated 23 signals at aromatic region. Besides, the signal of carbonyl group carbon is at 161.09 ppm. Moreover, the chemical structures of 2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}alkanoic acid (7a–m) were confirmed with their elemental analysis, 1HNMR and 13CNMR. 1HNMR spectra clarified that compounds 7a–h and 7j showed an apparent decrease in the number of aromatic signals to be integrated for 11 aromatic protons only; consequently, that explains the actual loss of 1H-benzotriazole as a good leaving group. In addition to, appearance of new signals in aliphatic region that vary from compound to the other. Compound 7a showed singlet signal in aliphatic region at 3.91 ppm integrated to 2 protons of (CH2) of glycine, while compound 7b illustrated 2 significant signals; one doublet signal at 1.43 ppm integrated to 3 protons of (CH3) group and the other quartette signal at the range of 4.40–4.46 ppm refer to 1 proton of (CH) at C2 of alanine amino acid. Furthermore, compound 7c revealed 3 considerable signals in aliphatic region; at 0.96 ppm that represents a large doublet signal integrated to 6 protons of 2 (CH3) groups, a multiplet signal from 1.17 to 1.25 ppm integrated to 1 proton corresponding to (CH) at C3 of valine beside a doublet signal at 4.28 ppm indicates the proton of (CH) at C2. Beyond, compound 7d showed doublet signal at 0.93 ppm corresponding to (CH3) group radiated from C3, a triplet signal at 1.31 ppm indicates the (CH3) group substituted at C4 of isoleucine, another multiplet signal at the range of 1.95–1.99 ppm performs (CH) at C3 beside a doublet signal at 4.38 ppm refer to (CH) at C2. Moreover, compound 7e exhibited a quartette signal at the range of 1.98–2.11 ppm of aliphatic region due to (CH2) at C3 of glutamic acid alongside a triplet signal at 2.40 ppm integrated to 2 protons of (CH2) at C4 and a triplet peak at 4.43 ppm due to (CH) at C2. As well, compound 7f revealed 3 signals indicate the 3 (CH2) groups of arginine amino acid, a multiplet signal appeared at 0.79–0.85 ppm corresponding to (CH2) at C4 besides, a quartette peak at 1.09–1.21 ppm due to (CH2) at C3, the triplet signal performs (CH2) at C5 of arginine become clear at 1.66 ppm. In addition to a triplet peak at 4.38 ppm corresponding to (CH) at C2 of arginine and an evident increase in the signals refer to NH groups due to presence of guanidine moiety. Too, compound 7g demoed a doublet signal at 3.13 ppm integrated to 2 protons corresponding to (CH2) at C3 of cysteine along with a triplet signal at 4.69 ppm due to (CH) at C2. Also, compound 7h displayed several peaks at aliphatic region; a quartette peak at 1.15–1.23 ppm corresponding to (CH2) at C3 of methionine, singlet signal at 2.08 ppm due to (–S–CH3) group and 2 triplet signals at 2.60 and 4.50 ppm represent the (CH2) at C4 and (CH) at C2 of methionine respectively. With a similar manner, compound 7j elucidated 4 significant signals at aliphatic region due to pyrrolidine ring of proline. These aliphatic signals appeared at the range of 1.18–1.28 ppm and 2.25–2.30 ppm beside 3.40 ppm referring to (CH2) of pyrrolidine ring at C4, C3 and C5 respectively. Besides, a triplet peak at 4.42 ppm corresponding to C2 of pyrrolidine. In contrast, compound 7i demonstrated an aromatic region with numerous signals integrated to 13 aromatic protons, this increase in the number of aromatic signals is owing to the presence of 2 singlet signals indicates (CH) at C5 and C2 of imidazole ring of histidine at 6.91 and 7.92 ppm respectively. Remarkably, compounds 7k–m revealed another increase in the number of aromatic signals to be integrated to 16 aromatic protons in compounds 7k,l and 15 aromatic protons in compound 7 m that could be explained by removal of 1H-benzotriazole and increment of an aromatic amino acid such as tryptophan, phenyl alanine and tyrosine. Interestingly, all synthesised novel N-acyl amino acids 7a–m show singlet signal represents the proton of (COOH) group at a range of 10.51–10.86 ppm except compound 7e that demoed 2 singlet signals at 10.51 and 12.48 ppm that demonstrates the presence of 2 carboxylic groups. Furthermore, 13CNMR spectrum of compounds 7a–h and 7j clarified a perspicuous decrease in the number of aromatic carbons to 17 carbons rather than the 23 carbon signals presented at N-acyl benzotriazole compound 6 that proves the replacement of 1H-benzotriazole by an aliphatic amino acid. In addition, an appearance of new signals in aliphatic region such as the signals appeared at 18.19 and 50.57 ppm of compound 7b representing C3 and C2 of alanine respectively. Moreover, compound 7e exhibited 3 additional aliphatic signals at 26.95, 30.57 and 53.91 ppm corresponding to C3, C4 and C2 of glutamic acid respectively. In contrast, compounds 7k–m showed a sudden increase in aromatic signals due to the addition of aromatic amino acid. Interestingly, the 13C NMR spectra of all prepared compounds demoed 1 signal corresponding to one carbonyl group except compound 7e that exerts 2 signals at 175.47 and 176.74 ppm referring to 2 carbonyl groups that explain the presence of glutamic acid.
Biological evaluation
Cytotoxic activity of synthesised compounds
The SRB colorimetric assay was employed to assess the synthesized compounds’ cytotoxic effects on several cancer cell lines ()Citation11–14. The synthesized compounds especially 7c, 7d, 7f, 7i, 7j and 7l inhibited the cellular proliferation of cancer cell lines at lower IC50 values. Compounds 7e, 7g and 7m inhibited the proliferation of cancer cells at higher IC50 in compression to above compounds. The highest IC50 to almost cancer cell lines were observed for compounds 6, 7a, 7h, 7k and 7b. Noticeably, the lowest IC50 values were observed on MCF-7 breast cancer line when treated with the tested compounds, especially compounds 7c, 7d, 7f, 7i, 7j and 7l. Furthermore, the effect of tested compounds was evaluated on the normal immortalized HPDE cell line. All synthesized compounds have no toxicity to the normal cells with higher IC50 exceeding 5.61 μM ().
Table 1. IC50 values of compound 6 and novel N-acyl amino acid conjugates 7a–m in different cell lines.
Inhibitory effect of tested compounds on human DHFR enzyme
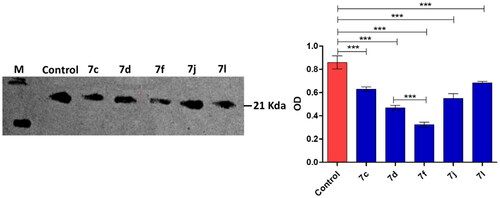
Compounds 7c, 7d, 7f, 7i, 7j and 7l inhibited DHFR at considerable low IC50 < 1 µM in compression to MTX the reference drug (IC50 = 5.61 µM) (). Then compounds 7e, 7g and 7m inhibited DHFR at IC50 range 1–5 µM which is lower than observed with the MTX. The highest DHFR (IC50) was observed for all compounds 6, 7a, 7h, 7k and in particular 7b. Stimulatingly, the DHFR inhibition by compounds 7c, 7d, 7f, 7j and 7l was in a great compliance with their cytotoxic effects on MCF-7 cancer cell lineCitation15. The efficient DHFR inhibition with considerable cytotoxic effects on cancer cell lines could elect the compound in order 7c, 7d, 7f, 7j and 7l to be possible anticancer candidates. For more investigation, the inhibitory effects of compounds 7c, 7d, 7f, 7j and 7l on the expression of DHFR in the MCF-7 cancer cell line were further western blot quantified (). Obviously, the nominated compound especially the compound 7f significantly diminished the DHFR expression that attests their anti-DHFR activities.
Figure 3. Western blot quantification anti-DHFR effects of compounds 7c, 7d, 7f, 7j and 7l in MCF-7 breast cancer cell.
The structure activity relationship clarified that the isosteric replacement of pteridine ring in MTX by pyrazolo[3,4-d]pyrimidine nucleus improve the DHFR inhibition activity in the majority of the prepared compounds. Consequently, compound 7e is considered the direct isostere of MTX with retention of 4-aminobenzoyl and glutamic acid moieties exhibits superior DHFR inhibition activity (IC50 = 1.83 µM) comparable to MTX (IC50 = 5.57 µM) ().
Figure 4. Isosteric approach of compound 7e and MTX along with DHFR inhibition IC50 (µM) values.
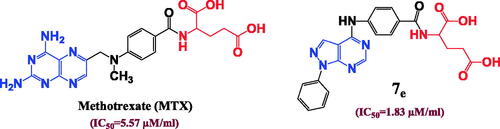
In a strangest manner, hydrophilic and hydrophobic nature of the conjugated amino acid is insignificant parameter as both compounds 7d with hydrophobic (Ile) amino acid and 7f with hydrophilic (Arg) amino acid elucidated near considerable DHFR inhibition activities (IC50= 0.43 and 0.31 µM), respectively and near cytotoxic activities against different cell lines. In addition, the aliphatic and aromatic nature of the inserted amino acid also has no remarkable effect on the DHFR inhibition activities since both compounds 7c and 7l have approximate IC50 values of 0.61 and 0.66 µM, respectively although compound 7c possess (Val) aliphatic amino acid while compound 7l enclose (Phe) aromatic one and with also approximate cytotoxic activities as in (). In consequence, the solubility parameter beside the aliphatic and aromatic nature of the inserted amino acid is not important for DHFR inhibition activity in compounds bearing pyrazolo[3,4-d]pyrimidine ring instead of pteridine. However, the isosteric replacement of pteridine ring by pyrazolo[3,4-d]pyrimidine revealed a considerable increase in the DHFR inhibition activity.
Figure 5. The effect of the structural differences between compounds 7c, 7d, 7f and 7l on their IC50 (µM) values.
Apoptosis induction in cancer cells
Induction of cancer cells apoptosis play the crucial role in diminishing their proliferation and metastasis, and so the anti-apoptotic effect indicates a possible anticancer activityCitation16,Citation17. Apoptotic process involves diverse metabolic reactions that can be activated or inhibited. It is well documented that suppression of cancer antiapoptotic proteins as well as activation proapoptotic proteins indicates efficient antitumor activityCitation11–14. In this direction, the most efficient anti-DHFR compound with the least cytotoxicity on MCF-7 cells "compound 7f” was selected for further anti-apoptotic activity evaluation.
Compound 7f induces the expression of proapoptotic proteins in MCF-7 breast cancer cell line
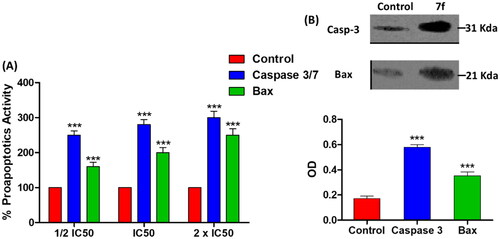
Caspases as well as Bax are proapoptotic proteins that are regularly triggered during apoptosis, and up-regulation of their expression in cancer cells indicates antitumor activityCitation11,Citation14. In this context, the effect of 7f at IC50 on the levels of caspase 3/7 and Bax proteins was evaluated. In dose dependent manner, the compound 7f significantly surge the release of caspase 3/7 and Bax proteins (). For further testing the colorimetric assays of caspases or Bax proteins, a western blot was performed to visualise and quantify their expressions. Compound 7f significantly increased the expression of both caspase 3 and Bax proteins (). The β-actin (42 KDa) was used as internal reference to validate the western blots. The levels of the expressed β-actin remain unaffected in the presence of different tested compounds (Supporting Information).
Figure 6. The compound 7f increases the levels of caspases in breast cancer cell line MCF-7. (A) Coulometric assay of caspase 3/7 and Bax levels, the data are presented as percentage change from untreated control. (B) Western blot gel of caspase-3 and Bax and quantification of the intensities of the bands in western blot.
Compound 7f diminishes the expression of antiapoptotic Bcl2 protein
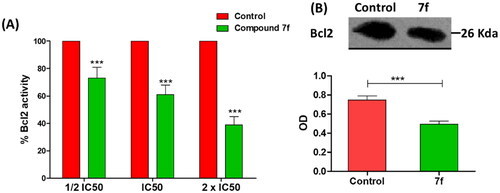
B-cell lymphoma-2 (Bcl-2) protein regulates the apoptosis; it is an antiapoptotic protein to facilitate the cancer cells survival under stressful circumstances via averting the proapoptotic signalsCitation11,Citation14. To attest the compound 7f apoptotic effect, Bcl-2 level was quantified in MCF-7 breast cancer cell line. Compound 7f significantly reduced the Bcl-2 production in a dose-dependent manner (). Furthermore, the effect of compound 7f at IC50 on the expression of Bcl-2 was visualised and quantified using western blot. Compound 7f significantly down-regulated the expression of Bcl-2 (). The β-actin (42 KDa) was used as internal reference to validate the western blots. The levels of the expressed β-actin remain unaffected in the presence of different tested compounds (Supporting Information).
Figure 7. (A) The compound 7f decreases the level of Bacl2 in breast cancer cell line MCF-7. (A) ELISA assay of Bcl2 level, the data are presented as percentage change from untreated control. (B) Western blot gel of Bcl2 and quantification of the intensities of the bands in western blot.
Flow cytometric cell cycle analysis
Cell cycle analysis was done for the compound 7f against MCF-7 breast cancer cell lineCitation11,Citation14. Clearly, compound 7f enhanced the accumulation proportion of cells at the pre-G1 phase from 2.19 to 41.36%, and the accumulation percentages in S phase from 31.87 to 46.36%. The decrease in the accumulation percentage of cells at G2/M phase from 12.01 to 1.5% after treatment with the compound indicated that the compound 7f arrested cell cycle at S phase (). Moreover, the percentage of cell apoptosis was markedly increased from 0.13% for control MCF-7 cells to 21.31% in treated cells (; ). The proportion of late apoptosis is greater than that of early, which evidences that compound 7f irreversibly induced apoptosis.
Figure 8. Flow cytometric cell cycle analysis upon treatment of MCF-7 breast cancer cell line with the compound 7f.
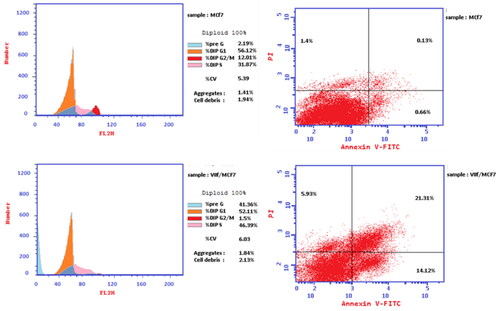
Table 2. Cell cycle analysis and apoptosis detection of the compound 7f.
Table 3. Apoptotic assay of the compound 7f.
Molecular modeling
The original ligand methotrexate (MTX) was extracted and docked into the active site of DHFR enzyme (pdb ID: 1U72)Citation18. Validation of docking algorithm was achieved by re-docking MTX into the active site of DHFR, which was found to retrieve the reported X-ray crystal structure binding mode of MTX, with root mean square difference (RMSD) between the top docking pose and original crystallographic geometry of 1.263 A°. A stick representation of side-chain residues Ile60, Leu22, Arg70, Val115, Phe31, Phe34 and Gln35 of DHFR (pdb code ID: 1U72) in contact with MTX normal ligand as showed in (). The crucial binding interactions attributable to MTX were; an important (HBD) effect due to 4-amino group of pteridine moiety with Val115 residue, besides the arene-H interaction of pteridine ring with Thr56 residue. In addition, there is a significant salt bridge caused by the γ-glutamate moiety of MTX with Arg70 accompanied with the (HBA) effect of both carbonyl and anionic carboxylate oxygens with Gln35 and Lys68 respectively. Unfortunately, previous studies of hDHFR have shown that certain mutations at Leu22, Phe31, Phe34, Gln35 and Arg70 residues could yield catalytically-active MTX-resistant mutantsCitation19. From this point, we investigated pyrazolo[3,4-d]pyrimidine derivatives 6 and 7a–m and subsequently flexibly docked them into the binding site of DHFR compared with MTX as mentioned in .
Figure 9. Two-dimensional (2 D) and three-dimensional (3 D) binding modes and residues involved in the energy minimised docking pose of the novel compounds 7f, 7d, 7l, 7k and MTX against DHFR (pdb ID: 1U72).
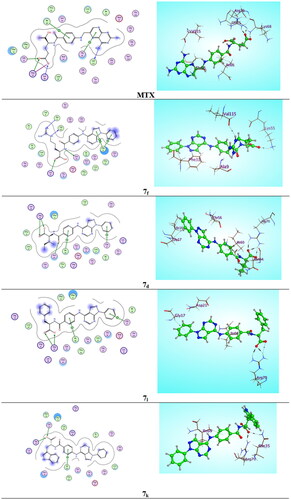
Table 4. Docking scores (kcal/mol) and calculated parameters of Lipinski rule of prepared compounds 6, 7a–m and methotrexate (MTX).
Parallel to the in vitro DHFR enzyme assay screening (IC50) results, compound 7f energy minimised pose elucidated binding energy of (S = −9.2584 kcal/mol) that is better than that of the normal ligand MTX (S = −8.3951 kcal/mol) due to presence of ᴨ-ᴨ* stacking interaction between pyrazole ring of pyrazolo[3,4-d]pyrimidine nucleus and Phe31 residue and arene-H interaction between 1-phenyl ring with the same Phe31 amino acid with bond lengths of 3.10 and 3.25 A° respectively, as well 2 (HBD) interactions were demonstrated by 2 NH groups of guanidine moiety with Ala9 residue and the key Val115 residue separately with bond energies of −0.8 and −1.4 Kcal/mol successively. Moreover, the docking results of the most stable conformer of compound 7d revealed scoring function of (−8.6795 Kcal/mol) that also exceed MTX as a result of considerable salt bridge between carboxylate group and the crucial amino acid Arg70 (2.49 A°) with binding energy from (−1.9 to −7.6 Kcal/mol), also there were 2 arene-H interactions between 2 phenyl rings with Gly17 and Ile60 beside, an important (HBA) interaction of carbonyl group with Asn64 (2.04 A°) through bonding energy of −1.8 Kcal/mol. Interestingly, the energy minimised conformer of compound 7 l established 2 binding interactions (S = − 8.7263 Kcal/mol) one of them as arene-H bond between 1-phenyl ring and Gly17 with bond length of (3.23 A°) and the other as the imperative salt bridge of the carboxylate moiety with the key Arg70 residue (bond energy from −0.8 to −7.6 kcal/mol). In atypical manner, compound 7k showed docking scoring function of (−9.0760 kcal/mol) that is better than that of MTX normal ligand (−8.3951 kcal/mol) due to the dual interaction effect of the carboxylate moiety with Arg70 residue by salt bridge and with Gln35 by (HBA) effect (−1.8 Kcal/mol), beside the arene-H interaction arise between phenyl ring and Ile60 (−0.7 Kcal/mol), however it exhibited inferior in vitro IC50 value of 7 µM (MTX = 5.57 µM) as presented in .
In addition, the parameters of Lipinski rule were calculated for all prepared novel compounds 6, 7a–m. Profitability, all prepared compounds obey Lipinski rule of five except compounds 7f due to presence of guanidine moiety of arginine amino acid that increase the number of (HBD) groups into 7 and the number of (HBA) groups into 12 as predicted in .
Conclusions
It can be concluded that new pyrazolo[3,4-d]pyrimidines bearing different amino acid conjugates (6, 7a–m) were prepared as efficient antifolate agents due to their structural similarities with methotrexate (MTX). It was found that pyrazolo[3,4-d]pyrimidines 7c, 7d, 7f, 7i, 7j and 7l inhibited DHFR at considerable lower IC50 values than MTX reference drug beside their significant cytotoxic effects on many MTX-resistant cancer cell lines. Pyrazolo[3,4-d]pyrimidine 7f bearing arginine amino acid is the most active antitumor agent against MCF-7 breast cancer cell line, DHFR inhibitor, as well it could arrest MCF-7 cells in the S-phase and induce apoptosis. Compound 7f was proven to induce the expression of proapoptotic caspases and Bax proteins in MCF-7 breast cancer cell line beside its ability to diminish the expression of antiapoptotic Bcl-2 protein. Molecular modelling studies revealed that compound 7f displayed binding energy of (S = −9.2584 kcal/mol) that is better than that of the normal ligand MTX (S = −8.3951 kcal/mol). Finally, compound 7f require further future investigation as antitumor drug against MTX-resistant cancers because of its potential DHFR inhibitory activity.
Experimental
Chemistry
All starting materials were purchased from Sigma-Aldrich (Germany). Melting points (°C) were determined with a Gallenkamp melting point apparatus (London, UK), and are uncorrected. 1HNMR and 13CNMR were performed in NMR department, Faculty of Science, Mansoura University, Mansoura City, Egypt. 1HNMR spectra were recorded on Varian Mercury-500 (500 MHz) (Palo Alto, CA, USA), Varian Gemini-300BB (300 MHz) (Foster City, CA, USA), and Bruker 400 MHz AV III (400 MHz) (Biocity’s, CA, USA) using dimethyl sulfoxide (DMSO)-d6 as a solvent and tetramethylsilane (TMS) as internal standard (chemical shift in δ, ppm). 13CNMR spectra were recorded on Varian Gemini-300BB (100 MHz) (Foster City, CA, USA). All reactions were monitored by thin-layer chromatography (TLC) using Silica gel 60 GF254 (E-Merck-Germany) and were visualised by iodine vapours or by UV-lamp at wavelength λ 254 nm.
General procedure for synthesis of {1H-benzo[d][1,2,3]triazol-1-yl}{4-[(1-phenyl-1H-pyrazolo-[3,4-d]pyrimidin-4-yl)-amino]phenyl}-methanone (6)
To (1.4 g, 12 mmol) benzotriazole dissolved in (50 ml) CH2Cl2, (0.26 ml) SOCl2 (3.6 mmol) were added dropwise. The mixture was stirred at room temperature for 30 min, followed by the addition of the carboxylic acid containing compound 5 (1 g, 3 mmol) and the reaction was allowed to stir for an additional 2 h at room temperature. The reaction mixture was dried, then dissolved in ethyl acetate followed by washing using dilute saturated Na2CO3 solution. The organic layer was dried over anhydrous sodium sulphate. Hexane (50 ml) was added to the filtrate, and then the solid obtained was dried under vacuum to give the N-acyl benzotriazole 6.
Yield: 92%. m.p. 307–309 °C; 1H NMR (500 MHz, DMSO-d6) δ: 4.00 (s, 1H, NH), 7.39 (t, J = 8.0 Hz, 2H, Ar-H), 7.59 (t, J = 8.0 Hz, 3H, Ar-H), 7.69 (d, J = 12.0 Hz, 5H, Ar-H), 8.23 (s, 2H, Ar-H), 8.62 (d, J = 12.0 Hz, 3H, Ar-H) ppm; 13CNMR (100 MHz, DMSO-d6) δ: 100.31(Ar-C), 113.85(Ar-C), 118.18(2 Ar-C), 119.87(Ar-C), 121.26(2 Ar-C), 124.76(Ar-C), 126.57(Ar-C), 127.60(2 Ar-C), 128.39(Ar-C), 129.36(3 Ar-C), 136.28(Ar-C), 137.49(Ar-C), 139.42(Ar-C), 143.01(Ar-C), 144.73(Ar-C), 152.58(Ar-C), 152.89(Ar-C), 154.70(Ar-C), 161.09(C = O) ppm. Anal. Calcd. for C24H16N8O: C, 66.66; H, 3.73; N, 25.91 Found: C, 66.74; H, 3.65; N, 26.07.
General procedure for synthesis of 2-{4-[(1-Phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]-benzamido}-alkanoic acid (7a–m)
Suitable amino acid (0.55 mmol) was dissolved in H2O (2 ml) using 0.13 ml triethyl amine (0.9 mmol), then it is added portionwise to the solution of synthesised novel N-acyl benzotriazole 6 (0.2 g, 0.5 mmol) in CH3CN (15 ml). The mixture was heated at 70 °C for 4–6 h until complete consumption of compound 6 as monitored by TLC. The solvent was evaporated under reduced pressure. The residue was then washed by 4 N HCl (10 ml), water (10 ml) and filtered then dried to give the purposed compounds 7a–m.
2-{4-[(1-Phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}acetic acid (7a)
Yield: 82%. m.p. 302–304 °C; 1H NMR (500 MHz, DMSO-d6) δ: 1.92 (s, 1H, NH), 3.91 (s, 2H, CH2), 4.01 (s, 1H, NH), 7.38 (t, J = 8.0 Hz, 1H, Ar-H), 7.58 (t, J = 8.0 Hz, 2H, Ar-H), 8.03 (d, J = 12.0 Hz, 4H, Ar-H), 8.22 (s, 2H, Ar-H), 8.65 (d, J = 20.0 Hz, 2H, Ar-H), 10.57 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 43.27 (CH2), 100.31(Ar-C),120.48 (2 Ar-C), 121.26 (2 Ar-C), 126.57 (Ar-C), 127.43 (2 Ar-C), 128.70 (Ar-C), 129.33 (2 Ar-C), 136.28 (Ar-C), 139.42 (Ar-C), 144.43 (Ar-C), 152.58 (Ar-C), 152.89 (Ar-C), 154.70 (Ar-C), 169.25 (C = O), 172.44 (COOH). Anal. Calcd. for C20H16N6O3: C, 61.85; H, 4.15; N, 21.64 Found: C, 61.97; H, 4.22; N, 21.80.
2-{4-[(1-Phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}propanoic acid (7b)
Yield: 89%. m.p. 311–313 °C; 1H NMR (500 MHz, DMSO-d6) δ: 1.43 (d, J = 4.0 Hz, 3H, CH3 C3), 1.94 (s, 1H, NH), 3.99 (s, 1H, NH), 4.40–4.46 (q, J = 8.0 Hz, 1H, CH C2), 7.38 (t, J = 8.0 Hz, 1H, Ar-H), 7.57 (t, J = 8.0 Hz, 2H, Ar-H), 8.02 (d, J = 8.0 Hz, 4H, Ar-H), 8.21 (s, 2H, Ar-H), 8.63 (d, J = 8.0 Hz, 2H, Ar-H), 10.51 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 18.19 (CH3, C3), 50.57 (CH, C2), 100.33 (Ar-C),119.92 (2 Ar-C),121.25 (2 Ar-C), 126.52 (Ar-C), 127.45 (2 Ar-C), 128.39 (Ar-C), 129.37 (2 Ar-C), 136.26 (Ar-C), 139.40 (Ar-C), 144.89 (Ar-C), 152.56 (Ar-C), 152.87 (Ar-C), 154.71 (Ar-C), 167.71(C = O), 175.52 (COOH) ppm. Anal. Calcd. for C21H18N6O3: C, 62.68; H, 4.51; N, 20.88 Found: C, 62.51; H, 4.71; N, 20.62.
3-Methyl-2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}butanoic acid (7c)
Yield: 80%. m.p. 324–326 °C; 1H NMR (500 MHz, DMSO-d6) δ: 0.96 (d, J = 24.0 Hz, 6H, 2 CH3), 1.17–1.25 (m, 1H, CH C3), 1.93 (s, 1H, NH), 4.03 (s, 1H, NH), 4.28 (d, J = 8.0 Hz, 1H, CH C2), 7.38 (t, J = 14.0 Hz, 1H, Ar-H), 7.59 (t, J = 12.0 Hz, 2H, Ar-H), 8.04 (d, J = 16.0 Hz, 4H, Ar-H), 8.22 (s, 2H, Ar-H), 8.66 (d, J = 16.0 Hz, 2H, Ar-H), 10.56 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 19.03 (2 CH3), 30.88 (CH, C3), 59.66 (CH, C2), 100.35 (Ar-C), 119.90 (2 Ar-C), 121.26(2 Ar-C), 126.55 (Ar-C), 127.47 (2 Ar-C), 128.34 (Ar-C), 129.37 (2 Ar-C), 136.25 (Ar-C), 139.40 (Ar-C), 144.90 (Ar-C), 152.55 (Ar-C), 152.87 (Ar-C), 154.72 (Ar-C), 167.85(C = O), 176.55 (COOH) ppm. Anal. Calcd. for C23H22N6O3: C, 64.17; H, 5.15; N, 19.52 Found: C, 64.33; H, 5.22; N, 19.60.
3-Methyl-2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}pentanoic acid (7d)
Yield: 74%. m.p. 338–3406 °C; 1H NMR (500 MHz, DMSO-d6) δ: 0.93 (d, J = 24.0 Hz, 3H, CH3 C3), 1.31 (t, J = 4.0 Hz, 3H, CH3 C4), 1.50–1.56 (m, 2H, CH2), 1.93 (s, 1H, NH), 1.95–1.99 (m, 1H, CH C3), 3.96 (s, 1H, NH), 4.38 (d, J = 5.0 Hz, 1H, CH C2), 7.39 (t, J = 8.0 Hz, 1H, Ar-H), 7.59 (t, J = 8.0 Hz, 2H, Ar-H), 8.05 (d, J = 8.0 Hz, 4H, Ar-H), 8.22 (s, 1H, Ar-H), 8.36 (s, 1H, Ar-H), 8.66 (d, J = 8.0 Hz, 2H, Ar-H), 10.53 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ:11.56 (CH3, C4), 16.27 (CH3, C3), 24.95 (CH2, C4), 35.26 (CH, C3), 58.62 (CH, C2), 100.31 (Ar-C), 121.24 (2 Ar-C), 121.55 (Ar-C), 122.25(Ar-C), 126.58 (Ar-C), 126.83 (Ar-C), 127.68 (Ar-C), 129.26 (2 Ar-C), 136.25 (Ar-C), 138.68 (Ar-C), 139.44 (Ar-C), 142.57 (Ar-C), 152.54 (Ar-C), 152.86 (Ar-C), 154.76 (Ar-C), 167.54(C = O), 176.17 (COOH) ppm. Anal. Calcd. for C24H24N6O3: C, 64.85; H, 5.44; N, 18.91 Found: C, 65.09; H, 5.58; N, 19.11.
2-{4-[(1-Phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}pentanedioic acid (7e)
Yield: 89%. m.p. 375–377 °C; 1H NMR (500 MHz, DMSO-d6) δ: 1.22 (s, 1H, NH), 1.98–2.11 (q, J = 7.0 Hz, 2H, CH2 C3), 2.40 (t, J = 8.0 Hz, 2H, CH2 C4), 4.00 (s, 1H, NH), 4.43 (t, J = 12.0 Hz, 1H, CH C2), 7.40 (t, J = 4.0 Hz, 1H, Ar-H), 7.60 (t, J = 8.0 Hz, 2H, Ar-H), 7.97 (d, J = 8.0 Hz, 2H, Ar-H), 8.04 (d, J = 12.0 Hz, 2H, Ar-H), 8.22 (s, 1H, Ar-H), 8.24 (s, 1H, Ar-H), 8.59 (d, J = 8.0 Hz, 1H, Ar-H), 8.65 (m, 1H, Ar-H), 10.51 (s, 1H, COOH), 12.48 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 26.95 (CH2, C3), 30.57 (CH2, C4), 53.91 (CH, C2), 100.31(Ar-C), 119.95 (2 Ar-C), 121.23 (2 Ar-C), 126.52 (Ar-C), 127.44 (2 Ar-C), 128.35 (Ar-C), 129.30 (2 Ar-C), 136.25 (Ar-C), 139.40 (Ar-C), 144.86 (Ar-C), 152.55 (Ar-C), 152.87 (Ar-C), 154.66 (Ar-C), 167.80 (C = O), 175.47 (COOH, C1), 176.74 (COOH, C5) ppm. Anal. Calcd. for C23H20N6O5: C, 60.00; H, 4.38; N, 18.25 Found: C, 60.17; H, 4.52; N, 18.40.
5-Guanidino-2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}pentanoic acid (7f)
Yield: 78%. m.p. 404–406 °C; 1H NMR (500 MHz, DMSO-d6) δ: 0.36 (s, 2H, NH2 guanidine), 0.57 (s, 1H, NH guanidine), 0.79–0.85 (m, 2H, CH2 C4), 1.09–1.21 (q, J = 16.0 Hz, 2H, CH2 C3), 1.66 (t, J = 18.0 Hz, 2H, CH2 C5), 1.90 (s, 1H, NH), 3.69 (s, 1H, NH guanidine), 4.00 (s, 1H, NH), 4.38 (t, J = 4.0 Hz, 1H, CH C2), 7.37 (t, J = 8.0 Hz, 1H, Ar-H), 7.57 (t, J = 6.0 Hz, 2H, Ar-H), 8.00 (d, J = 8.0 Hz, 2H, Ar-H), 8.06 (d, J = 8.0 Hz, 2H, Ar-H), 8.19 (s, 1H, Ar-H), 8.21 (s, 1H, Ar-H), 8.64 (d, J = 12.0 Hz, 2H, Ar-H), 10.56 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 25.83 (CH2, C4), 29.53 (CH2, C3), 41.88 (CH2, C5), 55.18 (CH, C2), 100.31 (Ar-C), 119.90 (2 Ar-C), 121.25 (2 Ar-C), 126.53 (Ar-C), 127.47 (2 Ar-C), 128.35 (Ar-C), 129.28 (2 Ar-C), 136.29 (Ar-C), 139.40 (Ar-C), 144.88 (Ar-C), 152.55 (Ar-C), 152.88 (Ar-C), 154.73 (Ar-C), 157.16 (C = NH), 167.80 (C = O), 175.47 (COOH) ppm. Anal. Calcd. for C24H25N9O3: C, 59.13; H, 5.17; N, 25.86 Found: C, 59.30; H, 5.12; N, 25.99.
3-Mercapto-2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}propanoic acid (7g)
Yield: 90%. m.p. 348–350 °C; 1H NMR (500 MHz, DMSO-d6) δ: 1.18 (s, 1H, SH), 1.93 (s, 1H, NH), 3.13 (d, J = 4.0 Hz, 2H, CH2 C3), 4.01 (s, 1H, NH), 4.69 (t, J = 12.0 Hz, 1H, CH C2), 7.38 (t, J = 8.0 Hz, 1H, Ar-H), 7.57 (t, J = 8.0 Hz, 2H, Ar-H), 8.01 (d, J = 8.0 Hz, 4H, Ar-H), 8.20 (s, 2H, Ar-H), 8.62 (d, J = 8.0 Hz, 2H, Ar-H), 10.54 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 28.23 (CH2, C3), 57.11 (CH, C2), 100.33 (Ar-H), 119.96 (2 Ar-H), 121.26 (2 Ar-H), 126.59 (Ar-H), 127.43 (2 Ar-H), 128.38 (Ar-H), 129.37 (2 Ar-H), 136.25 (Ar-H), 139.40 (Ar-H), 144.87 (Ar-H), 152.54 (Ar-H), 152.87 (Ar-H), 154.70 (Ar-H), 167.80 (C = O), 176.32 (COOH) ppm. Anal. Calcd. for C21H18N6O3S: C, 58.05; H, 4.18; N, 19.34 Found: C, 58.14; H, 4.32; N, 19.49.
4-(Methylthio)-2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}butanoic acid (7h)
Yield: 83%. m.p. 344–346 °C; 1HNMR (500 MHz, DMSO-d6) δ: 1.15–1.23 (q, J = 12.0 Hz, 2H, CH2 C3), 1.93 (s, 1H, NH), 2.08 (s, 3H, CH3), 2.60 (t, J = 6.0 Hz, 2H, CH2 C4), 4.00 (s, 1H, NH), 4.50 (t, J = 14.0 Hz, 1H, CH C2), 7.43 (t, J = 24.0 Hz, 1H, Ar-H), 7.58 (t, J = 20.0 Hz, 2H, Ar-H), 8.04 (d, J = 20.0 Hz, 4H, Ar-H), 8.22 (s, 2H, Ar-H), 8.65 (d, J = 12.0 Hz, 2H, Ar-H), 10.55 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 14.64 (CH3), 29.91 (CH2, C4), 31.61 (CH2, C3), 53.61 (CH, C2), 100.34 (Ar-H), 119.97 (2 Ar-H), 121.28 (2 Ar-H), 126.53 (Ar-H), 127.47 (2 Ar-H), 128.35 (Ar-H), 129.38 (2 Ar-H), 136.34 (Ar-H), 139.45 (Ar-H), 144.93 (Ar-H), 152.58 (Ar-H), 152.91 (Ar-H), 154.72 (Ar-H), 167.80 (C = O), 175.47 (COOH) ppm. Anal. Calcd. for C23H22N6O3S: C, 59.73; H, 4.79; N, 18.17 Found: C, 59.89; H, 4.90; N, 18.39.
3-(1h-Imidazol-4-yl)-2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}-propanoic acid (7i)
Yield: 82%. m.p. 381–383 °C; 1H NMR (500 MHz, DMSO-d6) δ: 1.16 (s, 1H, NH imidazole), 1.93 (s, 1H, NH), 3.13 (d, J = 12.0 Hz, 2H, CH2), 4.00 (s, 1H, NH), 4.62 (t, J = 12.0 Hz, 1H, CH C2), 6.91 (s, 1H, CH, imidazole C5), 7.37 (t, J = 20.0 Hz, 1H, Ar-H), 7.57 (t, J = 12.0 Hz, 2H, Ar-H), 7.92 (s, 1H, CH, imidazole C2), 8.03 (d, J = 16.0 Hz, 4H, Ar-H), 8.21 (s, 2H, Ar-H), 8.64 (d, J = 16.0 Hz, 2H, CH2), 10.56 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 29.09 (CH2, C3), 50.83 (CH, C2),100.31 (Ar-C),118.23 (imidazole C5),119.99 (2 Ar-H),121.26 (2 Ar-H),126.59 (Ar-H),127.49 (2 Ar-H),128.37 (Ar-H),129.34 (2 Ar-H),131.23 (imidazole C4),134.60 (imidazole C2), 136.33 (Ar-H), 139.47 (Ar-H), 144.91 (Ar-H), 152.53 (Ar-H), 152.89 (Ar-H), 154.72 (Ar-H), 167.80 (C = O), 174.39 (COOH) ppm. Anal. Calcd. for C24H20N8O3: C, 61.53; H, 4.30; N, 23.92 Found: C, 61.42; H, 4.21; N, 23.88.
1-{4-[(1-Phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzoyl}pyrrolidine-2-carboxylic acid (7j)
Yield: 77%. m.p. 342–344 °C; 1HNMR (500 MHz, DMSO-d6) δ: 1.18–1.28 (m, 2H, CH2, C4 pyrrolidine), 2.25–2.30 (q, 2H, J = 6.7 Hz, CH2, C3 pyrrolidine), 3.40 (t, J = 8.0 Hz, 2H, CH2, C5 pyrrolidine), 4.01 (s, 1H, NH), 4.42 (t, J = 8.0 Hz, 1H, CH, C2 pyrrolidine), 7.39 (t, J = 16.0 Hz, 1H, Ar-H), 7.59 (t, J = 18.0 Hz, 2H, Ar-H), 8.04 (d, J = 20.0 Hz, 4H, Ar-H), 8.22 (s, 2H, Ar-H), 8.66 (d, J = 20.0 Hz, 2H, Ar-H), 10.57 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 24.63 (C4, pyrrolidine), 29.06 (C3, pyrrolidine),45.62 (C5, pyrrolidine), 61.85 (C2, pyrrolidine), 100.31 (Ar-H), 119.65 (2 Ar-H), 121.28 (2 Ar-H), 126.58 (Ar-H), 127.40 (2 Ar-H), 129.36 (2 Ar-H), 129.72 (Ar-H), 136.38 (Ar-H), 139.40 (Ar-H), 144.40 (Ar-H), 152.58 (Ar-H), 152.86 (Ar-H), 154.76 (Ar-H), 170.60 (C = O), 175.14 (COOH) ppm. Anal. Calcd. for C23H20N6O3: C, 64.48; H, 4.71; N, 19.62 Found: C, 64.56; H, 4.86; N, 19.79.
3-(1h-Indol-3-yl)-2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}propanoic acid (7k)
Yield: 87%. m.p. 398–400 °C; 1HNMR (500 MHz, DMSO-d6) δ: 1.18 (s, 1H, NH), 3.33 (d, J = 40.0 Hz, 2H, CH2 C3), 4.01 (s, 1H, NH), 4.69 (t, J = 4.0 Hz, 1H, CH C2), 7.03 (d, J = 12.0 Hz, 1H, Ar-H), 7.38 (t, J = 12.0 Hz, 2H, Ar-H), 7.59 (t, J = 14.0 Hz, 3H, Ar-H), 7.92 (s, 1H, Ar-H), 8.04 (d, J = 12.0 Hz, 4H, Ar-H), 8.22 (s, 2H, Ar-H), 8.65 (d, J = 16.0 Hz, 3H, Ar-H), 10.56 (s, 1H, NH indole), 10.86 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 28.42 (CH2, C3), 51.98 (CH, C2), 100.32 (Ar-H), 108.54 (indole C3), 111.57 (Ar-H), 119.49 (Ar-H), 119.99 (2 Ar-H), 120.12 (Ar-C), 121.24 (2 Ar-H), 121.69 (Ar-H), 123.42 (indole C2), 126.52 (Ar-H), 127.47 (2 Ar-H), 127.95 (Ar-H), 128.37 (Ar-H), 129.37 (2 Ar-H), 136.28 (Ar-H), 137.29 (Ar-H), 139.40 (Ar-H), 144.94 (Ar-H), 152.56 (Ar-H), 152.90 (Ar-H), 154.73 (Ar-H), 167.79 (C = O), 174.39 (COOH) ppm. Anal. Calcd. for C29H23N7O3: C, 67.30; H, 4.48; N, 18.94 Found: C, 67.44; H, 4.60; N, 19.09.
3-Phenyl-2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}propanoic acid (7l)
Yield: 84%. m.p. 387–389 °C; 1H NMR (500 MHz, DMSO-d6) δ: 1.92 (s, 1H, NH), 3.25 (d, J = 4.0 Hz, 2H, CH2 C3), 4.00 (s, 1H, NH), 4.60 (t, J = 8.0 Hz, 1H, CH C2), 7.17 (t, J = 8.0 Hz, 1H, Ar-H), 7.26 (t, J = 8.0 Hz, 1H, Ar-H), 7.33 (d, J = 8.0 Hz, 1H, Ar-H), 7.38 (t, J = 8.0 Hz, 1H, Ar-H), 7.58 (t, J = 8.0 Hz, 3H, Ar-H), 7.87 (d, J = 8.0 Hz, 1H, Ar-H), 8.01 (d, J = 8.0 Hz, 2H, Ar-H), 8.07 (d, J = 8.0 Hz, 2H, Ar-H), 8.21 (s, 1H, Ar-H), 8.23 (s, 1H, Ar-H), 8.62 (d, J = 12.0 Hz, 1H, Ar-H), 8.67 (d, J = 8.0 Hz, 1H, Ar-H), 10.58 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 37.05 (CH2, C3), 55.97 (CH, C2), 100.35 (Ar-H), 119.94 (2 Ar-H), 121.28 (2 Ar-H), 126.55 (Ar-H), 127.15 (Ar-H), 127.51 (2 Ar-H), 128.37 (Ar-H), 129.05 (2 Ar-H), 129.34 (2 Ar-H), 129.38 (2 Ar-H), 136.28 (Ar-H), 137.68 (Ar-H), 139.42 (Ar-H), 144.90 (Ar-H), 152.54 (Ar-H), 152.92 (Ar-H), 154.70 (Ar-H), 167.82 (C = O), 174.49 (COOH) ppm. Anal. Calcd. for C27H22N6O3: C, 67.77; H, 4.63; N, 17.56 Found: C, 67.90; H, 4.86; N, 17.70.
3-(4-Hydroxyphenyl)-2-{4-[(1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino]benzamido}-propanoic acid (7m)
Yield: 80%. m.p. 401–403 °C; 1H NMR (500 MHz, DMSO-d6) δ: 1.24 (s, 1H, OH), 1.92 (s, 1H, NH), 3.00 (d, J = 4.0 Hz, 2H, CH2 C3), 4.01 (s, 1H, NH), 4.43 (t, J = 16.0 Hz, 1H, CH C2), 6.62 (d, J = 8.0 Hz, 1H, Ar-H), 7.07 (t, J = 6.0 Hz, 1H, Ar-H), 7.19 (t, J = 6.0 Hz, 2H, Ar-H), 7.48 (d, J = 8.0 Hz, 3H, Ar-H), 7.94 (d, J = 8.0 Hz, 4H, Ar-H), 8.21 (s, 2H, Ar-H), 8.65 (d, J = 11.0 Hz, 2H, Ar-H), 10.59 (s, 1H, COOH) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 37.07 (CH2, C3), 55.99 (CH, C2), 100.35 (Ar-H), 115.90 (2 Ar-H), 119.97 (2 Ar-H), 121.24 (2 Ar-H), 126.52 (Ar-H), 127.49 (2 Ar-H), 128.37 (Ar-H), 129.29 (2 Ar-H), 129.74 (Ar-H), 130.62 (2 Ar-H), 136.25 (Ar-H), 139.39 (Ar-H), 144.93 (Ar-H), 152.54 (Ar-H), 152.88 (Ar-H), 154.67 (Ar-H), 155.93 (Ar-H), 167.80 (C = O), 174.49 (COOH) ppm. Anal. Calcd. for C27H22N6O4: C, 65.58; H, 4.48; N, 16.99 Found: C, 65.69; H, 4.63; N, 17.14.
Biological evaluation
Cytotoxicity screening using SRB assay
Sulforhodamine B (SRB) assay was employed to assay in-vitro antitumor activities of all the synthesised compounds as describer previouslyCitation11–14. Six human cancer cell lines, human prostate cancer (PC-3), pancreatic human cancer cell lines (BxPC-3), colorectal carcinoma (HCT-116), human hepatocellular carcinoma (HepG-2), cervical carcinoma (HeLa), and mammary gland breast cancer (MCF-7), besides normal immortalised pancreatic cell line (HPDE) were used in the evaluation of antitumor activity. The cell lines were obtained from the American Type Culture Collection (Rockville, USA).
Elisa assay of human DHFR enzyme
Enzyme-linked immunosorbent assay (ELISA) assay technique was used to evaluate the anti-DHFR effects of all the synthesised compounds on as described previouslyCitation15. The DHFR ELISA kit (MyBioSource, MBS9312476 San Diego, CA, USA) was used according to the manufacturer instructions. Methotrexate (MTX) was used as positive control, and a standard curve was plotted to detect IC50. The test was repeated in triplicate, and the significance was assessed by one-way ANOVA test followed by Tukey’s posttest, where p < 0.05 was considered significance.
Molecular modeling
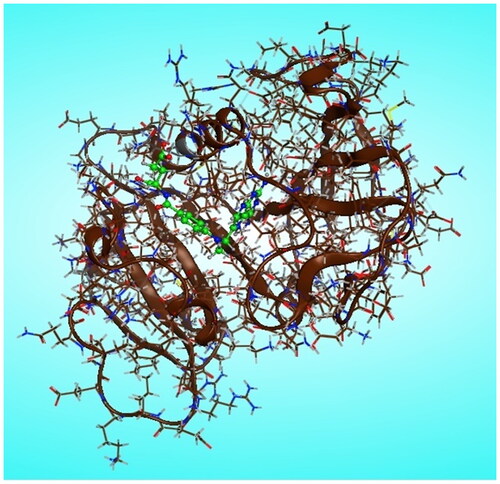
Molecular modelling calculations were carried out using molecular operating environment MOE version 2014.0901 (Chemical Computing Group Inc. software). The most stable conformers of newly synthesised pyrazolo[3,4-d]pyrimidine derivatives 6 and 7a–m were docked into the binding pocket of DHFR-binding domain which is in tertiary complex with dihydro-nicotinamide-adenine-dinucleotide phosphate (NADPH) and methotrexate (MTX), (pdb code ID: 1U72)Citation18. The three-dimensional structures of the novel prepared pyrazolo[3,4-d]pyrimidine derivatives, in their neutral forms, were built using the builder interface of the MOE software. Besides the referred enzymes were obtained from the research collaborator for structural bioinformatics Protein Data Bank (RCSB-PDB), where the hydrogens were added after releasing the backbone and side chain of amino acid residues into the secondary structures, and then the enzymes structure was subjected to a refinement protocol by automatic connect and type to correct the loosed bonds during X-ray crystallography followed by protein potential fixation where the constraints on the enzyme were gradually removed and minimised until the RMSD gradient was 0.01 kcal/mol A° as shown in . The active site of the utilised enzymes in this study was detected using a radius of 10.0 A° around MTX. Conformational analysis of the newly synthesised pyrazolo[3,4-d]pyrimidine compounds was performed using MMFF94 force field with root mean square (RMS) gradient of 0.01 kcal/mol A°. The flexible alignment of our novel compounds was done using the flexible alignment tool of the MOE program adjusting the energy cut off to 15 kcal/mol, and RMSD tolerance to 0.5. A complete study was done for each ligand to check its fitting in the active site of the enzyme and to identify the different binding interactions involved in its fitting using MOE program software.
Figure 10. Chain A of DHFR enzyme with (green) methotrexate/NADPH complex ligand in the active site.
Cell apoptosis assay
Flow cytometric analysis using Annexin-V-fluoresce in isothiocyanate (FITC) and propidium iodide (PI) staining kit (BD Pharmingen, San Diego, USA) was performed to assess the apoptosis. At 70% confluence, the cells were treated with compound 7f at different concentrations of (0–8 μmol/l) for 24 h. Then after trypsinization of cells, washing with PBS and centrifugation, the cells were mixed with FITC and PI in binding buffer. Becton Dickinson (Franklin Lakes, NJ, USA) was used to do the flow cytometry analysisCitation16,Citation17.
Assessment of the compound 7f effect on the caspase-3/7, Bax and Bcl2 activities
The apoptotic effect of compound 7f on breast cancer cell line MCF-7 were evaluated by quantification of Bax, caspase 3/7, and Bcl-2 using Bax ELISA Kit ab199080 (ABCAM, Cambridge, UK), Caspase-Glo 3/7 assay kits (Promega, Fitchburg, MA, USA), and Bcl-2 ELISA Kit ab119506 (ABCAM, Cambridge, UK), according the instructions of manufacturersCitation11,Citation14. The cells were treated with compounds 7f (½ IC50, IC50, or 2 ×IC50), and the activities were shown as a percentage change from the untreated control. The experiments were performed in triplicate applying two-way ANOVA test followed by Bonferroni posttest to evaluate the significance (p < 0.05).
Western blotting
The effect of compound 7f on the expression of DHFR, Casp3, Bax, and Bcl-2 proteins in MCF-7 cancer cells was evaluated by western blotting as previously performedCitation20–22. The cancer cells treated or untreated were collected were harvested and blotted on SDS-polyacrylamide gel and separated by Cleaver electrophoresis unit (Cleaver, UK), transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA) for 30 min using a Semi-dry Electroblotter (Biorad, USA) at 2.5 A and 25 V for 30 min. The membranes were blocked with 5% non-fat dry milk in TBS-T for 2 h at RT and then incubated overnight at 4 °C with anti-DHFR, anti-Bax, anti-Bcl-2, and anti-β-actin primary antibodies (abcam, Cambridge, UK). Finally, the horse radish peroxidase (HRP)-linked secondary antibody (Agilent, Santa Clara, CA, USA) was added for 1 h at room temperature. After washing, the chemiluminescent Western ECL substrate (Perkin Elmer, Waltham, MA, USA) was applied to the blot and then incubated for 1 min with a mixture of equal volumes from ECL solution A and ECL solution B. The chemiluminescent signals were captured using a CCD camera-based imager (Chemi Doc imager, Biorad, USA), and the bands intensities were then measured by Image J software program. Protein-sized markers were used in all gels to localise the gel transfer regions for specific proteins and determine the transfer efficiency. The experiments were conducted in triplicate. For colorimetric assays two-way ANOVA test followed by Bonferroni posttest were used, while for quantification expression on western blot, student-t-test was used (significance p < 0.05).
Supplemental Material
Download PDF (1.8 MB)Acknowledgements
The authors gratefully acknowledge DSR at King Abdulaziz University (KAU) for technical and financial support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Berman EM, Werbel LM. The renewed potential for folate antagonists in contemporary cancer chemotherapy. J Med Chem. 1991;34(2):479–485.
- Visentin M, Zhao R, Goldman ID. The antifolates. Hematol Oncol Clin North Am. 2012;26(3):629–648, ix.
- Wińska P, Widło Ł, Skierka K, Krzyśko A, Koronkiewicz M, Cieśla JM, Cieśla J, Bretner M. Simultaneous inhibition of protein kinase CK2 and dihydrofolate reductase results in synergistic effect on acute lymphoblastic leukemia cells. Anticancer Res. 2019;39(7):3531–3542.
- Ng C, Xiao Y, Lum BL, Han Y. Quantitative structure activity relationships of methotrexate and methotrexate analogues transported by the rat multispecific resistance-associated protein 2 (rMrp2). Eur J Pharm Sci. 2005;26(5):405–413.
- Walling J. From methotrexate to pemetrexed and beyond. A review of the pharmacodynamic and clinical properties of antifolates. Invest New Drugs. 2006;24(1):37–77.
- Gangjee A, Devraj R, McGuire JJ, Kisliuk RL. Effect of bridge region variation on antifolate and antitumor activity of classical 5-substituted 2,4-diaminofuro[2,3-d]pyrimidin. J Med Chem. 1995;38(19):3798–3805.
- Santos IF, Guedes GP, Mercante LA, Bernardino AMR, Vaz MGF. Synthesis, crystal structure and magnetism of three novel copper (II) complexes with pyrazole-based ligands. J Mol Struct. 2012;1011:99–104.
- Wang H-Q, Liu Z-J, Ding M-W. Versatile synthesis and fungicidal activity of 6-amino-3-alkylthio-1,5-dihydro-1-phenyl-pyrazolo[3,4-d]pyrimidin-4-one derivatives. Phosphorus Sulfur Silicon Relat Elem. 2004;179(10):2039–2050.
- Sugimoto O, Sudo M, Tanji K. Lithiation of 1H-pyrazolo[3,4-d]pyrimidine derivative using lithium alkanetellurolate. Tetrahedron Lett. 1999;40(11):2139–2140.
- Somakala K, Tariq S, Amir M. Synthesis, evaluation and docking of novel pyrazolo pyrimidines as potent p38α MAP kinase inhibitors with improved anti-inflammatory, ulcerogenic and TNF-α inhibitory properties. Bioorg Chem. 2019;87:550–559.
- Youns M, Askoura M, Abbas HA, Attia GH, Khayyat AN, Goda RM, Almalki AJ, Khafagy E-S, Hegazy WAH. Celastrol modulates multiple signaling pathways to inhibit proliferation of pancreatic cancer via DDIT3 and ATF3 up-regulation and RRM2 and MCM4 down-regulation. Onco Targets Ther. 2021;14:3849–3860.
- Askoura M, Abbas HA, Al Sadoun H, Abdulaal WH, Abu Lila AS, Almansour K, Alshammari F, Khafagy E-S, Ibrahim TS, Hegazy WAH. Elevated levels of IL-33, IL-17 and IL-25 indicate the progression from chronicity to hepatocellular carcinoma in hepatitis C virus patients. Pathogens. 2022;11(1):57.
- Abbas HA, Hegazy WAH. Targeting the virulence factors of Serratia marcescens by ambroxol. Rouman Arch Microbiol Immunol. 2017;76:27–32.
- Youns M, Abdel Halim Hegazy W. The natural flavonoid fisetin inhibits cellular proliferation of hepatic, colorectal, and pancreatic cancer cells through modulation of multiple signaling pathways. PLoS One. 2017;12(1):e0169335.
- Abdelaziz OA, El Husseiny WM, Selim KB, Eisa HM. Dihydrofolate reductase inhibition effect of 5-substituted pyrido[2,3-d]pyrimidines: synthesis, antitumor activity and molecular modeling study. Bioorg Chem. 2019;90:103076.
- Aldawsari MF, Alalaiwe A, Khafagy ES, Al Saqr A, Alshahrani SM, Alsulays BB, Alshehri S, Lila ASA, Rizvi SMD, Hegazy WAH. Efficacy of SPG-ODN 1826 nanovehicles in inducing M1 phenotype through TLR-9 activation in murine alveolar J774A.1 cells: plausible nano-immunotherapy for lung carcinoma. IJMS. 2021;22(13):6833.
- Ibrahim TS, Malebari AM, Mohamed MFA. Design, synthesis, in vitro anticancer evaluation and molecular modelling studies of 3,4,5-trimethoxyphenyl-based derivatives as Dual EGFR/HDAC hybrid inhibitors. Pharmaceuticals. 2021;14(11):1177.
- Moroy G, Sperandio O, Rielland S, Khemka S, Druart K, Goyal D, Perahia D, Miteva M. Sampling of conformational ensemble for virtual screening using molecular dynamics simulations and normal mode analysis. Future Med Chem. 2015;7(17):2317–2331.
- Fotoohi AK, Albertioni F. Mechanisms of antifolate resistance and methotrexate efficacy in leukemia cells. Anticancer Res. 2008;3:410–426.
- Askoura M, Almalki AJ, Lila ASA, Almansour K, Alshammari F, Khafagy E, Ibrahim TS, Hegazy WAH. Alteration of Salmonella enterica virulence and host pathogenesis through targeting sdiA by using the CRISPR-Cas9 system. Microorganisms. 2021;9(12):2564.
- Askoura M, Hegazy WAH. Ciprofloxacin interferes with Salmonella typhimurium intracellular survival and host virulence through repression of Salmonella pathogenicity island-2 (SPI-2) genes expression. Pathog Dis. 2020;78:1–12.
- Hegazy WAH, Abbas HA. Evaluation of the role of SsaV Salmonella pathogenicity island-2 dependent type III secretion system components on the virulence behavior of Salmonella enterica serovar Typhimurium. African Journal of Biotechnology. 2017;16:718–726.