
Abstract
New spiro-piperidine derivatives were synthesised via the eco-friendly ionic liquids in a one-pot fashion. The in vitro antileishmanial activity against Leishmania major promastigote and amastigote forms highlighted promising antileishmanial activity for most of the derivatives, with superior activity compared to miltefosine. The most active compounds 8a and 9a exhibited sub-micromolar range of activity, with IC50 values of 0.89 µM and 0.50 µM, respectively, compared to 8.08 µM of miltefosine. Furthermore, the antileishmanial activity reversal of these compounds via folic and folinic acids displayed comparable results to the positive control trimethoprim. This emphasises that their antileishmanial activity is through the antifolate mechanism via targeting DHFR and PTR1. The most active compounds showed superior selectivity and safety profile compared to miltefosine against VERO cells. Moreover, the docking experiments of 8a and 9a against Lm-PTR1 rationalised the observed in vitro activities. Molecular dynamics simulations confirmed a stable and high potential binding to Lm-PTR1.
Introduction
Leishmaniasis is a complex disease that is caused by more than 20 species of Leishmania and correlated to several clinical manifestations ranging from simple skin lesions around the bite site to fatal visceral formsCitation1,Citation2. More than one billion people are at risk of leishmaniasis in endemic areasCitation3,Citation4. Based on literature data, there is no effective and safe treatment for leishmaniasis for further developments. About 50 years ago pentavalent antimonials used as first-line drugs for the treatment of leishmaniasis diseases despite of high toxicityCitation1,Citation2. Besides, existing approved drugs for leishmania can cause various sever adverse effects like gastro-intestinal disturbance, hepatic and renal dysfunction, especially, co-infections of immunocompromised patients with leishmaniasis, e.g. HIV-leishmania co-infection, are fatalCitation1,Citation2.
Therefore, there is a continuing necessity for efforts to discover new antileishmanial agentsCitation5,Citation6, and able to overcome resistance mechanisms of leishmania. For the folate pathway, dihydrofolate Reductase (DHFR) and Pteridine reductase (PTR1) are validated targets for leishmaniaCitation7. Their main role is to reduce oxidised pteridines like biopterin and folate to active cofactors tetrahydrobiopterin (THB) and tetrahydrofolate (THF), respectively. However, many leishmania species developed resistance against dihydrofolate reductase-thymidylate synthase (DHFR-TS) inhibitorsCitation8,Citation9, owing to the presence of an alternative salvage pathway regulated by PTR1. Remarkably, PTR1 enzyme is overexpressed in strains exhibited antifolate resistance, consequently, offering the ways to evade DHFR-TS pathwayCitation10–12.
The piperidine ring is a known scaffold in many biologically active compoundsCitation13 and an important framework present in a large variety of natural productsCitation14,Citation15. Particularly, piperidin-4-ones are versatile building blocks due to the easy manipulation of the carbonyl group for the introduction of different substituents into the six-membered ringCitation16–18. Moreover, piperidones were reported to possess analgesicCitation19, anti-inflammatoryCitation19, central nervous system (CNS)Citation20, local anaesthetic, anticancerCitation21, and antimicrobial activity. Interestingly, some piperidine derivatives were reported to possess activities against some Neglected Tropical Diseases (NTDs), such as malariaCitation22, leishmania and trypanosomiasisCitation23,Citation24. The diverse biological actions are continuously attracting chemists to synthesise new piperidone molecules with diversified substituents to enhance their activities.
Interestingly, various spiro-compounds were reported to possess antiprotozoal and antileishmanial activities. For instance, compound A () is able to halt the replication of both promastigote and axenic amastigote forms of L. infantum in a dose-dependent mannerCitation25. Moreover, the spiro-compound BCitation26 acts as a catalytic inhibitor of the unusual bisubunit DNA topoisomerase IB of L. donovaniCitation26. Furthermore, other spiro-compounds were reported to display promising antileishmanial activities, such as CCitation27, as shown in .
Figure 1. Some reported spiro-compounds with antileishmanial activities A–C, and the target compounds D.
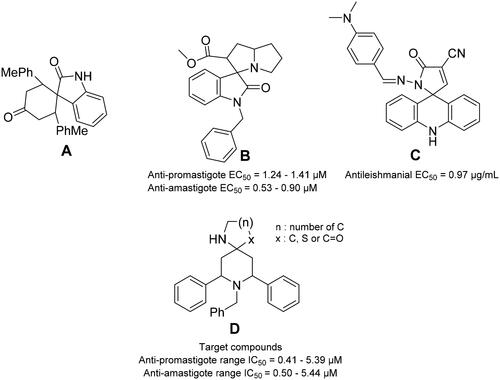
These reports encouraged us to build spiro-piperidine derivatives (D in ) and investigate their antileishmanial activities. Our synthetic strategy is based on the green and eco-friendly Ionic Liquids (ILs). ILs have been attracted considerable interest as eco-friendly, benign reaction media for a wide variety of organic reactions due to their unique properties as high thermal and chemical stability, non-flammable, solubility and easy recyclingCitation28. Then, the compounds were tested versus both promastigote and amastigote forms of Leishmania major. The reversal of the antileishmanial activity via folic and folinic acid confirmed the antifolate mechanism indicating PTR1 inhibition. Interestingly, the docking experiments and the molecular dynamics (MD) simulations on the putative leishmanial Lm-PTR1 target rationalised the observed antileishmanial activity of the most active compounds. Finally, the cytotoxicity of the most active compounds was evaluated using VERO cells reflecting their high safety profile.
Results and discussion
Chemistry
Herein, we report an efficient synthesis of 1-benzyl-2,6-diarylpiperidin-4-one 1a–d using acetone, aromatic aldehyde and benzylamine in the presence of piperidinium acetate as ionic liquid. Piperidin-one derivatives can be used as a building block of some fused and spiro piperidine derivatives (Scheme 1).
Scheme 1. Synthesis of 1-benzyl-2,6-diarylpiperidin-4-one 1a–d.
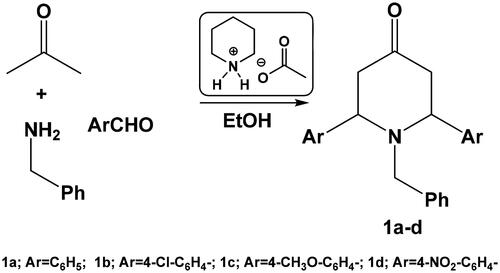
In order to optimise this reaction, we studied the variation of the amount of catalyst as well as the reaction solvent. The results of the optimisation study suggested that ethanol is the best solvent and that 10 mol% piperidinium acetate is the ideal catalyst concentration required for effective conversion. Thus, by reacting acetone, benzaldehyde and benzylamine (in a 1:2:1 ratio, respectively) in ethanol in the presence of 10 mol% piperidinium acetate, the corresponding 1-benzyl-2,6-diphenylpiperidin-4-one 1a was obtained ().
Table 1. Optimisation of reaction conditions of the reaction of acetone, benzaldehyde, and benzylamine.a
The IR spectrum of compound 1a as an example showed a sharp band at 1718 cm−1 for C = O group, where its 1H-NMR spectrum revealed a doublet at 3.01 ppm with coupling constant equals to 13.6 Hz, which could be assigned for two CH2 groups (C3 and C5), a triplet signal at 3.17 ppm which may be assigned for two CH groups (C2 and C6), a singlet 3.67 ppm for N–CH2– group and a multiplet between 6.91 and 7.59 ppm for aromatic protons.
The one-pot reaction of 1-benzyl-2,6-diarylpiperidin-4-one 1a–d with potassium cyanide and aniline in glacial acetic acid afforded 1-benzyl-2,6-diaryl-4-(phenylamino)piperidine-4-carbonitrile 2a–d (Scheme 2).
Scheme 2. Synthesis of 1-benzyl-2,6-diaryl-4-(phenylamino)piperidine-4-carbonitrile 2a–d.
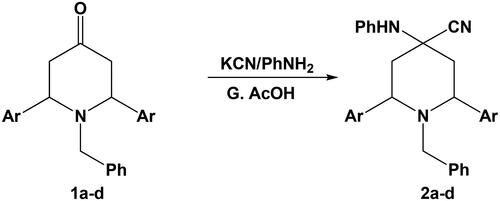
The structures of the obtained 4-phenylamino piperidine-1-carbonitrile derivatives 2a–d were characterised by their elemental and spectral data where the IR spectrum of compound 2a for example showed absorption maxima at 3367 cm−1 for NH group and 2223 cm−1 for CN group, where the 1H-NMR spectrum of this compound showed a singlet signal at 9.12 ppm for NH group.
On the other hand, reduction of compounds 2a–d with lithium aluminium hydride LAH in diethyl ether afforded the corresponding primary amines 3a–d, where acid hydrolysis of compounds 2a–d gave the expected amides 4a–d (Scheme 3).
Scheme 3. Reduction and acid hydrolysis of compounds 2a–d.
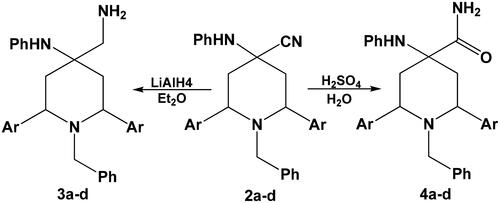
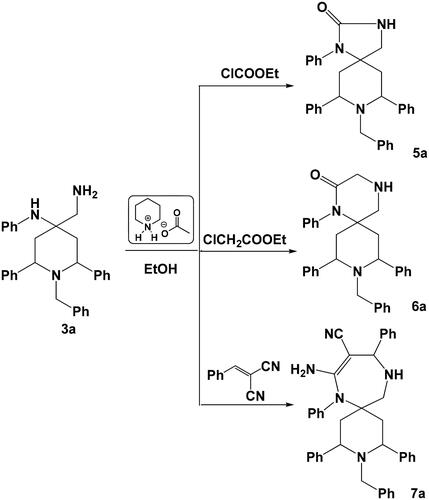
In an extension of our workCitation29 on the synthesis of spiro heterocycles, our strategy is how to use these compounds as building blocks for the synthesis of a variety of five-membered, six-membered and seven-membered rings spiro heterocycles. So when compound 3a was allowed to react with ethyl chloroformate, ethyl chloroacetate in ethanol in the presence of piperidinium acetate-IL, the corresponding five-membered ring spiro-compound 8-benzyl-1,7,9-triphenyl-1,3,8-triazaspiro[4.5]decan-2-one 5a and six-membered ring spiro-compound 9-benzyl-1,8,10-triphenyl-1,4,9-triazaspiro[5.5]undecan-2-one 6a were obtained. Where the reaction of compound 3a with 2-benzylidenemalononitrile under the same experimental conditions, gave the corresponding seven-membered ring spiro-compound 8-amino-3-benzyl-2,4,7,10-tetraphenyl-3,7,11-triazaspiro[5.6]dodec-8-ene-9-carbonitrile 7a (Scheme 4). Mechanism of formation of compound 7a was suggested to proceed via a preliminary nucleophilic attack of the primary amine group onto the activated ethylenic bond of 2-benzylidenemalononitrile followed by another nucleophilic attack of the other –NHPh group onto the nitrile group.
Scheme 4. Synthesis of spiro heterocycles 5a–7a.
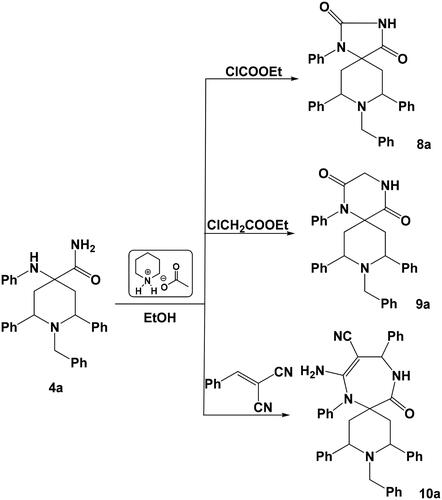
Similarly, the reaction of 1-benzyl-2,6-diphenyl-4-(phenylamino)piperidine-4-carboxamide 4a with ethyl formate, ethyl chloroactate or 2-benzylidenemalononitrile under the same experimental conditions (ethanol/piperidinium acetate-IL) afforded the corresponding spiro heterocycles 8a–10a, respectively (Scheme 5).
Scheme 5. Synthesis of spiro heterocycles 8a–10a.
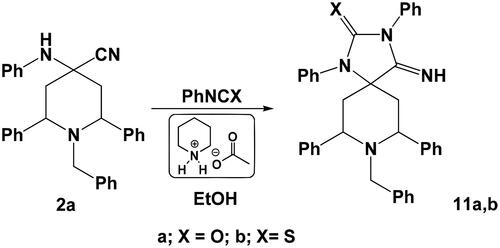
On the hand, the reaction of compound 2a with phenyl isocyanate or phenyl isothiocyantate in ethanol in the presence of piperidinium acetate-IL afforded the corresponding spiro heterocycles namely: 8-benzyl-7,9-diaryl-4-imino-1,3-diphenyl-1,3,8-triazaspiro[4.5]decan-2-one 11a or 8-benzyl-7,9-bis(4-chlorophenyl)-4-imino-1,3-diphenyl-1,3,8-triazaspiro[4.5]decane-2-thione 11 b, respectively (Scheme 6).
Scheme 6. Synthesis of spiro heterocycles 11a,b.
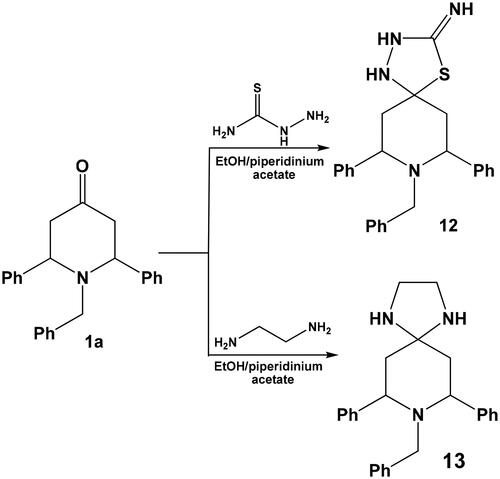
Finally, compound 1a was allowed to react with some bifunctional reagents viz, thiosemicarbazide or ethylenediamine in ethanol in the presence of piperidinium acetate-IL afforded the corresponding spiro heterocycles namely: 8-benzyl-7,9-diphenyl-4-thia-1,2,8-triazaspiro[4.5]decan-3-imine 12 and 8-benzyl-7,9-diphenyl-1,4,8-triazaspiro[4.5]decane 13, respectively (Scheme 7).
Scheme 7. Synthesis of spiro heterocycles 12 and 13.
Structures of the newly synthesised compounds were confirmed based upon their IR, 1H-NMR, 13C-MR, MS spectral data, and elemental analyses.
Biological evaluations
In vitro antileishmanial activity
All synthesised compounds were evaluated against L. major promastigote form. The most active compounds showing best antipromastigote activities were then evaluated against axenic amastigote form. All compounds showed inhibition for the promastigote form in a low micromolar range of activity (IC50: 0.4102–5.3974 µM), as shown in . Interestingly, all spiro piperidine derivatives (5a–14) exhibited superior activities to the standard miltefosin against both leishmanial forms. Remarkably, spiro piperidine derivatives namely 8a and 9a displayed promising antileishmanial activities superior to miltefosine in a sub-micromolar range against the amastigote form, 0.89 and 0.50 µM, respectively.
Table 2. Antipromastigote and antiamastigote activity (IC50) of the test compounds and the reference.
Reversal of antileishmanial activity of most active compounds by folic acid and folinic acid
To validate our design that the synthesised compounds possess their antileishmanial activity via the antifolate mechanism, we employed the approach reported by Mendoza-Martínez et al. for the two most active compounds 8a and 9aCitation30. The approach involves exposing the parasite to concentrations of the tested compounds above their IC50s after the addition of folic acid and folinic acid using trimethoprim as a positive control. Exposure to trimethoprim after addition of folic acid led to an increase in parasite survival up to nearly 100%. It is worthy to mention that folic acid competes for the active sites of both DHFR and PTR1 while folinic acid involves in DNA synthesis without any necessity to undergo activation. As seen in , reversal of antileishmanial effect of compounds 8a and 9a took place upon addition of folic acid with percentage parasite growth in the range of 72–87%.
Table 3. In vitro evaluation of folate pathway inhibition expressed as percentage survival.a
Based on that, we can determine that the greatest, if not all, of the antileishmanial activity of compounds 8a and 9a is attributed to the antifolate mechanism, by acting on DHFR-TS and PTR1. Addition of excess folic acid to parasitic cells after exposure to the test compounds had been performed to investigate its ability to reverse the DHFR and PTR1 inhibition. Test compounds exhibited reversibility of DHFR and PTR1 inhibition in a comparable fashion to that of trimethoprim.
In vitro toxicity evaluation
To verify the safety of the most active compounds 8a and 9a, they were checked for their cytotoxicity against African green monkey kidney cells (VERO cells) as reported earlierCitation31. Briefly the cells were incubated for 72 h with different dilutions of the selected compounds. The 50% cytotoxic concentration (CC50) values were calculated representing the concentration of compound required to kill 50% of the fibroblast cells. The selectivity indices were determined using the formula SI = CC50/IC50, against the respective activities as shown in . Interestingly, for 8a and 9a, the concentrations needed to inhibit viability of VERO cells (CC50) are at least 2 orders of magnitude higher than those required to inhibit the viability of promastigotes of leishmania parasite (IC50). Furthermore, both compounds displayed superior selectivity and safety profile compared to the standard miltefosine.
Table 4. CC50 values of the most active compounds against normal VERO cells and their selectivity index.
Molecular modelling
Molecular docking
The aim of this section is to rationalise the observed in vitro antileishmanial activity. Our investigation was devoted on the protein structure of Leishmania major PTR1 (LmPTR1) as a presumed target for the antifolate pathway because the co-crystal structure of Lm DHFR-TS enzyme is not determined yet.
The docking score distribution of the most active compounds 8a and 9a came in coherence with the observed in vitro antileishmanial activity.
Both 8a and 9a displayed comparable docking scores, with slight superiority for 9a, as seen in . Moreover, compounds 8a and 9a demonstrated superior scores compared to the reference trimethoprim (PTR1 inhibitor). Such observations came in agreement with the observed in vitro antileishmanial activity. Furthermore, this foresees advantageous binding towards PTR1 and thus rationalising the antifolate mechanism validated by the in vitro experiment (Section: Reversal of the antileishmanial activity via folic and folinic acid).
Table 5. Docking scores of the most active compounds against PTR1.
Perceiving the intermolecular interactions, the docking poses of 8a and 9a demonstrated comparable interactions, as shown in . For 8a pose, its triazaspiro[4.5]decane core is centralised between Phe113 and Val230, pointing its hydrophilic imidazolidine-2,4-dione moiety into the solvent exposed area, as seen in . However, the hydrophobic 1, 7-phenyl groups appeared to show favourable hydrophobic interactions with the side chain of Val230. Likewise, the 9-phenyl group displays pi-pi stacking with the side chain of Phe113 and favourable interactions with Arg17. Importantly, the piperidinyl group forms attractive ionic interaction via its cationic N with the phosphate group of the co-factor NADPH.
Figure 2. The docking poses of 8a and 9a poses as cyan sticks in the binding site of the Lm-PTR1 for (A) and (B), respectively, as 3 D and 2 D depictions. The Simon sticks representation is for NADPH co-factor (annotated as “NDP” in the 2 D depiction). Non-polar hydrogen atoms were omitted for clarity.
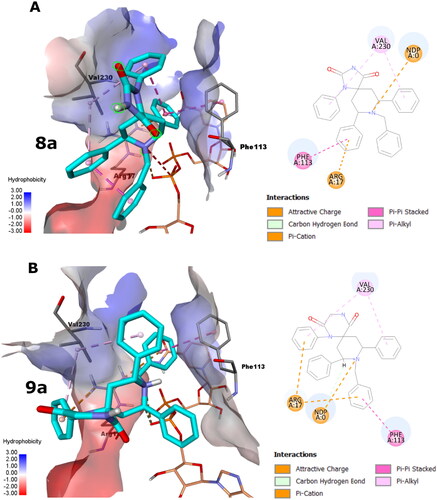
For the 9a pose, its triazaspiro[5.5]undecane core showed almost 90 degrees flip compared to 8a core accommodating for the ring size and topology differences, as shown in . Accordingly, likewise, its piperazine-2,5-dione moiety points towards the solvent exposed area. Like 8a pose, the hydrophobic 1, 8-phenyl groups of 9a appeared to show favourable hydrophobic interactions with the side chain of Val230 as well as favourable interactions with Arg17. Interestingly, the 10-benzyl group displays pi-pi stacking with the side chain of Phe113 and favourable interactions with Arg17. Again, the piperidinyl group demonstrates attractive ionic interaction via its cationic N with the phosphate group of the co-factor NADPH.
Generally, these observations augment the high potentiality of the 8a and 9a poses to block the catalytic activity of Lm-PTR1.
Molecular dynamics
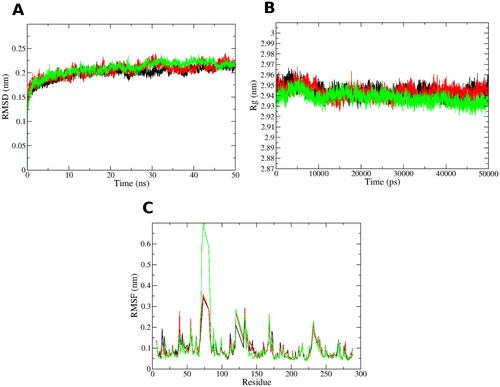
The 8a and 9a docking poses in PTR1 were subjected to 50 ns molecular dynamics (MD) simulations for evaluating the stability of its docked pose in a time-dependent manner in the binding site. Furthermore, another run was conducted for the unliganded PTR1 form, to account its dynamicity as a reference. This results in a total of three MD runs, 50 ns each. Root Mean Square Deviation (RMSD) is a measure of protein backbone stability during the simulation time. RMSD of the three systems () reach a converged state after 30 ns with a minor fluctuation with 0.025 nm range. This reflects appropriate stability of the protein structure during the three simulation runs. This is also in agreement with analysis obtained by the Radius of gyration (Rg) in . Rg is a measure of protein structure compactness during the simulation time. There is no great fluctuation in the Rg of the protein complexed with 8a and 9a compared to the unliganded structure since they display Rg range of 0.03 nm after 10 000 ps (10 ns). This gives an indication of the low conformational changes of the protein throughout the simulation, and hence, its stabilityCitation32,Citation33. Per residue root mean square fluctuation (RMSF) assesses the conformational changes that occur to each residue of the protein, as shown in . The structural loops exhibit the highest RMSF contemplated by the high free movement, especially in the region of residue 70–80. However, the key binding site amino acids (numbers: Arg17, Val230, Leu188, His241, Tyr191, Leu229, Met233, Phe113) show low RMSF and comparable fluctuation behaviour to all the three simulated protein systems. This emphasises the good binding of the complexed ligand 8a and 9a with minimal conformational changes in these residues comparable to the unliganded system.
Figure 3. MD simulations for the three systems, the unliganded leishmanial PTR1 (black line), 8a-PTR1 (red line - dark grey) and 9a-PTR1 (green line - pale grey) complex systems. (A) Root mean square deviation (RMSD) of the protein alpha carbon atoms across the 50 ns simulation. (B) Radius of gyration (Rg) for the PTR1 protein across the 50 ns simulation time. (C) Per residue root mean square fluctuation (RMSF).
Experimental
Chemistry
All melting points were determined on a Koffler melting point apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were recorded on a Bruker avance 400 MHz spectrometer using TMS as internal reference (chemical shifts in δ, ppm), and IR spectra were obtained on a Nicolet 710 FT-IR spectrometer (KBr, νmax in cm−1). Mass spectra were recorded on a GC-MSQP 1000EX Schimadzu at the Microanalytical laboratory, Cairo University, Cairo, Egypt. Elemental analyses were recorded on Vario El Fab-Nr elemental analyser (Cairo University).
General procedure for preparation of ionic liquid piperidinium acetate-IL
Piperidine (0.9 ml, 0.1 mol) was cooled in an ice bath with continuous stirring then was treated with acetic acid (0.6 g, 0.1 mol) drop-wise through a separating funnel. After reaction completion water was removed under reduced pressure using rotatory evaporator then the product was stored in a vacuum desiccator. White solid (13.5 g, 95%) mp 105–107 °C, 1H-NMR (400 MHz, δ, CDCl3): 1.55 (d, 2H, NH2), 1.66 (m, 2H, CH2), 1.82 (m. 4H, 2CH2), 2.22 (s, 3H, CH3), 3.04 (t, 4H, 2CH2). 13CMR (100 MHz, δ, CDCl3): 21.5, 23.0, 24.4, 45.8, 180.6.
General procedure for synthesis of 1-benzyl-2,6-diarylpiperidin-4-one 1a–d
A mixture of acetone (0.1 mol), aromatic aldehyde (2.0 mmol) in ethanol (4 ml) was treated with benzylamine (0.1 mol) and Piperidinium acetate-IL (30 mg) were added to a round-bottom flask equipped with a magnetic stir bar and condenser. The mixture was heated at 70 °C for the time specified in . The reaction progress was monitored by TLC (EtOAc/hexane = 2:8). After completion of the reaction, the mixture was cooled to room temperature then poured onto crushed ice. The formed solid was filtered, dried, and purified by crystallisation using ethanol as a solvent.
1-Benzyl-2,6-diphenylpiperidin-4-one 1a
Pale yellow solid: yield 81%, mp 189–191 °C; Anal. Calcd. for (C24H23NO, 341.44): C, 84.45; H, 6.74; N, 4.10. Found: C, 84.09; H, 6.23; N, 3.98%. IR (KBr, cm−1): 1718 (C = O); 1H NMR (400 MHz, δ, DMSO-d6): δ 3.02 (d, 4H, J = 13.6 Hz, 2CH2), 3.17 (t, 2H, J = 5.4 Hz, 2CH), 3.67 (s, 2H, N–CH2–), 6.91–7.59 (m, 15 H, CH-arom.); 13C NMR (100 MHz, DMSO-d6): δ 42.3, 44.1, 53.7, 126.3, 127.2, 127.8, 128.1, 128.7, 129.6, 130.2, 130.8, 180.1. MS (m/z): ESI, ([M+] 341.
1-Benzyl-2,6-bis(4-chlorophenyl)piperidin-4-one 1b
Yellow crystals, yield 89%, mp 211–213 °C; Anal. Calcd. for (C24H21Cl2NO, 410.33): C, 70.24; H, 5.12; N, 3.41, Cl, 17.31. Found: C, 70.02; H, 4.97; N, 3.16, Cl, 17.01%. IR (KBr, cm−1): 1717 (C = O); 1H NMR (400 MHz, δ, DMSO-d6): δ 3.04 (d, 4H, J = 13.6 Hz, 2CH2), 3.21 (t, 2H, J = 5.4 Hz, 2CH), 3.69 (s, 2H, N–CH2–), 6.90–7.76 (m, 13 H, CH-arom.); 13C NMR (100 MHz, DMSO-d6): δ 42.5, 44.3, 53.7, 126.2, 127.3, 127.9, 128.2, 128.7, 129.6, 130.7, 136.6, 180.8. MS (m/z): ESI, ([M + 2] 412.
1-Benzyl-2,6-bis(4-methoxyphenyl)piperidin-4-one 1c
White crystals, yield 92%, mp 252–254 °C; Anal. Calcd. for (C26H27NO3, 401.2): C, 77.78; H, 6.78; N, 3.49. Found: C, 77.50; H, 6.61; N, 3.22%. IR (KBr, cm−1): 1715 (C = O); 1H NMR (400 MHz, δ, DMSO-d6): δ 3.02 (d, 4H, J = 13.6 Hz, 2CH2), 3.20 (t, 2H, J = 5.4 Hz, 2CH), 3.63 (s, 2H, N–CH2–), 3.98 (s, 6H, 2OCH3), 6.95–7.68 (m, 13 H, CH-arom.); 13C NMR (100 MHz, DMSO-d6): δ 42.8, 44.5, 53.9, 61.8, 126.8, 127.4, 127.9, 128.3, 128.9, 129.6, 130.9, 138.7, 181.2.
1-Benzyl-2,6-bis(4-nitrophenyl)piperidin-4-one 1d
Brown powder, yield 89%, mp 270–272 °C; Anal. Calcd. for (C24H21N3O5, 431.44): C, 66.81; H, 4.91; N, 9.74. Found: C, 66.38; H, 4.61; N, 9.60%. IR (KBr, cm−1): 1721 (C = O); 1H NMR (400 MHz, δ, DMSO-d6): δ 3.05 (d, 4H, J = 13.6 Hz, 2CH2), 3.24 (t, 2H, J = 5.4 Hz, 2CH), 3.68 (s, 2H, N–CH2–), 6.92–7.78 (m, 13 H, CH-arom.); 13C NMR (100 MHz, DMSO-d6): δ 42.6, 44.5, 53.9, 126.7, 127.3, 127.8, 128.2, 128.6, 129.5, 130.5, 138.8, 181.4.
Synthesis of 1-benzyl-2,6-diaryl-4-(phenylamino)piperidine-4-carbonitrile 2a–d
In a two necked, round bottomed flask equipped with a reflux condenser and pressure equalising dropping funnel. The flask was charged with 1-benzyl-2,6-diarylpiperidone-4-one 1a–d (0.01 mol), aniline (3.72 g, 0.04 mol), solid KCN (2.60 g, 0.04 mol) and CH2Cl2 (50 ml). The mixture was cooled (∼ 5 °C, ice-acetone bath) and stirred magnetically. The dropping funnel was charged with AcOH (18.0 ml, 0.3 mol) which was added drop-wise, over ∼3 h period. The stirring was continued, and the mixture was gently heated at 45–50 °C (oil bath) for 24 h. After cooling to ∼20 °C, the contents were poured (hood, gas-mask) onto crushed ice (500 g), then partially neutralised with K2CO3 (40% solution) to pH ∼10. The formed solid was collected by filtration, washed thoroughly with water and then dried. The obtained 4-phenylaminopiperidine-4-carbonitrile 2a–d were crystallised from EtOH and kept under reduced pressure.
1-Benzyl-2,6-diphenyl-4-(phenylamino)piperidine-4-carbonitrile 2a
White crystals, yield 78%, m.p. 165–168 °C; Anal. Calcd. for (C31H29N3, 443.24): C, 83.94; H, 6.59; N, 9.47. Found: C, 83.66; H, 6.59; N, 9.34%. IR: 3245 (NH), 2227 (CN); 1H-NMR (400 MHz, δ, DMSO-d6): 1.92 (d, 4H, J = 10.8 Hz, 2CH2), 3.08 (t, 2H, J = 13.7 Hz, 2CH), 3.56 (s, 2H, N–CH2), 6.88–7.55 (m, 20 H, CH-arom.), 9.12 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 36.09, 49.27, 58.78, 62.58, 117.78, 126.93, 127.26, 128.96, 129.00, 129.31, 130.2, 130.8, 131.6, 132.1, 134.02, 138.00, 143.29.
1-Benzyl-2,6-bis(4-chlorophenyl)-4-(phenylamino)piperidine-4-carbonitrile 2b
Pale yellow needles, yield 80%, mp: 143–145 °C; Anal. Calcd. for (C31H27Cl2N3, 511.16): C, 72.65; H, 5.31; Cl, 13.84; N, 8.20. Found: C, 72.40; H, 5.10; Cl, 13.66; N, 8.02%. IR: 3252 (NH), 2221 (CN); 1H-NMR (400 MHz, δ, DMSO-d6): 1.93 (d, 4H, J = 10.8 Hz, 2CH2), 3.11 (t, 2H, J = 13.7 Hz, 2CH), 3.57 (s, 2H, N–CH2), 6.90–7.73 (m, 18 H, CH-arom.), 9.11 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 36.11, 49.30, 58.80, 62.62, 117.83, 126.96, 127.34, 128.70, 129.08, 129.48, 130.30, 130.81, 131.74, 132.32, 134.61, 138.06, 143.55.
1-Benzyl-2,6-bis(4-methoxyphenyl)-4-(phenylamino)piperidine-4-carbonitrile 2c
White crystals yield 82%, mp: 166–168 °C; Anal. Calcd. for (C33H33N3O2, 503.26): C, 78.70; H, 6.60; N, 8.34. Found: C, 78.52; H, 6.43; N, 8.05%. IR: 3252 (NH), 2220 (CN); 1H-NMR (400 MHz, δ, DMSO-d6): 1.91 (d, 4H, J = 10.8 Hz, 2CH2), 3.09 (t, 2H, J = 13.7 Hz, 2CH), 3.55 (s, 2H, N–CH2), 3.98 (s, 6H, 2OCH3), 6.91–7.65 (m, 18 H, CH-arom.), 9.11 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 36.01, 49.24, 57.80, 58.83, 62.45, 117.65, 126.88, 127.25, 128.65, 129.00, 129.43, 130.27, 130.68, 131.69, 132.23, 134.55, 138.01, 143.48.
1-Benzyl-2,6-bis(4-nitrophenyl)-4-(phenylamino)piperidine-4-carbonitrile 2d
Dark brown powder, yield 82%, mp: 166–168 °C; Anal. Calcd. for (C31H27N5O4, 533.21): C, 69.78; H, 5.10; N, 13.13. Found: C, 69.50; H, 4.96; N, 12.86%. IR: 3258 (NH), 2227 (CN); 1H-NMR (400 MHz, δ, DMSO-d6): 1.96 (d, 4H, J = 10.8 Hz, 2CH2), 3.13 (t, 2H, J = 13.7 Hz, 2CH), 3.61 (s, 2H, N–CH2), 6.91–7.77 (m, 18 H, CH-arom.), 9.16 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 36.13, 49.53, 58.92, 62.56, 117.87, 126.90, 127.33, 128.74, 129.10, 129.66, 130.35, 130.91, 131.82, 132.36, 134.68, 138.23, 143.66.
Synthesis of 4-(aminomethyl)- 4-(aminomethyl)-1-benzyl-2,6-diaryl-N-phenylpiperidin-4-amine 3a–d
The nitrile 2a–d (5 mmol) in ether was added to lithium aluminium hydride LAH (1.52 g, 40 mmoles) in ether (15 ml) and stirred at room temperature overnight. Sodium hydroxide (2.8 ml, 10% solution) was added at 0 °C and after 30 min, water (5 ml) was added. The formed precipitate was filtered and washed copiously with ether. The combined, washed and dried organic layers were evaporated “under vacuum” to give the desired products 3a–d.
4-(Aminomethyl)-1-benzyl-N,2,6-triphenylpiperidin-4-amine 3a
Yellow crystals, yield 77%, mp: 130–132 °C; Anal. Calcd. for (C31H33N3, 447.27): C, 83.18; H, 7.43; N, 9.39. Found: C, 82.97; H, 7.18; N, 9.11%. IR: 3318, 3258, 3223 (NH2+NH); 1H-NMR (400 MHz, δ, DMSO-d6): 1.52 (br, 2H, NH2), 1.88 (d, 4H, J = 10.5 Hz, 2CH2), 3.10 (t, 2H, J = 13.6 Hz, 2CH), 3.41 (s, 2H, CH2), 3.64 (s, 2H, N–CH2), 6.91–7.77 (m, 20 H, CH-arom.), 9.08 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 34.12, 44.43, 49.41, 58.76, 62.51, 126.85, 127.29, 128.71, 129.05, 129.58, 130.32, 130.85, 131.77, 132.33, 134.65, 138.20, 143.61.
4-(Aminomethyl)-1-benzyl-2,6-bis(4-chlorophenyl)-N-phenylpiperidin-4-amine
Pale yellow needles, yield 85%, mp: 145–148 °C; Anal. Calcd. for (C31H13Cl2N3, 515.19): C, 72.09; H, 6.05; N, 8.14, Cl, 13.73. Found: C, 71.90; H, 5.72; N, 7.78, Cl, 13.53%. IR: 3324, 3266, 3225 (NH2+NH); 1H-NMR (400 MHz, δ, DMSO-d6): 1.55 (br, 2H, NH2), 1.87 (d, 4H, J = 10.5 Hz, 2CH2), 3.14 (t, 2H, J = 13.7 Hz, 2CH), 3.42 (s, 2H, CH2), 3.65 (s, 2H, N–CH2), 6.93–7.77 (m, 18 H, CH-arom.), 9.11 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 34.33, 44.47, 49.45, 58.79, 62.55, 126.89, 127.33, 128.76, 129.07, 129.62, 130.327, 130.88, 131.75, 132.31, 134.60, 138.25, 144.64.
4-(Aminomethyl)-1-benzyl-2,6-bis(4-methoxyphenyl)-N-phenylpiperidin-4-amine 3c
White crystals yield 80%, mp: 158–160 °C; Anal. Calcd. for (C33H37N3O2, 507.29): C, 78.07; H, 7.35; N, 8.28. Found: C, 77.82; H, 7.01; N, 8.08%. IR: 3252, 3262, 3220 (NH2+NH); 1H-NMR (400 MHz, δ, DMSO-d6): 1.87 (d, 4H, J = 10.8 Hz, 2CH2), 3.10 (t, 2H, J = 13.7 Hz, 2CH), 3.40 (s, 2H, CH2), 3.62 (s, 2H, N–CH2), 3.97 (s, 6H, 2OCH3), 6.90–7.65 (m, 18 H, CH-arom.), 9.13 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 36.01, 44,18, 49.24, 57.80, 58.83, 62.45, 126.88, 127.25, 128.65, 129.00, 129.43, 130.27, 130.68, 131.69, 132.23, 134.55, 138.01, 143.48.
4-(Aminomethyl)-1-benzyl-2,6-bis(4-nitrophenyl)-N-phenylpiperidin-4-amine 3d
Yellowish-brown crystals yield 77%, mp: 184–187 °C; Anal. Calcd. for (C31H31N5O4, 537.61): C, 69.26; H, 5.81; N, 13.03. Found: C, 69.46; H, 4.90; N, 12.77%. IR: 3343, 3257. 3212 (NH2+NH); 1H-NMR (400 MHz, δ, DMSO-d6): 1.98 (d, 4H, J = 10.8 Hz, 2CH2), 3.16 (t, 2H, J = 13.7 Hz, 2CH), 3.42 (s, 2H, CH2), 3.67 (s, 2H, N–CH2), 6.96–7.79 (m, 18 H, CH-arom.), 9.15 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 36.25, 49.58, 57.97, 58.95, 62.58, 126.94, 127.39, 128.75, 129.11, 129.69, 130.38, 130.93, 131.86, 132.39, 134.65, 138.14, 144.01.
Synthesis of 1-benzyl-2,6-bis(diaryl)-4-(phenylamino)piperidine-4-carboxamide 4a–d
Method A: 4-Phenylamino piperidine-4-carbonitrile 2a–d (0.1 mol) was dissolved in conc. H2SO4 (50 ml) at ∼2 °C, in a single necked flask with a CaCl2 trapCitation34. The reaction mixture was left at room temperature overnight (24 h). Water was added (∼150 ml) to the precipitated dihydrogen sulphate of amide and then the reaction mixture was neutralised with Na2CO3. The precipitated free amides 4a–d were filtered off, washed with water and air dried.
Method B: Acidified kaolin (2% w/w) (150 mg) was added to a solution of 4-Phenylamino piperidine-4-carbonitrile 2a–d (4 mmol) in water (10 ml) and refluxed for 24 h. After completion of the reaction (as indicated by TLC), the reaction mixture was cooled to room temperature and neutralised with sodium hydroxide solution (4 N) to pH = 7 carefully. The reaction mixture was filtered and extracted with ethyl acetate (2 × 20 ml). The organic layer dried over sodium sulphate and evaporated. The obtained amides were crystallised in H2O-EtOH.
Preparation of the acidified kaolin with sulphuric acid (2% w/w) :
Kaolin (7.5 g) was treated with concentrated sulphuric acid (0.15 g, 0.08 ml) and stirred for 1 hCitation34. The prepared acidified kaolin (2% w/w), was stored for further applications.
1-Benzyl-2,6-diphenyl-4-(phenylamino)piperidine-4-carboxamide 4a
Pale yellow crystals, yield 72%, mp: 180–182 °C; Anal. Calcd. for (C31H31N3O, 461.25): C, 80.66; H, 6.77; N, 9.10. Found: C, 80.32; H, 6.42; N, 9.89%. IR: 3446, 3358, 3229 (NH2+NH), 1679 (C = O); 1H-NMR (400 MHz, δ, DMSO-d6): 1.92 (d, J = 11.8, 4H, 2CH2), 3.04 (t, 2H, J = 13.6 Hz, 2CH), 3.62 (s, 2H, N–CH2), 4.06 (s, 1H, NH), 5.34 (br, 2H, CONH2), 6.85–7.57 (m, 20 H, CH-arom.); 13C-NMR (100 MHz, DMSO-d6): 31.12, 44.43, 48.41, 62.85, 124.10, 126.80, 127.33, 128.68, 129.00, 129.67, 130.41, 130.98, 131.65, 132.12, 134.13, 138.65, 143.72, 178.66.
1-Benzyl-2,6-bis(4-chlorophenyl)-4-(phenylamino)piperidine-4-carboxamide 4b
Yellow needles, yield 72%, mp: 200–202 °C; Anal. Calcd. for (C31H29Cl2N3O, 530.49): C, 70.19; H, 5.51; N, 7.92, Cl, 13.37. Found: C, 69.87; H, 5.40; N, 7.54, Cl, 13.01%. IR: 3448, 3353, 3229 (NH2+NH), 1678 (C = O); 1H-NMR (400 MHz, δ, DMSO-d6): 1.92 (d, J = 11.8, 4H, 2CH2), 2.34 (t, 2H, J = 13.6 Hz, 2CH), 3.62 (s, 2H, N–CH2), 4.03 (s, 1H, NH), 5.34 (br, 2H, CONH2), 6.85–7.57 (m, 20 H, CH-arom.), 9.03 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 31.12, 44.43, 48.41, 57.82, 58.10, 62.85, 124.10, 126.80, 127.33, 128.68, 129.00, 129.67, 130.41, 130.98, 131.65, 132.12, 134.13, 138.65, 143.72, 178.66.
1-Benzyl-2,6-bis(4-methoxyphenyl)-4-(phenylamino)piperidine-4-carboxamide 4c
Pale yellow needles, yield 75%, mp: 193–195 °C; Anal. Calcd. for (C33H35N3O3, 521.27): C, 75.98; H, 6.76; N, 8.06. Found: C, 80.32; H, 6.42; N, 9.89%. IR: 3446, 3358, 3229 (NH2+NH), 1679 (C = O); 1H-NMR (400 MHz, δ, DMSO-d6): 1.93 (d, J = 11.8, 4H, 2CH2), 2.36 (t, 2H, J = 13.6 Hz, 2CH), 3.65 (s, 2H, N–CH2), 3.98 (s, 6H, 2OCH3), 4.04 (s, 1H, NH), 5.38 (br, 2H, CONH2), 6.80–7.62 (m, 18 H, CH-arom.); 13C-NMR (100 MHz, DMSO-d6): 31.13, 44.45, 48.40, 57.81, 58.15, 62.80, 63.44, 124.08, 126.77, 127.30, 128.55, 129.09, 129.61, 130.36, 130.76, 131.52, 132.07, 134.25, 138.04, 144.65, 178.04.
1-Benzyl-2,6-bis(4-nitrophenyl)-4-(phenylamino)piperidine-4-carboxamide 4d
Brown crystals yield 72%, mp: 178–180 °C; Anal. Calcd. for (C31H29N5O5, 551.22): C, 67.50; H, 5.30; N, 12.70. Found: C, 67.22; H, 5.12; N, 12.46%. IR: 3453, 3356, 3236 (NH2+NH), 1677 (C = O); 1H-NMR (400 MHz, δ, DMSO-d6): 1.95 (d, J = 11.8, 4H, 2CH2), 3.06 (t, 2H, J = 13.6 Hz, 2CH), 3.67 (s, 2H, N–CH2), 4.045 (s, 1H, NH), 5.37 (br, 2H, CONH2), 6.87–7.66 (m, 18 H, CH-arom.); 13C-NMR (100 MHz, DMSO-d6): 31.22, 44.48, 48.53, 57.87, 58.23, 62.81, 124.11, 126.80, 127.33, 128.59, 129.13, 129.67, 130.35, 130.78, 131.55, 132.12, 134.18, 138.13, 144.38, 178.10.
Synthesis of spiro heterocycles 5a–10a
A mixture of 4-(aminomethyl)-4-(aminomethyl)-1-benzyl-2,6-diaryl-N-phenylpiperidin-4-amine 3a–d or 1-benzyl-4-(phenylamino)-2,6-diarylpiperidine-4-carboxamide 4a–d (1.0 mmol) in ethanol (4 ml) at room temperature, ethyl chloroformate, ethyl chloroacetate or 2-benzylidenemalononitrile (1.0 mmol) and Piperidinium acetate-IL (30 mg) were added to a round-bottom flask equipped with a magnetic stir bar and condenser. The mixture was heated at 70 °C for 5 h and the reaction progress was monitored by TLC (EtOAc/hexane = 4:8). After completion of the reaction, the mixture was cooled to room temperature for 45 min and poured on crushed ice. Thus, acquired solid was filtered, dried, and purified by crystallisation using ethanol as a solvent.
8-Benzyl-1,7,9-triphenyl-1,3,8-triazaspiro[4.5]decan-2-one 5a
White solid, yield 70%, mp: 128–130 °C; Anal. Calcd. for (C32H31N3O, 473.25): C, 81.15; H, 6.60; N, 8.87. Found: C, 80.92; H, 6.33; N, 8.57%. IR: 3241 (NH), 1739 (C = O); 1H-NMR (400 MHz, δ, DMSO-d6): 1.90 (d, J = 11.2, 4H, 2CH2), 2.84 (t, 2H, J = 13.6 Hz, 2CH), 3.44 (s, 1H, CH2Imidazoline), 3.65 (s, 2H, N–CH2), 6.94–7.60 (m, 20 H, CH-arom.), 9.03 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 31.34, 44.78, 58.10, 62.87, 78.21 124.12, 126.65, 127.37, 128.68, 129.67, 130.41, 130.98, 131.65, 132.12, 134.13, 138.65, 143.33, 180.54.
9-Benzyl-1,8,10-triphenyl-1,4,9-triazaspiro[5.5]undecan-2-one 6a
Pale yellow solid, yield 74%, mp: 150–153 °C; Anal. Calcd. for (C33H33N3O, 487.26): C, 81.25; H, 6.82; N, 8.62. Found: C, 80.98; H, 6.48; N, 8.45%. IR: 3234 (NH), 1738 (C = O); 1H-NMR (400 MHz, δ, DMSO-d6): 1.92 (d, J = 11.2, 4H, 2CH2), 2.82 (t, 2H, J = 13.6 Hz, 2CH), 3.42 (s, 2H, CH2), 3.49, (s, 2H, CH2), 3.61 (s, 2H, N–CH2), 6.92–7.62 (m, 20 H, CH-arom.), 8.78 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 31.30, 44.04, 58.23, 62.81, 72.44, 78.27, 124.23, 126.51, 127.11, 128.42, 129.55, 130.31, 130.74, 131.52, 132.05, 134.22, 138.23, 143.08, 180.03.
8-Amino-3-benzyl-2,4,7,10-tetraphenyl-3,7,11-triazaspiro[5.6]dodec-8-ene-9-carbonitrile 7a
White solid, yield 62%, mp: > 300 °C; Anal. Calcd. for (C41H39N5, 601.32): C, 81.83; H, 6.53; N, 11.64. Found: C, 81.38; H, 6.433 N, 11.40%. IR: 3378, 3280, 3230 (NH2,NH); 1H-NMR (400 MHz, δ, DMSO-d6): 1.88 (d, J = 11.4, 4H, 2CH2), 2.33 (t, 2H, J = 13.6 Hz, 2CH), 3.43 (s, 1H, CH2Diazepine), 3.67 (s, 2H, N–CH2), 5.36 (s, 1H, CHDiazepine), 5.65 (br, 2H, NH2), 6.86–7.62 (m, 25 H, CH-arom.), 8.81 (s, 1H, NH).
8-Benzyl-1,7,9-triphenyl-1,3,8-triazaspiro[4.5]decane-2,4-dione 8a
Pale yellow solid, yield 68%, mp: 188–190 °C; Anal. Calcd. for (C32H29N3O2, 487.23): C, 78.82; H, 5.99; N, 8.62. Found: C, 78.55; H, 5.67; N, 8.37%. IR: 3223 (NH), 1739, 1707 (2 C = O); 1H-NMR (400 MHz, δ, DMSO-d6): 1.90 (d, J = 11.5, 4H, 2CH2), 3.03 (t, 2H, J = 13.6 Hz, 2CH), 3.66 (s, 2H, N–CH2), 6.98–7.65 (m, 20 H, CH-arom.), 8.67 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 31.50, 44.74, 62.85 78.20, 124.13, 126.62, 127.30, 128.49, 129.55, 130.33, 130.91, 131.51, 132.03, 134.09, 138.48, 143.21, 181.43, 182.55.
9-Benzyl-1,8,10-triphenyl-1,4,9-triazaspiro[5.5]undecane-2,5-dione 9a
White solid, yield 70%, mp: 201–203 °C; Anal. Calcd. for (C33H31N3O2, 501.24): C, 79.01; H, 6.23; N, 8.38. Found: C, 78.76; H, 6.02; N, 8.13%. IR: 3238 (NH), 1736, 1708 (2 C = O); 1H-NMR (400 MHz, δ, DMSO-d6): 1.91 (d, J = 11.3, 4H, 2CH2), 3.02 (t, 2H, J = 13.5 Hz, 2CH), 3.48, (s, 2H, CH2), 3.63 (s, 2H, N–CH2), 6.91–7.60 (m, 20 H, CH-arom.), 8.78 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 31.32, 44.10, 57.28, 58.26, 62.87, 72.44, 78.26, 124.20, 126.56, 127.14, 128.45, 129.50, 130.38, 130.67, 131.49, 132.10, 134.18, 138.23, 143.08, 180.03, 182.67.
8-Amino-3-benzyl-12-oxo-2,4,7,10-tetraphenyl-3,7,11-triazaspiro[5.6]dodec-8-ene-9-carbonitrile 10a
White solid, yield 62%, mp: > 300 °C; Anal. Calcd. for (C41H39N5, 601.32): C, 81.83; H, 6.53; N, 11.64. Found: C, 81.38; H, 6.43; N, 11.40%. IR: 3374, 3272, 3225 (NH2,NH); 1H-NMR (400 MHz, δ, DMSO-d6): 1.88 (d, J = 11.4, 2H, CH2), 2.33 (t, 2H, J = 13.6 Hz, 2CH), 3.65 (s, 2H, N–CH2), 5.32 (s, 1H, CHDiazepine), 5.65, br, 2H, NH2), 6.80–7.68 (m, 25 H, CH-arom.), 8.78 (s, 1H, NH).
Synthesis of 8-benzyl-4-imino-1,3,7,9-tetraphenyl-1,3,8-triazaspiro[4.5]decan-2-one 11a and 8-benzyl-4-imino-1,3,7,9-tetraphenyl-1,3,8-triazaspiro[4.5]decane-2-thione 11b
An equimolar mixture of 1-benzyl-2,6-diphenyl-4-(phenylamino)piperidine-4-carbonitrile 2a (0.001 mol) and phenyl isocyanate or phenylisothiocyanate (0.001 mol) was mixed in ethanol (10 ml) then was treated with piperidinium acetate-IL (30 mg). The reaction mixture was heated under reflux for 4–6 h, then left to cool. The formed precipitates were collected by filtration, washed thoroughly with water and then recrystallised from ethanol to give the corresponding spiro heterocycles.
8-Benzyl-4-imino-1,3,7,9-tetraphenyl-1,3,8-triazaspiro[4.5]decan-2-one 11a
Pale yellow crystals, yield 78%, mp: 180–183 °C; Anal. Calcd. for (C38H34N4O, 562.27): C, 81.11; H, 6.09; N, 9.96. Found: C, 80.87; H, 5.88; N, 9.59%. IR: 3310 (NH), 1705 (C = O); 1H-NMR (400 MHz, δ, DMSO-d6): 1.92 (d, J = 11.3, 4H, 2CH2), 3.03 (t, 2H, J = 13.5 Hz, 2CH), 3.61 (s, 2H, N–CH2), 6.90–7.68 (m, 25 H, CH-arom.), 9.15 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 31.32, 44.10, 62.87, 78.26, 121.32, 122.45, 123.80, 123.87, 124.20, 126.56, 127.14, 128.45, 129.50, 130.38, 130.67, 131.49, 132.10, 134.18, 138.23, 143.08, 154.23, 180.01.
8-Benzyl-4-imino-1,3,7,9-tetraphenyl-1,3,8-triazaspiro[4.5]decane-2-thione 11b
Yellow solid, yield 74%, mp: 168–170 °C; Anal. Calcd. for (C38H34N4S, 578.25): C, 78.86; H, 5.92; N, 9.68; S, 5.54. Found: C, 78.61; H, 5.67; N, 9.51; S, 5.28%. IR: 3310 (NH), 1225 (C = S); 1H-NMR (400 MHz, δ, DMSO-d6): 1.90 (d, J = 11.3, 4H, 2CH2), 3.04 (t, 2H, J = 13.5 Hz, 2CH), 3.64 (s, 2H, N–CH2), 6.92–7.65 (m, 25 H, CH-arom.), 8.87 (s, 1H, NH); 13C-NMR (100 MHz, DMSO-d6): 31.30, 44.04, 62.65, 78.18, 121.28, 122.41, 123.74, 123.82, 124.15, 126.53, 127.11, 128.40, 129.47, 130.33, 130.62, 131.41, 132.05, 134.11, 138.08, 143.01, 154.12, 178.45.
Synthesis of 8-benzyl-7,9-diphenyl-4-thia-1,2,8-triazaspiro[4.5]decan-3-imine 12 and 8-benzyl-7,9-diphenyl-1,4,8-triazaspiro[4.5]decane 13
In a round bottomed flask, 1-benzyl-2,6-diphenylpiperidin-4-one 1a (0.001 mol) and thiosemicarbazide or ethylenediamine (0.015 mol) was mixed in ethanol (10 ml) then was treated with piperidinium acetate-IL (30 mg). The reaction mixture was heated under reflux for 6 h, then left to cool. The formed precipitates were collected by filtration, washed thoroughly with water and then recrystallised from ethanol where the corresponding spiro heterocycles 12 and 13 were obtained.
8-Benzyl-7,9-diphenyl-4-thia-1,2,8-triazaspiro[4.5]decan-3-imine 12
Bright yellow crystals: yield 85%, mp 132–234 °C; Anal. Calcd. for (C25H26N4S, 414.19): C, 72.43; H, 6.32; N, 13.51; S, 7.73. Found: C, 72.11; H, 6.05; N, 13.20; S,7.46%. IR (KBr, cm−1): 3312, 3238, 3186 (3NH), 1638 (C = NH); 1H NMR (400 MHz, δ, DMSO-d6): δ 1.92 (d, 4H, J = 13.6 Hz, 2CH2), 3.03 (t, 2H, J = 5.4 Hz, 2 CH), 3.65 (s, 2H, N–CH2–), 4.43 (br, 1H, NH), 7.59–6.91 (m, 15 H, CH-arom.), 8.76 (br, 2H, 2NH); 13C NMR (100 MHz, DMSO-d6): δ 42.23, 44.32, 53.78, 76.21, 126.12, 127.26, 127.54, 128.21, 128.37, 129.26, 130.12, 130.23, 154.21.
8-Benzyl-7,9-diphenyl-1,4,8-triazaspiro[4.5]decane 13
Pale yellow solid: yield 83%, mp 209–211 °C; Anal. Calcd. for (C26H29N3, 383.24): C, 81.42; H, 7.62; N, 10.96. Found: C, 81.09; H, 7.34; N, 10.65%. IR (KBr, cm−1): 3228 (2NH); 1H NMR (400 MHz, δ, DMSO-d6): δ 1.90 (d, 4H, J = 13.6 Hz, 2CH2), 3.07 (t, 2H, J = 5.4 Hz, 2 CH), 4.13 (m, 4H, 2CH2Imidazoline), 5.98 (s, 2H, 2NHImidazoline), 6.92–7.56 (m, 15 H, CH-arom.); 13C NMR (100 MHz, DMSO-d6): δ 42.07, 43.09, 44.19, 62.34, 73.27, 126.23, 127.42, 127.18, 128.23, 128.44, 129.26, 130.12, 130.28, 154.55.
Biological evaluation
In vitro antileishmanial activity
We used both promastigote and amastigote forms of L. major strain for the in vitro evaluation. All experimental steps were carried out as reported earlierCitation35,Citation36. The value of the compound concentration causing 50% inhibition (IC50) was calculated. The software employed is Graph Pad Prism 6 software (GraphPad Software, San Diego, CA, USA). The coefficient of determination (R2) displayed good relationship range, more than 0.9.
Reversal of antileishmanial activity of most active compounds by folic acid and folinic acid
This experiment was performed on the in vitro growth assay for the promastigote and based on the previously published methodologyCitation30. All experimental steps were performed as reported earlierCitation35.
In vitro cytotoxicity testing
The most active compounds were conducted with different concentrations ranging from 0–100 μM in a 96-well plate 1 × 105 cells/well for 72 h at 37 °C incubator, with 95% humidity and 5% CO2. All experimental steps were conducted as reported earlierCitation35,Citation37.
Molecular modelling
Molecular docking
The X-ray structures of Lm PTR1 enzyme was extracted from the Protein Data Bank (PDB) with PDB ID: 2BFMCitation38. Redundant chains, non-essential ions, water molecules and ligands were discarded. The co-factor NADPH was kept. The search box around the X-ray co-crystal ligand was set 20 × 24 × 20 with grid box spacing of 1 Å, while the exhaustiveness option we set to 150. The protein PDB file was converted into PDBQT file by employing a python script (prepare_receptor4.py) provided by the MGLTools package (version 1.5.4)Citation39 for AutoDock Vina (version 1.1.2)Citation40 docking experiments.
The 3 D conformations of the compounds 8a and 9a were constructed by OpenBabelCitation41 then converted into PDBQT files for AutoDock Vina. The 3 D and 2 D depictions of the docking poses in the protein binding sites were generated via Discovery Studio Visualiser V21.1.0.20298Citation6.
Molecular dynamics
The molecular dynamics simulations were carried out as reported earlier in some proceduresCitation42,Citation43. Molecular dynamics simulations and systems build up were carried out using GROMACS 2020.3Citation44. The protein-ligand complex was solvated in a triclinic box of SPC216 with explicit water modelCitation45. System was then neutralised by NaCl molecules at 0.1 M concentration. Steepest descent minimisation algorithm was applied for system energy minimisation setting 10 kJ/mol and 50,000 steps as convergence criteria. NVT followed by NPT equilibration were completed for 500 ps each at 300 K temperature and 1 atm pressure. Then, a production run was carried out for 50 ns at NPT ensemble. The coordinates of the trajectory were saved each 10 ps time interval resulting in 5000 frames for the whole 50 ns simulation time. The V-rescale modified Berendsen thermostatCitation46 was used for temperature coupling for each equilibration run, while Berendsen couplingCitation47 was used for pressure coupling with 2 ps time constant for equilibration and production runs. However, Parrinello-Rahman pressure coupling schemeCitation48 was employed for pressure coupling for the production runs. A Verlet cut-off-scheme was used for searching neighbouring atoms and Van Der Waals calculations with cut-off and switch list distances of 1.2 and 1.0 nm, respectively. Particle Mesh Ewald methodCitation49 was used for the calculations long-range electrostatics within 1.2 nm. Bond lengths were constrained using the LINear Constraint Solver (LINCS) algorithmCitation50. CHARMM36 all-atom force fieldCitation51 was used for topology and parameter generation of the protein molecules, and SwissParam serverCitation52 was used for ligand parameterisation. For all simulations, a leap-frog integrator was used with a steps size of 2 fs. Different analysis metrics, such as root mean squared deviation (RMSD), radius of gyration (Rg) and root mean squared fluctuation (RMSF) were calculated via GROMCS and were plotted using XMGRACECitation53.
Conclusion
The synthesis conditions of some spiro-piperidine derivatives were optimised via the eco-friendly ionic liquids in a one-pot fashion in acceptable yields. The compounds were evaluated for their in vitro antileishmanial activity against Leishmania major promastigote and amastigote forms. Interestingly, the results showed that the tested compounds exhibited antiamastigote activity in a range of IC50 values from 0.50 to 5.44 μM, compared to miltefosine (8.08 μM). Besides, the antipromastigote activity revealed promising results from 0.41 to 5.39 μM, compared to miltefosine (7.89 μM). These results highlight compounds 8a and 9a to be the most potent with antiamastigote activity of 0.89 μM and 0.50 μM, respectively. Furthermore, reversal of antileishmanial activity of both 8a and 9a via folic and folinic acids demonstrated analogous results to the positive control Trimethoprim. This defines an antifolate mechanism of these compounds proposing both leishmanial DHFR-TS and PTR1 enzymes as putative targets.
The in vitro cytotoxicity test of the best candidates presented high selectivity indices emphasising their safety on mammalian cells. Molecular docking of 8a and 9a against the putative Lm-PTR1 targets rationalised the observed antileishmanial activity. Their docking poses displayed superior performances compared to the PTR1 inhibitor, trimethoprim. Remarkably, molecular dynamics simulations for 50 ns of the unliganded PTR1, 8a-PTR1 and 9a-PTR1 systems, emphasised the stable binding of these compounds with PTR1. These findings indicate that both 8a and 9a exert their antileishmanial activity via inhibiting leishmanial PTR1.As an outlook, these results represent a fruitful template to develop a focussed library of these spiro heterocycles bearing piperidine for targeting additional leishmanial/NTDs targets, such as trypanothione reductase, for enhancing synergistic actions and tacking resistance mechanisms.
Supplemental Material
Download PDF (987.9 KB)Acknowledgement
TMI would like to acknowledge Bibliotheca Alexandrina High-Performance Computing (BA-HPC) for granting access to perform the molecular dynamics simulations. We thank the the Deanship of Scientific Research at Umm Al-Qura University for the support offered via (grant code: 23UQU4290565DSR114).
Disclosure statement
The authors report no conflicts of interest.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- Goncalves GA, Spillere AR, das Neves GM, Kagami LP, von Poser GL, et al. Natural and synthetic coumarins as antileishmanial agents: a review. Eur J Med Chem. 2020;203:112514.
- Bekhit AA, El-Agroudy E, Helmy A, Ibrahim TM, Shavandi A, et al. Leishmania treatment and prevention: natural and synthesized drugs. Eur J Med Chem. 2018;160:229–244.
- Pigott DM, Bhatt S, Golding N, Duda KA, Battle KE, Brady OJ, Messina JP, Balard Y, Bastien P, Pratlong F, et al. Global distribution maps of the leishmaniases. eLife. 2014;3:e02851.
- World Health Organization (WHO). Leishmaniasis. 2020. Available from: https://www.who.int/health-topics/leishmaniasis#tab=tab_1.
- Ogutu B. Artemether and lumefantrine for the treatment of uncomplicated Plasmodium falciparum malaria in sub-Saharan Africa. Expert Opin Pharmacother. 2013;14:643–654.
- Nwaka S, Hudson A. Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov. 2006;5:941–955.
- Rajasekaran R, Chen YP. Potential therapeutic targets and the role of technology in developing novel antileishmanial drugs. Drug Discov Today. 2015;20:958–968.
- Vickers TJ, Beverley SM. Folate metabolic pathways in Leishmania. Essays Biochem. 2011;51:63–80.
- Nare B, Hardy LW, Beverley SM. The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J Biol Chem. 1997;272:13883–13891.
- Corona P, Gibellini F, Cavalli A, Saxena P, Carta A, et al. Structure-based selectivity optimization of piperidine–pteridine derivatives as potent Leishmania pteridine reductase inhibitors. J Med Chem. 2012;55:8318–8329.
- Dube D, Periwal V, Kumar M, Sharma S, Singh TP, et al. 3D-QSAR based pharmacophore modeling and virtual screening for identification of novel pteridine reductase inhibitors. J Mol Model. 2012;18:1701–1711.
- de Souza Moreira D, Ferreira RF, Murta SMF. Molecular characterization and functional analysis of pteridine reductase in wild-type and antimony-resistant Leishmania lines. Exp Parasitol. 2016;160:60–66.
- Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem. 2014;57:10257–10274.
- Bailey PD, Smith PD, Morgan KM, Rosair GM. The use of the aza-Diels–Alder reaction in the synthesis of pinidine and other piperidine alkaloids. Tetrahedron Lett . 2002;43(6):1071–1074.
- Amat M, Llor N, Hidalgo J, Escolano C, Bosch J. Enantioselective synthesis of piperidine, indolizidine, and quinolizidine alkaloids from a phenylglycinol-derived δ-lactam. J Org Chem. 2003;68:1919–1928.
- Tortolani DR, Poss MA. A convenient synthesis to N-aryl-substituted 4-piperidones. Org Lett. 1999;1(8):1261–1262.
- Weintraub PM, Sabol JS, Kane JM, Borcherding DR. Recent advances in the synthesis of piperidones and piperidines. Tetrahedron. 2003;59(17):2953–2989.
- Jagt RBC, de Vries JG, Feringa BL, Minnaard AJ. Enantioselective synthesis of 2-aryl-4-piperidones via rhodium/phosphoramidite-catalyzed conjugate addition of arylboroxines. Org Lett. 2005;7(12):2433–2435.
- Cong W, Sun Y, Sun Y-F, Yan W-B, Zhang Y-L, Gao Z-F, Wang C-H, Hou G-G, Zhang J-J. Trifluoromethyl-substituted 3,5-bis(arylidene)-4-piperidones as potential anti-hepatoma and anti-inflammation agents by inhibiting NF-кB activation. J Enzyme Inhib Med Chem. 2021;36(1):1621–1630.
- Ganellin CR, Spickett RGW. Compounds affecting the central nervous system. I. 4-Piperidones and related compounds. J Med Chem. 1965;8(5):619–625.
- Baracu I, Dobre V, Niculescu-Duvaz I. Potential anticancer agents. XXVI. Spin labelled nitrosoureas. J Prakt Chem. 1985;327(4):667–674.
- Meyers MJ, Anderson EJ, McNitt SA, Krenning TM, Singh M, Xu J, Zeng W, Qin L, Xu W, Zhao S, et al. Evaluation of spiropiperidine hydantoins as a novel class of antimalarial agents. Bioorg Med Chem. 2015;23(16):5144–5150.
- Veale CGL. Unpacking the pathogen box—an open source tool for fighting neglected tropical disease. Chem Med Chem. 2019;14(4):386–453.
- Battista T, Colotti G, Ilari A, Fiorillo A. Targeting trypanothione reductase, a key enzyme in the redox trypanosomatid metabolism, to develop new drugs against leishmaniasis and trypanosomiases. Molecules. 2020;25(8):1924.
- Scala A, Cordaro M, Grassi G, Piperno A, Barberi G, Cascio A, Risitano F. Direct synthesis of C3-mono-functionalized oxindoles from N-unprotected 2-oxindole and their antileishmanial activity. Bioorg Med Chem. 2014;22(3):1063–1069.
- Saha S, Acharya C, Pal U, Chowdhury SR, Sarkar K, Maiti NC, Jaisankar P, Majumder HK. A novel spirooxindole derivative inhibits the growth of Leishmania donovani parasites both in vitro and in vivo by targeting type IB topoisomerase. Antimicrob Agents Chemother. 2016;60(10):6281–6293.
- Almeida FS, Sousa GLS, Rocha JC, Ribeiro FF, de Oliveira MR, de Lima Grisi TCS, Araújo DAM, de C Nobre MS, Castro RN, Amaral IPG, et al. In vitro anti-Leishmania activity and molecular docking of spiro-acridine compounds as potential multitarget agents against Leishmania infantum. Bioorg Med Chem Lett. 2021;49:128289.
- Welton T. Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem Rev. 1999;99(8):2071–2084.
- Mohamed MAA, Moustafa HM, El‐Remaily M. An efficient synthesis of some new azaspirocycloalkane derivatives from 1-anilinocycloalkanecarboxamide. Chem Sci. 2014;5:1–6.
- Mendoza-Martínez C, Galindo-Sevilla N, Correa-Basurto J, Ugalde-Saldivar VM, Rodríguez-Delgado RG, Hernández-Pineda J, Padierna-Mota C, Flores-Alamo M, Hernández-Luis F. Antileishmanial activity of quinazoline derivatives: synthesis, docking screens, molecular dynamic simulations and electrochemical studies. Eur J Med Chem. 2015;92:314–331.
- Tonelli M, Gabriele E, Piazza F, Basilico N, Parapini S, Tasso B, Loddo R, Sparatore F, Sparatore A. Benzimidazole derivatives endowed with potent antileishmanial activity. J Enzyme Inhib Med Chem. 2018;33(1):210–226.
- Arba M, Wahyudi ST, Brunt DJ, Paradis N, Wu C. Mechanistic insight on the remdesivir binding to RNA-Dependent RNA polymerase (RdRp) of SARS-cov-2. Comput Biol Med. 2021;129:104156.
- Ismail MI, Ragab HM, Bekhit AA, Ibrahim TM. Targeting multiple conformations of SARS-CoV2 papain-like protease for drug repositioning: an in-silico study. Comput Biol Med. 2021;131:104295.
- Gordi Z, Eshghi H. Natural kaolin supported sulfuric acid as an efficient catalyst for selective hydrolysis of nitriles to amides. J Korean Chem Soc. 2011;55(4):715–718.
- Temraz MG, Elzahhar PA, Din A, Bekhit EA, Bekhit AA, Labib HF. Anti-leishmanial click modifiable thiosemicarbazones: design, synthesis, biological evaluation and in silico studies. Eur J Med Chem. 2018;151:585–600.
- Bekhit AA, Lodebo ET, Hymete A, Ragab HM, Bekhit SA, Amagase K, Batubara A, Abourehab MAS, Bekhit AE-DA, Ibrahim TM, et al. New pyrazolylpyrazoline derivatives as dual acting antimalarial-antileishamanial agents: synthesis, biological evaluation and molecular modelling simulations. J Enzyme Inhib Med Chem. 2022;37(1):2320–2333.
- Eldehna WM, Almahli H, Ibrahim TM, Fares M, Al-Warhi T, Boeckler FM, Bekhit AA, Abdel-Aziz HA. Synthesis, in vitro biological evaluation and in silico studies of certain arylnicotinic acids conjugated with aryl (thio) semicarbazides as a novel class of anti-leishmanial agents. Eur J Med Chem. 2019;179:335–346.
- Schüttelkopf AW, Hardy LW, Beverley SM, Hunter WN. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J Mol Biol. 2005;352(1):105–116.
- Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17(1):57–61.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461.
- O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open babel: an open chemical toolbox. J Cheminform. 2011;3:33.
- Elghoneimy LK, Ismail MI, Boeckler FM, Azzazy HME, Ibrahim TM. Facilitating SARS CoV-2 RNA-Dependent RNA polymerase (RdRp) drug discovery by the aid of HCV NS5B palm subdomain binders: In silico approaches and benchmarking. Comput Biol Med. 2021;134:104468.
- Bekhit AA, Nasralla SN, El-Agroudy EJ, Hamouda N, El-Fattah AA, Bekhit SA, Amagase K, Ibrahim TM. Investigation of the anti-inflammatory and analgesic activities of promising pyrazole derivative. Eur J Pharm Sci. 2022;168:106080.
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25.
- Mark P, Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J Phys Chem A. 2001;105(43):9954–9960.
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1):014101.
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690.
- Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190.
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092.
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472.
- Huang J, MacKerell AD Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34(25):2135–2145.
- Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem. 2011;32(11):2359–2368.
- Turner P. XMGRACE, version 5.1. 19. Beaverton (OR): Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology; 2005.