
Abstract
A series of 1H-benzo[f]chromene moieties (4a–z) were synthesised under Ultrasonic irradiation and confirmed with spectral analyses. Derivative 4i solely possessed an X-ray single crystal. The anti-proliferative efficacy of the desired molecules has been explored against three cancer cells: MCF-7, HCT-116, and HepG-2 with the cytotoxically active derivatives screened against MCF-7/ADR and normal cells HFL-1 and WI-38. Furthermore, compounds 4b–d, 4k, 4n, 4q, and 4w, which possessed good potency against MCF-7/ADR, were tested as permeability glycoprotein (P-glycoprotein [P-gp]) expression inhibitors. The attained data confirmed that 4b–d, 4q, and 4w exhibited strong expression inhibition against the P-gp alongside its cytotoxic effect on MCF-7/ADR. The western blot results and Rho123 accumulation assays showed that compounds 4b–d, 4q, and 4w effectively inhibited the P-gp expression and efflux function. Meanwhile, 4b–d, 4q, and 4w induced apoptosis and accumulation of the treated MCF-7/ADR cells in the G1 phase and 4k and 4n in the S phase of the cell cycle.
Graphical Abstract
Introduction
An efficient and green tool for organic reactions is to use the Ultrasound technique as a non-conventional energy source, which can provide a range of benefits, such as shorter reaction times, easier operation, and improved yields of pure productsCitation1–4. Benzochromene is one of the most attractive oxygen-incorporating heterocyclic scaffolds in the light of their unique natural characteristics and their biomedical performancesCitation5. Benzochromene derivatives are the most valuable pharmacological compounds, possessing a range of significant biological properties, such as antimicrobialCitation6–9, anti-inflammatory and analgesicCitation10, antiviral and central nerve system activitiesCitation11,Citation12, antioxidantCitation6,Citation13, hypolipidemicCitation14, anti-proliferativeCitation15, anticancerCitation16, anti-rheumaticCitation17, anti-tubercularCitation18,Citation19, Alzheimer’s preventativeCitation20, anti-obesityCitation21 effects, and agents. Similarly, various studies have reported their targeting of signalling pathways, which is critical for the inhibition of tumour cell proliferation and enables them as prospective primary candidates in the manufacture of antitumor agentsCitation22–27. For example, some benzochromene derivatives displayed inhibitory behaviour of the c-Src kinase with their antitumor ability and apoptotic effectsCitation28–31. Other benzochromene templates exhibited antitumor activities, trigger cell cycle arrest at M, S, and G2 stages, enhance the formation of caspases 3/7, and initiate apoptosis with diminished toxicity in tumour cells through the dual inhibition of topoisomerase I/II and in breast cancer xenograftsCitation27,Citation31–35. Alongside its anti-proliferative features, chromene derivatives exhibited a DNA binding ability, the inhibition of the Bcl-2 proteinCitation26 and a high potency for the hAChECitation36. One of the main molecular mechanisms shared in failure of chemotherapy is multi-drug resistance (MDR) Citation37,Citation38. The main cause of MDR is overexpression of the ABC transporters, such as permeability glycoprotein (P-glycoprotein [P-gp)), multidrug resistance protein 1 (MDR1), and ATP-binding cassette sub-family B member 1 (ABCB1), (DGAT-1), and (hCA XII) inhibitorsCitation38–40. In addition, P-gp/(ABCB1) plays a main role in the multidrug-resistant phenotype in cancer mediating MDRCitation41. Our continuous endeavours to develop powerful, novel oxygen-integrating heterocyclic molecules antitumor and antimicrobial agents are expanded upon by this current reportCitation42–51.
Aim of the work and rationale
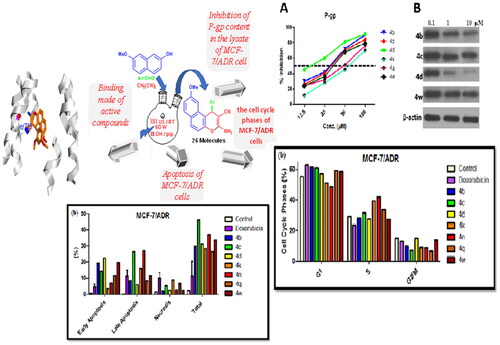
Herein, we report the synthesis of anti-proliferative 1H-benzo[f]chromene derivatives (a–z). In this work, we explore the potent and anti-proliferative effects of the substituents on the C-1 position of the phenyl-bearing moieties of the 1H-benzo[f]chromene derivatives. To achieve this goal, three tumour cell lines MCF-7 (breast cancer), HCT-116 (human colon cancer), and HepG-2 (hepatocellular carcinoma) were utilised to investigate the anti-proliferative activity of the desired molecules. Subsequently, the highly cytotoxically-active derivatives 4b–e, 4g, 4i, 4k, 4n, 4o, 4q, 4r, 4u, 4w, and 4z were subjected to further screenings against Adriamycin (ADR)-resistant human breast cancer cells (MCF-7/ADR) and the normal cell lines human foetal lung (HFL-1) alongside WI-38 human (diploid fibroblasts). Furthermore, compounds 4b–d, 4k, 4n, 4q, and 4w revealed good potency towards MCF-7/ADR cells and were examined for the inhibition of P-gp expression; meanwhile, 4b–d, 4q, and 4w displayed high potency against P-gp expression MDR in MCF-7/ADR and they are able to inhibit P-gp. Besides, western blot results and Rho123 accumulation assays showed that compounds 4b–d, 4q, and 4w effectively inhibited P-gp expression and efflux function, while compounds 4b–d, 4q, and 4w induced accumulation of the treated MCF-7/ADR cells in the G1 phase, and compounds 4k and 4n in the S phase of the cell cycle, as illustrated in .
Chart 1. Study rationale analysis of antitumor activities, MCF-7/ADR, P-gp inhibitor, apoptosis, and cell cycle analysis western blot and Rh123 results.

The rational design of the 9-methoxy-1H-benzo[f]chromene derivatives was based on the following considerations: (i) the 1H-benzo[f]chromene template, (ii) the existence of the efficacious 9-position substituent, (iii) the varying sorts of substituents located on the aryl moieties bound to the 9-methoxy-1H-benzo[f]chromene framework at the 1-position which emerges as a critical component in cytotoxic behaviour, and (iv) comparative analyses regarding the performances of the freshly prepared molecules and the formerly prepared molecules with a bromine atom at the 9-positionCitation24 as illustrated in .
The final feature in this rationale study revealed that the recent molecules 4b, 4g, 4n, 4q, 4u, and 4z possessed a remarkable influence regarding their behaviours against tumour cells, which had an elevated potency in comparison with the molecules 1–4Citation24.
Results and discussion
Chemistry
A series of substituted fused heterocyclic derivatives (4a–z) were synthesised using 7-methoxynaphthalen-2-ol (1) as a starting material. The attained molecules 4e, 4f, 4j, and 4r–u are described herein for the first time, while previous reported compounds 4a–d, 4g–i, 4k–q, and 4v–z have been acquired employing an ultrasound as a new synthetic strategy. The interaction of 1 with a number of aldehydes derivatives (2a–z) in the presence of and malononitrile (3) and absolute ethanol/piperidine solution has been achieved utilising 60 W of ultrasonic irradiation conditions at ambient temperature and allowed for the formation of β-enamionitriles containing 9-methoxy-1H-benzo[f]chromene motifs (4a–z) as shown in Scheme 1.
Scheme 1. Synthesis of 1H-benzo[f]chromene derivatives (4a–z).
![Scheme 1. Synthesis of 1H-benzo[f]chromene derivatives (4a–z).](/cms/asset/043c8976-755b-4596-a11f-987f1d845fae/ienz_a_2155814_sch0001_c.jpg)
The 1-position of compounds 4a–z is a chiral centre and all the reactions were controlled using TLC technique. The optical execution of 4a–z was detected utilising using a Carl Zeiss polarimeter and displayed zero rotation (i.e. optically inactive) due to their occurrence as a racemic (±) mixtureCitation52,Citation53, as shown in Scheme 1.
The structure’s identity of the novel molecules 4e, 4f, 4j, and 4r–u was substantiated via their IR, 1H NMR, 13C NMR, 13C NMR-APT, and MS data. Supporting evidences for the suggested structures comes from their infra-red spectra which exhibited at υ 3448–3402, 3338–3291, 3261–3200 cm−1 for an NH2 group and at υ 2204–2167 cm−1 for the CN group of compounds 4e, 4f, 4j, and 4r–u. The 1H NMR data of 4e, 4f, 4j, and 4r–u demonstrated singlet signals at δ 7.17–6.93 ppm that is corresponding to the amino protons, while signals at δ 6.06–5.26 are attributable for the methine protons. Moreover, the 13C NMR-APT of compound 4s as well as the MS spectrum, the single crystal X-ray analysis of compounds 4d, 4 g,Citation9,Citation54 and 4i provided a conclusive verification for the desired molecules (see Supplementary Materials 1S-100S).
Crystal data
illustrates the crystallographic analysis and the refinement information of molecule 4i with the chemical formula of C22H18N2O2. The asymmetric unit of compound 4i is containing one molecule as shown in . The length of all bonds and the angles are in normal rangesCitation55. In the crystal packing, , molecules of compound 4i were linked via two intermolecular hydrogen bonds ().
Chart 2. Rationale for designing target compounds.

Figure 1. 2D, 3D, and energy framework molecular packing of compound 4i.

Table 1. X-Ray experimental details for compounds 4i.
Table 2. Hydrogen-bond geometry (Å, °) for compound 4i.
The central pyran ring (O1–C1–C10–C11–C12–C13) is almost planar with the largest deviation from the mean plan of −0.013(2) at C10 atom, which connected to naphthalene ring and to a tolyl ring (). The dihedral angle between the main core ring and the tolyl is 73.51(7)°. The mean plan through the naphthalene ring (C8—C13) is nearly orthogonal to that of the tolyl ring as signified by the dihedral angle between them of 144.44(4)°. Also, there are two intermolecular hydrogen bonds interactions, N2—H2N1•••N1, given in .
Crystal explorer17Citation56 was used to investigate the Hirschfield-surface “HS” with their 2D-fingerprint surfaces “2D-FS” analysis, which displayed the intermolecular interactions in the crystal packing. HS was mapped over dnorm for 4i (). The HS-volume “HSV” and the surface area “HSA” are 433.89 Å3 and 382.56 Å2, respectively, which represented in red(−dnorm), blue(+dnorm), and white (normal dnorm) colour schemes. Negative, positive, and normal dnorm, are shorter, longer and equal contacts than van-der-Waals-radii, respectively. The decomposed 2D-HS in () showed the chief contribution in C–H and H–H contacts, which contribute the most (30.9%) to the total HSA. The lowest contributions are from C–C, C–N contacts, while C–O, O–H, and N–H displayed contributions at 2.7, 6.9, and 14.1 contacts. The strong intermolecular interactions appear as distinct spikes in the fingerprint plots.
Shape index (SI) and curvedness (CS) were used to study the curvature of the surface (). The SI sheds light on the stacking arrangement for 4i, which was used to identify the complementary cavities (red) and bumps (blue) where two molecular HS close to each other. The blue and red coloured triangular-shaped patterns represented the particular stacking arrangement of 4i. Molecule 4i displayed no significant triangular-patterns on SI map, which suggested the absence of the π–π interaction. The CS was represented by the function of the r.m.s. curvature. CS mapped was found to be in range of (−4.0–4.0) which categorised by bulky green-coloured regions and detached by deep blue boundaries. Since there is no flat surface seen on the CS plot, there is no planar stacking between the molecules.
The B3LYP/6-311G(d,p) function was employed for simulating the intermolecular interaction energies in the crystal to examine their stabilisation degree of the crystal lattice (). represents their energy framework as a cylinder-shaped, which displayed the relative-strength for the interaction energies and also give a clear view for their role in the stabilisation of the crystal packing. According to , it is clear that the dispersion force (−233.3 kJ/mol) plays a central role, which having a maximum energy of −233.3 kJ/mol, among the total interaction energy which is −244.7 kJ/mol.
Table 3. The energetic interaction in kJ/mol of the 4b at B3LYP/6-311G(d,p), R: Centroid distance; Etot: The total interaction energy; Eele: sum classical electrostatic and coulomb energy; Epol: polarisation energy, Edisp: dispersion energy; Erep; exchange repulsion energy.
Biological activity
Cell viability assay
According to previous reports regarding the cytotoxic features of a numerous array of benzochromene scaffolds, compounds 4a − z have been elected to explore their cytotoxic ability against three tumour cell lines, including HCT-116 (colon cancer), MCF-7 (breast cancer), and HepG-2 (hepatocellular cancer), and employing the 3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) colorimetric assay as described in literatureCitation57. Judges choice of such cell lines has been stimulated by the acknowledged antitumor performance of a substantial number of 1H-benzo[f]chromene and 4H-benzo[h]chromene molecules towards the asserted cell linesCitation18–35. The in-vitro assay has been demonstrated utilising different concentrations (0–50 µM), where Doxorubicin and Vinblastine have been selected to be the reference drugs. Moreover, benzochromene derivatives with highly cytotoxic behaviour, 4a − d, 4g, 4i, 4k, 4n, 4q, 4 u, and 4w, are subjected to further inspection against ADR-resistant human breast cancer cells (MCF-7/ADR) and the two normal cell lines, HFL-1 and human diploid fibroblasts (WI-38). The attained data were declared as growth inhibitory concentration (IC50) values, which represent the compounds concentrations required to produce a 50% inhibition of cell growth after 24 h of incubation, compared to the untreated controls. The results revealed potent growth inhibitory activity against MCF-7, HCT-116, and HepG-2 cancer cells as shown in . The results revealed potent growth inhibitory activity against MCF-7, HCT-116, and HepG-2 cancer cells; HFL-1 and WI-38 normal cell lines as shown in , and MCF-7/ADR cancer cell as shown in . Also, the results are illustrated in .
Figure 2. IC50 values expressed in (µM) of 1H-benzo[f]chromene derivatives (4a–z) against MCF-7, HCT, and HepG-2 tumour cells.
![Figure 2. IC50 values expressed in (µM) of 1H-benzo[f]chromene derivatives (4a–z) against MCF-7, HCT, and HepG-2 tumour cells.](/cms/asset/89147681-edbd-4149-9a1f-ee3802e9a9aa/ienz_a_2155814_f0002_c.jpg)
Figure 3. Dose-dependent cytotoxicity in MCF-7/ADR cells and the effect of varying concentrations of tested compounds 4a–4d, dg, 4i, 4k, 4n, 4q, 4u, and 4w on cell growth of MCF-7/ADR cells following exposure 24 h.

Table 4. Cytotoxic activity of the target compounds against MCF-7, HCT-116, HepG-2, MCF-7/ADR cancer cell lines and normal cell lines, HFL-1, WI-38.
divulges that the 1H-benzo[f]chromene molecules 4d, 4 g, 4i, 4k, 4n, 4o, 4q, 4r, 4u, 4w, and 4z exhibited potent anti-proliferative character towards MCF-7 cell line with IC50 in the range of 0.2–7.0 µM. Molecules 4b, 4d, 4g, 4i, 4n, and 4q displayed superior activity against HCT-116 with IC50 in the range of 1.3–2.9 µM. Meanwhile, compounds 4e, 4 g, 4q, and 4z possessed growth inhibitory ability towards HepG-2 cell line with IC50 1.5–4.9 µM. The remaining derivatives illustrated good, moderate, or inactive cytotoxic activity. Additionally, compounds 4a–e, 4 g, 4i, 4k, 4n, 4q, 4r, 4 u, 4w, 4y, and 4z demonstrated week growth inhibitory impact towards two normal cell lines, HFL-1 and WI-38 with IC50 values between 29.9 and 83.0 µg/mL. Furthermore, derivatives 4b–d, 4k, 4n, 4q, and 4w revealed good potency against MCF-7/ADR cell with IC50 values between 10.9 and 15.5 μM, as compared with Doxorubicin (IC50 = 18.6 μM), while compounds 4a, 4 g, 4i, and 4u are inactive against MCF-7/ADR cell with IC50 ranging from 19.6 to 42.8 μM. Finally, the remaining compounds exhibited fair cytotoxic behaviour towards the examined tumour cell types in comparison Doxorubicin.
P-glycoprotein-mediated multidrug resistance in MCF-7/ADR cell
P-gp pumps multiple types of drugs out of the cell, using energy generated from adenosine triphosphate (ATP), and confers MDR on cancer cellsCitation58. It is one of the noteworthy problems in malignant tumour clinical therapeuticsCitation59. The active compounds 4b–4d, 4k, 4q, and 4w against MCF-7/ADR lines were tested as possible inhibitory effect on P-gp inhibitors and the acquired results revealed that compounds 4b–4d, 4q, and 4w having 2-Cl, 3-Cl, 4-Cl, 3,4-Cl2, and 3,4-(OMe)2 substituents have good inhibitory potency against P-gp expression MDR in MCF-7/ADR with IC50 ranging from 13.5 to 45.3 μM as compared with Doxorubicin (IC50 = 50.9 μM), as shown in and .
Table 5. IC50 values of the active compounds 4b, 4c, 4d, 4q, and 4w against P-gp mediated MDR in MCF-7/ADR cells.
The effect of compounds 4b–d, 4k, and 4w on the inhibition of P-gp expression in MCF-7/ADR was also confirmed using western blot analysis as shown in . Compounds 4b–d, 4k, 4q, and 4w have strong cytotoxic effects on MCF-7/ADR cells; they also displayed excellent inhibitory performance on the P-gp content () with the exception of compound 4k. These results demonstrated that only compounds 4b–d and 4w possessed high inhibitory effect on the expression of P-gp which subsequently have a great potency in reversal the MDR in MCF-7/ADR, similar results have been reported previouslyCitation60,Citation61. Furthermore, the reversion of P-gp mediated multidrug resistance may be achieved by down-regulation of P-gp expression and or inhibition of P-gp efflux functionCitation62. For this reason, the effect of our synthesised drugs was tested for inhibitory potential of P-gp activity using a Rhodamine 123 Accumulation Assay (Rhodamine Competitive ELISA Kit) ().
Table 6. P-glycoprotein inhibitory potential (IC50 values) based on P-glycoprotein content in MCF-7/ADR cell lysate and rhodamine 123 accumulation assay.
The data in portrayed the (IC50) of Rhodamine 123 for evaluating the P-gp functional inhibition of compounds 4b–d, and 4w which ranging from 10.6 to 25.6 µM compared to 14.3 µM of the reference drug Verapamil. Compounds 4b–d and 4w had an impact on the restoration of the sensitivity to MCF-7/ADR cells by reducing not only the P-gp expression but also its function. On the other hand, compounds 4k and 4q exhibited cytotoxic activity against MDR which may be associated with different types of mechanisms such as genetic factors, growth factors, increased DNA repair capacityCitation63.
Cell cycle arrest in treated MCF-7/ADR cancer cells
Development of anticancer agents targeting cell cycle arrest represents an important therapeutic intervention in treating diseases like cancer. Cancer cells undergo unscheduled cell divisions by the down regulation of the four cell cycle stages (G1, S, G2, and M) Citation64,Citation65. P-gp breast cancer resistant proteins (BCRP) normally prevent intercellular drug accumulation, affecting the anticancer agent’s impact on the cell cycle arrestCitation66. Therefore, the effect of the most potent newly synthesised compounds 4b–d, 4k, 4n, 4q, and 4w on regulating cell cycle progression of MCF-7/ADR cancer cells cycle was analysed by the flow cytometry, exploiting the FACS Calibres (Becton Dickinson). The distribution of cells along the G1 (2n), G2/M (4n), and S (2n–4n) phases of the cycle was exhibited in the representative cell cycle distribution histogram of the stained DNA in . The MCF-7/ADR cancer cells were remedied with each derivative at its IC50 values for 24h, a controlled experiment with no treatment done.
The outcomes of the cell cycle progression showed that all the tested compounds have expressed a significant increased percentage of 10–15% (60–65%) of cells at the G1 phase in comparison to the (55%) control cells expect for compounds 4k and 4n which show increased percentages 40% and 43%, respectively, in S phase compared to the control (30%). In addition, these results were accompanied by a considerable decrease in percentage at the G2/M phases compared to the untreated control cells (). The cell cycle evaluation presented that the tested derivatives significantly arrested the cells’ progression by restricting the G1 and S phases.
Apoptosis induction in MCF-7/ADR cancer cells
Several lines of evidence indicate that many anticancer compounds exerted their effects by blocking the cell cycle progression, by inducing apoptosis, or the combined effect of bothCitation67. Also, P-gp inhibits apoptosis by preventing the release of cytochrome c which is mediated by the intrinsic mitochondrial pathwayCitation68. To further assess the pivotal relation of the newly synthesised MCF-7/ADR anticancer compounds and apoptosis, phosphatidylserine (PS) translocation to the cell membrane as a marker for apoptosis was measured by the means of the Annexin V/PI double staining flow cytometric assayCitation69. The representative dot plots of the double-stained MCF-7/ADR cells after treatment with the diverse examined compounds were displayed in .
Figure 4. A. Inhibition of P-gp content in the lysate of MCF-7/ADR cell using varying conc. (12.5–100 µM) of tested compounds 4b–4d, 4k, 4q, and 4w follows exposure 48 h as determined by ELISA. B. Western blot analysis of P-gp expression in MCF-7/ADR cells after treatment with 0.1, 1.0, and 10.0 µM of compounds 4b–4d, and 4w for 48 h, the β-actin was used as a control.

Figure 5. Effects of compounds 4 b–d, 4k, 4n, 4q, and 4w on the cell cycle phases of MCF-7/ADR cells. (a) Representative histograms of the DNA content distribution of cells were incubated with IC50 values for 24 h and stained with propidium iodide (PI). Their DNA content was analysed by the fluorescence flow cytometry. (b) The percentage of MCF-7/ADR cells in the G1, S, and G2/M phases after incubation with tested compounds (IC50 value) for 24 h. The data are expressed as the mean ± SD of three independent experiments in triplicate.

Unlike necrosis, which was not observed in all the results, all treated cells illustrated up to 40% in total apoptosis in evaluation against 30% and 2% of the Doxorubicin and untreated cells respectively. Moreover, all the tested compounds showed early (Annexin V positive, PI negative) as well as late (Annexin V positive, PI positive) apoptosis for all the treated cells (). Our results proposed that the induction of MCF-7/ADR cytotoxicity occurs via mechanisms associated with apoptosis with no obvious negative effects of the P-gp.
Structure–activity relationship SAR
The initial SAR investigations performed were centred on the impact of substituting hydrogen atoms on the phenyl group at 1-position of the 1H-benzo[f]chromene platform by halogen atoms, methyl or methoxy groups and methoxy group at 9-position as illustrate in Scheme 2. The implementation of the halogen or methoxy monosubstituted for the first series (4a–j) reduced their activities against MCF-7 cell line (IC50 in the range of 0.6–69.2 µM) as compared with Vinblastine. This behaviour could be attributed to the effect of the grafting of a lipophilic electron withdrawing or electron donating groups on the phenyl group at 1-position of the 1H-benzo[f]chromene moiety. Meanwhile, the second series with disubstituted halogens atoms or methoxy groups (4k–w) revealed a fluctuation in their antiproliferative activities with a simultaneous variation in the position and size of the disubstituted halogens or methoxy. Among the third series with the trisubstituents, compounds (4x–z), only compound (4z) possessed adequate activity in assessment with Vinblastine.
Scheme 2. Schematic representation of structure–activity relationship.

Regarding the influence of the substituent’s groups of the desired molecules from the first series on their potency against HCT-116 cell lines, the halogenated or methylated monosubstituent (4a–j) exhibited diminished IC50 in the range of 1.9–27.7 µM values, while for compounds (4k–w) the activities were in the range of 1.3–96.5 µM. Furthermore, through the trisubstituents of the third series (4x–z), only compound (4z) exhibited activity approximate to Vinblastine, while the other derivatives were inactive, intimating that the disubstituent demonstrated superior potency in evaluation with the mono- and tri-substituent analogues.
In addition, selected derivatives of the three series demonstrated superior potency against HepG-2 cell lines. For instance, compounds 4q, 4g, 4z, 4e, and 4j had IC50 values in the range of 1.5–5.3 µM in appraisal with Vinblastine and Doxorubicin. Also, compounds 4b–d, 4k, 4n, 4q, and 4w possessed good potency against resistant cell strains (MCF-7/ADR) with IC50 in the range of 10.9–15.5 μM. Such results tentatively suggested that grafting a lipophilic electron withdrawing with moderate size (difluoro or dichloro substituents) is more beneficial than other substituents for the activity. Furthermore, compounds 4a–e, 4g, 4i, 4k, 4n, 4q, 4r, 4u, 4w, 4 y, and 4z have been screened against two normal cell lines, HFL-1 and WI-38 and displayed IC50 ranging from 29.9 to 83.0 µM, which confirm their inadequate performance against these control cell lines. Finally, we can deduce that the position and the type of the substituent on the phenyl group at the 1-position of the 1H-benzo[f]chromene moiety played a vital role in its antitumor activity.
Molecular docking
To account for the most profiling compounds 4b–d, 4q, and 4w against P-gp, molecular docking was conducted to investigate their possible interactions (PDB code 3G60)Citation70 and theoretical model was utilised in all docking experiment. I-TASSERCitation71 was used to generate Human P-gp, and then used AMBER force field to optimise their model. Ramachandran plots were obtained which showed that model is similar to that one gained from the experimental mouse structure, which reported in the protein data bank. We also predicted the crystal structure of mouse P-gp (code 3G60) by I-TASSER. In this docking analysis we used the P-gp structure and translocation of the substrate pore as a rigid object in the docking procedure. However, since the ligand-binding pose of P-gp (a facing inward closed contact to apo conformation) was employed, the docking data give insight in the suitable complex geometry to ATP hydrolysis. To validate the accuracy of the docking analysis we redocked the reference cyclic peptide “QZ59-RRR” bound (PDB code 3G60), and compared with original geometry for QZ59-RRR. The original QZ59-RRR was docked into the experimentally determined structure of mouse P-gp with a high accuracy with a RMSD value of 1.78 Å. To gain further validation for the docking experiment, the inhibition constant (Ki) and bioactivity factor also examined as ligand efficiency (LE) were calculated (). All investigated compounds and reference molecules appeared in acceptable range as listed in .
Table 7. The binding affinity for compounds 4a–z and QZ59-RRR in (kcal/mol) against P-gp.
The docking experiment has been accomplished by Glide’s module®. The binding free energies ΔG are listed in . The original-inhibitors have been suitably fitted into their own binding sites for their crystal structures. The QZ59-RRR capped the 3G60 binding pocket through bounded with and formed the same H-bond with Gn721 and Ser725. All compounds 4b-d, 4q, and 4w and reference molecule (Doxorubicin) docked fruitfully into active sites in the same manner as original inhibitor through formation H-bond with Gln721 ().
Figure 6. Apoptosis of MCF-7/ADR cells treated with compounds 4b–d, 4k, 4n, 4q, and 4w. (a) The dot plot of the Annexin V/PI stained cells, treated with the indicated drugs. (b) The apoptosis percentage of MCF-7/ADR cells after incubation with tested compounds (IC50 value) for 24 h. The data are expressed as the mean ± SD of three independent experiments in triplicate.

Figure 7. Binding mode of most active compounds, reference molecule (Doxorubicin), and QZ59-RRR into P-glycoprotein (PDB: 3G60). H-bonding represented in blue lines.

From , the most potential antiproliferative compounds 4b–d, 4k, 4q, and 4w showed high promising binding affinity compared to QZ59-RRR for the investigated P-gp (PDB: 3G60).
Compound 4d showed the highest binding affinity ΔG = −11.313 kcal/mol against all investigated compounds and reference inhibitor, which explain the highest P-gp-inhibition activity (IC50 = 13.5 μM), through blocked active site by the formation of the H-bond with vital Gln 721. Besides, the other compounds 4b, 4c, 4k, 4q, and 4w showed higher ΔG than QZ59-RRR, which arranged in decreasing order 4c< 4b < 4q < 4w< 4k < QZ59-RRR (). Compounds 4b, 4c, 4k, and 4q were occupied the binding pocket with the same behaviour of QZ59-RRR through engaged Gln721, only compound 4w showed high stability in binding pocket through the interaction with important Ser725 ().
Conclusions
In continuation with our preceding endeavours in cultivating novel efficacious molecules, this report explores the design and synthesis of a series of oxygen-incorporating heterocyclic derivatives (4a–z) with the integration of a benzochromene moiety. Upon the evaluation of these derivatives for their cytotoxic behaviours against MCF-7, HCT-116, HepG-2, and MCF-7/ADR tumour cell lines in comparison with the standard reference drugs Vinblastine, and Doxorubicin, only molecules exhibiting cytotoxically active characteristics were selected for additional assessment against the representative tumour cell line ADR-resistant human breast cancer cells (MCF-7/ADR)) and two healthy cell lines (HFL-1 and human diploid fibroblasts (WI-38). Additionally, molecules 4b–d, 4k, 4n, 4q, and 4w which possessed good potency against MCF-7/ADR cell comparable to that of Doxorubicin were tested to show the possible inhibitory effect on P-gp expression and the obtained data concluded that 4b–d, 4q, and 4w possessed good inhibitory potency against P-gp expression in MCF-7/ADR cells. Furthermore, the western blot results and the Rh123 accumulation assays showed that compounds 4b–d, and 4w effectively inhibited P-gp expression and efflux function while, compounds 4b–d, 4k, 4n, 4q, and 4w induced accumulation of the treated MCF-7/ADR cells in the G1 phase and 4k and 4n additionally in the S phase of the cell cycle. Lastly, the SARs investigations of the molecules corroborated that the variation within the substitution on the phenyl ring of the 1H-benzo[f]chromene scaffold alongside the incidence of the methoxy moiety at the 9-position elevates molecular capabilities against these diverse cell lines.
Experimental section
Materials and equipments
All chemicals were purchased from Sigma-Aldrich Chemical Co. (Sigma-Aldrich Corp., St. Louis, MO). All melting points were measured with a Stuart Scientific Co. Ltd apparatus are uncorrected. The IR spectra were recorded on a KBr disc on a Jasco FT/IR 460 plus spectrophotometer. The 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were measured on BRUKER AV 500 MHz spectrometer in DMSO-d6 as a solvent, using tetramethylsilane (TMS) as an internal standard, and chemical shifts were expressed as δ (ppm). 13C-NMR spectra were obtained using distortion-free enhancement by polarisation transfer (DEPT) and the attached proton test (APT). The Ultrasonic apparatus used is Fisher Scientific UL TRASONIC CLEANER FS220. The mass spectra were determined on a Shimadzu GC/MS-QP5050A spectrometer. Elemental analysis was carried out at the Regional Centre for Mycology and Biotechnology (RCMP), Al-Azhar University, Cairo, Egypt, and the results were within ± 0.25%. Reaction courses and product mixtures were routinely monitored by thin-layer chromatography (TLC) on silica gel precoated F254 Merck plates.
General procedure for synthesis of 1H-benzo[f]chromene derivatives (4a–z)
A mixture of 7-methoxynaphthalen-2-ol (1) (0.01 mol), different aromatic aldehydes (2a–z) (0.01 mol), malononitrile (3) (0.01 mol), and piperidine (0.5 ml) in ethanol (30 ml) was equipped with Ultrasonic probe under the power of 60 W at room temperature. After completion of the reaction, as indicated by TLC (n-hexane/ethyl acetate 1:3), the reaction mixture was cooled to ambient temperature and the precipitated solid was filtered off, washed with methanol, and was recrystallised from ethanol. The physical and spectral data of compounds 4a–z are as follows:
3-Amino-1–(4-fluorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4a)
Pale yellow crystals; yield 92%; m.p. 267–268 °C (Literature procedure, reflux condition, yield 89%; m.p. 265–266 °C) Citation72.
3-Amino-1–(2-chlorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4b)
Colourless needles; yield 90%; m.p. 266–267 °C (Literature procedure, reflux condition, yield 82%; m.p. 265–266 °C) Citation28.
3-Amino-1–(3-chlorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4c)
Colourless needles; yield 89%; m.p. 262–263 °C (Literature procedure, reflux condition, yield 86%; m.p. 260–261 °C) Citation28.
3-Amino-1–(4-chlorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4d)
Colourless needles; yield 92%; m.p. 257–258 °C (Literature procedure, reflux condition, yield 89%; m.p. 257–258 °C) Citation72.
3-Amino-1–(2-bromophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4e)
Colourless crystals; yield 90%; m.p. 273–275 °C; IR (KBr) υ (cm−1): 3423, 3338, 3202 (NH2), 2180 (CN); 1H NMR δ: 7.89–6.83 (m, 11H, aromatic and NH2), 5.54 (s, 1H, H-1), 3.72 (s, 3H, OMe); 13C NMR δ: 160.14 (C-3), 158.69 (C-9), 148.03 (C-4a), 144.76 (C-1, Ar), 135.99 (C-3, Ar), 134.10 (C-6a), 132.99 (C-6, Ar), 132.10 (C-10a), 130.65 (C-4, Ar), 129.94 (C-5, Ar and C-6), 129.26 (C-10), 126.42 (C-8), 122.54 (C-7), 120.19 (C-2, Ar), 117.49 (C-10b), 114.56 (CN), 56.93 (C-2), 102.79 (C-5), 55.76 (CH3), 35.76 (C-1); MS m/z (%): 408 (M++2, 16.92), 406 (M+, 1754) with a base peak at 203 (100); Anal. Calcd for C21H15BrN2O2 (407.26): C, 61.93; H, 3.71; N, 6.88. Found: C, 62.00; H, 3.78; N, 6.95%.
3-Amino-1–(3-bromophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4f)
Pale yellow crystals; yield 93%; m.p. 263–265 °C; IR (KBr) υ (cm−1): 3438, 3322, 3200 (NH2), 2176 (CN); 1H NMR δ: 7.88–6.69 (m, 11H, aromatic and NH2), 5.36 (s, 1H, H-1), 3.75 (s, 3H, OMe); 13C NMR δ: 160.26 (C-3), 158.53 (C-9), 148.90 (C-4a), 147.76 (C-1, Ar), 131.98 (C-10a), 131.38 (C-2, Ar), 130.45 (C-5, Ar), 129.97 (C-4, Ar), 128.78 (C-6, Ar and C-6), 126.81 (C-7), 126.42 (C-6a), 122.24 (C-3, Ar), 120.82 (C-10b), 117.41 (C-5), 114.69 (C-8), 114.50 (CN), 103.53 (C-10), 57.75 (C-2), 55.55 (CH3), 37.77 (C-1); MS m/z (%): 408 (M++2, 38.57), 406 (M+, 39.59) with a base peak at 337 (100); Anal. Calcd for C21H15BrN2O2 (407.26): C, 61.93; H, 3.71; N, 6.88. Found: C, 61.83; H, 3.65; N, 6.81%.
3-Amino-1–(4-bromophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4g)
Colourless crystals; yield 90%; m.p. 252–253 °C (Literature procedure, reflux condition, yield 87%; m.p. 251–252 °C) Citation72.
3-Amino-1–(4-iodophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4h)
Yellow crystals; yield 93%; m.p. 227–228 °C (Literature procedure, reflux condition, yield 89%; m.p. 226–227 °C) Citation28.
3-Amino-1–(4-methylphenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4i)
Yellow crystals; yield 94%; m.p. 245–246 °C (Literature procedure, reflux condition, yield 88%; m.p. 244–245 °C) Citation72.
3-Amino-1–(4-trifluoromethylphenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4j)
Yellow crystals; yield 91%; m.p. 212–214 °C; IR (KBr) υ (cm−1): 3402, 3337, 3252 (NH2), 2204 (CN); 1H NMR δ: 8.96–7.02 (m, 11H, aromatic and NH2), 5.17 (s, 1H, H-1), 3.75 (s, 3H, OMe); MS m/z (%): 396 (M+, 22.79) with a base peak at 70 (100); Anal. Calcd for C22H15F3N2O2 (396.36): C, 66.67; H, 3.81; N, 7.07. Found: C, 66.75; H, 3.88; N, 7.13%.
3-Amino-1–(2,4-difluorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4k)
Yellow crystals; yield 89%; m.p. 255–256 °C (Literature procedure, reflux condition, yield 87%; m.p. 255–256 °C) Citation28.
3-Amino-1–(2,6-difluorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4l)
Colourless crystals; yield 91%; m.p. 305–306 °C (Literature procedure, reflux condition, yield 88%; m.p. 305–306 °C) Citation28.
3-Amino-1–(2,3-dichlorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4m)
Colourless crystals; yield 93%; m.p. 310–311 °C (Literature procedure, reflux condition, yield 87%; m.p. 310–311 °C) Citation28.
3-Amino-1–(2,4-dichlorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4n)
Colourless crystals; yield 90%; m.p. 261–262 °C (Literature procedure, reflux condition, yield 87%; m.p. 260–261 °C) Citation28.
3-Amino-1–(2,5-dichlorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4o)
Colourless crystals; yield 90%; m.p. 268–269 °C (Literature procedure, reflux condition, yield 82%; m.p. 268–269 °C) Citation28.
3-Amino-1–(2,6-dichlorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4p)
Colourless crystals; yield 88%; m.p. 321–322 °C (Literature procedure, reflux condition, yield 86%; m.p. 320–321 °C) Citation28.
3-Amino-1–(3,4-dichlorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4q)
Colourless crystals; yield 90%; m.p. 255–256 °C (Literature procedure, reflux condition, yield 86%; m.p. 255–256 °C) Citation28.
3-Amino-1–(2,5-dibromophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4r)
Pale yellow crystals; yield 89%; m.p. 260–262 °C; IR (KBr) υ (cm−1): 3410, 3291, 3209 (NH2), 2191 (CN); 1H NMR δ: 7.92–6.79 (m, 10H, aromatic and NH2), 5.53 (s, 1H, H-1), 3.72 (s, 3H, OMe); 13C NMR δ: 160.22 (C-3), 158.82 (C-9), 148.04 (C-4a), 146.94 (C-1, Ar), 135.27 (C-6, Ar), 132.82 (C-10a), 132.30 (C-3, Ar), 131.91 (C-4, Ar), 130.87 (C-6), 130.38 (C-7), 126.42 (C-6a), 122.01 (C-5, Ar), 121.60 (C-10b, C-2, Ar), 117.65 (C-5), 114.60 (CN), 113.54 (C-8), 102.61 (C-10), 56.22 (C-2), 55.77 (CH3), 31.91 (C-1); MS m/z (%):488 (M++4, 8.13), 486 (M++2, 15.53), 484 (M+, 8.85) with a base peak at 89 (100); Anal. Calcd for C21H14Br2N2O2 (486.16): C, 51.88; H, 2.90; N, 5.76. Found: C, 51.80; H, 2.82; N, 5.70%.
3-Amino-1–(3,5-dibromophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4s)
Pale yellow crystals; yield 90%; m.p. 268–270 °C; IR (KBr) υ (cm−1): 34105, 3295, 3212 (NH2), 2196 (CN); 1 H NMR δ: 7.92–7.03 (m, 10H, aromatic and NH2), 5.46 (s, 1H, H-1), 3.81 (s, 3H, OMe); 13C NMR δ: 160.45 (C-3), 158.72 (C-9), 150.79 (C-4a), 147.91 (C-1, Ar), 132.24 (C-10a), 131.91 (C-2,6, Ar), 130.65 (C-4, Ar), 130.12 (C-6), 129.74 (C-7), 126.47 (C-6a), 123.18 (C-3,5, Ar), 120.67 (C-10b), 117.61 (C-5), 114.59 (CN), 114.08 (C-8), 103.41 (C-10), 57.20 (C-2), 55.65 (CH3), 32.24 (C-1). In 13C NMR-APT spectrum: CH, CH3 [positive (up)] and CH2, Cq [negative (down)], revealed the following signals at δ: 160.45 (C-3 ↓), 158.72 (C-9 ↓), 150.79 (C-4a ↓), 147.91 (C-1, Ar ↓), 132.24 (C-10a ↓), 131.91 (C-2,6, Ar ↑), 130.65 (C-4, Ar ↓), 130.12 (C-6 ↑), 129.74 (C-7 ↑), 126.47 (C-6a ↓), 123.18 (C-3,5, Ar ↓), 120.67 (C-10b ↓), 117.61 (C-5 ↑), 114.59 (CN ↓), 114.08 (C-8 ↑), 103.41 (C-10 ↑), 57.20 (C-2 ↓), 55.65 (CH3 ↑), 32.24 (C-1 ↑); MS m/z (%): 488 (M++4, 18.83), 486 (M++2, 37.81), 484 (M+, 19.82) with a base peak at 176 (100); Anal. Calcd for C21H14Br2N2O2 (486.16): C, 51.88; H, 2.90; N, 5.76. Found: C, 51.96; H, 2.97; N, 5.84%.
3-Amino-1–(2-chloro-6-fulorophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4t)
Pale yellow crystals; yield 94%; m.p. 292–294 °C; IR (KBr) υ (cm−1): 3448, 3328, 3256 (NH2), 2174 (CN); 1H NMR δ: 7.96–7.02 (m, 10H, aromatic and NH2), 5.17 (s, 1H, H-1); MS m/z (%): 382 (M++2, 15.20), 380 (M+, 23.99) with a base peak at 102 (100); Anal. Calcd for C21H14ClFN2O2 (380.80): C, 66.24; H, 3.71; N, 7.36. Found: C, 66.19; H, 3.65; N, 7.30%.
3-Amino-1–(2-fluoro-6-trifuloromethylphenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4u)
Pale yellow crystals; yield 90%; m.p. 237–239 °C; IR (KBr) υ (cm−1): 3443, 3325, 3261 (NH2), 2200 (CN); 1H NMR δ: 7.86–6.96 (m, 10H, aromatic and NH2), 5.55 (s, 1H, H-1), 3.70 (s, 3H, OMe); 13C NMR δ: 160.70 (C-3), 158.65 (C-2, Ar), 153.65 (C-9), 147.98 (C-4a), 137.11 (C-10a), 136.82 (C-6, Ar), 133.82 (C-4, Ar), 131.82 (C-6), 130.64 (C-1, Ar), 129.99 (C-7), 126.28 (C-6a), 122.28 (CF3); 120.49 (C-10b, C-3,5, Ar), 117.63 (C-5), 114.45 (CN), 112.43 (C-8), 102.43 (C-10), 58.65 (C-2), 55.40 (CH3), 33.20 (C-1); MS m/z (%): 414 (M+, 31.03) with a base peak at 106 (100); Anal. Calcd for C22H14F4N2O2 (414.35): C, 63.77; H, 3.41; N, 6.76. Found: C, 63.71; H, 3.03; N, 6.70%.
3-Amino-1–(2,4-dimethoxyphenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4v)
Pale yellow crystals; yield 89%; m.p. 218–220 °C (Literature procedure, reflux condition, yield 80%; m.p. 205–206 °C)Citation72 IR (KBr) υ (cm−1): 3444, 3328, 3267 (NH2), 2199 (CN); 1H NMR δ: 7.78–6.26 (m, 10H, aromatic and NH2), 5.36 (s, 1H, H-1), 3.81 (s, 3H, OMe); 13C NMR δ: 160.38 (C-3), 159.58 (C-2, Ar), 158.40 (C-4, Ar), 156.90 (C-9), 147.86 (C-4a), 132.18 (C-10a), 130.43 (C-6, Ar), 129.98 (C-6), 129.04 (C-7), 126.35 (C-6a), 126.24 (C-10b), 121.044 (C-5), 117.24 (C-8), 115.77 (CN), 114.49 (C-1, Ar), 106.27 (C-10), 102.64 (C-,5, Ar), 98.53 (C-3, Ar), 57.91 (C-2), 56.37 (CH3), 55.49 (CH3), 55.29 (CH3), 31.23 (C-1); MS m/z (%): 388 (M+, 37.08) with a base peak at 102 (100); Anal. Calcd for C23H20N2O4 (388.42): C, 71.12; H, 5.19; N, 7.21. Found: C, 71.19; H, 5.25; N, 7.27%.
3-Amino-1–(3,4-dimethoxyphenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4w)
Yellow crystals; yield 88%; m.p. 253–255 °C (Literature procedure, reflux condition, yield 79%; m.p. 195–196 °C)Citation51 IR (KBr) υ (cm −1): 3365, 3324, 3293 (NH2), 2191 (CN); 1H NMR δ: 7.86–6.56 (m, 10H, aromatic and NH2), 5.21 (s, 1H, H-1), 3.74 (s, 3H, OMe), 3.65 (s, 6H, 2OMe); 13C NMR δ: 159.98 (C-3), 158.36 (C-9), 148.97 (C-4a), 147.84 (C-3, Ar), 147.39 (C-4, Ar), 138.93 (C-10a), 132.23 (C-1, Ar), 130.38 (C-6), 129.37 (C-7), 126.41 (C-6a), 121.09 (C-6, Ar), 119.75 (C-10b), 117.28 (C-5), 115.59 (C-8), 114.48 (CN), 112.62 (C-,5, Ar), 111.80 (C-2, Ar), 103.69 (C-10), 58.63 (C-2), 55.90 (CH3), 55.54 (CH3), 32.23 (C-1); MS m/z (%): 388 (M+, 37.08) with a base peak at 106 (100); Anal. Calcd for C23H20N2O4 (388.42): C, 71.12; H, 5.19; N, 7.21. Found: C, 71.07; H, 5.13; N, 7.15%.
3-Amino-1–(2,3,4-trimethoxyphenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4x)
Colourless crystals; yield 89%; m.p. 239–240 °C (Literature procedure, reflux condition, yield 84%; m.p. 239–240 °C) Citation28.
3-Amino-1–(3,4,5-trimethoxyphenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4y)
Colourless crystals; yield 89%; m.p. 260–261 °C (Literature procedure, reflux condition, yield 84%; m.p. 259–260 °C) Citation28.
3-Amino-1–(3,5-dibromo-2-methoxyphenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile (4z)
Colourless crystals; yield 92%; m.p. 263–264 °C (Literature procedure, reflux condition, yield 86%; m.p. 263–264 °CCitation28).
X-ray crystallography analysis
The compounds 4i was obtained as single crystals by slow evaporation from ethanol solution of the pure compound at room temperature. Data were collected on a Bruker APEX-II D8 Venture area diffractometre, equipped with graphite monochromatic Mo Kα and Cu Kα radiations at 293 (2) K. Cell refinement and data reduction were carried out by Bruker SAINT. SHELXTL-2018/3Citation73,Citation74 was used to solve structure. The final refinement was carried out by full-matrix least-squares techniques with anisotropic thermal data for nonhydrogen atoms on F. CCDC 2132096 contain the Supplementary crystallographic data for this compound can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Biological screening
Cell culture
The tumour cell lines MCF-7, HepG-2, and PC-3 were obtained from the American Type Culture Collection (ATCC, Rockville, MD). The cells were grown on RPMI-1640 medium supplemented with 10% inactivated foetal calf serum and 50 µg/mL gentamycin. The cells were maintained at 37 °C in a humidified atmosphere with 5% CO2 and were subculture two to three times a week.
Cytotoxicity evaluation using viability assay
The cytotoxic activity was appraised, using the 3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) colorimetric assay, as reported previouslyCitation57 and ELISA assay for MCF-7/ADRCitation75.
Analysis of P-glycoprotein
The content of P-gp in the MCF-7/ADR cell lysates after incubation with varying conc. (12.5–100 µM) of tested compounds 4b, 4c, 4d, 4q, and 4w following exposure for 48 h. was determined using commercial human P-gp ELISA Kit (MBS2506188, MyBioSource Inc., San Diego, CA). Absorption was recorded at 450 nm with a Spectramax Gemini fluorescence microplate reader (Molecular Devices, Sunnyvale, CA) Citation76.
Rhodamine 123 accumulation assay
P-gp activity was determined by measuring intracellular accumulation of Rhodamine 123 in MCF-7/ADR cells in the absence or presence of compounds 4b–4d, 4k, and 4w, according to commercial Rhodamine Competitive ELISA Kit (AKR-5142, Cell Biolabs Inc., San Diego, CA) which provides a convenient method for the detection of total Rhodamine in extracts from cellsCitation77. Briefly, resistant cancer cells were harvested, washed twice, and counted. After dilution to 1 × 106 cells/mL in 6 well plate each well, 1 mL of fresh media containing different concentrations of compounds 4b–4d, 4k, and 4w were added and incubated at 37 °C for 4 h in an atmosphere containing 5% CO2. Subsequently, 5.25 μM of Rho123 was added to each well and the wells were incubated for another 30 min at 37 °C. Finally, cells were washed, lysed as described before and intracellular quantification levels of rhodamine 123 were further analysed according to ELISA protocol Kit. Absorbance at 450 nm of each well was measured using Spectramax Gemini fluorescence microplate reader (Molecular Devices, Sunnyvale, CA). The total content of Rhodamine in each sample was determined by comparison with a Rhodamine standard curve.
Western blot analysis
Cellular protein extracts of cell lysates and western blotting were prepared after treatment of MCF-7/ADR cells with different compounds 4b–4d and 4w conc. (0.1, 1.0, and 10 μM) for 48 h as already describedCitation78. Cells were washed twice with ice-cold phosphate-buffered saline and total cell lysates were collected in sodium dodecyl sulphate (SDS) sample buffer. Cell lysates, containing equal amounts of protein, were separated by SDS–polyacrylamide gel electrophoresis (PAGE) and transferred to Hybond enhanced chemiluminescence nitrocellulose membrane (Amersham Biosciences, NJ). After being blocked in 5% non-fat milk in Tris-buffered saline with 0.1% Tween 20 (pH 7.6), membranes were incubated with the appropriate primary antibodies at 4 °C, overnight, and exposed to the appropriate secondary antibody for 3 h at room temperature. Finally, enzyme-linked chemiluminescence was visualised according to the ECL kit (Thermal Fisher, Waltham, MA) protocol. β-Actin was used to confirm equal loading in each lane in the samples prepared from cell lysates.
Cell cycle assay
Cell cycle arrest and distribution were done using Propidium Iodide Flow Cytometry Kit (ab139418, Abcam, Cambridge, UK) as previously describedCitation79. Briefly, MCF-7/ADR cancer cells at 1 × 104 cells were cultured in 60-mm dishes in the presence of various tested compounds with a concentration equal to the IC50 value for 24 h. Cells were collected and washed with PBS, fixed with precooled 70% ethanol at 4 °C. Staining went along in PBS containing 40 μg/ml RNase A and 10 μg/mL propidium iodide (PI) in the dark for 15 min. The DNA content in each cell nucleus was determined by a FACS Calibur flow cytometer (BD Biosciences, Franklin Lakes, NJ). Finally, Cell cycle phase distribution was analysed using Cell Quest Pro software (BD Biosciences) showing collected PI fluorescence intensity on FL2.
Annexin V-FITC apoptosis assay
Apoptosis assay was performed with an Annexin V-FITC/PI double staining apoptosis detection kit (K101, Biovison, Milpitas, CA) using a flow cytometerCitation80. MCF-7/ADR cancer cells treated with different newly synthesised compounds (IC50 value) were harvested by trypsinization, washed twice with 4 °C PBS, and re-suspended in binding buffer. Annexin V-FITC and PI solutions were then added to stain the cells before analysis by flow cytometry A minimum of 10,000 cells per sample were acquired. Annexin V-FITC binding (FL1) and PI (FL2) were analysed using Cell Quest Pro software (BD Biosciences).
Statistics
All data were expressed as the means ± standard deviation (SD), from at least three independent experiments with similar results. Statistical analysis and figures were performed by Graph Pad Prism version 5.01 (Graph Pad software, San Diego, CA).
Molecule docking
The docking experiment was employed according to previous workCitation81, the 3D model of P-gp was obtained from Swiss-ModelCitation82. Finally, the outcomes for docking were achieved by PyMol softwareCitation83.
Supplemental Material
Download PDF (4.5 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Liu YQ, Li LH, Yang L, Li HY. A novel, stereoselective and practical protocol for the synthesis of 4β-aminopodophyllotoxins. Chem Pap. 2010;64(4):533–536.
- Mečiarová M, Poláčková V, Toma Š. The effect of microwave and ultrasonic irradiation on the reactivity of benzaldehydes under Al2O3, Ba (OH)2 and K2CO3 catalysis. Chem Pap. 2002;56(3):208–213.
- Mečiarová M, Toma Š, Babiak P. Effect of ultrasound on one-pot conversion of alcohols to nitro and azido compounds. Chem Pap. 2004;58(2):104–108.
- Tabatabaeian K, Mamaghani M, Mahmoodi NO, Khorshidi A. Ultrasonic-assisted ruthenium-catalyzed oxidation of aromatic and heteroaromatic compounds. Catal Commun. 2008;9(3):416–420.
- Slobbe P, Ruijter E, Orru RV. Recent applications of multicomponent reactions in medicinal chemistry. Med Chem Commun. 2012;3(10):1189–1218.
- Batran RZ, Dawood DH, El-Seginy SA, et al. Coumarinyl pyranopyrimidines as new neuropeptide S receptor antagonists; design, synthesis, homology and molecular docking. Bioorg Chem. 2017;75:274–290.
- Kamdar NR, Haveliwala DD, Mistry PT, Patel SK. Synthesis and evaluation of in-vitro antitubercular activity and antimicrobial activity of some novel 4H-chromeno[2,3-d]pyrimidine via 2-amino-4-phenyl-4H-chromene-3-carbonitriles. Med Chem Res. 2011;20(7):854–864.
- Foroumadi A, Emami S, Sorkhi M, et al. Chromene-based synthetic chalcones as potent antileishmanial agents: synthesis and biological activity. Chem Biol Drug Des. 2010;75(6):590–596.
- Singh G, Sharma A, Kaur H, Ishar M. Chromanyl-isoxazolidines as antibacterial agents: synthesis, biological evaluation, quantitative structure activity relationship, and molecular docking studies. Chem Biol Drug Des. 2016;87(2):213–223.
- Rajanarendar E, Reddy MN, Krishna SR, et al. Design, synthesis, antimicrobial, anti-inflammatory and analgesic activity of novel isoxazolyl pyrimido[4,5-b]quinolines and isoxazolyl chromeno[2,3-d]pyrimidin-4-ones. Eur J Med Chem. 2012;55:273–283.
- Eberhardt L, Kumar K, Waldmann H. Exploring and exploiting biologically relevant chemical space. Curr Drug Targets. 2011;12(11):1531–1546.
- Ali TE, Ibrahim MA, Abdel-Kariem SM. Synthesis of biologically active 4-oxo-4 H-chromene derivatives containing sulfur-nitrogen heterocycles. Phosphorus Sulf Silicon. 2009;184(9):2358–2392.
- Lasemi Z, Azimi R, Azizi Amiri M. Efficient synthesis of 9, 10-dihydropyrano [2,3-h] chromene-2,8-dione derivatives in ionic liquid and the study of their antioxidant activity. Nat Prod Res. 2017;31(1):1–6.
- Sashidhara KV, Kumar M, Modukuri RK, et al. Discovery and synthesis of novel substituted benzocoumarins as orally active lipid modulating agents. Bioorg Med Chem Lett. 2011;21(22):6709–6713.
- Gupta S, Maurya P, Upadhyay A, et al. Synthesis and bio-evaluation of indole-chalcone based benzopyrans as promising antiligase and antiproliferative agents. Eur J Med Chem. 2018;143:1981–1996.
- Kumar MS, Singh J, Manna SK, et al. Diversity oriented synthesis of chromene-xanthene hybrids as anti-breast cancer agents. Bioorg Med Chem Lett. 2018;28(4):778–782.
- Smith CW, Bailey JM, Billingham ME, et al. The anti-rheumatic potential of a series of 2,4-disubstituted-4H-naphtho[1,2-b]pyran-3-carbonitriles. Bioorg Med Chem Lett. 1995;5(23):2783–2788.
- Kamdar NR, Haveliwala DD, Mistry PT, Patel SK. Design, synthesis and in vitro evaluation of antitubercular and antimicrobial activity of some novel pyranopyrimidines. Eur J Med Chem. 2010;45(11):5056–5063.
- Termentzi A, Khouri I, Gaslonde T, et al. Synthesis, biological activity, and evaluation of the mode of action of novel antitubercular benzofurobenzopyrans substituted on A ring. Eur J Med Chem. 2010;45(12):5833–5847.
- Dgachi Y, Bautista-Aguilera O, Benchekroun M, et al. Synthesis and biological evaluation of benzochromenopyrimidinones as cholinesterase inhibitors and potent antioxidant, non-hepatotoxic agents for Alzheimer’s disease. Molecules. 2016;21(5):634.
- Ahn J, Lee H, Jang J, Kim S, Ha T. Anti-obesity effects of glabridin-rich supercritical carbon dioxide extract of licorice in high-fat-fed obese mice. Food Chem Toxicol. 2013;51:439–445.
- Ahmed HE, El-Nassag MA, Hassan AH, et al. Introducing novel potent anticancer agents of 1H-benzo[f]- chromene scaffolds, targeting c-Src kinase enzyme with MDA-MB-231 cell line anti-invasion effect. J Enzyme Inhib Med Chem. 2018;33(1):1074–1088.
- Fouda AM, Okasha RM, Alblewi FF, et al. A proficient microwave synthesis with structure elucidation and the exploitation of the biological behavior of the newly halogenated 3-amino-1H-benzo[f]chromene molecules, targeting dual inhibition of topoisomerase II and microtubules. Bioorg Chem. 2020;95:103549.
- Fouda AM, Assiri MA, Mora A, et al. Microwave synthesis of novel halogenated β-enaminonitriles linked 9-bromo-1H-benzo[f]chromene moieties: induces cell cycle arrest and apoptosis in human cancer cells via dual inhibition of topoisomerase I and II. Bioorg Chem. 2019;93:103289.
- Gorle S, Maddila S, Maddila SN, et al. Synthesis, molecular docking study and in vitro anticancer activity of tetrazole linked benzochromene derivatives. Anticancer Agents Med Chem. 2017;17(3):464–470.
- Ahagh MH, Dehghan G, Mehdipour M, et al. Synthesis, characterization, anti-proliferative properties and DNA binding of benzochromene derivatives: increased Bax/Bcl-2 ratio and caspase-dependent apoptosis in colorectal cancer cell line. Bioorg Chem. 2019;93:103329.
- Elgaafary M, Fouda AM, Mohamed HM, et al. Synthesis of β-enaminonitrile-linked 8-methoxy-1H-benzo[f]chromene moieties and analysis of their antitumor mechanisms. Front Chem. 2021;9:759148.
- Elgaafary M, Lehner J, Fouda AM, et al. Synthesis and evaluation of antitumor activity of 9-methoxy-1H-benzo[f]- chromene derivatives. Bioorg Chem. 2021;116:105402.
- Rafinejad A, Fallah-Tafti A, Tiwari R, et al. 4-Aryl-4H-naphthopyrans derivatives: one-pot synthesis, evaluation of Src kinase inhibitory and anti-proliferative activities. Daru. 2012;20(1):100– 107.
- Kheirollahi A, Pordeli M, Safavi M, et al. Cytotoxic and apoptotic effects of synthetic benzochromene derivatives on human cancer cell lines. Naunyn Schmiedebergs Arch Pharmacol. 2014;387(12):1199–1208.
- El-Agrody AM, Abd El-Mawgoud HK, Fouda AM, Khattab ES. Synthesis, in-vitro cytotoxicity of 4H-benzo[h]chromene derivatives and structure–activity relationships of 4-aryl group and 3-, 7-positions. Chem Pap. 2016;70(9):1279–1292.
- Afifi TH, Okasha RM, Ahmed HE, et al. Structure-activity relationships and molecular docking studies of chromene and chromene based azo chromophores: a novel series of potent antimicrobial and anticancer agents. EXCLI J. 2017;16:868–902.
- Halawa AH, Fouda AM, Al-Dies AAM, El-Agrody AM. Synthesis, biological evaluation and molecular docking studies of 4H-benzo[h]chromenes, 7H-benzo[h]- chromeno[2,3-d]pyrimidines as antitumor agents. Lett Drug Des Discov. 2016;13(1):77–88.
- El-Agrody AM, Fouda AM, Khattab ES. Halogenated 2-amino-4H-benzo [h] chromene derivatives as antitumor agents and the relationship between lipophilicity and antitumor activity. Med Chem Res. 2017;26(4):691–700.
- Alblewi FF, Okasha RM, Eskandrani AA, et al. Design and synthesis of novel heterocyclic-based 4H-benzo[h]chromene moieties: targeting antitumor caspase 3/7 activities and cell cycle analysis. Molecules. 2019;24(6):1060.
- Piazzi L, Cavalli A, Belluti F, et al. Extensive SAR and computational studies of 3-{4-[(benzylmethylamino)methyl]phenyl}-6,7-dimethoxy-2-H-2-chromenone (AP2238) derivatives. J Med Chem. 2007;50(17):4250–4254.
- Teodori E, Braconi L, Bua S, et al. Dual P-glycoprotein and CA XII inhibitors: a new strategy to reverse the P-gp mediated multidrug resistance (MDR) in cancer cells. Molecules. 2020;25(7):1748.
- Braconi L, Teodori E, Riganti C, et al. New dual P-glycoprotein (P-gp) and human carbonic anhydrase XII (hCA XII) inhibitors as multidrug resistance (MDR) reversers in cancer cells, cancer cells. J Med Chem. 2022;65(21):14655–14672.
- Wang S, Wang SQ, Teng QX, et al. Structure-based design, synthesis, and biological evaluation of new triazolo[1,5-a]pyrimidine derivatives as highly potent and orally active ABCB1 modulators. J Med Chem. 2020;63(24):15979–15996. −
- Hong DJ, Jung SH, Kim J, et al. Synthesis and biological evaluation of novel thienopyrimidine derivatives as diacylglycerol acyltransferase 1 (DGAT-1) inhibitors. J Enzyme Inhib Med Chem. 2020;35(1):227–234.
- Callaghan R, Luk F, Bebawy M. Inhibition of the multidrug resistance P-glycoprotein: time for a change of strategy? Drug Metab Dispos. 2014;42(4):623–631.
- El-Agrody AM, Al-Ghamdi AM. Synthesis of certain novel 4H-pyrano[3,2-h]quinoline derivatives. Arkivoc. 2011;2011(11):134–146.
- Al-Sehemi AG, Irfan A, El-Agrody AM. Synthesis, characterization and DFT study of 4H-benzo[h]chromene derivatives. J Mol Struct . 2012;1018:171–175.
- El-Agrody AM, Al-Dies AA, Fouda AM. Microwave assisted synthesis of 2-amino-6-methoxy-4H-benzo[h]chromene derivatives. Eur J Chem. 2014;5(1):133–137.
- El-Wahab AH, Mohamed HM, El-Agrody AM, et al. Synthesis and biological screening of 4-benzyl-2H-phthalazine derivatives. Pharmaceuticals. 2011;4(8):1158–1170.
- Abd-El-Aziz AS, Shipman PO, Neeland EG, et al. Benzo [f]-and Benzo[h] coumarin-containing poly (methyl methacrylate) s and poly (methyl methacrylate) s with pendant coumarin-containing azo dyes. Macromol Chem Phys. 2008;209(1):84–103.
- El-Agrody MA, Khattab SAM, Fouda A. Synthesis, structure-activity relationship (SAR) studies on some 4-Aryl-4H-chromenes and relationship between lipophilicity and antitumor activity. Lett Drug Design Discov. 2014;11(10):1167–1176.
- El-Agrody AM, Khattab ES, Fouda AM, Al-Ghamdi AM. Synthesis and antitumor activities of certain novel 2-amino-9-(4-halostyryl)-4H-pyrano[3,2-h]quinoline derivatives. Med Chem Res. 2012;21(12):4200–4213.
- Mohamed HM, Abd EL-Wahab AH, El-Agrody AM, et al. Synthesis and characterization of new diiodocoumarin derivatives with promising antimicrobial activities. Beilstein J Org Chem. 2011;7(1):1688–1696.
- Halawa AH, Elaasser MM, El Kerdawy AM, et al. activities, molecular docking and structure–activity relationship of novel synthesized 4H-chromene, and 5H-chromeno[2,3-d]pyrimidine candidates. Med Chem Res. 2017;26(10):2624–2638.
- El-Agrody AM, Ali FM, Eid FA, et al. Synthesis and antimicrobial activity of thioxopyrimidines and related derivatives. Phosphorus Sulf Silicon Relat Element. 2006;181(4):839–864.
- Okasha RM, Albalawi FF, Afifi TH, et al. Structural characterization and antimicrobial activities of 7H-benzo[h]chromeno[2,3-d]- pyrimidine and 14H-benzo[h]chromeno[3,2-e][1,2,4]triazolo[1,5-c]pyrimidine derivatives. Molecules. 2016;21(11):1450.
- El Gaafary M, Syrovets T, Mohamed HM, et al. Synthesis, cytotoxic activity, crystal structure, DFT studies and molecular docking of 3-amino-1-(2,5-dichlorophenyl)-8-methoxy-1H-benzo[f]chromene-2-carbonitrile. Crystals. 2021;11(2):184.
- Mohamed HM, Amr AE, El-Agrody AM, et al. Crystal structure of 3-amino-1-(4-bromophenyl)-9-methoxy-1H-benzo[f]chromene-2-carbonitrile, C21H15BrN2O2. Zeitschrift für Kristallographie-new crystal structures. 2017;232(4):561–563.
- Allen FH, Kennard O, Watson DG, et al. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J Chem Soc Perkin Trans 2. 1987;(12):S1–S19.
- Spackman PR, Turner MJ, McKinnon JJ, et al. CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J Appl Crystallogr. 2021;54(Pt 3):1006–1011.
- Furukawa T, Kubota T, Suto A, et al. Clinical usefulness of chemosensitivity testing using the MTT assay. J Surg Oncol. 1991;48(3):188–193.
- Mi Y, Lou L. ZD6474 reverses multidrug resistance by directly inhibiting the function of P-glycoprotein. Br J Cancer. 2007;97(7):934–940.
- Waghray D, Zhang Q. Inhibit or evade multidrug resistance P-glycoprotein in cancer treatment: miniperspective. J Med Chem. 2018;61(12):5108–5121.
- Li YS, Yang X, Zhao DS, et al. Design, synthesis and bioactivity study on 5-phenylfuran derivatives as potent reversal agents against P-glycoprotein-mediated multidrug resistance in MCF-7/ADR cell. Eur J Med Chem. 2021;216:113336.
- Li S, Zhao Q, Wang B, et al. Quercetin reversed MDR in breast cancer cells through down-regulating P-gp expression and eliminating cancer stem cells mediated by YB‐1 nuclear translocation. Phytother Res. 2018;32(8):1530–1536.
- Luqmani YA. Mechanisms of drug resistance in cancer chemotherapy. Med Princ Pract. 2005;14(1):35–48.
- Dallavalle S, Dobričić V, Lazzarato L, et al. Improvement of conventional anti-cancer drugs as new tools against multidrug resistant tumors. Drug Resist Updat. 2020;50:100682.
- Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005;23(36):9408–9421.
- Williams GH, Stoeber K. The cell cycle and cancer. J Pathol. 2012;226(2):352–364.
- De U, Chun P, Choi WS, Lee BM, Kim ND, Moon HR, Jung JH, Kim HS. A novel anthracene derivative, MHY412, induces apoptosis in doxorubicin-resistant MCF-7/Adr human breast cancer cells through cell cycle arrest and down regulation of P-glycoprotein expression. Int J Oncol. 2014;44(1):167–176.
- Kim R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer. 2005;103(8):1551–1560.
- Tainton KM, Smyth MJ, Jackson JT, et al. Mutational analysis of P-glycoprotein: suppression of caspase activation in the absence of ATP-dependent drug efflux. Cell Death Differ. 2004;11(9):1028–1037.
- Fadok VA, Voelker DR, Campbell PA, et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148(7):2207–2216.
- Aller SG, Yu J, Ward A, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323(5922):1718–1722.
- Yang J, Yan R, Roy A, et al. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8.
- Mohamed HM, Fouda AM, Khattab ES, et al. Synthesis, in-vitro cytotoxicity of 1H-benzo[f]chromene derivatives and structure–activity relationships of the 1-aryl group and 9-position. Z Naturforsch C J Biosci. 2017;72(5-6):161–171.
- Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64(Pt 1):112–122.
- Sheldrick GM. SHELXTL-PC (version 5.1). Madison (WI): Siemens Analytical Instruments Inc.; 1997.
- Chaiyarit S, Thongboonkerd V. Comparative analyses of cell disruption methods for mitochondrial isolation in high-throughput proteomics study. Anal Biochem. 2009;394(2):249–258.
- Shchulkin AV, Abalenikhina YV, Erokhina PD, et al. The role of P-glycoprotein in decreasing cell membranes permeability during oxidative stress. Biochemistry (Mosc). 2021;86(2):197–206.
- Jouan E, Le Vée M, Mayati A, et al. Evaluation of P-glycoprotein inhibitory potential using a rhodamine 123 accumulation assay. Pharmaceutics. 2016;8(2):12.
- Tang X, Gu X, Ai H, et al. Synthesis and evaluation of nitric oxide-releasing DDB derivatives as potential Pgp-mediated MDR reversal agents in MCF-7/Adr cells. Bioorg Med Chem Lett. 2012;22(2):801–805.
- Vindeløv LL, Christensen IJ, Nissen NI. Standardization of high-resolution flow cytometric DNA analysis by the simultaneous use of chicken and trout red blood cells as internal reference standards. Cytometry. 1983;3(5):328–331.
- Zhang G, Gurtu V, Kain SR, Yan G. Early detection of apoptosis using a fluorescent conjugate of annexin V. Biotechniques. 1997;23(3):525–531.
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749.
- Lagares LM, Minovski N, Alfonso AYC, et al. Homology modeling of the human p-glycoprotein (ABCB1) and insights into ligand binding through molecular docking studies. Int J Mol Sci. 2020;21(11):4058.
- Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24(5):417–422.