
Abstract
A new series of piperine-carboximidamide hybrids VIa-k was developed as a new cytotoxic agent targeting EGFR, BRAF, and CDK2. The antiproliferative effect against four cancer cells was investigated against erlotinib. Hybrids VIc, VIf, VIg, VIi, and VIk have the highest antiproliferative activity. Compounds VIc, VIf, VIg, VIi, and VIk inhibited EGFR with IC50 values ranging from 96 to 127 nM. Compounds VIf and VIk had the most potent inhibitory activity as BRAFV600E (IC50 = 49 and 40 nM, respectively) and were discovered to be potent inhibitors of cancer cell proliferation (GI50 = 44 and 35 nM against four cancer cell lines, respectively). Compound VIk, the most effective derivative as an antiproliferative agent, demonstrated potent anti-CDK2 action with an IC50 value of 12 nM, which is 1.5-fold more potent than the reference dinaciclib. Finally, VIc, VIf, and VIk have a high capacity to inhibit LOX-IMVI cell line survival.
Graphical Abstract

A series of piperine-carboximidamide hybrids VIa-k was designed and synthesised.
The new compounds were evaluated as antiproliferative agent targeting EGFR, BRAF, and CDK2.
Antiproliferative activities were evaluated against four human cancer cell lines.
Molecular docking studies were carried out against EGFR, mutated BRAF and CDK2-TK.
In silico ADME/pharmacokinetic analysis was considered.
HIGHLIGHTS
Introduction
Because cancer is one of the world’s top causes of death, identifying and developing new effective anticancer drugs is one of the most difficult concerns in drug developmentCitation1–5.
A single-targeted treatment method that results in chemotherapy resistance has recently been extensively describedCitation6. This problem is addressed by a combination treatment that has received clinical approvalCitation7. Despite the fact that combination therapy offers the potential for extra and potentially synergistic benefits, it typically results in unanticipated side effects such as increased toxicity. Dual or multi-target medications with lower drug interaction risk, improved pharmacokinetics (PK), and safety profiles might be utilised as an alternative to combination therapy. A dual or multiple target kinases can also help to minimise poor patient adherence, off-target effects, medication interactions, and excessive manufacturing costsCitation8.
Natural products are a major constant source of lead medicines. Natural product-based developments, including anticancer medicines such as docetaxel, topotecan, and etoposide have been reportedCitation9,Citation10. Natural products derived from bacteria, fungi, marine organisms, plants, and animals, as well as natural product-inspired compounds, have shown promising results in clinical studies, including anticancer treatmentsCitation11. Natural product-derived derivatives are thought to account for more than half of anticancer medicines; around 74% of anticancer drugs are either natural or natural product-inspired compoundsCitation12.
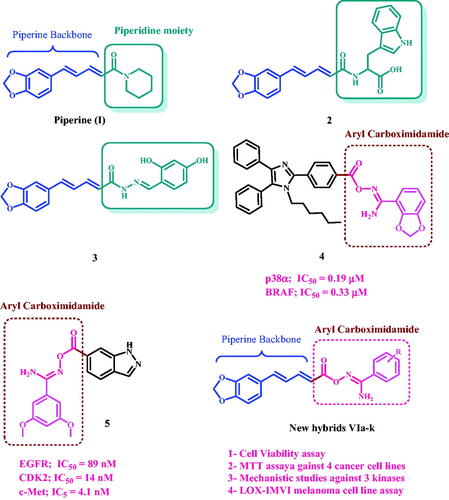
Piperine I, , is found in black pepper powder fruit and is widely utilised as a food flavour in a variety of cultures, as well as in many traditional food preservation systems and traditional treatmentsCitation13. Piperine has been shown to have anticancer effect through a variety of pathways. The substitution of the piperidine moiety with other pharmacophoric groups has been shown to increase activity and potency, as demonstrated by compounds 2 and 3Citation9,Citation14.
Figure 1. Structures of piperine, compounds 2–5, and newly designed compounds VIa-k.
Amidoxime-containing compounds, on the other hand, demonstrated strong pharmacological effects, with anticancer activity topping the listCitation15–18. The amidoxime moiety has been included as carboxylic and ester group bioisosteres, with the goal of developing prodrugs with enhanced pharmacokinetic and pharmacodynamic propertiesCitation19. Furthermore, amidoxime moiety has demonstrated its capacity to release NO in vivo for the purposes of developing nitric oxide-releasing prodrugsCitation20.
We recently reported on the design, synthesis, and in vitro assessment of a novel series of aryl carboximidamides as dual p38α/BRAFV600E inhibitors. The results revealed that the presence of the carboximidamide moiety is essential for activity, and the best activity correlates with compound 4, . Compound 4 showed significant antiproliferative action against a panel of cancer cell lines. It has an IC50 of 0.33 µM and 0.19 µM against BRAFV600E and p38α, respectivelyCitation21.
Another novel series in which the indazole moiety is substituted at the 6-position with aryl and or/heteroaryl carboximidamide moiety was developed in our labCitation22. Compound 5 () was the most active synthetic derivative, with a GI50 value of 0.77 µM against the tested four cancer cell lines, compared to the control doxorubicin (GI50 = 1.10 µM). Compound 5 inhibited EGFR, CDK2, and c-Met with IC50 values of 89 nM, 14 nM, and 4.1 nM, respectively. Furthermore, Compound 5 induced apoptosis by increasing cytochrome C levels and activating the intrinsic apoptotic pathway.
Motivated by the antiproliferative effects of piperine (I), compounds 2 and 3, as well as the promising antiproliferative action of aryl carboximidamide derivatives 4 and 5, we present here the design, synthesis, and antiproliferative action of a new hybrid scaffold VIa-k (), in which the aryl carboximidamide pharmacophore was bound to the piperine backbone using molecular hybridisation approach in order to obtain potent antiproliferative agents. The new hybrids will be tested against four cancer cell lines using the MTT assay to determine their IC50. The most effective hybrids will be tested further for mechanistic action against EGFR, BRAF, and CDK2. Furthermore, the most potent derivatives as BRAF inhibitors will be tested for their inhibitory action against the LOX-IMVI melanoma cell line, which has BRAFV600E kinase overexpression. Finally, docking studies were performed on the most active derivatives against the active sites of EGFR, BRAF, and CDK2.
Results and discussion
Chemistry
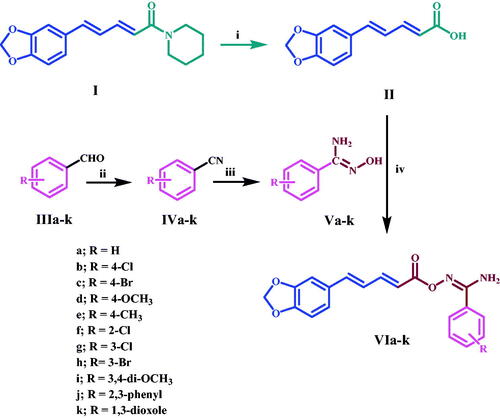
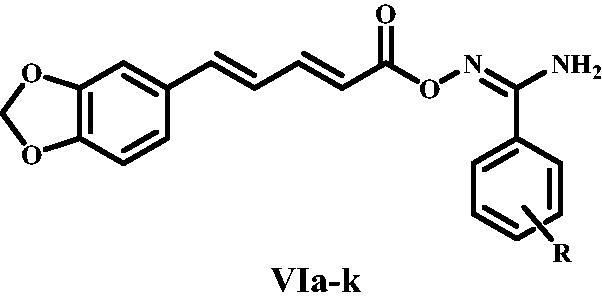
Scheme 1 depicts the synthetic steps for the key intermediates II, Va-k, and target compounds VIa-k. Piperine I was extracted, purified, and crystallised from natural sources using the methods describedCitation23. Piperic acid II was synthesised by hydrolysing piperine I with alcoholic KOHCitation24, as shown in Scheme 1. Amidoximes Va-l were synthesised as described25 and then coupled with piperic acid II using CDI at room temperature for 3 h to obtain the target compounds VIa-k. 1H NMR, 13C NMR, and elemental microanalyses were used to confirm all of the aryl carboximidamides VIa-k. The IR spectrum of VId, a prominent example of this series, shows the presence of peaks at 3498 and 3381 (NH2), 1760 (C = O), and 1622 (C = N). The 1H NMR spectrum of this compound revealed the appearance of new two doublet signals with J = 8.5, 8.7 Hz, corresponding to para disubstituted pattern of amidoxime moiety at δ 7.69 and δ 7 ppm, respectively, as well as the appearance of new broad singlet NH2 at δ 6.76 ppm and finally the appearance of singlet signal of methoxy (OCH3) group at δ 3.8 ppm. The presence of a new carbonyl (C = O) group signal at 161.4 ppm in the 13C NMR spectra of this molecule confirmed the earlier observations, as did the appearance of a methoxy (OCH3) group signal at 55.7 ppm.
Scheme 1: Synthesis of target compounds VIa-k. Reagents and reaction conditions: i) KOH, Ethanol, reflux 12 h; ii) NH3, THF, I2, r.t. 3 h, 65–80%; iii) NH2OH, methanol, NaHCO3, reflux 3–5 h, 80–90%; iv) CDI, Acetonitrile, r.t. 3 h.
Biology
In vitro anticancer activity
Cell viability assay
A human mammary gland epithelial (MCF-10A) normal cell line was used to perform the viability test. The viability of compounds VIa-k was determined using the MTT assay after 4 days of incubation with MCF-10A cellsCitation26,Citation27. The results showed that none of the compounds tested were cytotoxic, and that majority of the compounds tested at 50 µM had cell viability more than 85%.
Antiproliferative activity
Compounds VIa-k were investigated for antiproliferative efficacy against four human cancer cell lines using the MTT assay and erlotinib as the reference drug: Panc-1 (pancreatic cancer cell line), MCF-7 (breast cancer cell line), HT-29 (colon cancer cell line), and A-549 (epithelial cancer cell line)Citation28,Citation29. shows the median inhibitory concentration (IC50) calculated using Graph Pad Prism software (Graph Pad Software, San Diego, CA, USA). The average (GI50) versus the four cancer cell lines was chosen for ease of manipulation.
Table 1. Antiproliferative activity of compounds VIa-k and Erlotinib.
In general, compounds VIa-k demonstrated strong antiproliferative activity with GI50 values ranging from 35 nM to 100 nM when compared to the reference erlotinib, which had a GI50 of 33 nM. The data in reveal that VIa-k were more effective against epithelial cancer cell line (A-549) than breast cancer cell line (MCF-7).
The 1,3-benzodioxole derivative VIk (R = 1,3-dioxole) was the most potent of the synthesised derivatives, with a GI50 value of 35 nM, which was comparable to erlotinib and even more potent than erlotinib in MCF-7 cell line with IC50 = 35 nM, while for erlotinib IC50 = 40 nM.
Compound VIi (R = 3,4-di-OMe) ranks second in activity against the cell lines examined, with a GI50 of 39 nM. The number of the methoxy groups appears to be important for the antiproliferative action. Compound VId (R = 4-OMe), for example, has a GI50 of 54 nM, which is about 1.4-fold less potent than the dimethoxy derivative VIi. Furthermore, compound VIj (R = 2,3-Ph) has a GI50 of 83 nM, which is 2-times lower than VIi, indicating that the dimethoxy moiety is the most well tolerated.
Compound VIf (R = 2-Cl) demonstrated promising antiproliferative activity against the four cancer cell lines, with a GI50 of 44 nM. The antiproliferative action appears to be strongly influenced by the position and/or type of halogen atom. The GI50 of compounds VIb (R = 4-Cl) and VIg (R = 3-Cl) was 57 nM and 47 nM, respectively, indicating that when the chlorine atom is the substituent, position 2 on the phenyl group is the optimum for activity. Furthermore, compound VIc (R = 4-Br) had a GI50 of 50 nM, which is more potent than compound VIb (R = 4-Cl), which had a GI50 of 57 nM, demonstrating that the bromine atom is more tolerated than the chlorine one in terms of antiproliferative action.
The unsubstituted derivative, compound VIa (R = H), was the least potent of all synthesised derivatives, with a GI50 of 100 nM, approximately three times less active as erlotinib, highlighting the relevance of phenyl moiety substitution for antiproliferative action.
EGFR inhibitory activity
The EGFR-TK testCitation30 was used to evaluate the inhibitory potency of the most potent derivatives VIc, VIf, VIg, VIi, and VIk against EGFR, and the outcomes are displayed in . Compounds VIc, VIf, VIg, VIi, and VIk inhibited EGFR with IC50 values ranging from 96 to 127 nM. According to the findings, all the tested derivatives were less potent than erlotinib (IC50 = 80 ± 5 nM). The 3-chloro phenyl derivative VIg (R = 3-Cl) was the most potent of all synthesised derivatives, with an IC50 value of 96 nM, which was 1.2-fold lower than erlotinib. The results of the EGFR inhibitory assay test indicate that the EGFR may be a potential target for the antiproliferative effect of some compounds.
Table 2. Effects of compounds VIc, VIf, VIg, VIi, Vik, Erlotinib, Vemurafenib, and Dinaciclib on EGFR, BRAFV600E, and CDK2.
BRAFv600e assay
An in vitro study was conducted to evaluate the anti-BRAFV600E of VIc, VIf, VIg, VIi, and VIkCitation31. The enzyme assay revealed that the tested five compounds strongly inhibited BRAFV600E, with IC50 values ranging from 40 to 72 nM (See ). In all cases, the IC50 of the investigated compounds are higher than that of vemurafenib (IC50 = 30 ± 3). Compounds VIf and VIk had the most potent inhibitory activity as BRAFV600E (IC50 = 49 and 40 nM, respectively) and were discovered to be potent inhibitors of cancer cell proliferation (GI50 = 44 and 35 nM, respectively). According to the study’s findings, the examined compounds have substantial antiproliferative activity and are effective at inhibiting BRAFV600E.
Cdk2 inhibitory assay
Compounds VIc, VIf, VIg, VIi, and VIk were further investigated for their potential to inhibit the CDK2 enzymeCitation32. The IC50 values are shown in . According to the results, all compounds inhibited CDK2 with IC50 values ranging from 12 nM to 23 nM in comparison to the reference dinaciclib (IC50 = 20 nM). Compound VIk, the most effective derivative as an antiproliferative agent, demonstrated potent anti-CDK2 action with an IC50 value of 12 nM, which is 1.5-fold more potent than the reference dinaciclib. On the other hand, compounds VIf, VIg, and VIi exhibited significant activity against CDK2 (IC50 = 21, 20, and 17 nM) being equipotent to dinaciclib. The results of this assay show that the examined compounds have substantial antiproliferative activity and are effective at inhibiting both CDK2 and BRAFV600E.
Lox-IMVI melanoma cell line cytotoxicity assay
The MTT cytotoxicity assay was used to assess the anticancer activity of VIc, VIf, and VIk on the LOX-IMVI melanoma cell line, which comprises BRAFV600E kinase overexpressionCitation33. The compounds were evaluated at 5-dose concentrations to ascertain their IC50 values, with staurosporine acting as a control. reveals that the derivatives VIc, VIf, and VIk have a high capacity to inhibit LOX-IMVI cell line survival, with IC50 values ranging from 1.05 µM to 1.40 µM. In all cases, the tested compounds were at least 5 times more effective than the reference Staurosporine, which had an IC50 of 7.10 µM. Compounds VIf and VIf demonstrated promising cytotoxic activity against LOX-IMVI melanoma cell line with IC50 values of 1.10 and 1.05 μM, respectively.
Table 3. IC50 cytotoxicity of compounds VIc, VIf, and VIk against LOXIMVI melanoma cell line.
Molecular docking simulations
To determine the binding affinity and mode of inhibition of the most active derivatives against potential cellular targets of this class of compounds, we ran in-silico molecular docking simulations for compounds VIc, VIf, VIg, VIi, and VIk against EGFR, mutated BRAF, and CDK2 proteins. The results were extremely promising, particularly with compounds VIg, VIi, and VIk against all the cellular targets used in this study.
Starting with the RSCB deposited crystal structure of the EGFR protein having Erlotinib as a co-crystallised ligand (PDB ID: 1M17), simulations revealed good docking score (S = −5.84 to −6.72 kcal/mol) with all test compounds as listed in Table S1 (supporting information). Visual inspections of each test molecule’s docking poses revealed numerous interactions with various amino acid residues lining the EGFR active site ( and Figure S1). Using compound VIg as a model compound (highest docking score; S = −6.72 kcal/mol) to examine its docking poses and gain an understanding of the mode of inhibition of this class of compounds within the EGFR protein, we observed H-bond interactions with PRO770 and GLY772 amino acid residues, as shown in .
Figure 2. Schematic representation of docking interactions of compound VIg within EGFR (PDB ID: 1M17) showing two H-bond interactions with PRO 770 and GLY772 as blue-coloured arrows.
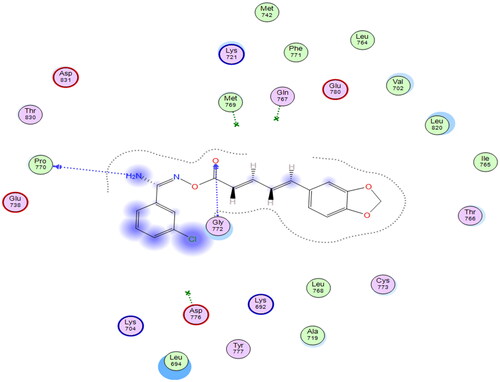
Although the docking scores of the test molecules were very promising, their binding interactions with various amino acid residues lining the EGFR active site remained random, and none of them managed to show a binding interaction with key amino acids (GLN767 and MET769) as did co-crystallised ligand. All these findings demonstrated that the EGFR protein is not the best active site to explain the mode of inhibition of such compounds as potential anti-proliferative agents.
Docking into BRAF and CDK2 active sites, on the other hand, revealed a significant common bonding interaction between test molecules and the amino acid residues lining these two active sites, as well as at least one or more bonding interactions with key amino acid residues.
Additionally, MTT assay results on the LOX-IMVI melanoma cell line, which is known for being enriched with BRAFV600E kinases, showed promising inhibitory results (5-times better than reference compound; staurosporine). As a result of all these intriguing findings regarding CDK2 and BRAFV600E proteins, we chose to investigate the potential mode of interactions within the active sites of these two proteins.
As shown in Table S1, compounds VIc, VIf, VIg, VIi, and VIk showed good and comparable docking score and RMSD values to that of co-crystallised ligands. Compound VIk showed the best docking score (S) within its congeners
As shown in , compound VIk managed to have three strong binding interactions in the form of H-bond donor and acceptor bonds, which stabilised its structure within the active site of BRAFV600E kinase protein, which was likely also seen with compound VIf (as shown in Figure S2), confirming how such class of compound could inhibit BRAFV600E protein.
Figure 3. Schematic representation of docking interactions of compound VIk within both BRAFV600E (PDB ID: 4MNF) and CDK2 (PDB ID: 1PYE): (a,b) 2 D Interactions of VIk within 4MNF and 1PYE; respectively; (c) Presumptive binding modes of VIk within 4MNF active site as 3 D diagram: showing three H-bond interactions with Asn 580 and Asp 594 coloured in red-colour.
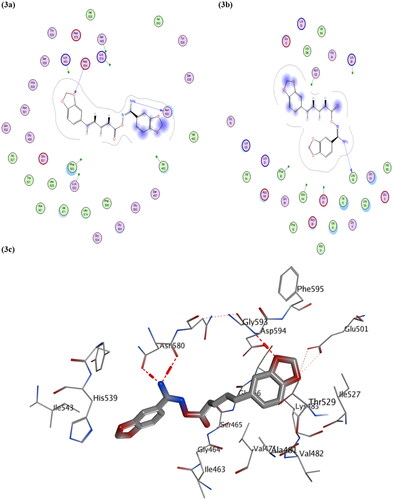
On the other hand, visual inspections of the best docking poses obtained with molecular docking of both compounds VIf and VIk within active site of CDK2 protein, revealed their achievement in having H-bonding interactions with two key amino acid residues GLU81 and LEU83 as did the co-crystallised ligand (as shown in and Figure S3). Additionally, compound VIc showed two H-bonding interactions with another two amino acid residues (ASN 132 and LYS89, as shown in Figure S3) rather than key amino acid residues, and this could explain its lower potency compared to its congers VIf and VIk against CDK2 protein.
To summarise, molecular docking simulations revealed a good docking score of such class of compounds within three active sites used in this study, as well as a good idea about the mode of inhibition of these compounds within BRAFV600E and CDK2 active sites.
ADME parameters and drug-likeness computational analysis
From our efforts to investigate bioavailability of such class of compounds, we measured physicochemical descriptors, pharmacokinetics, drug-like nature, and medicinal chemistry friendliness of all newly synthesised 11 compounds via SwissADME free web toolCitation34.
Starting with role of five (Lipinski’s RO5)Citation36,Citation37, all newly synthesised derivatives VIa-k have values for physicochemical within required ranges of candidate drugs, as shown in Table S2 (supporting information). Briefly, having H-bonding centres (either acceptor or donor) helps in enhancing water solubility and H-bond formation with various amino acid residues lining target active sites, in the same way, having acceptable number of rotating bonds helps in adaptation and flexibility alignment of such molecules within target active sites. Additionally, having a partition coefficient (i.e. lipophilicity parameter) within −0.5 to ≤ 5, and both TPSA and MR values below 140 and 130; respectively, generally indicates good probability of penetration through cell membranes, gastrointestinal penetration, and hence bioavailability. Fortunately, all the newly synthesised were found to not a substantial substrate for permeability glycoprotein (which is also known as multidrug resistance protein which is responsible for efflux of drugs out of cells and hence reduce their pharmacological efficacy) and having Abbott oral bioavailability score above zero, indicating their high probability of medicinal impact and biological activities in clinical trialsCitation38.
Finally, all the compounds pass the five medicinal chemistry filters (Lipinski, Ghose, Veber, Egan, and Muegge) and Pan-assay of interference compounds (PAINS), indicating their specificity for target proteins affected by them and being a drug-candidate for drug discovery and development, as shown in Table S2.
Conclusion
In the search for multi-targeted antiproliferative agents, a novel series of piperine-carboximidamide hybrids was designed and synthesised as EGFR, BRAFV600E, and CDK2 inhibitors. The new hybrids demonstrated promising antiproliferative properties. Some new hybrids exhibited strong EGFR and/or BRAFV600E inhibitory activity. Furthermore, some potent antiproliferative derivatives outperformed the reference drug against CDK2. Docking simulations within the active sites of BRAFV600E and CDK2 provided insight into the potential mode of inhibition of these compounds. Furthermore, the new class of compounds follows Lipinski’s rule of five, as well as passing all medicinal chemistry filters and having an acceptable bioavailability profile. Following optimisation, these novel hybrids could be considered as potential anticancer agents.
Materials and methods
Chemistry
General Details: See Appendix A
Plant material
Black Pepper seeds were purchased from the local market and grounded to a powder then kept in dry container. They were ground to 40 mesh and stored in airtight container until further use.
Extraction, isolation and purification of piperine alkaloid from black Pepper
Extraction
The sample of black pepper (100 g) was refluxed with 300 ml of dichloromethane for 2 h. in a round bottom flaskCitation23. Attach a water condenser to the top of the flask and allow water to run through it to condense the CH2Cl2 vapours. After cooling the flask, use vacuum filtration with a Büchner funnel and filter paper to filter out the pepper grounds. Wash the grounds with 50 ml CH2Cl2. The filtrate resulting from the extraction solutions were concentrated to dryness using rotary evaporator until a dark brown oil is left.
Thin layer chromatography
Thin layer chromatography experiment were carried out on commercial silica coated plates using hexane: ethyl acetate 7:3 (v/v) as a mobile phase (Rf = 0.36 for piperine)
Preparative silica gel chromatography
Adsorption of the extract on activated silica gel. Prepared sample of the extract was subjected to a silica gel column (200 g) and eluted with hexane: ethyl acetate system in a gradient manner (increasing polarity) from 15 to 40%. Fractions were collected and monitored by TLC method using hexane: ethyl acetate 7:3 (v/v) as a solvent system and concentrated H2SO4 was used as spraying reagent to identify those fractions containing the target compound.
Crystallisation of piperine
The piperine-containing fractions identified by TLC were pooled and the volatiles removed under reduced pressure using rotary evaporator to provide a thick orange oil placed in an ice bath for cooling. The mass of the thus obtained material, being highly enriched in piperine. 50 ml of cold diethyl ether was added with continuous stirring. After stirring for 5 min, the solvent was evaporated again by using rotary evaporator. The oil was placed in an ice bath for it to cool down. 50 ml of cold diethyl ether was added again and left in the refrigerator for about 24 h. After 24 h, the yellowish piperine precipitates were collected and washed with small volumes of cold diethyl ether. Two more re-crystallisation steps were carried out to rise the purity of the piperine.
Compounds II and Va-k were prepared as previously describedCitation24,Citation25.
General method for the synthesis of compounds via-k
The N,N'-carbonyl diimidazole CDI (2.29 mmol, 2eq.) was added to a stirred solution of piperic acid (II) (1.14 mmol, 1eq.) in dry acetonitrile (10 ml) and stirred for 1 h at room temperature. The appropriate amidoximes Va-l were then added to the resulting mixture and stirred for another 10 h. Following the completion of the reaction (monitored by TLC), The precipitate VIa-k that developed was filtered and then crystallised from acetonitrile.
N'-{[(2E,4E)-5-(2H-1,3-Benzodioxol-5-yl)penta-2,4-dienoyl]oxy}benzene carboximidamide (via)
Yield: 0.27 g (70%), yellow solid, m.p: 175–177 °C, Rf: 0.26 (Hexane: Ethyl acetate, 2:1, v/v); 1H NMR (400 MHz, δ ppm DMSO-d6): 7.73 (d, J = 7.2 Hz, 2H, Ar-H), 7.55–7.44 (m, 5H, Ar-H), 7.29 (s, 1H, Ar-H), 7.04–7.02 (m, 4H, CH = CH), 6.87 (s, broad, 2H, NH2), 6.06 (s, 2H, O-CH2-O); 13C NMR (100 MHz, δ ppm DMSO-d6): 164.35, 156.49, 148.23, 148.01, 144.78, 140.34, 131.79, 130.52, 128.35, 126.76, 124.99, 123.29, 118.96, 108.53, 105.77, 101.41. Anal. Calc. (%) for C19H16N2O4: C, 67.85; H, 4.80; N, 8.33; Found: C, 67.86; H, 4.87; N, 8.46.
N'-{[(2E,4E)-5-(2H-1,3-Benzodioxol-5-yl)penta-2,4-dienoyl]oxy}-4-chlorobenzene-1-carboximidamide (VIb)
Yield: 0.08 g (35%), yellow solid, m.p: 157–160 °C, Rf. 0.28 (Hexane: Ethyl acetate, 2:1, v/v); 1H NMR (400 MHz, δ ppm DMSO-d6): 8.55 (s, 1H, Ar-H), 7.82 (d, J = 8.6 Hz, 2H, Ar-H), 7.72–7.65 (m, 3H, Ar-H), 7.45 (d, J = 8.5 Hz, 1H, Ar-H), 7.25 (t, J = 7.7 Hz, 2H, CH = CH), 7.14–7.06 (m, 2H, CH = CH), 7.1 (s, broad, NH2), 6.09 (s, 2H, O-CH2-O); 13C NMR (100 MHz, δ ppm DMSO-d6): 162.00, 156.64, 152.26, 149.18, 148.85, 148.13, 143.62, 136.88, 135.88, 130.59, 129.99, 129.34, 128.64, 128.53, 128.12, 127.83, 127.15, 124.51, 124.02, 117.78, 116.64, 108.73, 105.90, 101.60. Anal. Calc. (%) for C19H15ClN2O4: C, 61.55; H, 4.08; N, 7.56. Found: C, 61.86; H, 4.20; N, 7.77.
N'-{[(2E,4E)-5-(2H-1,3-Benzodioxol-5-yl)penta-2,4-dienoyl]oxy}-4-bromobenzene-1-carboximidamide (VIc)
Yield: 0.12 g (42.1%), yellow solid, m.p: 159–161 °C, Rf: 0.29 (Hexane: Ethyl acetate, 2:1, v/v); IR (FTIR): νmax (cm−1) 3491,3358 (NH2), 3134, 2919 (CH), 1769 (C = O), 1624, 1590 (C = N and C = C), 834 (Ar-CH bending); 1H NMR (400 MHz, δ ppm DMSO-d6): 8.55 (s, 1H, Ar-H), 7.82 (s, 1H, Ar-H), 7.82–7.74 (m, 1H, Ar-H), 7.26 (t, J = 7.27 Hz, 2H, CH = CH), 7.14–7.08 (m, 4H, Ar-H), 7.00 (s, 2H, CH = CH), 6.97 (s, broad, 2H, NH2), 6.09 (s, 2H, O-CH2-O). 13C NMR (100 MHz, δ ppm DMSO-d6) : 161.99, 156.67, 152.27, 149.13, 148.86, 148.14, 143.59, 136.92, 132.15, 131.45, 131.02, 130.37, 130.10, 128.88, 127.96, 127.38, 124.49, 124.11, 117.80, 116.63, 108.72, 105.91, 101.59. Anal. Calc. (%) for C19H15BrN2O4: C, 54.96; H, 3.64; N, 6.75. Found: C, 55.12; H, 3.75; N, 6.87.
N'-{[(2E,4E)-5-(2H-1,3-Benzodioxol-5-yl)penta-2,4-dienoyl]oxy}-4-methoxybenzene-1-carboximidamide (VId)
Yield: 0.24 g (72.7%), yellow solid, m.p: 145–148 °C, Rf: 0.29 (Hexane: Ethyl acetate, 2:1, v/v); IR (FTIR): νmax (cm−1) 3498,3381 (NH2), 3134, 2978 (CH), 1760 (C = O), 1683, 1588 (C = N and C = C), 827 (Ar-CH bending); 1H NMR (400 MHz, δ ppm DMSO-d6): 8.55 (s, 1H, Ar-H), 7.82 (s, 1H, Ar-H), 7.77 (d, J = 10.9 Hz, 1H, Ar-H), 7.69 (d, J = 8.5 Hz, 2H, Ar-H), 7.14–7.06 (m, 4H, CH = CH), 7.00 (d, J = 8.7 Hz, 2H, Ar-H), 6.76 (s, broad, 2H, NH2), 6.09 (s, 2H, O-CH2-O), 3.8 (s, 3H, O-CH3). 13C NMR (100 MHz, δ ppm DMSO-d6) : 161.50, 157.61, 152.98, 149.64, 144.08, 128.68, 124.99, 124.43, 123.68, 118.23, 117.20, 114.23, 109.22, 106.35, 102.10, 55.84. Anal. Calc. (%) for C20H18N2O5: C, 65.57; H, 4.95; N, 7.65. Found: C, 65.43; H, 5.10; N, 7.77.
N'-{[(2E,4E)-5-(2H-1,3-Benzodioxol-5-yl)penta-2,4-dienoyl]oxy}-4-methylbenzene-1-carboximidamide (VIe)
Yield: 0.20 g (50%), yellow solid, m.p: 185–188 °C, Rf: 0.24 (Hexane: Ethyl acetate, 2:1, v/v); 1H NMR (400 MHz, δ ppm DMSO-d6): 7.63 (d, J = 7.76 Hz, 2H, Ar-H), 7.53–7.46 (m, 1H, CH = CH), 7.28 (s, 1H, Ar-H), 7.25 (d, J = 7.76 Hz, 2H, Ar-H), 7.00 (d, J = 4.36 Hz, 2H, CH = CH) , 7.00 (d, J = 4.36 Hz, 1H, Ar-H) , 6.93 (d, J = 7.96 Hz, 1H, Ar-H), 6.81 (s, broad, 2H, NH2), 6.18 (d, J = 15.4 Hz, 1H, CH = CH), 6.00 (s, 2H, O-CH2-O), 2.50 (s, 3H, Ar-CH3). 13C NMR (100 MHz, δ ppm DMSO-d6) : 164.40, 156.35, 148.22, 148.06, 144.70, 140.28, 140.15, 130.50, 128.86, 126.65, 125.00, 123.29, 119.01, 108.52, 105.78, 101.40, 20.94. Anal. Calc. (%) for C20H18N2O4: C, 68.56; H, 5.18; N, 8.00. Found: C, 68.80; H, 5.30; N, 8.17.
N'-{[(2E,4E)-5-(2H-1,3-benzodioxol-5-yl)penta-2,4-dienoyl]oxy}-2-chlorobenzene-1-carboximidamide (VIf)
Yield: 0.134 g (63%), yellow solid, m.p: 165–167 °C, Rf: 0.26 (Hexane: Ethyl acetate, 2:1, v/v); IR (FTIR): νmax (cm−1) 3491,3358 (NH2), 3134, 2919 (CH), 1769 (C = O), 1624, 1590 (C = N and C = C), 834 (Ar-CH bending); 1H NMR (400 MHz, δ ppm DMSO-d6): 8.54 (s, 1H, Ar-H), 7.57–7.47 (m, 4H, Ar-H), 7.43 (d, J = 7.56 Hz, 1H, Ar-H), 7.26 (d, J = 8.2 Hz, 1H, Ar-H), 7.25 (d, J = 15.4 Hz, 1H, CH = CH) , 7.14–7.07 (m, 3H, CH = CH), 6.97 (s, broad, 2H, NH2), 6.08 (s, 2H, O-CH2-O). 13C NMR (100 MHz, δ ppm DMSO-d6) : 162.03, 156.68, 152.22, 149.17, 148.84, 148.13, 143.57, 136.92, 132.12, 131.12, 130.50, 129.89, 129.50, 126.97, 124.44, 123.57, 117.99, 116.46, 108.79, 105.87, 101.42. Anal. Calc. (%) for C19H15ClN2O4: C, 61.55; H, 4.08; N, 7.56. Found: C, 61.67; H, 4.23; N, 7.77.
N'-{[(2E,4E)-5-(2H-1,3-benzodioxol-5-yl)penta-2,4-dienoyl]oxy}-3-chlorobenzene-1-carboximidamide (VIg)
Yield: 0.08 g (44.4%), yellow solid, m.p: 163–165 °C, Rf: 0.26 (Hexane: Ethyl acetate, 2:1, v/v); 1H NMR (400 MHz, δ ppm DMSO-d6): 8.28 (s, 1H, Ar-H), 7.65–7.59 (m, 2H, Ar-H), 7.15–7.06 (m,2H, CH = CH), 7.08 (s, broad, 2H, NH2), 6.95 (d, J = 7.6 Hz, 1H, Ar-H), 6.08 (s, 2H, O-CH2-O). 13C NMR (100 MHz, δ ppm DMSO-d6) :, 162.33, 149.95, 149.35, 144.26, 130.96, 130.66, 124.96, 124.36, 118.28, 117.06, 109.70, 106.28, 101.87, Anal. Calc. (%) for C19H15ClN2O4: C, 61.55; H, 4.08; N, 7.56. Found: C, 61.43; H, 4.26; N, 7.83.
N'-{[(2E,4E)-5-(2H-1,3-benzodioxol-5-yl)penta-2,4-dienoyl]oxy}-3-bromobenzene-1-carboximidamide (VIh)
Yield: 0.34 g (72.34%), yellow solid, m.p: 141–143 °C, Rf: 0.28 (Hexane: Ethyl acetate, 2:1, v/v); IR (FTIR): νmax (cm−1) 3486, 3348 (NH2), 3134, 2916 (CH), 1778 (C = O), 1622, 1589 (C = N and C = C), 988, 830, 761 (Ar-CH bending); 1H NMR (400 MHz, δ ppm DMSO-d6): 8.55 (s, 1H, Ar-H), 7.92 (s, 2H, Ar-H), 7.75 (t, J = 6.74 Hz, 4H, Ar-H), 7.45 (t, J = 7.9 Hz, 1H, Ar-H), 7.45 (t, J = 7.9 Hz, 1H, CH = CH), 7.27 (t, J = 7.7 Hz, 1H, Ar-H), 7.27 (t, J = 7.7 Hz, 1H, CH = CH) , 7.13–7.07 (m, 2H, CH = CH), 6.98 (s, broad, 2H, NH2), 6.09 (s, 2H, O-CH2-O). 13C NMR (100 MHz, δ ppm DMSO-d6) : 162.33, 156.64, 152.46, 149.65, 148.36, 143.95, 133.85, 131.27, 130.36, 129.67, 126.25, 124.96, 124.36, 122.08, 118.28, 117.37, 109.17, 106.28, 101.87. Anal. Calc. (%) for C19H15BrN2O4: C, 54.96; H, 3.64; N, 6.75. Found: C, 55.06; H, 3.79; N, 6.97.
N'-{[(2E,4E)-5-(2H-1,3-benzodioxol-5-yl)penta-2,4-dienoyl]oxy}-3,4-dimethoxybenzene-1-carboximidamide (VIi)
Yield: 0.134 g (37.2%), yellow solid, m.p: 147–149 °C, Rf: 0.24 (Hexane: Ethyl acetate, 2:1, v/v); IR (FTIR): νmax (cm−1) 3490, 3378 (NH2), 3079, 2938 (CH), 1757 (C = O), 1623, 1585 (C = N and C = C), 766 (Ar-CH bending); 1H NMR (400 MHz, δ ppm DMSO-d6): 8.56 (s, 1H, Ar-H), 7.36–7.29 (m, 4H, Ar-H), 7.25 (d, J = 7.33 Hz, 1H, Ar-H), 7.15–7.08 (m, 2H, CH = CH), 7.03 (d, J = 8.34 Hz, 2H, CH = CH), 6.81 (s, broad, 2H, NH2), 6.09 (s, 2H, O-CH2-O), 3.81 (s, 6H, -(OCH3)2) . 13C NMR (100 MHz, δ ppm DMSO-d6) : 162.14, 158.69, 157.38, 152.55, 151.32, 150.63, 149.40, 148.71, 148.10, 125.69, 123.16, 119.63, 117.71, 116.79, 111.95, 111.03, 110.04, 109.12, 55.70 . Anal. Calc. (%) for C21H20N2O6: C, 63.63; H, 5.09; N, 7.07. Found: C, 63.50; H, 5.29; N, 7.17.
N'-{[(2E,4E)-5-(2H-1,3-benzodioxol-5-yl)penta-2,4-dienoyl]oxy} naphthalene -1-carboximidamide (VIj)
Yield: 0.148 g (70%), yellow solid, m.p: 170–172 °C, Rf: 0.26 (Hexane: Ethyl acetate, 2:1, v/v); 1H NMR (400 MHz, δ ppm DMSO-d6): 8.56 (s, 1H, Ar-H), 8.00 (m, 2H, Ar-H), 7.59 (m, 5H, Ar-H), 7.31 (s, 1H, Ar-H), 7.07 (m, 3H, CH = CH), 7.05 (s, broad, 2H, NH2), 6.95 (d, J = 8.0 Hz, 1H, Ar-H), 6.22 (d, J = 15.3 Hz, 1H, CH = CH), 6.08 (s, 2H, O-CH2-O). 13C NMR (100 MHz, δ ppm DMSO-d6) : 164.84, 157.55, 148.66, 145.25, 140.76, 133.47, 131.27, 130.05, 128.46, 127.47, 127.17, 126.56, 126.25, 125.57, 123.98, 119.57, 109.02, 106.25, 101.87 . Anal. Calc. (%) for C23H18N2O4: C, 71.49; H, 4.70; N, 7.25. Found: C, 71.55; H, 4.83; N, 7.47.
N'-{[(2E,4E)-5-(2H-1,3-Benzodioxol-5-yl)penta-2,4-dienoyl]oxy}-2H-naphtho[1,2-d][1,3]dioxole-8-carboximidamide (VIk)
Yield: 0.09 g (70%), yellow solid, m.p: 166–168 °C, Rf: 0.27 (Hexane: Ethyl acetate, 2:1, v/v); 1H NMR (400 MHz, δ ppm DMSO-d6): 8.41 (s, 1H, Ar-H), 7.79 (s, 1H, Ar-H), 7.4–7.3 (m, 2H, Ar-H), 7.00 (s, broad, 2H, NH2), 6.95(dd, J = 8.3 Hz, 7.7 Hz, 2H, Ar-H), 6.74–6.82 (m, 2H, CH = CH), 6.68 (d, J = 8 Hz, 1H, CH = CH), 6.19 (d, J = 15.4 Hz, 1H, CH = CH), 6.06 (s, 2H, O-CH2-O), 5.93 (s, 2H, O-CH2-O). 13C NMR (100 MHz, δ ppm DMSO-d6) : 164.46, 162.16, 158.71, 156.40, 150.04, 149.12, 147.81, 147.20, 144.59, 141.44, 140.44, 135.00, 130.54, 125.40, 125.01, 123.18, 121.57, 120.95, 119.03, 118.04, 110.36, 108.83, 108.14, 107.83, 105.60, 102.07, 101.58, 100.77 . Anal. Calc. (%) for C24H18N2O6: C, 66.97; H, 4.22; N, 6.51. Found: C, 67.17; H, 4.40; N, 6.65.
Biology
Cell viability testing and IC50 determination
MTT assay
The MTT assay was used to assess how the synthetic compounds impacted the viability of mammary epithelial cells (MCF-10A)Citation26,Citation27. Appendix A.
Antiproliferative test
The MTT assay was performed using various cell lines in accordance with previously reported proceduresCitation28,Citation29 to investigate the antiproliferative potential of VIa-k. Refer to Appendix A.
EGFR inhibitory assay
The EGFR-TK testCitation30 was used to evaluate the inhibitory potency of the most potent derivatives VIc, VIf, VIg, VIi, and VIk against EGFR. See Appendix A.
BRAF kinase assay
An in vitro studyCitation31 was conducted to evaluate the anti-BRAFV600E of VIc, VIf, VIg, VIi, and Vik. See Appendix A.
CDK2-TK assay
Compounds VIc, VIf, VIg, VIi, and VIk were further investigated for their potential to inhibit the CDK2 enzymeCitation32. See Appendix A.
LOX-IMVI melanoma cell line cytotoxicity assay
The MTT cytotoxicity assay was used to assess the anticancer activity of VIc, VIf, and VIk on the LOX-IMVI melanoma cell line, which comprises BRAFV600E kinase overexpressionCitation33. Refer to Appendix A.
In-Silico molecular docking simulations
Molecular Operating Environment (MOE® 2014.0901) software was used to evaluate and examine possible interactions of test molecules (VIc, VIf, VIg, VIi, and VIk) within crystal structure of 3 target proteins: Epidermal growth factor receptor (EGFR; PDB ID: 1M17), BRAFV600E kinase (PDB ID: 4MNF) and cyclin-dependent kinase (CDK2; PDB ID: 1PYE) obtained from RSCB protein data bankCitation34 and results in the form of docking score (S; kcal/mol), docking accuracy expressed as root-mean square deviation (RMSD; Å) and binding interactions with various amino acid residues lining active site (as listed in Table S1). Preparation of structural formulas of test molecules and structure of target proteins were performed as reported elsewhereCitation35. Validation of prepared protein structures was done via re-docking of co-crystallised ligands with their protein crystal-structure obtained from RSCB protein data bank and their docking score and RMSD values were within acceptable range for running docking simulations within target proteins (as listed in Table S1). Docking simulations were performed as docking protocol reported elsewhere and results were reported in Table S1Citation35. Visual inspection of produced docking poses (10 poses/molecule) for binding interactions with various amino acid residues lining active site of both 4MNF and 1PYE crystal structure and were listed in Table S1 and represented as 2 D and 3 D diagrams in , Figures S1–S3.
ADME parameters and drug-likeness computational analysis
SwissADME free web tool for prediction of physicochemical parametersCitation34, pharmacokinetics, and drug-likeness of small molecules, was used to measure such parameters of compounds VIa-VIk and results were listed in Table S2. Also, the medicinal chemistry filters (Lipinski, Ghose, Veber, Egan, and Muegge) were applied on all test compounds and none disobey such filters. PAINS (Pan-Assay of Interference Compounds) were used to examine specificity of such class of compounds as possible anticancer agents and all compounds gave zero score as shown in Table S2.
Supplemental Material
Download PDF (1.3 MB)Disclosure statement
The author reported no potential conflicts of interest(s).
Additional information
Funding
References
- Zha G-F, Qin H-L, Youssif BGM, Amjad MW, Raja MAG, Abdelazeem AH, Bukhari SNA. Discovery of potential anticancer multi-targeted ligustrazine based cyclohexanone and oxime analogs overcoming the cancer multidrug resistance. Eur J Med Chem. 2017;135:34–48.
- Hisham M, Youssif BGM, Osman EEA, Hayallah AM, Abdel-Aziz M. Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur J Med Chem. 2019;176:117–128.
- Youssif BGM, Abdelrahman MH, Abdelazeem AH, Abdelgawad MA, Ibrahim HM, Salem OIA, Mohamed MFA, Treambleau L, Bukhari SNA. Design, synthesis, mechanistic and histopathological studies of small-molecules of novel indole-2-carboxamides and pyrazino[1,2-a]indol-1(2H)-ones as potential anticancer agents effecting the reactive oxygen species production. Eur J Med Chem. 2018;146:260–273.
- Abou-Zied HA, Youssif BGM, Mohamed MFA, Hayallah AM, Abdel-Aziz M. Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg Chem. 2019;89:102997.
- El-Sherief HAM, Youssif BGM, Abbas Bukhari SN, Abdelazeem AH, Abdel-Aziz M, Abdel-Rahman HM. Synthesis, anticancer activity and molecular modeling studies of 1,2,4-triazole derivatives as EGFR inhibitors. Eur J Med Chem. 2018;156:774–789.
- Hughes D, Andersson DI. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet. 2015;16(8):459–471.
- Lehár J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, Rickles RJ, Short GF, Staunton JE, Jin X, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27(7):659–666.
- Shah KN, Bhatt R, Rotow J, Rohrberg J, Olivas V, Wang VE, Hemmati G, Martins MM, Maynard A, Kuhn J, et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat Med. 2019;25(1):111–118.
- Umadevi P, Deepti D, Venugopal DVR. Synthesis, anticancer and antibacterial activities of piperine analogs. Med Chem Res. 2013;22(11):5466–5471.
- Deng Y, Sriwiriyajan S, Tedasen A, Hiransai P, Graidist P. Anti-cancer effects of Piper nigrum via inducing multiple molecular signaling in vivo and in vitro. J Ethnopharmacol. 2016;188:87–95.
- Katz L, Baltz RH. Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol. 2016;43(2–3):155–176.
- Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981-2002. J Nat Prod. 2003;66(7):1022–1037.
- Ali Y, Alam MS, Hamid H, Husain A, Bano S, Dhulap A, Kharbanda C, Nazreen S, Haider S. Design, synthesis, and biological evaluation of piperic acid triazolyl derivatives as potent anti-inflammatory agent. Eur J Med Chem. 2015;92:490–500.
- Tantawy AH, Meng X-G, Marzouk AA, Fouad A, Abdelazeem AH, Youssif BGM, Jiang H, Wang M-Q. Structure-based design, synthesis, and biological evaluation of novel piperine–resveratrol hybrids as antiproliferative agents targeting SIRT-2. RSC Adv. 2021;11(41):25738–25751.
- Baraldi PG, Bovero A, Fruttarolo F, Preti D, Tabrizi MA, Pavani MG, Romagnoli R. DNA minor groove binders as potential antitumor and antimicrobial agents. Med Res Rev. 2004;24(4):475–528.
- Karaaslan C. Synthesis and structure elucidation of new benzimidazole amidoxime derivatives. Turk J of Pharm Sci. 2020;17(1):108–114.
- Gobis K, Foks H, Kȩdzia A, et al. Studies on pyrazine derivatives. XLVII. Synthesis and antibacterial activity of novel pyrazine derivatives with amidoxime moiety. Acta Pol Pharm - Drug Res. 2006;63:39–45.
- Huang M-R, Hsu Y-L, Lin T-C, Cheng T-J, Li L-W, Tseng Y-W, Chou Y-S, Liu J-H, Pan S-H, Fang J-M, et al. Structure-guided development of purine amide, hydroxamate, and amidoxime for the inhibition of non-small cell lung cancer. Eur J Med Chem. 2019;181:111551.
- Ningaiah S, Bhadraiah UK, Keshavamurthy S, Javarasetty C. Novel pyrazoline amidoxime and their 1, 2, 4-oxadiazole analogues: synthesis and pharmacological screening. Bioorg Med Chem Lett. 2013;23(16):4532–4539.
- Burgaud JL, Ongini E, Del Soldato P. Nitric oxide-releasing drugs: a novel class of effective and safe therapeutic agents. Ann N Y Acad Sci. 2002; 962(1):360–371.
- Youssif BGM, Gouda AM, Moustafa AH, Abdelhamid AA, Gomaa HAM, Kamal I, Marzouk AA. Design and synthesis of new triarylimidazole derivatives as dual inhibitors of BRAFV600E/p38α with potential antiproliferative activity. J Mol Struct. 2022;1253:132218.
- Obaid Arhema Frejat F, Zhai H, Cao Y, Wang L, Mostafa YA, Gomaa HAM, Youssif BGM, Wu C. Novel indazole derivatives as potent apoptotic antiproliferative agents by multi-targeted mechanism: synthesis and biological evaluation. Bioorg Chem. 2022;126:105922.
- Shingate PN, Dongre PP, Kannur DM. New method development for extraction and isolation of piperine from black pepper. IJPSR. 2013;4:3165–3170.
- Wang Y, Yao Y, Liu J, Wu L, Liu T, Cui J, Lee DY-W. Synthesis and biological activity of piperine derivatives as potential PPARγ agonists. Drug Des Devel Ther. 2020;14:2069–2078.
- Youssif BGM, Mohamed MFA, Al-Sanea MM, Moustafa AH, Abdelhamid AA, Gomaa HAM. Novel aryl carboximidamide and 3-aryl-1,2,4-oxadiazole analogues of naproxen as dual selective COX-2/15-LOX inhibitors: design, synthesis and docking studies. Bioorg Chem. 2019;85:577–584.
- Abdelbaset MS, Abdel-Aziz M, Abuo-Rahma GEA et al. Novel quinoline derivatives carrying nitrones/oximes nitric oxide donors: design, synthesis, antiproliferative and caspase-3 activation activities. Arch Pharm. 2019;352:1800270.
- Al-Wahaibi LH, Gouda AM, Abou-Ghadir OF, Salem OIA, Ali AT, Farghaly HS, Abdelrahman MH, Trembleau L, Abdu-Allah HHM, Youssif BGM, et al. Design and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg Chem. 2020;104:104260.
- Mohassab AM, Hassan HA, Abdelhamid D, Gouda AM, Youssif BGM, Tateishi H, Fujita M, Otsuka M, Abdel-Aziz M. Design and synthesis of novel quinoline/chalcone/1,2,4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAFV600E kinases. Bioorg Chem. 2021;106:104510.
- Marzouk AA, Abdel-Aziz SA, Abdelrahman KS, Wanas AS, Gouda AM, Youssif BGM, Abdel-Aziz M. Design and synthesis of new 1,6-dihydropyrimidin-2-thio derivatives targeting VEGFR-2: molecular docking and antiproliferative evaluation. Bioorg Chem. 2020;102:104090.
- Mohamed FAM, Gomaa HAM, Hendawy OM, Ali AT, Farghaly HS, Gouda AM, Abdelazeem AH, Abdelrahman MH, Trembleau L, Youssif BGM, et al. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg Chem. 2021;112:104960.
- El-Sherief HAM, Youssif BGM, Bukhari SNA, Abdel-Aziz M, Abdel-Rahman HM. Novel 1,2,4-triazole derivatives as potential anticancer agents: design, synthesis, molecular docking and mechanistic studies. Bioorg Chem. 2018;76:314–325.
- Mekheimer RA, Allam SMR, Al-Sheikh MA, Moustafa MS, Al-Mousawi SM, Mostafa YA, Youssif BGM, Gomaa HAM, Hayallah AM, Abdelaziz M, et al. Discovery of new pyrimido[5,4-c]quinolines as potential antiproliferative agents with multitarget actions: rapid synthesis, docking, and ADME studies. Bioorg Chem. 2022;121:105693.
- Gomaa HAM, Shaker ME, Alzarea SI, Hendawy OM, Mohamed FAM, Gouda AM, Ali AT, Morcoss MM, Abdelrahman MH, Trembleau L, et al. Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg Chem. 2022;120:105616.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717.
- Abdel-Wahab NM, Gomaa AA, Mostafa YA, et al. Diterpenoids profile of the marine sponge Chelonaplysilla erecta and candidacy as potential antitumor drugs investigated by molecular docking and pharmacokinetic studies. Nat Prod Res. 2022;36(20):1–5.
- Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44(1):235–249.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997;23(1-3):3–25.
- Martin YC. A bioavailability score. J Med Chem. 2005;48(9):3164–3170.