
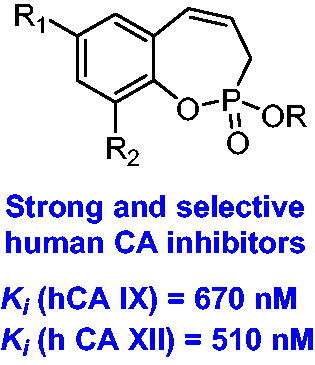
Abstract
The synthesis of 3H-1,2-benzoxaphosphepine 2-oxides and evaluation of their inhibitory activity against human carbonic anhydrase (hCA) isoforms I, II, IX, and XII are described. The target compounds were obtained via a concise synthesis from commercial salicylaldehydes and displayed low to sub-micromolar inhibition levels against the tumour-associated isoforms hCA IX and XII. All obtained benzoxaphosphepine 2-oxides possess remarkable selectivity for inhibition of hCA IX/XII over the off-target cytosolic hCA isoforms I and II, whose inhibition may lead to side effects.
Graphical Abstract
Introduction
Carbonic anhydrases (CA, EC 4.2.1.1) are a superfamily of metalloenzymes present across all kingdoms of lifeCitation1. These enzymes catalyse a simple yet essential physiological reaction – the reversible hydration of carbon dioxideCitation2. To date, 15 different human CA (hCA) isoforms have been identified, out of which hCA IX and XII isoforms are highly overexpressed in different tumour types and may contribute in the survival and progression of tumour cells by regulating intra- and extracellular pHCitation2–6. Therefore, the development of selective hCA IX/XII inhibitors is a potential strategy for designing anti-tumour agents.
Due to the high degree of structural homology and sequence similarities within the active site of the hCA isoforms, the design and development of isoform-selective hCA inhibitors pose a challengeCitation7. A variety of compounds have been reported as potent and selective inhibitors of tumour-associated isoforms hCA IX and XII including coumarinsCitation8–11, thiocoumarinsCitation8,Citation11, sulphocoumarinsCitation8,Citation12–15, as well as their congeners, homosulphocoumarins (3H-1,2-benzoxathiepine 2,2-dioxides)Citation16. In this work, attention was drawn to phosphorus, as phosphorus-containing molecules display a multitude of biological activities relevant in medicinal chemistryCitation17. Additionally, several groups have shown the use of organophosphorus compounds as CA inhibitorsCitation18.
Considering isosteric relationship between sulphonyl derivatives and phosphonatesCitation19, our research group designed and synthesised a series of benzoxaphosphepine 2-oxides pursuing the development of new classes of selective CA inhibitors. These compounds showed interesting inhibitory activity against hCA IX and XII. Moreover, the results of current study demonstrate the bioisosteric utility of the cyclic phosphonate moiety in the design of novel CA inhibitors.
Materials and methods
Chemistry
The air- or moisture-sensitive reactions were performed under argon atmosphere using dry glassware. Toluene was freshly distilled from Na prior to use. DCM and NEt3 were distilled from CaH2. Other reagents, starting materials and solvents were purchased from commercial sources and used as received. TLC was performed on silica gel plates (60 F254) and visualised under UV light (254 and 365 nm). Melting points were determined on an OptiMelt MPA100 apparatus. IR spectra were recorded on a Shimadzu FTIR IR Prestige-21 spectrophotometer. 1H, 13C, and 31P NMR spectra were recorded on a Bruker Avance Neo 400 MHz spectrometer. The chemical shifts (δ) were reported in parts per million (ppm) relative to the residual solvent peak as an internal reference (DMSO-d6: 1H 2.50, 13C 39.52; CDCl3: 1H 7.26, 13C 77.16; C6D6: 1H 7.16, 13C 128.06). 31P shifts were referenced externally to H3PO4. The coupling constants (J) were expressed in Hertz (Hz). HRMS was performed on a Q-TOF Micro mass spectrometer.
General procedure for the synthesis of vinylphenols 2
To a stirred solution of MePPh3Br (2.3 eq) in dry THF (3 ml/1 mmol of MePPh3Br) was added t-BuOK (2.35 eq) in small portions over 20 min. The reaction mixture was stirred under inert atmosphere for 2 h at rt. The corresponding hydroxybenzaldehyde 1 (1.0 eq) was added, and the mixture was kept stirring at rt for another 18 h. The reaction mixture was treated with sat. aq NH4Cl (25 ml) and then was diluted with Et2O (3 ml/1 mmol of MePPh3Br). The organic layer was washed with water (2 × 40 ml) and brine (2 × 40 ml), dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (PE/EtOAc 4:1).
2-Vinylphenol (2a)
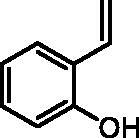 By following the general procedure, 2a was prepared from MePPh3Br (13.46 g, 37.7 mmol), t-BuOK (4.32 g, 38.5 mmol), and 2-hydroxybenzaldehyde (1a) (2.00 g, 16.4 mmol) as a yellowish oil (1.71 g, 87%)Citation16a. 1H NMR (400 MHz, CDCl3) δ = 5.34–5.38 (m, 2H), 5.75 (dd, 1H, J = 17.8, 1.4 Hz), 6.80 (dd, 1H, J = 8.1, 1.1 Hz), 6.89–7.01 (m, 2H), 7.12–7.17 (m, 1H), 7.40 (dd, 1H, J = 7.7, 1.7 Hz) ppm. 13C NMR (101 MHz, CDCl3) δ = 115.8, 116.0, 121.0, 125.0, 127.4, 129.0, 131.6, 153.0 ppm.
By following the general procedure, 2a was prepared from MePPh3Br (13.46 g, 37.7 mmol), t-BuOK (4.32 g, 38.5 mmol), and 2-hydroxybenzaldehyde (1a) (2.00 g, 16.4 mmol) as a yellowish oil (1.71 g, 87%)Citation16a. 1H NMR (400 MHz, CDCl3) δ = 5.34–5.38 (m, 2H), 5.75 (dd, 1H, J = 17.8, 1.4 Hz), 6.80 (dd, 1H, J = 8.1, 1.1 Hz), 6.89–7.01 (m, 2H), 7.12–7.17 (m, 1H), 7.40 (dd, 1H, J = 7.7, 1.7 Hz) ppm. 13C NMR (101 MHz, CDCl3) δ = 115.8, 116.0, 121.0, 125.0, 127.4, 129.0, 131.6, 153.0 ppm.
4-Iodo-2-vinylphenol (2b)
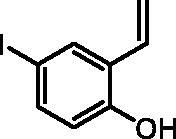 By following the general procedure, 2b was prepared from MePPh3Br (16.56 g, 46.4 mmol), t-BuOK (5.32 g, 47.4 mmol), and 2-hydroxy-5-iodobenzaldehyde (1b) (5.00 g, 20.2 mmol) as a yellowish solid (4.17 g, 84%)Citation16b. 1H NMR (400 MHz, CDCl3) δ = 5.23 (dd, 1H, J = 11.3, 1.3 Hz), 5.80 (dd, 1H, J = 17.6, 1.3 Hz), 6.67 (d, 1H, J = 8.6 Hz), 6.77–6.87 (m, 1H), 7.37 (dd, 1H, J = 8.6, 2.4 Hz), 7.70 (d, 1H, J = 2.4 Hz), 9.94 (s, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ = 81.4, 115.1, 118.4, 126.9, 130.4, 134.4, 137.0, 154.6 ppm.
By following the general procedure, 2b was prepared from MePPh3Br (16.56 g, 46.4 mmol), t-BuOK (5.32 g, 47.4 mmol), and 2-hydroxy-5-iodobenzaldehyde (1b) (5.00 g, 20.2 mmol) as a yellowish solid (4.17 g, 84%)Citation16b. 1H NMR (400 MHz, CDCl3) δ = 5.23 (dd, 1H, J = 11.3, 1.3 Hz), 5.80 (dd, 1H, J = 17.6, 1.3 Hz), 6.67 (d, 1H, J = 8.6 Hz), 6.77–6.87 (m, 1H), 7.37 (dd, 1H, J = 8.6, 2.4 Hz), 7.70 (d, 1H, J = 2.4 Hz), 9.94 (s, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ = 81.4, 115.1, 118.4, 126.9, 130.4, 134.4, 137.0, 154.6 ppm.
4-Bromo-2-vinylphenol (2c)
 By following the general procedure, 2c was prepared from MePPh3Br (8.17 g, 22.9 mmol), t-BuOK (2.62 g, 23.4 mmol), and 5-bromo-2-hydroxybenzaldehyde (1c) (2.00 g, 10 mmol) as a yellowish solid (1.74 g, 88%)Citation16a. 1H NMR (400 MHz, CDCl3) δ = 4.98 (s, 1H), 5.40 (dd, 1H, J = 11.3, 1.0 Hz), 5.74 (dd, 1H, J = 17.8, 1.0 Hz), 6.68 (d, 1H, J = 8.6 Hz), 6.85 (dd, 1H, J = 17.8, 11.3 Hz), 7.23 (dd, 1H, J = 8.6, 2.4 Hz), 7.49 (d, 1H, J = 2.4 Hz) ppm.
By following the general procedure, 2c was prepared from MePPh3Br (8.17 g, 22.9 mmol), t-BuOK (2.62 g, 23.4 mmol), and 5-bromo-2-hydroxybenzaldehyde (1c) (2.00 g, 10 mmol) as a yellowish solid (1.74 g, 88%)Citation16a. 1H NMR (400 MHz, CDCl3) δ = 4.98 (s, 1H), 5.40 (dd, 1H, J = 11.3, 1.0 Hz), 5.74 (dd, 1H, J = 17.8, 1.0 Hz), 6.68 (d, 1H, J = 8.6 Hz), 6.85 (dd, 1H, J = 17.8, 11.3 Hz), 7.23 (dd, 1H, J = 8.6, 2.4 Hz), 7.49 (d, 1H, J = 2.4 Hz) ppm.
2-Bromo-6-vinylphenol (2d)
 By following the general procedure, 2d was prepared from MePPh3Br (16.35 g, 45.8 mmol), t-BuOK (5.25 g, 46.8 mmol), and 3-bromo-2-hydroxybenzaldehyde (1d) (4.00 g, 19.9 mmol) as a yellowish solid (3.05 g, 77%)Citation16b. 1H NMR (400 MHz, CDCl3) δ = 5.34 (dd, 1H, J = 11.2, 1.3 Hz), 5.72 (s, 1H), 5.79 (dd, 1H, J = 17.7, 1.3 Hz), 6.77–6.82 (m, 1H), 7.00 (dd, 1H, J = 17.7, 11.2 Hz), 7.35–7.41 (m, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ = 111.2, 116.2, 121.6, 126.2, 126.5, 131.1, 131.3, 149.6 ppm.
By following the general procedure, 2d was prepared from MePPh3Br (16.35 g, 45.8 mmol), t-BuOK (5.25 g, 46.8 mmol), and 3-bromo-2-hydroxybenzaldehyde (1d) (4.00 g, 19.9 mmol) as a yellowish solid (3.05 g, 77%)Citation16b. 1H NMR (400 MHz, CDCl3) δ = 5.34 (dd, 1H, J = 11.2, 1.3 Hz), 5.72 (s, 1H), 5.79 (dd, 1H, J = 17.7, 1.3 Hz), 6.77–6.82 (m, 1H), 7.00 (dd, 1H, J = 17.7, 11.2 Hz), 7.35–7.41 (m, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ = 111.2, 116.2, 121.6, 126.2, 126.5, 131.1, 131.3, 149.6 ppm.
2-Methoxy-6-vinylphenol (2e)
 By following the general procedure, 2e was prepared from MePPh3Br (2.70 g, 7.6 mmol), t-BuOK (0.87 g, 7.7 mmol), and 2-hydroxy-3-methoxybenzaldehyde (1e) (0.50 g, 3.3 mmol) as a yellowish solid (0.40 g, 81%)Citation20. 1H NMR (400 MHz, CDCl3) δ = 3.89 (s, 3H), 5.33 (dd, 1H, J = 11.2, 1.5 Hz), 5.83 (dd, 1H, J = 17.8, 1.5 Hz), 5.93–5.94 (m, 1H), 6.76–6.80 (m, 1H), 6.81–6.86 (m, 1H), 7.00–7.11 (m, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ = 56.2, 109.7, 114.9, 118.9, 119.5, 124.1, 131.2, 143.4, 146.8 ppm.
By following the general procedure, 2e was prepared from MePPh3Br (2.70 g, 7.6 mmol), t-BuOK (0.87 g, 7.7 mmol), and 2-hydroxy-3-methoxybenzaldehyde (1e) (0.50 g, 3.3 mmol) as a yellowish solid (0.40 g, 81%)Citation20. 1H NMR (400 MHz, CDCl3) δ = 3.89 (s, 3H), 5.33 (dd, 1H, J = 11.2, 1.5 Hz), 5.83 (dd, 1H, J = 17.8, 1.5 Hz), 5.93–5.94 (m, 1H), 6.76–6.80 (m, 1H), 6.81–6.86 (m, 1H), 7.00–7.11 (m, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ = 56.2, 109.7, 114.9, 118.9, 119.5, 124.1, 131.2, 143.4, 146.8 ppm.
2,4-Dichloro-6-vinylphenol (2f)
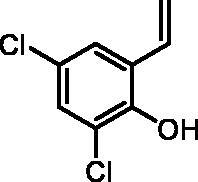 By following the general procedure, 2f was prepared from MePPh3Br (2.15 g, 6.0 mmol), t-BuOK (0.69 g, 6.2 mmol), and 3,5-dichloro-2-hydroxybenzaldehyde (1f) (0.50 g, 2.6 mmol) as a yellowish solid (0.38 g, 76%)Citation21. 1H NMR (400 MHz, CDCl3) δ = 5.39 (dd, 1H, J = 11.2, 1.0 Hz), 5.70–5.72 (m, 1H), 5.80 (d, 1H, J = 17.7, 1.0 Hz), 6.92 (dd, 1H, J = 17.7, 11.2 Hz), 7.23 (d, 1H, J = 2.5 Hz), 7.32–7.34 (m, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ = 117.3, 121.0, 125.5, 125.6, 127.2, 127.4, 130.1, 147.5 ppm.
By following the general procedure, 2f was prepared from MePPh3Br (2.15 g, 6.0 mmol), t-BuOK (0.69 g, 6.2 mmol), and 3,5-dichloro-2-hydroxybenzaldehyde (1f) (0.50 g, 2.6 mmol) as a yellowish solid (0.38 g, 76%)Citation21. 1H NMR (400 MHz, CDCl3) δ = 5.39 (dd, 1H, J = 11.2, 1.0 Hz), 5.70–5.72 (m, 1H), 5.80 (d, 1H, J = 17.7, 1.0 Hz), 6.92 (dd, 1H, J = 17.7, 11.2 Hz), 7.23 (d, 1H, J = 2.5 Hz), 7.32–7.34 (m, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ = 117.3, 121.0, 125.5, 125.6, 127.2, 127.4, 130.1, 147.5 ppm.
4-Nitro-2-vinylphenol (2g)
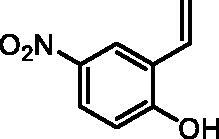 By following the general procedure, 2g was prepared from MePPh3Br (9.83 g, 27.5 mmol), t-BuOK (3.16 g, 28.1 mmol), and 2-hydroxy-5-nitrobenzaldehyde (1g) (2.00 g, 11 mmol). The solution of nitrobenzaldehyde 1g in THF (20 ml) was added at −78 °C. The title product was obtained as a yellow solid (1.70 g, 86%)Citation22. 1H NMR (400 MHz, DMSO-d6) δ = 5.39 (dd, 1H, J = 11.3, 1.2 Hz), 5.98 (dd, 1H, J = 17.8, 1.2 Hz), 6.92 (dd, 1H, J = 17.8, 11.3 Hz), 7.01 (d, 1H, J = 9.0 Hz), 8.02 (dd, 1H, J = 9.0, 2.9 Hz), 8.28 (d, 1H, J = 2.9 Hz), 11.32 (s, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 116.1, 116.9, 122.3, 124.7, 124.8, 130.1, 139.9, 161.0 ppm.
By following the general procedure, 2g was prepared from MePPh3Br (9.83 g, 27.5 mmol), t-BuOK (3.16 g, 28.1 mmol), and 2-hydroxy-5-nitrobenzaldehyde (1g) (2.00 g, 11 mmol). The solution of nitrobenzaldehyde 1g in THF (20 ml) was added at −78 °C. The title product was obtained as a yellow solid (1.70 g, 86%)Citation22. 1H NMR (400 MHz, DMSO-d6) δ = 5.39 (dd, 1H, J = 11.3, 1.2 Hz), 5.98 (dd, 1H, J = 17.8, 1.2 Hz), 6.92 (dd, 1H, J = 17.8, 11.3 Hz), 7.01 (d, 1H, J = 9.0 Hz), 8.02 (dd, 1H, J = 9.0, 2.9 Hz), 8.28 (d, 1H, J = 2.9 Hz), 11.32 (s, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 116.1, 116.9, 122.3, 124.7, 124.8, 130.1, 139.9, 161.0 ppm.
Diethyl allylphosphonate (S1)
 Triethylphosphite (31.0 ml, 180.5 mmol) and allyl bromide (18.7 ml, 216.7 mmol) were stirred and heated for 24 h at 70 °C. After evaporation of the remaining allyl bromide, the residue was distilled in vacuo (∼4 mbar) to afford product S1 as a colourless liquid (29.00 g, 90%), b.p. 60–62 °C/4 mbarCitation23. 31P NMR (162 MHz, DMSO-d6) δ = 26.81 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.22 (t, 6H, J = 7.1 Hz), 2.62 (dt, 1H, J = 7.3, 1.3 Hz), 2.67 (dt, 1H, J = 7.3, 1.3 Hz), 3.93–4.06 (m, 4H), 5.10–5.26 (m, 2H), 5.63–5.73 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 5.7 Hz), 30.6 (d, JP,C = 136 Hz), 61.2 (d, JP,C = 6.3 Hz), 119.5 (d, JP,C = 14.2 Hz), 128.3 (d, JP,C = 10.9 Hz) ppm.
Triethylphosphite (31.0 ml, 180.5 mmol) and allyl bromide (18.7 ml, 216.7 mmol) were stirred and heated for 24 h at 70 °C. After evaporation of the remaining allyl bromide, the residue was distilled in vacuo (∼4 mbar) to afford product S1 as a colourless liquid (29.00 g, 90%), b.p. 60–62 °C/4 mbarCitation23. 31P NMR (162 MHz, DMSO-d6) δ = 26.81 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.22 (t, 6H, J = 7.1 Hz), 2.62 (dt, 1H, J = 7.3, 1.3 Hz), 2.67 (dt, 1H, J = 7.3, 1.3 Hz), 3.93–4.06 (m, 4H), 5.10–5.26 (m, 2H), 5.63–5.73 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 5.7 Hz), 30.6 (d, JP,C = 136 Hz), 61.2 (d, JP,C = 6.3 Hz), 119.5 (d, JP,C = 14.2 Hz), 128.3 (d, JP,C = 10.9 Hz) ppm.
Ethyl allylphosphonochloridate (3)
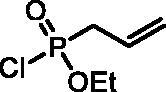 Diethyl allylphosphonate(S1) (28.6 g, 160.5 mmol) was dissolved in dry DCM (200 ml). The solution was cooled down to 0 °C, and oxalyl chloride (49.0 ml, 0.562 mol) was added dropwise. The reaction mixture was stirred for 16 h at rt. After evaporation of the solvent and remaining oxalyl chloride, the residue was distilled in vacuo (∼4 mbar) to afford product 3 as a colourless liquid (21.64 g, 80%), b.p. 88–90 °C/4 mbarCitation23. 31P NMR (162 MHz, DMSO-d6) δ = 39.17 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.37 (t, 3H, J = 7.1 Hz), 2.93 (dt, 1H, J = 7.3, 1.2 Hz), 2.99 (dt, 1H, J = 7.3, 1.2 Hz), 4.16–4.37 (m, 2H), 5.26–5.36 (m, 2H), 5.72–5.86 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 15.9 (d, JP,C = 7.0 Hz), 39.1 (d, JP,C = 123 Hz), 63.5 (d, JP,C = 8.4 Hz), 122.2 (d, JP,C = 16.8 Hz), 125.4 (d, JP,C = 12.8 Hz) ppm.
Diethyl allylphosphonate(S1) (28.6 g, 160.5 mmol) was dissolved in dry DCM (200 ml). The solution was cooled down to 0 °C, and oxalyl chloride (49.0 ml, 0.562 mol) was added dropwise. The reaction mixture was stirred for 16 h at rt. After evaporation of the solvent and remaining oxalyl chloride, the residue was distilled in vacuo (∼4 mbar) to afford product 3 as a colourless liquid (21.64 g, 80%), b.p. 88–90 °C/4 mbarCitation23. 31P NMR (162 MHz, DMSO-d6) δ = 39.17 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.37 (t, 3H, J = 7.1 Hz), 2.93 (dt, 1H, J = 7.3, 1.2 Hz), 2.99 (dt, 1H, J = 7.3, 1.2 Hz), 4.16–4.37 (m, 2H), 5.26–5.36 (m, 2H), 5.72–5.86 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 15.9 (d, JP,C = 7.0 Hz), 39.1 (d, JP,C = 123 Hz), 63.5 (d, JP,C = 8.4 Hz), 122.2 (d, JP,C = 16.8 Hz), 125.4 (d, JP,C = 12.8 Hz) ppm.
General procedure for the synthesis of diolefins 4
The corresponding vinylphenol 2 (1.0 eq) was dissolved in dry DCM (10 ml/1 mmol of 2). After cooling down the solution to 0 °C, ethyl allylphosphonochloridate (3) (1.2 eq) and NEt3 (1.25 eq) were added. The reaction mixture was stirred under inert atmosphere at rt for 18 h. Water (30 ml) was added, and the mixture was extracted with EtOAc (3 × 40 ml). The combined organic layers were washed with brine (2 × 40 ml), dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (PE/EtOAc 1.5:1).
Ethyl (2-vinylphenyl) allylphosphonate (4a)
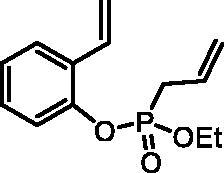 By following the general procedure, 4a was prepared from 2-vinylphenol (2a) (0.38 g, 3.16 mmol), ethyl allylphosphonochloridate (3) (0.56 ml, 3.79 mmol), and NEt3 (0.55 ml, 3.95 mmol) as a colourless oil (0.48 g, 60%). IR (thin film, cm−1): 1260 (P = O), 1219 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 24.63 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.19 (t, 3H, J = 7.0 Hz), 2.84–2.97 (m, 2H), 4.00–4.16 (m, 2H), 5.19–5.32 (m, 2H), 5.36 (dd, 1H, J = 11.2, 0.9 Hz), 5.71–5.83 (m, 1H), 5.86 (dd, 1H, J = 17.7, 0.9 Hz), 6.96 (dd, 1H, J = 11.7, 11.2 Hz), 7.16–7.22 (m, 1H), 7.26–7.33 (m, 2H), 7.67 (d, 1H, J = 7.7 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 5.8 Hz), 31.0 (d, JP,C = 138 Hz), 62.6 (d, JP,C = 6.8 Hz), 116.3, 120.4 (d, JP,C = 14.6 Hz), 120.9 (d, JP,C = 2.8 Hz), 125.0, 126.3, 127.5 (d, JP,C = 11.4 Hz), 128.8 (d, JP,C = 5.0 Hz), 129.1, 130.2, 147.4 (d, JP,C = 8.9 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H18O3P: 253.0994, found 253.1003.
By following the general procedure, 4a was prepared from 2-vinylphenol (2a) (0.38 g, 3.16 mmol), ethyl allylphosphonochloridate (3) (0.56 ml, 3.79 mmol), and NEt3 (0.55 ml, 3.95 mmol) as a colourless oil (0.48 g, 60%). IR (thin film, cm−1): 1260 (P = O), 1219 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 24.63 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.19 (t, 3H, J = 7.0 Hz), 2.84–2.97 (m, 2H), 4.00–4.16 (m, 2H), 5.19–5.32 (m, 2H), 5.36 (dd, 1H, J = 11.2, 0.9 Hz), 5.71–5.83 (m, 1H), 5.86 (dd, 1H, J = 17.7, 0.9 Hz), 6.96 (dd, 1H, J = 11.7, 11.2 Hz), 7.16–7.22 (m, 1H), 7.26–7.33 (m, 2H), 7.67 (d, 1H, J = 7.7 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 5.8 Hz), 31.0 (d, JP,C = 138 Hz), 62.6 (d, JP,C = 6.8 Hz), 116.3, 120.4 (d, JP,C = 14.6 Hz), 120.9 (d, JP,C = 2.8 Hz), 125.0, 126.3, 127.5 (d, JP,C = 11.4 Hz), 128.8 (d, JP,C = 5.0 Hz), 129.1, 130.2, 147.4 (d, JP,C = 8.9 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H18O3P: 253.0994, found 253.1003.
Ethyl (4-iodo-2-vinylphenyl) allylphosphonate (4b)
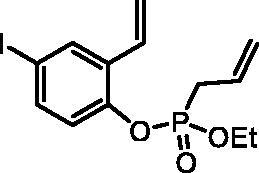 By following the general procedure, 4b was prepared from 4-iodo-2-vinylphenol (2b) (2.50 g, 10.2 mmol), ethyl allylphosphonochloridate (3) (1.81 ml, 12.2 mmol), and NEt3 (1.77 ml, 12.7 mmol) as a colourless oil (3.53 g, 92%). IR (thin film, cm−1): 1265 (P = O), 1220 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 25.01 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.19 (t, 3H, J = 7.1 Hz), 2.90 (dt, 1H, J = 7.3, 1.2 Hz), 2.96 (dt, 1H, J = 7.3, 1.2 Hz), 4.00–4.16 (m, 2H), 5.19–5.32 (m, 2H), 5.40 (dd, 1H, J = 11.2, 0.7 Hz), 5.69–5.83 (m, 1H), 5.93 (dd, 1H, J = 17.7, 0.7 Hz), 6.84 (dd, 1H, J = 17.7, 11.2 Hz), 7.11 (dd, 1H, J = 8.6, 1.3 Hz), 7.63 (dd, 1H, J = 8.6, 2.2 Hz), 7.98 (d, 1H, J = 2.2 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 5.6 Hz), 30.9 (d, JP,C = 137 Hz), 62.7 (d, JP,C = 6.7 Hz), 89.6 (d, JP,C = 1.4 Hz), 117.9, 120.5 (d, JP,C = 15.0 Hz), 123.2 (d, JP,C = 2.7 Hz), 127.3 (d, JP,C = 11.6 Hz), 128.8, 131.3 (d, JP,C = 5.3 Hz), 132.2 (d, JP,C = 2.6 Hz), 134.7, 137.5, 147.3 (d, JP,C = 9.0 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H17O3PI: 378.9960, found 378.9966.
By following the general procedure, 4b was prepared from 4-iodo-2-vinylphenol (2b) (2.50 g, 10.2 mmol), ethyl allylphosphonochloridate (3) (1.81 ml, 12.2 mmol), and NEt3 (1.77 ml, 12.7 mmol) as a colourless oil (3.53 g, 92%). IR (thin film, cm−1): 1265 (P = O), 1220 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 25.01 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.19 (t, 3H, J = 7.1 Hz), 2.90 (dt, 1H, J = 7.3, 1.2 Hz), 2.96 (dt, 1H, J = 7.3, 1.2 Hz), 4.00–4.16 (m, 2H), 5.19–5.32 (m, 2H), 5.40 (dd, 1H, J = 11.2, 0.7 Hz), 5.69–5.83 (m, 1H), 5.93 (dd, 1H, J = 17.7, 0.7 Hz), 6.84 (dd, 1H, J = 17.7, 11.2 Hz), 7.11 (dd, 1H, J = 8.6, 1.3 Hz), 7.63 (dd, 1H, J = 8.6, 2.2 Hz), 7.98 (d, 1H, J = 2.2 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 5.6 Hz), 30.9 (d, JP,C = 137 Hz), 62.7 (d, JP,C = 6.7 Hz), 89.6 (d, JP,C = 1.4 Hz), 117.9, 120.5 (d, JP,C = 15.0 Hz), 123.2 (d, JP,C = 2.7 Hz), 127.3 (d, JP,C = 11.6 Hz), 128.8, 131.3 (d, JP,C = 5.3 Hz), 132.2 (d, JP,C = 2.6 Hz), 134.7, 137.5, 147.3 (d, JP,C = 9.0 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H17O3PI: 378.9960, found 378.9966.
Ethyl (4-bromo-2-vinylphenyl) allylphosphonate (4c)
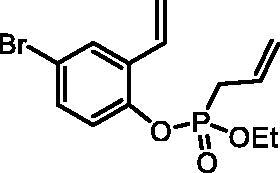 By following the general procedure, 4c was prepared from 4-bromo-2-vinylphenol (2c) (1.63 g, 8.19 mmol), ethyl allylphosphonochloridate (3) (1.46 ml, 9.83 mmol), and NEt3 (1.42 ml, 10.2 mmol) as a colourless oil (1.71 g, 63%). IR (thin film, cm−1): 1266 (P = O), 1224 (P = O), 1174 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 25.10 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.16–1.22 (m, 3H), 2.91 (dt, 1H, J = 7.3, 1.2 Hz), 2.97 (dt, 1H, J = 7.3, 1.2 Hz), 3.99–4.17 (m, 2H), 5.19–5.32 (m, 2H), 5.43 (dd, 1H, J = 11.2, 0.8 Hz), 5.70–5.83 (m, 1H), 5.97 (dd, 1H, J = 17.7, 0.8 Hz), 6.87 (dd, 1H, J = 17.7, 11.2 Hz), 7.26 (dd, 1H, J = 8.7, 1.3 Hz), 7.48 (dd, 1H, J = 8.7, 2.5 Hz), 7.86 (d, 1H, J = 2.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 6.0 Hz), 30.9 (d, JP,C = 137 Hz), 62.8 (d, JP,C = 6.7 Hz), 117.3 (d, JP,C = 1.5 Hz), 118.2, 120.5 (d, JP,C = 15.0 Hz), 123.0 (d, JP,C = 2.8 Hz), 127.3 (d, JP,C = 11.8 Hz), 128.8 (d, JP,C = 9.7 Hz), 131.1 (d, JP,C = 5.3 Hz), 131.6, 146.6 (d, JP,C = 8.9 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H17O3PBr: 331.0099, found 331.0103.
By following the general procedure, 4c was prepared from 4-bromo-2-vinylphenol (2c) (1.63 g, 8.19 mmol), ethyl allylphosphonochloridate (3) (1.46 ml, 9.83 mmol), and NEt3 (1.42 ml, 10.2 mmol) as a colourless oil (1.71 g, 63%). IR (thin film, cm−1): 1266 (P = O), 1224 (P = O), 1174 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 25.10 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.16–1.22 (m, 3H), 2.91 (dt, 1H, J = 7.3, 1.2 Hz), 2.97 (dt, 1H, J = 7.3, 1.2 Hz), 3.99–4.17 (m, 2H), 5.19–5.32 (m, 2H), 5.43 (dd, 1H, J = 11.2, 0.8 Hz), 5.70–5.83 (m, 1H), 5.97 (dd, 1H, J = 17.7, 0.8 Hz), 6.87 (dd, 1H, J = 17.7, 11.2 Hz), 7.26 (dd, 1H, J = 8.7, 1.3 Hz), 7.48 (dd, 1H, J = 8.7, 2.5 Hz), 7.86 (d, 1H, J = 2.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 6.0 Hz), 30.9 (d, JP,C = 137 Hz), 62.8 (d, JP,C = 6.7 Hz), 117.3 (d, JP,C = 1.5 Hz), 118.2, 120.5 (d, JP,C = 15.0 Hz), 123.0 (d, JP,C = 2.8 Hz), 127.3 (d, JP,C = 11.8 Hz), 128.8 (d, JP,C = 9.7 Hz), 131.1 (d, JP,C = 5.3 Hz), 131.6, 146.6 (d, JP,C = 8.9 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H17O3PBr: 331.0099, found 331.0103.
2-Bromo-6-vinylphenyl ethyl allylphosphonate (4d)
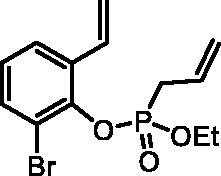 By following the general procedure, 4d was prepared from 2-bromo-6-vinylphenol (2d) (1.00 g, 5.02 mmol), ethyl allylphosphonochloridate (3) (0.89 ml, 6.03 mmol), and NEt3 (0.87 ml, 6.28 mmol) as a colourless oil (1.28 g, 77%). IR (thin film, cm−1): 1262 (P = O), 1219 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 25.07 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.16 (dt, 3H, J = 7.0, 0.4 Hz), 3.00–3.10 (m, 2H), 3.96–4.15 (m, 2H), 5.22–5.37 (m, 2H), 5.42 (dd, 1H, J = 11.1, 0.9 Hz), 5.77–5.92 (m, 2H), 7.06 (dd, 1H, J = 17.7, 11.1 Hz), 7.13–7.19 (m, 1H), 7.61 (dd, 1H, J = 7.9, 1.5 Hz), 7.69 (dd, 1H, J = 7.9, 1.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.0 (d, JP,C = 6.0 Hz), 32.1 (d, JP,C = 139 Hz), 63.1 (d, JP,C = 6.9 Hz), 116.5 (d, JP,C = 4.0 Hz), 117.6, 120.5 (d, JP,C = 15.2 Hz), 125.6 (d, JP,C = 1.5 Hz), 126.7, 127.4 (d, JP,C = 11.4 Hz), 130.7, 132.2 (d, JP,C = 2.8 Hz), 132.8 (d, JP,C = 1.5 Hz), 145.0 (d, JP,C = 10.7 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H17O3PBr: 331.0099, found 331.0092.
By following the general procedure, 4d was prepared from 2-bromo-6-vinylphenol (2d) (1.00 g, 5.02 mmol), ethyl allylphosphonochloridate (3) (0.89 ml, 6.03 mmol), and NEt3 (0.87 ml, 6.28 mmol) as a colourless oil (1.28 g, 77%). IR (thin film, cm−1): 1262 (P = O), 1219 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 25.07 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.16 (dt, 3H, J = 7.0, 0.4 Hz), 3.00–3.10 (m, 2H), 3.96–4.15 (m, 2H), 5.22–5.37 (m, 2H), 5.42 (dd, 1H, J = 11.1, 0.9 Hz), 5.77–5.92 (m, 2H), 7.06 (dd, 1H, J = 17.7, 11.1 Hz), 7.13–7.19 (m, 1H), 7.61 (dd, 1H, J = 7.9, 1.5 Hz), 7.69 (dd, 1H, J = 7.9, 1.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.0 (d, JP,C = 6.0 Hz), 32.1 (d, JP,C = 139 Hz), 63.1 (d, JP,C = 6.9 Hz), 116.5 (d, JP,C = 4.0 Hz), 117.6, 120.5 (d, JP,C = 15.2 Hz), 125.6 (d, JP,C = 1.5 Hz), 126.7, 127.4 (d, JP,C = 11.4 Hz), 130.7, 132.2 (d, JP,C = 2.8 Hz), 132.8 (d, JP,C = 1.5 Hz), 145.0 (d, JP,C = 10.7 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H17O3PBr: 331.0099, found 331.0092.
Ethyl (2-methoxy-6-vinylphenyl) allylphosphonate (4e)
 By following the general procedure, 4e was prepared from 2-methoxy-6-vinylphenol (2e) (0.32 g, 2.13 mmol), ethyl allylphosphonochloridate (3) (0.38 ml, 2.56 mmol), and NEt3 (0.37 ml, 2.66 mmol) as a colourless oil (0.40 g, 66%). IR (thin film, cm−1): 1274 (P = O), 1179 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 24.86 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.19–1.24 (m, 3H), 2.86–2.96 (m, 2H), 3.82 (s, 3H), 3.96–4.18 (m, 2H), 5.18–5.31 (m, 2H), 5.37 (dd, 1H, J = 11.1, 1.1 Hz), 5.74–5.88 (m, 2H), 6.97 (dd, 1H, J = 17.7, 11.1 Hz), 7.04 (dd, 1H, J = 8.1, 1.5 Hz), 7.11–7.17 (m, 1H), 7.22 (dd, 1H, J = 7.9, 1.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 6.1 Hz), 31.8 (d, JP,C = 139 Hz), 55.9, 62.1 (d, JP,C = 7.0 Hz), 112.2, 116.6, 117.3, 119.9 (d, JP,C = 15.0 Hz), 125.2, 128.1 (d, JP,C = 11.5 Hz), 130.4, 130.5 (d, JP,C = 3.5 Hz), 137.1 (d, JP,C = 9.6 Hz), 151.2 (d, JP,C = 3.0 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C14H20O4P: 283.1099, found 283.1105.
By following the general procedure, 4e was prepared from 2-methoxy-6-vinylphenol (2e) (0.32 g, 2.13 mmol), ethyl allylphosphonochloridate (3) (0.38 ml, 2.56 mmol), and NEt3 (0.37 ml, 2.66 mmol) as a colourless oil (0.40 g, 66%). IR (thin film, cm−1): 1274 (P = O), 1179 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 24.86 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.19–1.24 (m, 3H), 2.86–2.96 (m, 2H), 3.82 (s, 3H), 3.96–4.18 (m, 2H), 5.18–5.31 (m, 2H), 5.37 (dd, 1H, J = 11.1, 1.1 Hz), 5.74–5.88 (m, 2H), 6.97 (dd, 1H, J = 17.7, 11.1 Hz), 7.04 (dd, 1H, J = 8.1, 1.5 Hz), 7.11–7.17 (m, 1H), 7.22 (dd, 1H, J = 7.9, 1.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 6.1 Hz), 31.8 (d, JP,C = 139 Hz), 55.9, 62.1 (d, JP,C = 7.0 Hz), 112.2, 116.6, 117.3, 119.9 (d, JP,C = 15.0 Hz), 125.2, 128.1 (d, JP,C = 11.5 Hz), 130.4, 130.5 (d, JP,C = 3.5 Hz), 137.1 (d, JP,C = 9.6 Hz), 151.2 (d, JP,C = 3.0 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C14H20O4P: 283.1099, found 283.1105.
2,4-Dichloro-6-vinylphenyl ethyl allylphosphonate (4f)
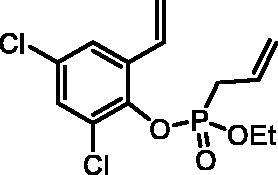 By following the general procedure, 4f was prepared from 2,4-dichloro-6-vinylphenol (2f) (0.80 g, 4.23 mmol), ethyl allylphosphonochloridate (3) (0.75 ml, 5.08 mmol), and NEt3 (0.74 ml, 5.29 mmol) as a colourless oil (1.17 g, 86%). IR (thin film, cm−1): 1262 (P = O), 1217 (P = O). 31P NMR (162 MHz, C6D6) δ = 24.33 ppm. 1H NMR (400 MHz, C6D6) δ = 0.88 (dt, 3H, J = 7.1, 0.4 Hz), 2.63–2.73 (m, 2H), 3.76–3.98 (m, 2H), 5.00–5.11 (m, 3H), 5.27 (d, 1H, J = 17.6 Hz), 5.76–5.89 (m, 1H), 7.03 (d, 1H, J = 2.5 Hz), 7.19–7.21 (m, 1H), 7.25 (dd, 1H, J = 17.6, 11.0 Hz) ppm. 13C NMR (101 MHz, C6D6) δ = 16.3 (d, JP,C = 5.7 Hz), 33.0 (d, JP,C = 141 Hz), 63.4 (d, JP,C = 7.0 Hz), 118.0, 120.4 (d, JP,C = 15.0 Hz), 125.0 (d, JP,C = 1.9 Hz), 127.5 (d, JP,C = 11.5 Hz), 128.9 (d, JP,C = 3.7 Hz), 129.4, 130.8, 130.9 (d, JP,C = 2.1 Hz), 134.3 (d, JP,C = 3.0 Hz), 143.8 (d, JP,C = 10.7 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H16O3PCl2: 321.0214, found 321.0233.
By following the general procedure, 4f was prepared from 2,4-dichloro-6-vinylphenol (2f) (0.80 g, 4.23 mmol), ethyl allylphosphonochloridate (3) (0.75 ml, 5.08 mmol), and NEt3 (0.74 ml, 5.29 mmol) as a colourless oil (1.17 g, 86%). IR (thin film, cm−1): 1262 (P = O), 1217 (P = O). 31P NMR (162 MHz, C6D6) δ = 24.33 ppm. 1H NMR (400 MHz, C6D6) δ = 0.88 (dt, 3H, J = 7.1, 0.4 Hz), 2.63–2.73 (m, 2H), 3.76–3.98 (m, 2H), 5.00–5.11 (m, 3H), 5.27 (d, 1H, J = 17.6 Hz), 5.76–5.89 (m, 1H), 7.03 (d, 1H, J = 2.5 Hz), 7.19–7.21 (m, 1H), 7.25 (dd, 1H, J = 17.6, 11.0 Hz) ppm. 13C NMR (101 MHz, C6D6) δ = 16.3 (d, JP,C = 5.7 Hz), 33.0 (d, JP,C = 141 Hz), 63.4 (d, JP,C = 7.0 Hz), 118.0, 120.4 (d, JP,C = 15.0 Hz), 125.0 (d, JP,C = 1.9 Hz), 127.5 (d, JP,C = 11.5 Hz), 128.9 (d, JP,C = 3.7 Hz), 129.4, 130.8, 130.9 (d, JP,C = 2.1 Hz), 134.3 (d, JP,C = 3.0 Hz), 143.8 (d, JP,C = 10.7 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H16O3PCl2: 321.0214, found 321.0233.
Ethyl (4-nitro-2-vinylphenyl) allylphosphonate (4 g)
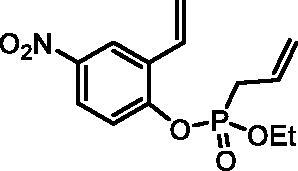 By following the general procedure, 4 g was prepared from 4-nitro-2-vinylphenol (2 g) (1.00 g, 6.06 mmol), ethyl allylphosphonochloridate (3) (1.08 ml, 7.27 mmol), and NEt3 (1.05 ml, 7.57 mmol) as a yellowish oil (1.70 g, 94%). IR (thin film, cm−1): 1273 (P = O), 1232 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 25.61 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.22 (dt, 3H, J = 7.1, 0.3 Hz), 3.00 (dt, 1H, J = 7.3, 1.2 Hz), 3.06 (dt, 1H, J = 7.3, 1.2 Hz), 4.06–4.22 (m, 2H), 5.21–5.35 (m, 2H), 5.54–5.58 (m, 1H), 5.71–5.85 (m, 1H), 6.11 (dd, 1H, J = 17.7, 0.6 Hz), 6.96 (dd, 1H, J = 17.7, 11.2 Hz), 7.59 (dd, 1H, J = 9.1, 1.2 Hz), 8.19 (dd, 1H, J = 9.1, 2.9 Hz), 8.45–8.48 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 5.8 Hz), 30.9 (d, JP,C = 137 Hz), 63.1 (d, JP,C = 6.9 Hz), 119.6, 120.8 (d, JP,C = 15.0 Hz), 121.7 (d, JP,C = 3.0 Hz), 121.8, 124.2, 127.0 (d, JP,C = 11.7 Hz), 128.6, 129.9 (d, JP,C = 5.5 Hz), 144.3, 152.0 (d, JP,C = 8.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H17NO5P: 298.0844, found 298.0858.
By following the general procedure, 4 g was prepared from 4-nitro-2-vinylphenol (2 g) (1.00 g, 6.06 mmol), ethyl allylphosphonochloridate (3) (1.08 ml, 7.27 mmol), and NEt3 (1.05 ml, 7.57 mmol) as a yellowish oil (1.70 g, 94%). IR (thin film, cm−1): 1273 (P = O), 1232 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 25.61 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.22 (dt, 3H, J = 7.1, 0.3 Hz), 3.00 (dt, 1H, J = 7.3, 1.2 Hz), 3.06 (dt, 1H, J = 7.3, 1.2 Hz), 4.06–4.22 (m, 2H), 5.21–5.35 (m, 2H), 5.54–5.58 (m, 1H), 5.71–5.85 (m, 1H), 6.11 (dd, 1H, J = 17.7, 0.6 Hz), 6.96 (dd, 1H, J = 17.7, 11.2 Hz), 7.59 (dd, 1H, J = 9.1, 1.2 Hz), 8.19 (dd, 1H, J = 9.1, 2.9 Hz), 8.45–8.48 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 5.8 Hz), 30.9 (d, JP,C = 137 Hz), 63.1 (d, JP,C = 6.9 Hz), 119.6, 120.8 (d, JP,C = 15.0 Hz), 121.7 (d, JP,C = 3.0 Hz), 121.8, 124.2, 127.0 (d, JP,C = 11.7 Hz), 128.6, 129.9 (d, JP,C = 5.5 Hz), 144.3, 152.0 (d, JP,C = 8.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H17NO5P: 298.0844, found 298.0858.
General procedure for the synthesis of 2-ethoxy-3H-1,2-benzoxaphosphepine 2-oxides 6
The corresponding diolefin 4 (1.0 eq) was dissolved in dry, degassed PhMe (18 ml/1 mmol of 4). The solution was sparged with argon followed by addition of ruthenium catalyst 5 (CAS: 254972–49-1) (5 mol%). The reaction mixture was stirred at 70 °C for 4 h, then cooled down to rt, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (EtOAc 100%).
2-Ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6a)
 By following the general procedure, 6a was prepared from ethyl (2-vinylphenyl) allylphosphonate (4a) (0.43 g, 1.70 mmol), and ruthenium catalyst 5 (81 mg, 0.085 mmol) as a greenish dense oil (0.28 g, 74%). IR (thin film, cm−1): 1265 (P = O), 1203 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 40.00 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.28 (t, 3H, J = 7.1 Hz), 2.62–2.88 (m, 2H), 4.14–4.23 (m, 2H), 5.92–6.04 (m, 1H), 6.71 (dd, 1H, J = 10.8, 5.3 Hz), 7.17–7.27 (m, 2H), 7.31–7.41 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.3 (d, JP,C = 5.9 Hz), 25.5 (d, JP,C = 125 Hz), 62.2 (d, JP,C = 6.9 Hz), 121.6 (d, JP,C = 3.4 Hz), 122.7 (d, JP,C = 12.2 Hz), 125.0, 127.6 (d, JP,C = 1.1 Hz), 129.4, 129.5, 129.6 130.6, 147.5 (d, JP,C = 8.3 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H14O3P: 225.0681, found 225.0692.
By following the general procedure, 6a was prepared from ethyl (2-vinylphenyl) allylphosphonate (4a) (0.43 g, 1.70 mmol), and ruthenium catalyst 5 (81 mg, 0.085 mmol) as a greenish dense oil (0.28 g, 74%). IR (thin film, cm−1): 1265 (P = O), 1203 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 40.00 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.28 (t, 3H, J = 7.1 Hz), 2.62–2.88 (m, 2H), 4.14–4.23 (m, 2H), 5.92–6.04 (m, 1H), 6.71 (dd, 1H, J = 10.8, 5.3 Hz), 7.17–7.27 (m, 2H), 7.31–7.41 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.3 (d, JP,C = 5.9 Hz), 25.5 (d, JP,C = 125 Hz), 62.2 (d, JP,C = 6.9 Hz), 121.6 (d, JP,C = 3.4 Hz), 122.7 (d, JP,C = 12.2 Hz), 125.0, 127.6 (d, JP,C = 1.1 Hz), 129.4, 129.5, 129.6 130.6, 147.5 (d, JP,C = 8.3 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H14O3P: 225.0681, found 225.0692.
2-Ethoxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6b)
 By following the general procedure, 6b was prepared from ethyl (4-iodo-2-vinylphenyl) allylphosphonate (4b) (3.50 g, 9.26 mmol), and ruthenium catalyst 5 (438 mg, 0.46 mmol) as a greenish dense oil (2.46 g, 76%). IR (thin film, cm−1): 1265 (P = O), 1173 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 39.83 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.27 (t, 3H, J = 7.1 Hz), 2.66–2.92 (m, 2H), 4.13–4.22 (m, 2H), 5.95–6.07 (m, 1H), 6.63–6.70 (m, 1H), 6.99–7.04 (m, 1H), 7.68 (dd, 1H, J = 8.5, 2.2 Hz), 7.71 (d, 1H, J = 2.2 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 5.6 Hz), 25.5 (d, JP,C = 125 Hz), 62.4 (d, JP,C = 6.7 Hz), 89.3 (d, JP,C = 1.4 Hz), 123.8, 123.9, 124.0, 128.3 (d, JP,C = 8.7 Hz), 130.2, 137.9, 138.8, 147.4 (d, JP,C = 8.0 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H13O3PI: 350.9647, found 350.9659.
By following the general procedure, 6b was prepared from ethyl (4-iodo-2-vinylphenyl) allylphosphonate (4b) (3.50 g, 9.26 mmol), and ruthenium catalyst 5 (438 mg, 0.46 mmol) as a greenish dense oil (2.46 g, 76%). IR (thin film, cm−1): 1265 (P = O), 1173 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 39.83 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.27 (t, 3H, J = 7.1 Hz), 2.66–2.92 (m, 2H), 4.13–4.22 (m, 2H), 5.95–6.07 (m, 1H), 6.63–6.70 (m, 1H), 6.99–7.04 (m, 1H), 7.68 (dd, 1H, J = 8.5, 2.2 Hz), 7.71 (d, 1H, J = 2.2 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 5.6 Hz), 25.5 (d, JP,C = 125 Hz), 62.4 (d, JP,C = 6.7 Hz), 89.3 (d, JP,C = 1.4 Hz), 123.8, 123.9, 124.0, 128.3 (d, JP,C = 8.7 Hz), 130.2, 137.9, 138.8, 147.4 (d, JP,C = 8.0 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H13O3PI: 350.9647, found 350.9659.
7-Bromo-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6c)
 By following the general procedure, 6c was prepared from ethyl (4-bromo-2-vinylphenyl) allylphosphonate (4c) (0.92 g, 2.78 mmol), and ruthenium catalyst 5 (132 mg, 0.14 mmol) as a greenish dense oil (0.53 g, 63%). IR (thin film, cm−1): 1274 (P = O), 1220 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 44.68 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.27 (t, 3H, J = 7.1 Hz), 2.68–2.93 (m, 2H), 4.13–4.23 (m, 2H), 5.97–6.10 (m, 1H), 6.65–6.71 (m, 1H), 7.15–7.19 (m, 1H), 7.54 (dd, 1H, J = 8.6, 2.5 Hz), 7.58 (d, 1H, J = 2.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 5.4 Hz), 25.4 (d, JP,C = 125 Hz), 62.5 (d, JP,C = 7.0 Hz), 116.9 (d, JP,C = 1.5 Hz), 123.8 (d, JP,C = 3.5 Hz), 124.1 (d, JP,C = 12.3 Hz), 128.4 (d, JP,C = 9.0 Hz), 130.0, 132.1, 132.9, 146.7 (d, JP,C = 7.8 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H13O3PBr: 302.9786, found 302.9791.
By following the general procedure, 6c was prepared from ethyl (4-bromo-2-vinylphenyl) allylphosphonate (4c) (0.92 g, 2.78 mmol), and ruthenium catalyst 5 (132 mg, 0.14 mmol) as a greenish dense oil (0.53 g, 63%). IR (thin film, cm−1): 1274 (P = O), 1220 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 44.68 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.27 (t, 3H, J = 7.1 Hz), 2.68–2.93 (m, 2H), 4.13–4.23 (m, 2H), 5.97–6.10 (m, 1H), 6.65–6.71 (m, 1H), 7.15–7.19 (m, 1H), 7.54 (dd, 1H, J = 8.6, 2.5 Hz), 7.58 (d, 1H, J = 2.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 5.4 Hz), 25.4 (d, JP,C = 125 Hz), 62.5 (d, JP,C = 7.0 Hz), 116.9 (d, JP,C = 1.5 Hz), 123.8 (d, JP,C = 3.5 Hz), 124.1 (d, JP,C = 12.3 Hz), 128.4 (d, JP,C = 9.0 Hz), 130.0, 132.1, 132.9, 146.7 (d, JP,C = 7.8 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H13O3PBr: 302.9786, found 302.9791.
9-Bromo-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6d)
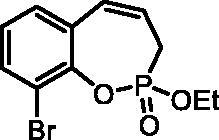 By following the general procedure, 6d was prepared from 2-bromo-6-vinylphenyl ethyl allylphosphonate (4d) (2.00 g, 6.04 mmol), and ruthenium catalyst 5 (286 mg, 0.30 mmol) as a greenish dense oil (1.58 g, 86%). IR (thin film, cm−1): 1268 (P = O), 1232 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 39.08 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.29 (t, 3H, J = 7.1 Hz), 2.70–2.83 (m, 1H), 2.87–3.00 (m, 1H), 4.18–4.32 (m, 2H), 5.95–6.08 (m, 1H), 6.67–6.74 (m, 1H), 7.17 (td, 1H, J = 7.8, 0.6 Hz), 7.34 (dd, 1H, J = 7.8, 1.6 Hz), 7.68 (dd, 1H, J = 7.8, 1.6 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 6.1 Hz), 25.8 (d, JP,C = 126 Hz), 62.8 (d, JP,C = 7.0 Hz), 115.4 (d, JP,C = 3.8 Hz), 123.4 (d, JP,C = 12.2 Hz), 126.1, 129.1 (d, JP,C = 9.0 Hz), 129.3, 130.4, 132.7, 144.2 (d, JP,C = 7.8 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H13O3PBr: 302.9786, found 302.9795.
By following the general procedure, 6d was prepared from 2-bromo-6-vinylphenyl ethyl allylphosphonate (4d) (2.00 g, 6.04 mmol), and ruthenium catalyst 5 (286 mg, 0.30 mmol) as a greenish dense oil (1.58 g, 86%). IR (thin film, cm−1): 1268 (P = O), 1232 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 39.08 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.29 (t, 3H, J = 7.1 Hz), 2.70–2.83 (m, 1H), 2.87–3.00 (m, 1H), 4.18–4.32 (m, 2H), 5.95–6.08 (m, 1H), 6.67–6.74 (m, 1H), 7.17 (td, 1H, J = 7.8, 0.6 Hz), 7.34 (dd, 1H, J = 7.8, 1.6 Hz), 7.68 (dd, 1H, J = 7.8, 1.6 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 6.1 Hz), 25.8 (d, JP,C = 126 Hz), 62.8 (d, JP,C = 7.0 Hz), 115.4 (d, JP,C = 3.8 Hz), 123.4 (d, JP,C = 12.2 Hz), 126.1, 129.1 (d, JP,C = 9.0 Hz), 129.3, 130.4, 132.7, 144.2 (d, JP,C = 7.8 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H13O3PBr: 302.9786, found 302.9795.
2-Ethoxy-9-methoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6e)
 By following the general procedure, 6e was prepared from ethyl (2-methoxy-6-vinylphenyl) allylphosphonate (4e) (315 mg, 1.12 mmol), and ruthenium catalyst 5 (53 mg, 0.056 mmol) as a greenish dense oil (0.23 g, 81%). IR (thin film, cm−1): 1270 (P = O), 1244 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 41.74 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.26 (t, 3H, J = 7.1 Hz), 2.50–2.63 (m, 1H), 2.79–2.91 (m, 1H), 3.84 (s, 3H), 4.13–4.22 (m, 2H), 5.92–6.04 (m, 1H), 6.65–6.72 (m, 1H), 6.86 (dd, 1H, J = 7.8, 1.4 Hz), 7.08 (dd, 1H, J = 8.2, 1.4 Hz), 7.14–7.20 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 6.1 Hz), 25.4 (d, JP,C = 127 Hz), 55.9, 62.0 (d, JP,C = 6.9 Hz), 112.0, 121.3, 122.9 (d, JP,C = 12.2 Hz), 125.0, 128.7, 129.6 (d, JP,C = 8.8 Hz), 136.6 (d, JP,C = 8.4 Hz), 151.2 (d, JP,C = 3.1 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C12H16O4P: 255.0786, found 255.0800.
By following the general procedure, 6e was prepared from ethyl (2-methoxy-6-vinylphenyl) allylphosphonate (4e) (315 mg, 1.12 mmol), and ruthenium catalyst 5 (53 mg, 0.056 mmol) as a greenish dense oil (0.23 g, 81%). IR (thin film, cm−1): 1270 (P = O), 1244 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 41.74 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.26 (t, 3H, J = 7.1 Hz), 2.50–2.63 (m, 1H), 2.79–2.91 (m, 1H), 3.84 (s, 3H), 4.13–4.22 (m, 2H), 5.92–6.04 (m, 1H), 6.65–6.72 (m, 1H), 6.86 (dd, 1H, J = 7.8, 1.4 Hz), 7.08 (dd, 1H, J = 8.2, 1.4 Hz), 7.14–7.20 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 6.1 Hz), 25.4 (d, JP,C = 127 Hz), 55.9, 62.0 (d, JP,C = 6.9 Hz), 112.0, 121.3, 122.9 (d, JP,C = 12.2 Hz), 125.0, 128.7, 129.6 (d, JP,C = 8.8 Hz), 136.6 (d, JP,C = 8.4 Hz), 151.2 (d, JP,C = 3.1 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C12H16O4P: 255.0786, found 255.0800.
7,9-Dichloro-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6f)
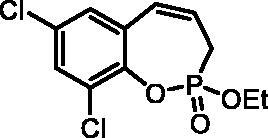 By following the general procedure, 6f was prepared from 2,4-dichloro-6-vinylphenyl ethyl allylphosphonate (4f) (0.70 g, 2.18 mmol), and ruthenium catalyst 5 (103 mg, 0.109 mmol) as a greenish dense oil (0.46 g, 72%). IR (thin film, cm−1): 1276 (P = O), 1242 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 40.02 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.29 (t, 3H, J = 7.1 Hz), 2.76–3.05 (m, 2H), 4.15–4.31 (m, 2H), 6.02–6.15 (m, 1H), 6.66–6.72 (m, 1H), 7.46 (d, 1H, J = 2.6 Hz), 7.73 (d, 1H, J = 2.6 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 6.0 Hz), 25.7 (d, JP,C = 126 Hz), 62.9 (d, JP,C = 6.9 Hz), 125.0 (d, JP,C = 12.2 Hz), 126.8 (d, JP,C = 3.6 Hz), 128.0 (d, JP,C = 9.2 Hz), 128.8, 128.9, 129.1, 130.8, 142.3 (d, JP,C = 7.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H12O3PCl2: 292.9901, found 292.9908.
By following the general procedure, 6f was prepared from 2,4-dichloro-6-vinylphenyl ethyl allylphosphonate (4f) (0.70 g, 2.18 mmol), and ruthenium catalyst 5 (103 mg, 0.109 mmol) as a greenish dense oil (0.46 g, 72%). IR (thin film, cm−1): 1276 (P = O), 1242 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 40.02 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.29 (t, 3H, J = 7.1 Hz), 2.76–3.05 (m, 2H), 4.15–4.31 (m, 2H), 6.02–6.15 (m, 1H), 6.66–6.72 (m, 1H), 7.46 (d, 1H, J = 2.6 Hz), 7.73 (d, 1H, J = 2.6 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 6.0 Hz), 25.7 (d, JP,C = 126 Hz), 62.9 (d, JP,C = 6.9 Hz), 125.0 (d, JP,C = 12.2 Hz), 126.8 (d, JP,C = 3.6 Hz), 128.0 (d, JP,C = 9.2 Hz), 128.8, 128.9, 129.1, 130.8, 142.3 (d, JP,C = 7.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H12O3PCl2: 292.9901, found 292.9908.
2-Ethoxy-7-nitro-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6 g)
 By following the general procedure, 6 g was prepared from ethyl (4-nitro-2-vinylphenyl) allylphosphonate (4 g) (1.85 g, 6.22 mmol), and ruthenium catalyst 5 (295 mg, 0.31 mmol) as a brown dense oil (1.12 g, 67%). IR (thin film, cm−1): 1278 (P = O), 1233 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 38.90 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.29 (t, 3H, J = 7.1 Hz), 2.78–3.03 (m, 2H), 4.18–4.28 (m, 2H), 6.06–6.19 (m, 1H), 6.81–6.88 (m, 1H), 7.44–7.48 (m, 1H), 8.22 (dd, 1H, J = 8.9, 2.8 Hz), 8.30 (d, 1H, J = 2.8 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 5.8 Hz), 25.7 (d, JP,C = 124 Hz), 62.8 (d, JP,C = 6.8 Hz), 123.2 (d, JP,C = 3.8 Hz), 124.6, 125.0 (d, JP,C = 12.4 Hz), 126.4, 128.2 (d, JP,C = 9.2 Hz), 128.8, 144.1, 151.9 (d, JP,C = 8.0 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H13NO5P: 270.0531, found 270.0539.
By following the general procedure, 6 g was prepared from ethyl (4-nitro-2-vinylphenyl) allylphosphonate (4 g) (1.85 g, 6.22 mmol), and ruthenium catalyst 5 (295 mg, 0.31 mmol) as a brown dense oil (1.12 g, 67%). IR (thin film, cm−1): 1278 (P = O), 1233 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 38.90 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.29 (t, 3H, J = 7.1 Hz), 2.78–3.03 (m, 2H), 4.18–4.28 (m, 2H), 6.06–6.19 (m, 1H), 6.81–6.88 (m, 1H), 7.44–7.48 (m, 1H), 8.22 (dd, 1H, J = 8.9, 2.8 Hz), 8.30 (d, 1H, J = 2.8 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 5.8 Hz), 25.7 (d, JP,C = 124 Hz), 62.8 (d, JP,C = 6.8 Hz), 123.2 (d, JP,C = 3.8 Hz), 124.6, 125.0 (d, JP,C = 12.4 Hz), 126.4, 128.2 (d, JP,C = 9.2 Hz), 128.8, 144.1, 151.9 (d, JP,C = 8.0 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H13NO5P: 270.0531, found 270.0539.
General procedure for the synthesis of 2-hydroxy-3H-1,2-benzoxaphosphepine 2-oxides 7
The corresponding ethoxy derivative 6 (1.0 eq) was dissolved in dry DCM (20 ml/1 mmol of 6), then TMSBr (6.0 eq) was added dropwise. The reaction mixture was stirred under inert atmosphere at rt for 24 h. The volatiles were removed in vacuo, and the residue was treated with MeOH (15 ml/1 mmol of 6), concentrated, purified by column chromatography on silica gel (EtOAc 100%). Products were recrystallised from EtOAc.
2-Hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide(7a)
 By following the general procedure, 7a was prepared from 2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6a) (0.32 g, 1.43 mmol) and TMSBr (1.12 ml, 8.56 mmol) as a white solid (0.25 g, 88%). Mp: 128–129 °C. IR (KBr, cm−1): 2487 (O = P-OH), 2203 (O = P-OH), 1665 (O = P-OH), 1258 (P = O), 1223 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.64 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.57 (dd, 1H, J = 6.7, 1.0 Hz), 2.62 (dd, 1H, J = 6.7, 1.0 Hz), 5.88–6.01 (m, 1H), 6.64 (dd, 1H, J = 10.8, 5.0 Hz), 7.09–7.14 (m, 1H), 7.17–7.23 (m, 1H), 7.27–7.37 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.1 (d, JP,C = 125 Hz), 121.8 (d, JP,C = 3.4 Hz), 123.6 (d, JP,C = 12.2 Hz), 124.5, 127.9, 129.1 (d, JP,C = 8.4 Hz), 129.2, 130.6, 147.9 (d, JP,C = 7.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H10O3P: 197.0368, found 197.0371.
By following the general procedure, 7a was prepared from 2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6a) (0.32 g, 1.43 mmol) and TMSBr (1.12 ml, 8.56 mmol) as a white solid (0.25 g, 88%). Mp: 128–129 °C. IR (KBr, cm−1): 2487 (O = P-OH), 2203 (O = P-OH), 1665 (O = P-OH), 1258 (P = O), 1223 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.64 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.57 (dd, 1H, J = 6.7, 1.0 Hz), 2.62 (dd, 1H, J = 6.7, 1.0 Hz), 5.88–6.01 (m, 1H), 6.64 (dd, 1H, J = 10.8, 5.0 Hz), 7.09–7.14 (m, 1H), 7.17–7.23 (m, 1H), 7.27–7.37 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.1 (d, JP,C = 125 Hz), 121.8 (d, JP,C = 3.4 Hz), 123.6 (d, JP,C = 12.2 Hz), 124.5, 127.9, 129.1 (d, JP,C = 8.4 Hz), 129.2, 130.6, 147.9 (d, JP,C = 7.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H10O3P: 197.0368, found 197.0371.
2-Hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7b)
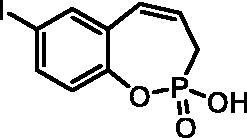 By following the general procedure, 7b was prepared from 2-ethoxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6b) (2.22 g, 6.34 mmol) and TMSBr (4.98 ml, 38.0 mmol) as a white solid (1.66 g, 81%). Mp: 193–194 °C.IR (KBr, cm−1): 2490 (O = P-OH), 2198 (O = P-OH), 1652 (O = P-OH), 1259 (P = O), 1217 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.14 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.59 (dd, 1H, J = 6.7, 1.0 Hz), 2.65 (dd, 1H, J = 6.7, 1.0 Hz), 5.91–6.03 (m, 1H), 6.56–6.62 (m, 1H), 6.92 (dd, 1H, J = 8.4, 1.1 Hz), 7.64 (dd, 1H, J = 8.4, 2.2 Hz), 7.68 (d, 1H, J = 2.2 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.1 (d, JP,C = 125 Hz), 88.6 (d, JP,C = 1.5 Hz), 124.2 (d, JP,C = 3.2 Hz), 124.9 (d, JP,C = 12.2 Hz), 127.8 (d, JP,C = 8.4 Hz), 130.6, 137.6, 138.7, 147.9 (d, JP,C = 7.4 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H9O3PI: 322.9334, found 322.9345.
By following the general procedure, 7b was prepared from 2-ethoxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6b) (2.22 g, 6.34 mmol) and TMSBr (4.98 ml, 38.0 mmol) as a white solid (1.66 g, 81%). Mp: 193–194 °C.IR (KBr, cm−1): 2490 (O = P-OH), 2198 (O = P-OH), 1652 (O = P-OH), 1259 (P = O), 1217 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.14 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.59 (dd, 1H, J = 6.7, 1.0 Hz), 2.65 (dd, 1H, J = 6.7, 1.0 Hz), 5.91–6.03 (m, 1H), 6.56–6.62 (m, 1H), 6.92 (dd, 1H, J = 8.4, 1.1 Hz), 7.64 (dd, 1H, J = 8.4, 2.2 Hz), 7.68 (d, 1H, J = 2.2 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.1 (d, JP,C = 125 Hz), 88.6 (d, JP,C = 1.5 Hz), 124.2 (d, JP,C = 3.2 Hz), 124.9 (d, JP,C = 12.2 Hz), 127.8 (d, JP,C = 8.4 Hz), 130.6, 137.6, 138.7, 147.9 (d, JP,C = 7.4 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H9O3PI: 322.9334, found 322.9345.
7-Bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7c)
 By following the general procedure, 7c was prepared from 7-bromo-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6c) (0.31 g, 1.02 mmol) and TMSBr (0.80 ml, 6.14 mmol) as a white solid (0.23 g, 82%). Mp: 163–164 °C. IR (KBr, cm−1): 1652 (O = P-OH), 1224 (P = O), 1206 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.27 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.61 (dd, 1H, J = 6.7, 0.9 Hz), 2.66 (dd, 1H, J = 6.7, 0.9 Hz), 5.93–6.06 (m, 1H), 6.58–6.64 (m, 1H), 7.07 (dd, 1H, J = 8.6, 0.9 Hz), 7.50 (dd, 1H, J = 8.6, 2.5 Hz), 7.54 (d, 1H, J = 2.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.1 (d, JP,C = 125 Hz), 116.3 (d, JP,C = 1.5 Hz), 124.0 (d, JP,C = 3.4 Hz), 125.1 (d, JP,C = 12.0 Hz), 127.9 (d, JP,C = 8.6 Hz), 130.3, 131.8, 132.8, 147.2 (d, JP,C = 7.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H9O3PBr: 274.9473, found 274.9470.
By following the general procedure, 7c was prepared from 7-bromo-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6c) (0.31 g, 1.02 mmol) and TMSBr (0.80 ml, 6.14 mmol) as a white solid (0.23 g, 82%). Mp: 163–164 °C. IR (KBr, cm−1): 1652 (O = P-OH), 1224 (P = O), 1206 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.27 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.61 (dd, 1H, J = 6.7, 0.9 Hz), 2.66 (dd, 1H, J = 6.7, 0.9 Hz), 5.93–6.06 (m, 1H), 6.58–6.64 (m, 1H), 7.07 (dd, 1H, J = 8.6, 0.9 Hz), 7.50 (dd, 1H, J = 8.6, 2.5 Hz), 7.54 (d, 1H, J = 2.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.1 (d, JP,C = 125 Hz), 116.3 (d, JP,C = 1.5 Hz), 124.0 (d, JP,C = 3.4 Hz), 125.1 (d, JP,C = 12.0 Hz), 127.9 (d, JP,C = 8.6 Hz), 130.3, 131.8, 132.8, 147.2 (d, JP,C = 7.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H9O3PBr: 274.9473, found 274.9470.
9-Bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7d)
 By following the general procedure, 7d was prepared from 9-bromo-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6d) (0.60 g, 1.98 mmol) and TMSBr (1.55 ml, 11.9 mmol) as a white solid (0.49 g, 90%). Mp: 180–181 °C. IR (KBr, cm−1): 2545 (O = P-OH), 2125 (O = P-OH), 1214 (P = O), 1210 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.44 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.63 (dd, 1H, J = 6.6, 0.9 Hz), 2.68 (dd, 1H, J = 6.6, 0.9 Hz), 5.92–6.05 (m, 1H), 6.61–6.67 (m, 1H), 7.09–7.14 (m, 1H), 7.30 (dd, 1H, J = 7.8, 1.3 Hz), 7.64 (dd, 1H, J = 7.9, 1.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 125 Hz), 115.9 (d, JP,C = 3.8 Hz), 124.6 (d, JP,C = 12.1 Hz), 125.5, 128.6 (d, JP,C = 8.6 Hz), 129.7, 130.2, 132.4, 144.9 (d, JP,C = 7.4 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H9O3PBr: 274.9473, found 274.9473.
By following the general procedure, 7d was prepared from 9-bromo-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6d) (0.60 g, 1.98 mmol) and TMSBr (1.55 ml, 11.9 mmol) as a white solid (0.49 g, 90%). Mp: 180–181 °C. IR (KBr, cm−1): 2545 (O = P-OH), 2125 (O = P-OH), 1214 (P = O), 1210 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.44 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.63 (dd, 1H, J = 6.6, 0.9 Hz), 2.68 (dd, 1H, J = 6.6, 0.9 Hz), 5.92–6.05 (m, 1H), 6.61–6.67 (m, 1H), 7.09–7.14 (m, 1H), 7.30 (dd, 1H, J = 7.8, 1.3 Hz), 7.64 (dd, 1H, J = 7.9, 1.5 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 125 Hz), 115.9 (d, JP,C = 3.8 Hz), 124.6 (d, JP,C = 12.1 Hz), 125.5, 128.6 (d, JP,C = 8.6 Hz), 129.7, 130.2, 132.4, 144.9 (d, JP,C = 7.4 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H9O3PBr: 274.9473, found 274.9473.
2-Hydroxy-9-methoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7e)
 By following the general procedure, 7e was prepared from 2-ethoxy-9-methoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6e) (185 mg, 0.73 mmol) and TMSBr (0.57 ml, 4.37 mmol) as a white solid (120 mg, 73%). Mp: 200–201 °C. IR (KBr, cm−1): 2527 (O = P-OH), 2224 (O=P-OH), 1275 (P = O), 1256 (P = O).31P NMR (162 MHz, DMSO-d6) δ = 37.38 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.56 (dd, 1H, J = 6.6, 0.9 Hz), 2.61 (dd, 1H, J = 6.6, 0.9 Hz), 3.80 (s, 3H), 5.87–5.99 (m, 1H), 6.59 (dd, 1H, J = 10.9, 4.8 Hz), 6.82 (dd, 1H, J = 7.7, 1.3 Hz), 7.01–7.05 (m, 1H), 7.08–7.14 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 126 Hz), 55.9, 112.0, 121.6, 123.7 (d, JP,C = 12.2 Hz), 124.4, 128.9, 129.0, 129.1, 137.2 (d, JP,C = 7.6 Hz), 151.5 (d, JP,C = 3.3 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C10H12O4P: 227.0473, found 227.0477.
By following the general procedure, 7e was prepared from 2-ethoxy-9-methoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6e) (185 mg, 0.73 mmol) and TMSBr (0.57 ml, 4.37 mmol) as a white solid (120 mg, 73%). Mp: 200–201 °C. IR (KBr, cm−1): 2527 (O = P-OH), 2224 (O=P-OH), 1275 (P = O), 1256 (P = O).31P NMR (162 MHz, DMSO-d6) δ = 37.38 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.56 (dd, 1H, J = 6.6, 0.9 Hz), 2.61 (dd, 1H, J = 6.6, 0.9 Hz), 3.80 (s, 3H), 5.87–5.99 (m, 1H), 6.59 (dd, 1H, J = 10.9, 4.8 Hz), 6.82 (dd, 1H, J = 7.7, 1.3 Hz), 7.01–7.05 (m, 1H), 7.08–7.14 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 126 Hz), 55.9, 112.0, 121.6, 123.7 (d, JP,C = 12.2 Hz), 124.4, 128.9, 129.0, 129.1, 137.2 (d, JP,C = 7.6 Hz), 151.5 (d, JP,C = 3.3 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C10H12O4P: 227.0473, found 227.0477.
7,9-Dichloro-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7f)

By following the general procedure, 7f was prepared from 7,9-dichloro-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6f) (0.37 g, 1.26 mmol) and TMSBr (1.00 ml, 7.57 mmol) as a white solid (0.27 g, 81%). Mp: 192–193 °C. IR (KBr, cm−1): 2522 (O = P-OH), 2219 (O = P-OH), 1230 (P = O), 1155 (P = O).31P NMR (162 MHz, DMSO-d6) δ = 36.50 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.68 (dd, 1H, J = 6.7, 0.9 Hz), 2.73 (dd, 1H, J = 6.7, 0.9 Hz), 5.99–6.11 (m, 1H), 6.62 (dd, 1H, J = 11.1, 4.9 Hz), 7.41 (d, 1H, J = 2.6 Hz), 7.66 (d, 1H, J = 2.6 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 125 Hz), 126.1 (d, JP,C = 12.2 Hz), 127.2 (d, JP,C = 3.8 Hz), 127.5 (d, JP,C = 8.6 Hz), 128.2, 128.5, 128.9, 131.2, 143.0 (d, JP,C = 7.4 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H8O3PCl2: 264.9588, found 264.9595.
2-Hydroxy-7-nitro-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7 g)
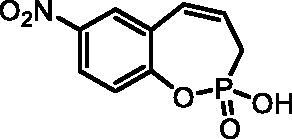 By following the general procedure, 7 g was prepared from 2-ethoxy-7-nitro-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6 g) (0.33 g, 1.23 mmol) and TMSBr (0.96 ml, 7.36 mmol) as a white solid (0.21 g, 71%). Mp: 183–184 °C. IR (KBr, cm−1): 2558 (O = P-OH), 2263 (O = P-OH), 1262 (P = O), 1221 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 39.26 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.69 (dd, 1H, J = 6.6, 0.9 Hz), 2.74 (dd, 1H, J = 6.6, 0.9 Hz), 6.02–6.15 (m, 1H), 6.73–6.80 (m, 1H), 7.32–7.36 (m, 1H), 8.18 (dd, 1H, J = 8.9, 2.9 Hz), 8.25 (d, 1H, J = 2.9 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.4 (d, JP,C = 125 Hz), 123.2 (d, JP,C = 3.6 Hz), 124.3, 126.0 (d, JP,C = 12.0 Hz), 126.3, 127.7 (d, JP,C = 9.0 Hz), 129.1, 143.7, 152.7 (d, JP,C = 7.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H9NO5P: 242.0218, found 242.0226.
By following the general procedure, 7 g was prepared from 2-ethoxy-7-nitro-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6 g) (0.33 g, 1.23 mmol) and TMSBr (0.96 ml, 7.36 mmol) as a white solid (0.21 g, 71%). Mp: 183–184 °C. IR (KBr, cm−1): 2558 (O = P-OH), 2263 (O = P-OH), 1262 (P = O), 1221 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 39.26 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.69 (dd, 1H, J = 6.6, 0.9 Hz), 2.74 (dd, 1H, J = 6.6, 0.9 Hz), 6.02–6.15 (m, 1H), 6.73–6.80 (m, 1H), 7.32–7.36 (m, 1H), 8.18 (dd, 1H, J = 8.9, 2.9 Hz), 8.25 (d, 1H, J = 2.9 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.4 (d, JP,C = 125 Hz), 123.2 (d, JP,C = 3.6 Hz), 124.3, 126.0 (d, JP,C = 12.0 Hz), 126.3, 127.7 (d, JP,C = 9.0 Hz), 129.1, 143.7, 152.7 (d, JP,C = 7.6 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C9H9NO5P: 242.0218, found 242.0226.
Carbonic anhydrase inhibition assay
The CA-catalysed CO2 hydration activity was assayed by using an applied photophysics stopped-flow apparatusCitation24. Phenol red (0.2 mM) was used as indicator following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The indicator worked at the absorbance maximum of 557 nm, with 20 mM HEPES buffer (pH 7.4) and 20 mM NaClO4 for maintaining constant ionic strength. For the determination of the kinetic parameters and inhibition constants, the CO2 concentrations were varied from 1.7 to 17 mM. For each inhibitor, at least six traces of the initial 5–10% of the reaction were used for determining the initial velocity. The uncatalysed rates were determined in the same fashion and subtracted from the total observed rates. The stock solutions of inhibitor were prepared as 1 mM solutions in distilled, deionised water. Afterwards, dilutions down to 0.01 nM were prepared in distilled and deionised water. Inhibitor and enzyme were preincubated together for 6 h at room temperature in order to allow for the formation of the enzyme–inhibitor complex. The inhibition constants were acquired by the non-linear least squares method using PRISM 3 and the Cheng–Prusoff equation, whereas the kinetic parameters of uninhibited enzymes were obtained from Lineweaver–Burk plots and represent the mean from at least three different determinations. All CA isoforms were recombinant, obtained in-house as reported earlierCitation25–27.
Results and discussion
Chemistry
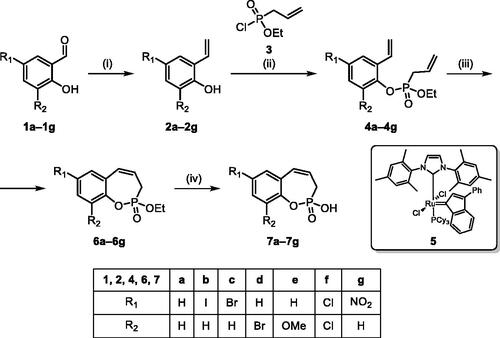
The synthetic strategy for the synthesis of 3H-1,2-benzoxaphosphepine 2-oxides is outlined in Scheme 1. The synthesis commenced with the Wittig reaction of commercially available 2-hydroxybenzaldehydes 1, which provided olefins 2 in high yields. In the following step, compounds 2 were treated with ethyl allylphosphonochloridate (3, the reagent was prepared according to the literature procedureCitation23) to give diolefins 4 in good to excellent yields. These key intermediates 4 were successfully cyclised by ring-closing metathesis, utilising commercially accessible Ru-based catalyst 5. The reaction furnished corresponding cyclic ethyl phosphonates 6 in good yields. Finally, compounds 6 were treated with TMSBr to afford hydroxy derivatives 7 in very good yields.
Scheme 1. Reagents and conditions: (i) MePPh3Br, t-BuOK, THF, rt, 18 h, 76–88%; (ii) NEt3, DCM, 0 °C to rt, 18 h, 60–94%; (iii) 5, PhMe, 70 °C, 4 h, 63–86%; (iv) TMSBr, DCM, rt, 24 h, 71–90%.
Carbonic anhydrase inhibition
The newly synthesised compounds 6 and 7 were evaluated for their CA inhibition activity by using the stopped-flow CO2 hydrase assayCitation24. The study was carried out against four human CA isoforms – the ubiquitous cytosolic CA I and II as well as trans-membrane tumour-associated CA IX and XIICitation1–7. The clinically used acetazolamide (AAZ) was used as the reference drug. The results of this study are shown in , and the following inferences could be drawn:
Table 1. Inhibition data of compounds 6–7 and the standard inhibitor acetazolamide (AAZ) against human CA isoforms I, II, IX and XII.
All synthesised benzoxaphosphepine2-oxide derivatives 6–7 have no inhibitory activity towards cytosolic isoforms hCA I and hCA II (KI > 100 µM), whose inhibition in most cases is undesirable, as hCA I and II isoforms are found in many tissues of the organismCitation1,Citation2,Citation7. It should be mentioned that AAZ is a highly effective inhibitor of all the four hCA isoforms considered here, which explains the many side effects of that drugCitation28,Citation29.
The tumour-associated hCA IX isoform was inhibited by all synthesised compounds 6–7 with inhibition constants in the sub-micromolar to low micromolar range (KI: 0.67–11.3 µM). The compound 7g was found to be the most potent hCA IX inhibitor among tested compounds with KI = 0.67 µM.
The other tumour-associated isoform hCA XII was also notably inhibited by all the synthesised derivatives 6–7 with KI values in the low micromolar and sub-micromolar range (KI: 0.51–7.4 µM). Among all tested compounds, compound 7a was the most effective inhibitor against hCA XII with KI = 0.51 µM.
Overall, hydroxy derivatives 7 showed slightly higher inhibition potency against tumour-associated hCA IX and XII than the corresponding ethoxy derivatives 6. In the case of hydroxy derivatives 7, the range of KI values was found to be from 0.67 to 2.5 µM for hCA IX and from 0.51 to 1.8 µM for hCA XII. Regarding ethoxy derivatives 6, the range of KI values was from 0.76 to 11.3 µM for hCA IX and from 0.95 to 7.4 µM for hCA XII.
Albeit the efficacy of the synthesised compounds 6–7 was lower in comparison to the reference drug AAZ, these compounds displayed desirable isoform-selective inhibition activity for tumour-associated isoforms hCA IX and hCA XII. The establishing of the selectivity is necessary to prevent possible side effects from inhibition of cytosolic hCA I and II isoformsCitation7,Citation28,Citation29.
Conclusions
Herein we report the synthesis of novel benzoxaphosphepine 2-oxide derivatives as a new class of tumour-associated CA IX and XII inhibitors. These compounds were investigated against four human CA isoforms with pharmacological applications (hCA I, hCA II, hCA IX, and hCA XII). All tested compounds exhibited selective inhibition of the tumour-associated hCA isoforms IX and XII with activities in the sub-micromolar or low micromolar range, whereas the off-target cytosolic isoforms hCA I and II were not significantly inhibited by these compounds. Considering that hCA IX and XII are implicated in processes connected to tumourigenesisCitation2–6, present findings give an insight towards development of new selective anti-tumour agents.
Disclosure statement
No potential competing interest was reported by all authors except CTS. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Supuran CT. Carbonic anhydrases and metabolism. Metabolites. 2018;21:25.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J. 2016;473(14):2023–2032.
- (a) Singh S, Lomelino CL, Mboge MY, Frost SC, McKenna R. Cancer drug development of carbonic anhydrase inhibitors beyond the active site. Molecules. 2018;23:1045. (b) Angeli A, Carta F, Nocentini A, Winum JY, Zalubovskis R, Akdemir A, Onnis V, Eldehna WM, Capasso C, Simone G, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites. 2020;10:412.
- (a) Winum J-Y, Rami M, Scozzafava A, Montero JL, Supuran C. Carbonic anhydrase IX: a new druggable target for the design of antitumor agents. Med Res Rev. 2008;28:445–463. (b) McDonald PC, Winum J-Y, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget. 2012;3:84–97. (c) McDonald PC, Chafe SC, Supuran CT, Dedhar S. Cancer therapeutic targeting of hypoxia induced carbonic anhydrase ix: from bench to bedside. Cancers. 2022;14:3297.
- Watson PH, Chia SK, Wykoff CC, Han C, Leek RD, Sly WS, Gatter KC, Ratcliffe P, Harris AL. Carbonic anhydrase XII is a marker of good prognosis in invasive breast carcinoma. Br J Cancer. 2003;88(7):1065–1070.
- (a) Ozensoy Guler O, De Simone G, Supuran CT. Drug design studies of the novel antitumor targets carbonic anhydrase IX and XII. Curr Med Chem. 2010;17:1516–1526. (b) Kciuk M, Gielecińska A, Mujwar S, Mojzych M, Marciniak B, Drozda R, Kontek R. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J Enzyme Inhib Med Chem. 2022;37:1278–1298.
- (a) Alterio V, Di Fiore A, D’Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev. 2012;112:4421–4468. (b) Aggarwal M, Kondeti B, McKenna R. Insights towards sulfonamide drug specificity in α-carbonic anhydrases. Bioorg Med Chem. 2013;21:1526–1533.
- Žalubovskis R. In a search for selective inhibitors of carbonic anhydrases: coumarin and its bioisosteres – synthesis and derivatization. Chem Heterocycl Comp. 2015;51(7):607–612.
- (a) Maresca A, Temperini C, Vu H, Pham NB, Poulsen S-A, Scozzafava A, Quinn RJ, Supuran CT. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc. 2009;131:3057–3062. (b) Supuran CT. Coumarin carbonic anhydrase inhibitors from natural sources. J Enzyme Inhib Med Chem. 2020;35:1462–1470.
- (a) Bonneau A, Maresca A, Winum J-Y, Supuran CT. Metronidazole-coumarin conjugates and 3-cyano-7-hydroxy-coumarin act as isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2013;28:397–401. (b) Sharma A, Tiwari M, Supuran CT. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J Enzyme Inhib Med Chem. 2014;29:292–296.
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem. 2010;53(1):335–344.
- (a) Tars K, Vullo D, Kazaks A, Leitans J, Lends A, Grandane A, Zalubovskis R, Scozzafava A, Supuran CT. Sulfocoumarins as dual inhibitors of human carbonic anhydrase isoforms IX/XII and of human thioredoxin reductase. J Med Chem. 2013;56(1):293–300. (b) Krasavin M, Žalubovskis R, Grandāne A, Domračeva I, Zhmurov P, Supuran CT. Sulfocoumarins as dual inhibitors of human carbonic anhydrase isoforms IX/XII and of human thioredoxin reductase. J Enzyme Inhib Med Chem. 2020;35:506–510.
- (a) Tanc M, Carta F, Bozdag M, Scozzafava A, Supuran CT. 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg Med Chem. 2013;21:4502–4510. (b) Nocentini A, Ceruso M, Carta F, Supuran CT. 7-Aryl-triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrase IX and XII. J Enzyme Inhib Med Chem. 2016;31:1226–1233.
- (a) Grandane A, Tanc M, Di Cesare Mannelli L, Carta F, Ghelardini C, Žalubovskis R, Supuran CT. 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem. 2015;58:3975–3983. (b) Grandane A, Tanc M, Žalubovskis R, Supuran CT. Synthesis of 6-aryl-substituted sulfocoumarins and investigation of their carbonic anhydrase inhibitory action. Bioorg Med Chem. 2015;23:1430–1436. (c) Grandane A, Tanc M, Zalubovskis R, Supuran CT. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett. 2014;24:1256–1260. (d) Grandane A, Tanc M, Zalubovskis R, Supuran CT. Synthesis of 6-tetrazolyl-substituted sulfocoumarins acting as highly potent and selective inhibitors of the tumor-associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem. 2014;22:1522–1528.
- Podolski-Renić A, Dinić J, Stanković T, Jovanović M, Ramović A, Pustenko A, Žalubovskis R, Pešić M. Sulfocoumarins, specific carbonic anhydrase IX and XII inhibitors, interact with cancer multidrug resistant phenotype through pH regulation and reverse P-glycoprotein mediated resistance. Eur J Pharm Sci. 2019;138:105012.
- (a) Pustenko A, Stepanovs D, Žalubovskis R, Vullo D, Kazaks A, Leitans J, Tars K, Supuran CT. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2017;32:767–775. (b) Pustenko A, Nocentini A, Balašova A, Alafeefy A, Krasavin M, Žalubovskis R, Supuran CT. Aryl derivatives of 3H-1,2-benzoxathiepine 2,2-dioxide as carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2020;35:245–254. (c) Pustenko A, Nocentini A, Balašova A, Krasavin M, Žalubovskis R, Supuran CT. 7-Acylamino-3H-1,2-benzoxathiepine 2,2-dioxides as new isoform-selective carbonic anhydrase IX and XII inhibitors. J Enzyme Inhib Med Chem. 2020;35:650–656.
- (a) Rodriguez JB, Gallo-Rodriguez C. The role of the phosphorus atom in drug design. Chem Med Chem. 2019;14:190–216. (b) Ceradini D, Shubin K. New methods for the synthesis of phosphono-δ-lactones (microreview). Chem Heterocycl Compd. 2021;57:1167–1169. (c) Balašova A, Žalubovskis R. Synthetic methods toward phosphacoumarins (microreview). Chem Heterocycl Comp. 2022;58:310–312.
- (a) Bonnac L, Innocenti, A, Winum J-Y, Casini A, Montero JL, Scozzafava A, Barragan V, Supuran CT. Carbonic anhydrase inhibitors: aliphatic N-phosphorylated sulfamates—a novel zinc-anchoring group leading to nanomolar inhibitors. J Enzyme Inhib Med Chem. 2004;19:275–278. (b) Winum J-Y, Innocenti A, Gagnard V, Montero JL, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase inhibitors. Interaction of isozymes I, II, IV, V, and IX with organic phosphates and phosphonates. Bioorg Med Chem Lett. 2005;15:1683–1686. (c) Gülçin İ, Trofimov B, Kaya R, Taslimi P, Sobenina L, Schmidt E, Petrova O, Malysheva S, Gusarova N, Farzaliyev V, et al. Synthesis of nitrogen, phosphorus, selenium and sulfur-containing heterocyclic compounds – Determination of their carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase and α-glycosidase inhibition properties. Bioorg Chem. 2020;103:104171. (d) Nocentini A, Alterio V, Bua S, Micheli L, Esposito D, Buonanno M, Bartolucci G, Osman SM, ALOthman ZA, Cirilli R, et al. Phenyl(thio)phosphon(amid)ate benzenesulfonamides as potent and selective inhibitors of human carbonic anhydrases II and VII counteract allodynia in a mouse model of oxaliplatin-induced neuropathy. J Med Chem. 2020;63:5185–5200.
- Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54(8):2529–2591.
- Hirschbeck V, Fleischer I. Synthesis of benzofuranones via palladium-catalyzed intramolecular alkoxycarbonylation of alkenylphenols. Chemistry. 2018;24(12):2854–2857.
- Zhang Y, Sigman MS. Development of a general Pd(II)-catalyzed intermolecular hydroalkoxylation reaction of vinylphenols by using a sacrificial alcohol as the hydride source. Org Lett. 2006;8(24):5557–5560.
- Fleischer I, Pospech J. Brønsted acid-catalyzed hydroarylation of activated olefins. RSC Adv. 2015;5(1):493–496.
- Fourgeaud P, Midrier C, Vors J-P, Volle J-N, Pirat J-L, Virieux D. Oxaphospholene and oxaphosphinene heterocycles via RCM using unsymmetrical phosphonates or functional phosphinates. Tetrahedron. 2010;66(3):758–764.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- (a) Supuran CT, Ilies MA, Scozzafava A. Carbonic anhydrase inhibitors. Part 29. Interaction of isozymes I, II and IV with benzolamide-like derivatives. Bioorg Med Chem. 1998;33:739–752. (b) Sentürk M, Gülçin I, Daştan A, Küfrevioğlu OI, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem. 2009;17:3207–3211.
- Leitans J, Kazaks A, Balode A, Ivanova J, Zalubovskis R, Supuran CT, Tars K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem. 2015;58(22):9004–9009.
- (a) Nocentini A, Angeli A, Carta F, Winum J-Y, Zalubovskis R, Carradori S, Capasso C, Donald WA, Supuran CT. N-substituted and ring opened saccharin derivatives selectively inhibit transmembrane, tumor-associated carbonic anhydrases IX and XII. J Enzyme Inhib Med Chem. 2021;36(1):561–580. (b) Pustenko A, Nocentini A, Gratteri P, Bonardi A, Vozny I, Žalubovskis R, Supuran CT. The antibiotic furagin and its derivatives are isoform-selective human carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2020;35:1011–1020. (c) Ivanova J, Carta F, Vullo D, Leitans J, Kazaks A, Tars K, Žalubovskis R, Supuran CT. N-substituted and ring opened saccharin derivatives selectively inhibit transmembrane, tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem. 2017;25:3583–3589.
- Schmickl CN, Owens RL, Orr JE, Edwards BA, Malhotra A. Side effects of acetazolamide: a systematic review and meta-analysis assessing overall risk and dose dependence. BMJ Open Resp Res. 2020;7(1):e000557.
- Bonardi A, Nocentini A, Bua S, Combs J, Lomelino C, Andring J, Lucarini L, Sgambellone S, Masini E, McKenna R, et al. Sulfonamide inhibitors of human carbonic anhydrases designed through a three-tails approach: improving ligand/isoform matching and selectivity of action. J Med Chem. 2020;63(13):7422–7444.