
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In this study, we report a series of newly synthesised sulphonamides of aziridine-2-carboxylic acid (Az-COOH) ester and amide analogues as potent protein disulphide isomerase (PDI, EC 5.3.4.1) inhibitors. The inhibitory activity on PDI was determined against recombinant human PDIA1 and PDIA3 proteins using an insulin reduction assay. These compounds in low micromolar to low nanomolar concentrations showed the effective in vitro inhibitory properties of PDIA1 with weaker effects on PDIA3. Complexes of 15N- and 15N,13C- uniformly labelled recombinant human PDIA1a with two PDIA1 inhibitors were produced and investigated by a protein nuclear magnetic resonance (NMR) spectroscopy. It was found that both C53 and C56 of the PDIA1 enzyme were involved in covalent binding. Finally, in a range of pharmacological studies, we demonstrated that investigated compounds displayed anti-cancer and anti-thrombotic activity. These findings demonstrate that sulphonamides of Az-COOH derivatives are promising candidates for the development of novel anti-cancer and anti-thrombotic agents.
Keywords:
Introduction
Aziridines, in general, are alkylating agents useful as anti-cancer medicines (thiotepa, mitomycin C, etc.). It is believed that protonated forms of aziridines mainly alkylate nucleobasesCitation1. Furthermore, aziridines also react with the cysteine thiol group of proteins, and this reaction is highly specific at slightly alkaline pH values. However, in aqueous solutions, the competing side reaction is hydrolysisCitation2. Electron withdrawing substituents decrease the electron density on aziridine ring atoms, leading to a lower reactivity of such aziridine derivatives towards nucleic acids and increasing their selectivity for thiolate anionsCitation3.
Activated aziridines, such as aziridine-2-carboxylic acid (Az-COOH), have been suggested as promising cysteine protease inhibitors. It was found that the aziridine moiety of Az-COOH is substantially more reactive towards cysteine’s thiol groups than activated double bonds (e.g. N-ethylmaleimide, or halides such as alpha-iodopropionic acid or chloroacetic acid). Markedly, the amide of Az-COOH (Az-CONH2) as a cysteine proteinase inhibitor is magnitudes of order less potent than Az-COOHCitation4. This observation reflects the lower reactivity of Az-CONH2 towards the thiol group of cysteine compared to the methyl ester of Az-COOH (Az-COOMe). Surprisingly, among a number of known cytostatic derivatives of Az-COOH, Az-CONH2 was selected and approved in the USSR as an active pharmaceutical ingredient (API) of an anti-cancer immunomodulator with the brand name Leakadine®Citation5.
Recently, it was demonstrated that Az-CONH2 in water solutions selectively alkylates the thiol group of cysteineCitation6, but the molecular target of Az-CONH2 in the human body was not clarified. In contrast to the majority of aziridines, Az-CONH2 does not have mutagenic potential (i.e. it does not attack nucleic acids). Therefore, it is highly possible that the biochemical target of Az-CONH2 is one or more cysteine-containing enzymes, such as the protein disulphide isomerases (PDIs). This class of enzymes consists of around 22 proteins that act as reductases, oxidases, isomerases as well as chaperonsCitation7. Nearly all PDIs have the thioredoxin-like unit CXXC in their catalytic centres. PDIA1, PDIA3, PDIA4, and PDIA6 are the most frequently expressed representatives of this class of enzyme. Each of them has at least two catalytic domains with a CGHC sequence in their active siteCitation8. PDIA1, in particular, is over-expressed in brain, lymphoma, kidney, ovarian, prostate, lung, and several other cancers. It was demonstrated that increased PDIA1 positively correlates with the progression of various cancersCitation9. Another difference of cancer cells in contrast to normal ones is their ability to secrete endoplasmic reticulum located PDIs into the extracellular matrixCitation10. Furthermore, we recently demonstrated that extracellular PDIA1 plays an important role in regulating the adhesion of cancer cells to the endothelium and their transendothelial migrationCitation11. Therefore, inhibitors of PDIA1 may have cytostatic potential even if they do not penetrate across the cell membrane. To test the hypothesis that Leakadine-like compounds may act as PDI inhibitors in relation to major isoforms of PDIs, such as PDIA1 and PDIA3, we decided to synthesise an API of Leakadine®, namely Az-CONH2 as well as a small library of Az-COOH derivatives, test these compounds as possible PDIA1 and PDIA3 inhibitors and examine their anti-cancer effects. In addition, because PDIA1 and PDIA3 were suggested to represent a valid target for anti-thrombotic therapyCitation12, we tested some of the developed PDI inhibitors as anti-thrombotic agents.
Materials and methods
Chemistry
1H NMR and 13C spectra of the PDI inhibitors were acquired on a Varian Inova 400 MR spectrometer (400 MHz). The solvent was CDCl3. 1H NMR chemical shifts were reported in parts per million (ppm) relative to the internal standard TMS. CD2Cl2 carbon atoms served as internal standard 13C NMR spectra (CD2Cl2: 54.0 ppm). The coupling constants (J) are expressed in Hertz (Hz). Melting points were determined on an OptiMelt apparatus and were not corrected. All reagents and solvents were purchased from commercial suppliers and used without further purification. LC-MS analyses were performed on an Acquity UPLC with a Waters 3100 mass detector.
All protein nuclear magnetic resonance (NMR) spectra were acquired at 298.15 K on a 600 MHz Bruker Avance Neo spectrometer equipped with a 5-mm 1H/19F {13C/15N} Quadruple-Resonance cryoprobe with z-gradients. 3D HNCA, 3D HNCO, 3D HN(CA)CO, and CBCA(CO)NH spectra were acquired for the assignment of the backbone and CB of the CGHC motif. A 3D [1H,1H]-NOESY-15N-HSQC spectrum was acquired at a mixing time of 120 ms. Chemical shifts were referenced internally to the residual water signal at 4.70 ppm relative to 4,4-dimethyl-4-silapentane-1-sulphonic acid (DSS). All acquired spectra were transformed in TopSpin version 4.0 software (Bruker). The assignment was performed in CARA 1.9.1Citation13.
Protein–ligand complexes were prepared by addition of the corresponding ligand from a 62.5 mM DMSO-d6 stock solution in four equal portions to achieve the following protein–ligand ratios: 1:0.5, 1:1, 1:1.5, and 1:2. The final DMSO-d6 amount did not exceed 2% (v/v). The chemical shift perturbation, d, was calculated from 2D 1H-15N-HSQC spectra by EquationEquation (1)(1)
(1) Citation14:
(1)
(1)
where δH and δN were the chemical shift differences for the apo and bound forms for the proton and nitrogen atoms, respectively. The scaling factor, α, was set to 0.1.
Synthesis of starting materials
6-(Dimethylamino)-5-formylnaphthalene-1-sulphonyl chloride

6-(Dimethylamino)naphthalene-1-sulphonic acid (1.0 g, 3.98 mmol) was transferred into a 50 ml round-bottom flask. Then, CH2Cl2 (20 ml) was added, followed by DMF (0.4 ml), and finally, oxalyl dichloride (2.0 g, 15.74 mmol) was added dropwise. The resulting solution was allowed to react with stirring for 24 h at room temperature, and the reaction mixture was then quenched by adding 50 ml of ice/salt. The solution was extracted twice with 10 ml of CH2Cl2 and the organic layers combined and dried over Na2SO4. The solvent was evaporated under vacuum. The resulting 6-(dimethylamino)-5-formylnaphthalene-1-sulphonyl chloride (0.83 g 70%) was used during the next stage without additional purification. The 1H-NMR spectrum (400 MHz, CDCl3, HMDSO) was as follows: δ 10.30 (s, 1H), 9.55 (dt, J = 8.7, 1.0 Hz, 1H), 8.87 (dd, J = 9.7, 0.9 Hz, 1H), 8.18 (dd, J = 7.6, 1.1 Hz, 1H), 7.66 (dd, J = 8.7, 7.6 Hz, 1H), 7.60 (d, J = 9.7 Hz, 1H), 3.22 (s, 6H).
The same method was used for the preparation of 6-(dimethylamino)-5-formylnaphthalene-2-sulphonyl chloride. 
The 1H-NMR spectrum (400 MHz, CDCl3, HMDSO) was as follows: δ 10.26 (s, 1H), 8.91 (d, J = 9.1 Hz, 1H), 8.11 (d, J = 9.1 Hz, 1H), 8.11 (m, 1H), 7.75 (dd, J = 9.1, 2.0 Hz, 1H), 7.51 (d, J = 9.1 Hz, 1H), 7.41 (d, J = 9.1 Hz, 1H), 3.13 (s, 6H).
5-Chloro-6-(methylamino)naphthalene-2-sulphonyl chloride
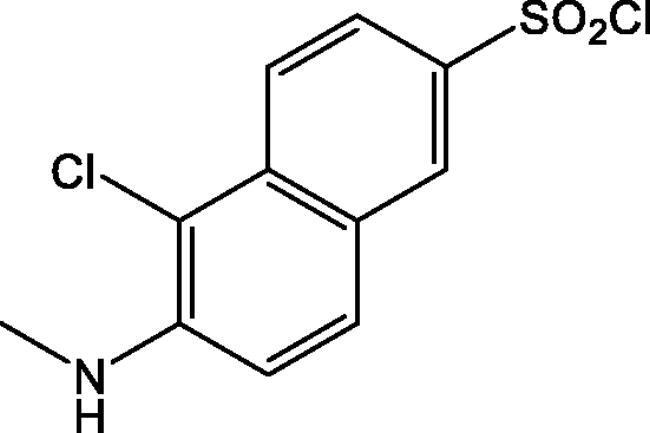 To a suspension of 6-(dimethylamino)naphthalene-2-sulphonic acid (1.0 g, 3.98 mmol) in POCl3 (5 ml) was slowly added PCl5 (3.7 g, 17.8 mmol). The resulting mixture was heated at 50 °C for 5 h before it was allowed to cool to room temperature and poured onto crushed ice. The aqueous mixture was stirred vigorously at 0 °C for 40 min. The product was extracted twice with 40 ml of CH2Cl2 and the organic layers combined and dried over Na2SO4. The solvent was evaporated under vacuum. The resulting 5-chloro-6-(methylamino)naphthalene-2-sulphonyl chloride was purified by chromatography (silica gel, petroleum ether/ethyl acetate 4:1). Yield 0.35 g (30%). The 1H-NMR spectrum (400 MHz, CDCl3, HMDSO) was as follows: δ 8.24 (d, J = 2.0 Hz, 1H), 8.15 (dt, J = 9.2, 0.6 Hz, 1H), 7.95 (dd, J = 9.2, 2.0 Hz, 1H), 7.86 (d, J = 9.0 Hz, 1H), 7.22 (d, J = 9.0 Hz, 1H), 3.11 (s, 3H).
To a suspension of 6-(dimethylamino)naphthalene-2-sulphonic acid (1.0 g, 3.98 mmol) in POCl3 (5 ml) was slowly added PCl5 (3.7 g, 17.8 mmol). The resulting mixture was heated at 50 °C for 5 h before it was allowed to cool to room temperature and poured onto crushed ice. The aqueous mixture was stirred vigorously at 0 °C for 40 min. The product was extracted twice with 40 ml of CH2Cl2 and the organic layers combined and dried over Na2SO4. The solvent was evaporated under vacuum. The resulting 5-chloro-6-(methylamino)naphthalene-2-sulphonyl chloride was purified by chromatography (silica gel, petroleum ether/ethyl acetate 4:1). Yield 0.35 g (30%). The 1H-NMR spectrum (400 MHz, CDCl3, HMDSO) was as follows: δ 8.24 (d, J = 2.0 Hz, 1H), 8.15 (dt, J = 9.2, 0.6 Hz, 1H), 7.95 (dd, J = 9.2, 2.0 Hz, 1H), 7.86 (d, J = 9.0 Hz, 1H), 7.22 (d, J = 9.0 Hz, 1H), 3.11 (s, 3H).
Preparation of known compounds (C-3314, C-3262, C-3273, C-3263, C-3272, C-3612)
1-(p-tolylsulphonyl)aziridine-2-carbonitrile (C-3314) was prepared as described earlierCitation15. 1-(p-tolylsulphonyl)aziridine-2-carbaldehyde (C-3262) was prepared according to the procedure of Lapinsky and BergmeierCitation16. 1–(4-Butylphenyl)sulphonylaziridine-2-carbaldehyde (C-3273) was synthesised by using the same method. 1-[1-(p-tolylsulphonyl)aziridin-2-yl]ethanone (C-3263) was prepared as described by Smith and KimCitation17. 1-[1–(4-Butylphenyl)sulphonylaziridin-2-yl]ethanone (C-3272) was prepared using the same method. Lithium 1-tosylaziridine-2-carboxylate (C-3612) was synthesised according to a method developed by Baldwin et al.Citation18.
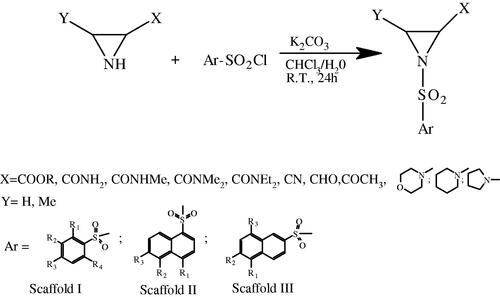
General procedure for the synthesis of novel sulphonamides (Scaffolds I–III)
Aromatic sulphonic acid chloride (1 mmol) was added with stirring to the solution of the appropriated aziridine (1.1 mmol) and K2CO3 (2 mmol) in a mixture of 1 ml CHCl3 + 1 ml water. The mixture was stirred for 24 h at room temperature, and the product was then extracted with CHCl3 and the solution dried over MgSO4. The solvent was evaporated. The product was purified by liquid chromatography (silica gel, petroleum ether/ethyl acetate 4:1= >1:2) to give the corresponding aziridine aromatic N-sulphonamide.
Determination of PDI inhibiting activity of compounds on enzymatic insulin reduction assay
The PDIA1 and PDIA3 activity was measured as an increase of disulphide bond reduction of human insulin in the presence of dithiothreitol (DTT) causing aggregation of its β-chain, a process that can be followed by turbidimetry. Briefly, the assay mixture for 96-well plates was prepared by dissolving in 0.1 mM phosphate buffer (pH 7.6) 6 µg/ml (96 nM) of PDIA1 (Escherichia coli recombinant human protein; MyBioSource) or 12 µg/ml (205 nM) of PDIA3 (E. coli recombinant human protein; MyBioSource), 2 mM EDTA and 0.08 mM DTT. Stock solutions of test compounds were freshly prepared in DMSO and subsequently diluted to maintain the final DMSO concentration in the assay mixture below 1%. Calculated amounts of compounds solutions were added into test wells and the reaction was started by the addition of insulin (human, recombinant; Sigma) and 0.08 mM of DTT. The final concentration of insulin and DTT in the assay mixture was 0.15 and 0.16 mM, respectively. The reaction rate was monitored at 650 nm on a Microplate Reader Infinite® M1000 PRO for 60 min at 37 °C. Turbidity values for the wells containing only PDIA1 (background values) were subtracted from the turbidity values of the wells containing PDIA1 + tested compound. The PDIA1 activity decrease in the presence of compound was calculated by the EquationEquation (2)(2)
(2) :
(2)
(2)
where OD means optical density (OD)Citation19,Citation20.
Protein expression and purification
The recombinant plasmid pET-28a(+)-PDIA1a coding thioredoxin 1 domain (residues 18–137) of human PDIA1 (P07237) was designed based on Kemmink et al.Citation21,Citation22 and ordered from BioCat (GmbH). The plasmid was transformed into BL21(DE3) E. coli cells to produce PDIA1a with an N-terminal His6-tag followed by a TEV-cleavage site. Bacterial colonies were selected on Luria–Bertani (LB) medium agar plates supplemented with 50 μg/ml kanamycin.
For the production of unlabelled-PDIA1a, 1 l of LB medium supplemented with 50 mg/l kanamycin was inoculated with 1% (v/v) of overnight culture and grown at 37 °C to an OD of 0.4 at A600. Afterwards, the temperature was reduced to 20 °C and the cells grown to an OD600 of 0.6–0.7. Isopropyl-β-D-1-thiogalactopyranoside (IPTG) was added at a final concentration of 1.0 mM to induce protein expression. Cells were harvested by centrifugation after 16 h of expression for 15 min at 4 °C and 7400 × g and stored at −20 °C before purification. The final yield was 84 mg protein from 1 l of LB.
15N- and 15N,13C-uniformly labelled PDIA1a were produced in M9 minimal medium containing 1 g/l 15NH4Cl and 2 g/l [13C6]-α-D-(+)-glucose (Cambridge Isotope Laboratories, Tewksbury, MA, USA) as sole sources of nitrogen and carbon, respectively. In the case of mono-labelled PDIA1a, non-labelled α-D-(+)-glucose was used at a concentration of 4 g/l. A single-cell colony was inoculated into 5 ml of LB medium supplemented with 50 mg/l kanamycin, day culture, and grown at 37 °C for 6–8 h to an OD600 > 1.0. Afterwards, 1% (v/v) of day culture was inoculated into 10 ml of M9 medium in the presence of 50 mg/l kanamycin, night culture, and grown at 37 °C overnight. On the next morning, 1% (v/v) of night culture was used to inoculate 1 l of M9, and the cells were then treated similarly as described above for unlabelled PDIA1a. The expression time after induction was prolonged to 20–24 h. The final yield was 54 and 44 mg of protein from 1 l of M9 for 15N-labelled and 15N,13C-labelled cells, respectively.
The cell pellets were re-suspended in buffer A (20 mM Tris–HCl pH 7.5, 500 mM NaCl, 20 mM imidazole, 2 mM DTT) at a 1-to-10 ratio and lysed by ultrasonication. The supernatant was isolated by centrifugation for 45 min at 4 °C and 27,000 × g. Afterwards, the solution was filtered through a 0.22 μm filter and applied onto a 5 ml HisTrap™ HP (Cytiva, Marlborough, MA) column pre-equilibrated in buffer A. The bound protein was eluted by applying a linear gradient of buffer B (20 mM Tris–HCl pH 7.5, 500 mM NaCl, 500 mM imidazole, 2 mM DTT) from 0% to 100% for 15 column volumes. The fractions containing PDIA1a were poured together. Specific protein batches were dialysed against buffer C (20 mM Tris–HCl pH 7.5, 2 mM DTT) overnight at 4oC, and the His6-tag was then cleaved by TEV-protease and protein-purified on a HisTrap™ HP (Cytiva) column using the same buffers A and B. Then, cleaved and non-cleaved PDIA1a batches were treated similarly: concentrated to ∼2 ml using Amicon ultra-centrifugal filters with a 10 kDa molecular weight cut-off and injected onto a HiLoad™ 16/600 Superdex™ 75 pg (Cytiva) column pre-equilibrated in buffer D (10 mM AcOH-NaOH pH 5.1, 50 mM NaCl, 2 mM DTT). All purification steps were analysed by SDS-PAGE gel-electrophoresis. Fractions containing pure protein were combined and concentrated to 0.5–1.0 mM. The remaining protein was aliquoted, fast-frozen in liquid nitrogen and placed at −80 °C for long-term storage. Each aliquot was thawed on ice before use.
The protein portion was dialysed overnight at 4 °C against the same buffer C but without DTT prior protein–ligand binding experiments. Each ligand was added to the protein in four portions, increasing the protein:ligand ratio from 1:0.5 to 1:2.NMR samples were prepared by the addition of 7% (v/v) D2O and 0.03% (v/v) NaN3 to 15N- or 15N,13C-PDIA1a solution. The protein concentration varied from 0.15 to 1.0 mM depending on the type of experiment.
Cell culture and measurement of cancer cell viability
Cell lines
Human connective tissue fibrosarcoma (HT-1080, ATCC® CCL-121™), human colon adenocarcinoma (CaCo-2, ATCC® HTB-37™), human breast adenocarcinoma (MDA-MB-231, ATCC® HTB-26™) and human breast adenocarcinoma (MCF-7, ATCC® HTB-22™) cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% or 20% fetal bovine serum for CaCo-2. The cultures were cultivated at 37 °C with 5% CO2, 95% air and complete humidity. All cell lines were regularly tested for Mycoplasma contamination.
MTT assay
HT-1080, CaCo-2, MDA-MB-231, and MCF-7 were grown to ∼90% confluence and were detached using 0.05% trypsin/EDTA and counted. Cells were plated at optimal density for the logarithmic phase of growth. These cells were then re-suspended at a concentration of 5000 cells/well and transferred onto 96-well plates. For background absorption, some wells remained cell free (i.e. as a blank control). Cells were incubated for 24 h to allow cells to adhere to the bottom of wells, then medium containing serial dilutions of the investigated compounds was added to the cells (n = 6), which were then further incubated for 48 h. Control cells were incubated with media containing DMSO. After incubation, living cell determination with MTT assays was performedCitation23. Briefly, the culture medium was removed from each well and replaced with fresh medium with MTT (0.2 mg/ml). After 3 h, the MTT solution was removed and replaced with 200 µl DMSO and 25 µl Sorenson’s glycine buffer (glycine 0.1 M, NaCl 0.1 M, pH 10.5 with 0.1 NaOH). The plate was further shaken for 15 min at room temperature, and the optical density (OD) of the wells was determined using a multichannel spectrophotometer (Thermo Scientific Multiskan EX, Waltham, MA) at 540 nm. The quantity of the control cells was determined in calculations for 100%. The percentage of surviving cells was calculated according to the formula 100 ×T/C, where T is the OD of the test well after 48 h exposure to the compound, and C is the OD for the control cell wells after 48 h. All compounds were tested in three repeats for each tumour cell line. The IC50 values as the averages of three determinations were calculated from the best fit (R2 > 0.95) of the Hill slope curve to the experimental data using a nonlinear regression analysis in Graph Pad Prism version 3 (GraphPad Software, Inc., La Jolla, CA, USA).
Basal cytotoxicity test
A neutral red uptake (NRU) assay was performed according to the standard protocol of StokesCitation24 modified by the NICEATM-ECVAM validation studyCitation25,Citation26. The NRU cytotoxicity assay procedure is based on the ability of viable cells to incorporate and bind neutral red, a supravital dye. Balb/c 3T3 (Mouse Swiss Albino embryo fibroblast) cells (9000 cells/well) were placed into 96-well plates for 24 h in DMEM medium containing 5% foetal bovine serum and then exposed to the test compound over a range of eight concentrations (1, 3, 10, 31, 100, 316, and 1000 µg/ml) for 24 h. Untreated cells were used as a control. After 24 h, the medium was removed from all plates. Then, 250 μl of neutral red solution were added (0.05 mg/ml NR in DMEM 24 h pre-incubated at 37 °C and then filtered before use through a 0.22 µm syringe filter). Plates were incubated for 3 h, and then the cells were washed three times with PBS. The dye within viable cells was released by extraction with a mixture of acetic acid, ethanol and water (1:50:49). The absorbance of neutral red was measured using a spectrophotometer multiplate reader (TECAN, Infinite M1000) at 540 nm. The OD was calculated using EquationEquation (3)(3)
(3) :
(3)
(3)
The IC50 values were calculated using the programme Graph Pad Prism® version 3.0.
Estimation of LD50 from IC50 values
Data from the in vitro tests were used for estimating the starting dose for acute oral systemic toxicity tests in rodents. The in vivo starting dose is an estimated LD50 value calculated by inserting the in vitro IC50 value into a regression formula: log LD50 (mM/kg) = 0.439 log IC50 (mM) + 0.621Citation26. The value is recalculated to mg/kg, and compounds are evaluated in accordance to the Globally Harmonised System of Classification and Labelling of Chemicals (GHS) with five toxicities categoriesCitation27:
category 1: LD50 ≤ 5 mg/kg (highly toxic);
category 2: 5 < LD50 ≤ 50 mg/kg (moderately toxic);
category 3: 50 < LD50 ≤ 300 mg/kg (slightly toxic);
category 4: 300 < LD50 ≤ 2 000 mg/kg (practically non-toxic);
category 5: LD50 > 2000 mg/kg (unclassified).
Estimation of acute toxicity
The acute toxicity for the selected compounds was determined using a fixed-dose procedure according to the OECD test guidelineCitation27 and in accordance with good laboratory practice principles. The study was approved by the Local Ethical Committee for Animal Experiments in Poznań (No. 60/2015 – Appendix 1). In the study, adult female Wistar rats, young, mature, nulliparous, and non-pregnant at the age of 10 weeks were used. The weight of animals did not exceed ± 20% of the mean weight of all dosed animals. Rats were randomly selected for the study. The analysed compounds were suspended in 0.5% methylcellulose solution. As recommended, if there was no information on the toxicity of the compounds, the initial dose in the sighting study was 300 mg/kg b.w. Because mortality or toxicity was not observed, the test was repeated with a higher dose (2000 mg/kg b.w.) with a 24-h gap between dosing. Based on the results from the sighting study, the main study dose was 2000 mg/kg b.w. administrated to four animals by gavage. After treatment, the animals were observed daily for 14 days. During the experiment, general and detailed observations were provided. The signs observed were piloerection, changes in the eye and mucous membranes, physical changes associated with central nervous, autonomic, cardiovascular and respiratory systems, pattern of behaviour and somatomotor activity, direct attention to tremors, convulsions, salivation, diarrhoea, lethargy, sleep and coma. After 14 days of observation, the animals were sacrificed, and post-mortem examinations of the external surface of the body, all orifices, cranial, thoracic, and abdominal cavities together with their contents and detailed macroscopic examinations were performed.
The acute toxicity test provides information on the hazardous properties and allows the compounds to be ranked and classified according to the GHS for the classification of chemicals which cause acute toxicityCitation27.
The influence of selected PDI A1 inhibitors on the clonogenic potential, cell cycle, and cell death of human colon cancer cell lines
For the purpose of these tests, the IC50 of the tested compounds (C-3251 and control compounds: CCF642 and LOC14) was determined in the 72 h proliferation inhibition test using an sulphorhodamine B (SRB) assay (as described previouslyCitation28). To confirm the anti-cancer effects observed in the various cancer cell lines, selected PDIA1 inhibitors were tested in a long-term 3D colony formation assay of human colorectal carcinoma cell lines (HT-29 and CaCo-2). For this long-term colony formation assay, the HT-29 (ATCC) and CaCo-2 (ATCC) cell lines were maintained as follows: HT-29 in RPMI-1640 with HEPES + OPTI-MEM (1:1) and CaCo-2 in Eagle’s medium (all from IIET, Wroclaw, Poland), and both culture media were supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate (both from Sigma-Aldrich Chemie GmbH, Steinheim, Germany) and fetal bovine serum: 5% HT-29 (GE Healthcare), 20% CaCo-2 (Sigma-Aldrich Chemie GmbH, Steinheim, Germany), 100 U/ml penicillin, 100 μg/ml streptomycin (both from Polfa Tarchomin S.A., Warsaw, Poland). Both cell lines were grown at 37 °C in a humidified atmosphere with 5% CO2.
The cells were treated with tested compounds in the doses of IC50 (72 h assay) for each compound and collected after three more days. The viable cells were counted using a haemocytometer (trypan blue exclusion method) and seeded in triplicate at a density of 5 × 102 cells/3 ml and 2.5 × 102 cells/3 ml (9.5 cm2) for CaCo-2 and HT-29, respectively. The dishes had been pre-coated with poly-L-lysine/PBS (0.001%; Sigma-Aldrich) and washed twice with PBS (with Ca2+ and Mg2+). After 2 weeks, the colonies were fixed and stained with 1% crystal violet/ethanol (Sigma-Aldrich), documented with a Sony Alpha 300 camera (Sony) and counted manually using ImageJ 1.47 version software (National Institutes of Health, Bethesda, MD, USA). The surviving fraction (SF) was then calculatedCitation29. Cell cycle and cell death analyses were performed as previously describedCitation30. Compounds were tested five times independently. The obtained results were analysed using BD FACSDiva 6.2 version software (Becton Dickinson, Franklin Lakes, NJ, USA). The caspase-3/7 activity assay was performed as previously describedCitation30, based on 48 h of incubation with the compounds. The compounds were tested in triplicate in a single experiment, and each experiment was repeated three times independently. The results were normalised to the protein content using the SRB method and reported as mean relative caspase-3/7 activity compared to the control sample ± SD.
Anti-thrombotic effects of selected PDIA1 inhibitors in vivo in rat model of arterial thrombosis
To confirm the anti-thrombotic activity of PDIA1 inhibitors, the selected compounds were tested in vivo in the rat model of arterial thrombosis. Wistar rats were anaesthetized with pentobarbital (40 mg/kg, i.p.) and placed in a supine position on a heated (37 °C) operating table. Arterial thrombosis was induced by electrical stimulation of the right common carotid artery, as previously describedCitation31. Briefly, the anode, a stainless-steel L-shaped wire, was inserted under the artery and connected to a constant current generator. The cathode was attached subcutaneously to the hind limb. The artery was stimulated (1 mA) for 10 min. Fifty-five minutes after the beginning of stimulation, the segment of the common carotid artery containing the formed thrombus was dissected and opened lengthwise, and the thrombus was completely removed and air-dried at room temperature for 24 h. The thrombus was then weighed in a blinded manner.
Results and discussion
Synthesis and characterisation of the new 1-arylsulphonyl-2-aziridinecarboxylic acid derivatives (Scaffolds I–III)
The aim of this work was to verify the concept that 1-arylsulphonyl- Az-COOH derivatives (Scaffolds I–III) may have PDI inhibiting activity and, therefore, represent promising scaffolds for the design of novel anti-cancer and anti-thrombotic medicines. To test this hypothesis, the synthesis of novel compounds (Scaffolds I–III) was performed as shown in Scheme 1.
Scheme 1. Synthetic pathway of 1-arylsulfonyl-aziridine-2-carboxylic acid derivatives (Scaffolds I–III, for definition of substituents see ).
According to Scheme 1, these compounds () were prepared by acylation of the appropriate derivative of Az-COOH, such as esters (Az-COOR), unsubstituted amides (Az-CONH2), mono-substituted amides (Az-CONHR), disubstituted amides (Az-CONR2), nitrile (Az-CN), carbaldehyde (Az-CHO) or methylketone (Az-COCH3) with corresponding aromatic sulphonic acid chloride in CHCl3/H2O as the solvent in the presence of K2CO3 as the base and isolated from the reaction mixture by flash chromatography with nearby quantitative yields. The synthesised compounds were characterised by 1H, 13C NMR and LC-MS spectroscopy (Supplemental materials, Table S1). All compounds did have HRMS m/z signals corresponding to the calculated mass peaks. The presence of an aziridine ring was confirmed by the characteristic proton resonances of the aziridine ring system (2-H, dd (1H) at δ = 2.35–2.81 (J = 4.1–4.4 Hz and J = 6.8–7.7 Hz), as well as 3-H dd (1H) at δ = 2.35–2.81 (J = 4.1–4.4 Hz) and dd (1H) at δ= 2.41–2.95 (J = 6.8 − 7.7 Hz). The expected aromatic proton resonance was observed in the corresponding aromatic region typical for substituted phenyl and naphthalene ring systems, respectively. Signals of proton resonance generated from substituents in the aromatic ring system, as well in the aziridine moiety, were in accordance with the assumed structures. The chemical shifts of carbon atoms in 13C-NMR corresponded to the expected values.
Table 1. PDIA1 inhibiting activity of benzene sulphonamides of aziridine-2-carboxylic acid derivatives (Scaffold I).
Table 2. PDIA1 inhibiting activity of 1-naphthalene sulphonamides of aziridine-2-carboxylic acid derivatives (Scaffold II).
Table 3. PDIA1 inhibiting activity of 2-naphthalene sulphonamides of aziridine-2-carboxylic acid derivatives (Scaffold III).
Protein disulphide isomerase A1 (PDIA1) inhibition
We previously demonstrated that 2-carbamoylaziridine (Az-CONH2, the API of Leakadine®) in water solutions reacts with the thiol group of thiophenol and cysteine as wellCitation6. To support our hypothesis that Leakadine® may have PDIA1 inhibiting activity, we tested this medicine as a possible PDIA1 inhibitor. For this purpose, we used an optimised insulin turbidity assay19,Citation20. This assay is based on the fact that PDIA1 reduces both –S-S- bonds of the insulin molecule causing the aggregation of its beta-chain and an increase of the reaction mixture turbidity at 650 nm. To supply reducing equivalents for supporting this reaction, DTT is recommended to be used. Unfortunately, covalent binders of thiol groups may react with DTT; moreover, DTT may also reduce S-S bridges in the insulin molecule too. Therefore, we optimised this assay to test our compounds and determined that the optimal final concentration of DTT in the assay’s mixture was 0.16 mM, whereby 0.08 mM of DTT had to be added into the assay mixture during preincubation with PDIA1, and then supplementation of DTT to the final concentration of 0.16 mM was carried out simultaneously by adding the insulin. The buffer system consisting of 100 mM potassium phosphate and 2 mM EDTA buffer with pH = 7.6 was selected as optimal (for experimental details, see Supplementary Materials S).
Using these reaction conditions for the insulin turbidity assay, we found that Az-CONH2 was a very weak inhibitor of PDIA1. Thus, preincubation of PDIA1 with 1 mM Leakadine® for one hour inhibits PDIA1 activity by approximately 30%.
We speculated that the double-activated aziridine ring system must be more sensitive to the nucleophilic attack of SH-groups of cysteine in the PDIA1 catalytic centre when compared with monoactivated derivatives of Az-COOH, for example, Az-CONH2. Therefore, we decided to synthesise and to test a small library of N-activated Az-COOH. For this purpose, we prepared N-aryl sulphonamides of Az-COOH derivatives (Scaffolds I–III) and tested them as potential PDIA1 inhibitors in an insulin turbidity assayCitation19,Citation20 (Scheme 1, ). It is known that the catalytic a and a’ domains of PDIA1 (referred to as PDIA1a and PDIA1a’) to some extend operate independently of each otherCitation32. Therefore, we expected that aryl sulphonamides would preferably attack the catalytic domain ‘a’ of the N-terminal, which in the presence of reducing equivalents is typically in the reduced form. Therefore, we expressed a recombinant PDIA1a domain as well and compared the catalytic activity of the PDIA1a domain with the full-length recombinant protein PDIA1 in the absence and presence of inhibitors of PDIA1 in an optimised (see above) insulin turbidity assayCitation33–37. In the assay mixture consisting of 0.1 mM of potassium phosphate buffer (pH 7.6), 2 mM EDTA, 0.16 mM DTT and 0.15 mM insulin, the final concentration of both proteins tested was 8 µg/ml. We found that the PDIA1a domain’s S-S cleavage rate was approximately 1.4-fold lower when compared with full-length PDIA1. Using both enzymes, we also determined the IC50 values for PDIA1 and PDIA1a of the known irreversible inhibitors of PDIA1, namely PACMA31 and CCF642. We found that the corresponding IC50 values of PACMA31 were 8.4 ± 0.4 and 18.3 ± 0.7 µM, and for CCF642, the IC50 values were 16.6 ± 0.8 and 13.0 ± 0.6 µM for PDIA1 and PDIA1a, respectively. For our selected test compound, C-3290 insulin reduction IC50 values were 0.6 ± 0.03 and 1.1 ± 0.05 µM for PDIA1 and PDIA1a, respectively. Hence, the aryl sulphonamide C-3290 does indeed block the PDIA1a domain.
The preference of the PDIA1a domain might have been related to its stability. Indeed, if stored at −20 °C, PDIA1a was more robust compared to the full-length PDIA1 and maintained its insulin-reducing activity for >10 months.
Our results demonstrated that for the testing of covalent inhibitors of PDIA1, the PDIA1a domain (rather than the full-length protein) can also be used. Unfortunately, according to the literature data, the activity of the known PDIA1 inhibitors was mainly determined on the full-length PDIA1. Therefore, to compare the activity of our novel aryl-sulphonamides of Az-COOH (Scaffolds I–III) with that of the previously described inhibitors, we decided to test all synthesised compounds on the full-length PDIA1.
We demonstrated that aromatic sulphonamides of Az-COOH derivatives (Scaffolds I–III) inhibited PDIA1 activity. Moreover, we found that N-sulphonylation with aromatic sulphonyl chlorides substantially increased the reactivity of Az-COOH derivatives towards the catalytic centre of PDIA1. Among the aromatic sulphonamides of Az-COOH, in the majority of cases, the naphthalene 1- and 2-sulphonamides (Scaffolds II and III) were more active inhibitors when compared to benzene sulphonamides (Scaffold I). We also found that esters of Az-COOH benzene sulphonamides were typically stronger inhibitors compared to the corresponding amides (C-3251 > C-3532; C-3212 > C-3216; C-3256 > C-3324; C-3304 > C-3578; C-3161 > C-3220, C-3353 > C-3402 and C-3290 > C-3591. The notable exception was the p-alkyl derivatives of Az-COOH benzene sulphonamides, where the activity of amides was equivalent to esters as PDIA1 inhibitors or even slightly better (C-3389 > C-3287). The same was true for the sulphonamides of methyl-(Az-CONHMe) and dimethyl-amides (Az-CONMe2) (C-3380 > C-3287 and C-3380 > C-3369), respectively. We found that among the 1-benzene sulphonamides of Az-CONR2, the volume of the amide group played a critical role. Thus, the PDIA1 inhibiting activity of the tested compounds decreased in following order: NMe2 > N-morpholinyl > N-piperidinyl > N-pyrrolidinyl > N,N-diethyl (C-3369 > C-3517 > C-3520 > C-3511 > C-3546). Substitution of the 4-H-atom of benzene ring by fluorine (C-3576) or chlorine (C-3296) atoms, MeO (C-3294), NMe2 (C-3343), MeCONH (C-3308), NO2 (C-3212) as well as an NH2 (C-3218) group lowered the inhibiting activity of benzene sulphonamides towards PDIA1. In contrast, replacement of 4-H by a 4-CF3 group improved the activity (C-3256 > C-3251).
In the case of benzosulphonamides of Az-COOMe (Scaffold I), replacing the 2-H and 4-H atoms of the benzene ring for the fluorine (C-3353 and C-3403) did not alter the PDIA1 inhibiting activity of the parent compound (C-3251) but slightly decreased it in the case of Az-CONH2 derivatives (C-3402 and C-3402). Increasing the lipophilicity elevated the activity of benzene sulphonamide. Thus, replacement of the 3-Me group on the benzene ring (C-3303) for the 3-CF3 (C-3305) resulted in a twofold increase of PDIA1 inhibiting activity (C-3305 > C-3303). Substitution of 4-H by a CH3-group was well tolerated (C-3161), but analogous replacement of 3-H (C-3303) or 2-H (C-3220) impaired the PDIA1 blocking activity, which decreased further in the following order: 4-Me > 4H > 3-Me > 2-Me (C-3161 > C-3251 > C-3303 > C-3220). 2,4,6-trimethyl substitution of the benzene ring did not substantially alter the PDIA1 inhibiting activity (C-3304). However, installing a 4-phenyl group upregulated the inhibiting capacity of the compound by one order of magnitude (C-3291 > C-3251).
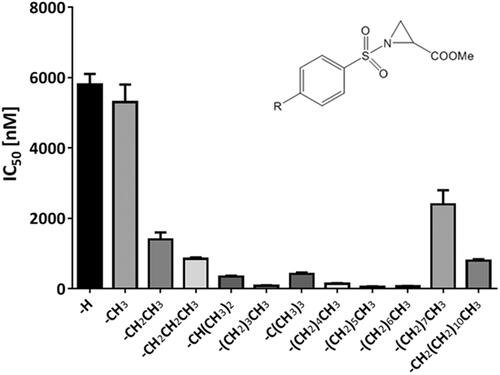
Based on this finding, we concluded that the p-position of benzene sulphonamides of Az-COOH derivatives was suitable for substitution with bulky lipophilic substituents. Therefore, we systematically introduced alkyl substituents in this p-position of the benzene sulphonamides ring and found a good correlation between increasing numbers of carbon atoms in the p-alkyl substituent with PDIA1 inhibition. This correlation was valid for alkyl substituents with a maximum of six carbon atoms in the chain. Seven and more carbon atoms containing alkyl chains were not as well tolerated because the PDIA1 inhibition was weaker as compared to when six carbon atoms were present in the chain (.
Figure 1. PDIA1 inhibiting activity of p-substituted benzosulphonamides of Az-COOMe with aliphatic chain of various lengths (from C1 to C12).
Next, we synthesised 1- and 2-naphthalene sulphonamides of Az-COOH derivatives (Scaffolds II and III, and ). We found that naphthalene sulphonamides were moderate inhibitors of PDIA1 reductase activity in an insulin turbidity assay. In the majority of cases, mono-substituted 2-naphthalene sulphonamides (Scaffold III) were slightly more active when compared to the corresponding 1-naphthalene sulphonamides (Scaffold II) (C-3292 > C-3311; C-3373 > C-3365; C-3399 > C-3326; C-3400 > C-3371; C-3399 > C-3383, except C-3398 < C-3377. In the case of double substitution, the derivatives of 1-naphthalene sulphonamides (Scaffold II) were more active inhibitors than the 2-naphthalene sulphonamides (Scaffold III) (C-3376 > C-3390).
Among the 1-naphthalene sulphonamides of Az-COOMe, substitution with an NMe2 group in ring positions 4 or 6 improved the inhibiting activity slightly but impaired it significantly if the NMe2 group was placed at position 5 of the naphthalene ring: C-3383 (4-NMe2) > C-3375 (6-NMe2) > C-3311 (H) > C-3326 (5-NMe2). In the sulphonamides of Az-CONMe2, replacement of a hydrogen atom with an NMe2- group increased the inhibition of PDIA1: C-3377 (6-NMe2) > C-3371 (5-NMe2) > C-3385 (4-NMe2) > C-3311 (H). The 5-CHO- group improved the activity of PDIA1 inhibitors (C-3376 > C-3375). The sulphonamides of Az-COOMe were slightly better inhibitors than the corresponding derivatives of Az-CONMe2 (C-3311 > C3365; C-3336 > C-3366; C-3383 > C-3385). Nevertheless, the Az-COOMe derivative C-3326 was a weaker inhibitor of PDIA1 compared to the corresponding dimethylamide C-3371.
Replacement of the naphthalene 5-H atom improved the activity of Scaffold III towards PDIA1 (C-3399 > C-3292; C-3400 > C-3373), but analogue substitutions at C-6 or C-8 decreased the activity of the corresponding compounds (C-3292 > C-3398; C-3373 > C-3398 and C-3292 > C-3427, respectively).
Installing an additional 5-CHO-substituent in the naphthalene ring of the 2-naphthalene sulphonamides of Az-COOMe did not change the activity (C-3390∼C-3397). In general, mono-substituted sulphonamides of Az-COOMe were typically more active than the corresponding Az-CONMe2 derivatives (C-3292 > C-3373; C-3397 > C-3398; C-3399 > C-3400).
The highest activity among the synthesised 2-naphthalene sulphonamides of Az-COOH towards PDIA1 was found for 5-NMe2 (IC50 = 0.12 µM) and decreased in the following order: C-3399 > C-3391 > C-3400 > C-3292 > C-3397 > C-3390 > C-3427 > C-3373 > C-3329 > C-3327 > C-3459.
Protein disulphide isomerase A3 (PDIA3) inhibition
To evaluate the selectivity of synthesised compounds towards PDIA1, we also tested all synthesised compounds as potential PDIA3 inhibitors (). We found that PDIA3 was less sensitive against the inhibitory action of Az-COOH sulphonamides as compared with the PDIA1. Thus, among the compounds of Scaffold I, the IC50 values for PDIA3 ranged from 5.4 to >200 µM. The corresponding naphthalene 1-sulphonamides (Scaffold II) were more active PDIA3 inhibitors with IC values from 2.8 to 93 µM, as compared with naphthalene-2 sulphonamides with IC values from 4.0 to >200 µM for (Scaffold III). Hence, the benzene sulphonamides of Az-COOH derivatives are better PDIA1 inhibitors with the highest activity among the group of 4-alkyl-benzenesulphonamides of Az-COOH, which represent selective PDIA1 inhibitors with a nanomolar range of IC50 values (from 30 to 50 nM for 4-hexyl-benzenesulphonamides of Az-COOH C-3287, C-3389, C-3380, C-3369 to 80–85 nM for 4-Butyryl and 4-Pentyl substituted derivatives of Scaffold I (C-3257 and C-3368). For these compounds, the ratio of IC50 for PDIA1/IC50 for PDIA3 was 0.0004–0.0035. In fact, nearly all sulphonamides of the Az-COOH derivatives appeared to be more effective PDIA1 inhibitors than PDIA3 inhibitors; however, for some of the compounds, the relative inhibition of PDIA1 vs. PDIA3 was similar (C-3212, C-3216, C-3537, C-3273, C-3517 and C-3594). For these compounds, the ratio of IC50 for PDIA1/IC50 for PDIA3 was 0.55:1.28. This result reflects the different catalytic site environments in PDIA1 and PDIA3 and points out that among the aromatic sulphonamides of Az-COOH derivatives described above were agents with relatively selective PDIA1 blocking activity discovered as well as compounds with non-selective PDIA1 and PDIA3 blocking mechanism of action were identified.
NMR data of PDIA1a domain binding experiments
As the PDIA1 protein has two catalytic centres, we decided to clarify whether one (or both) are targeted by our novel PDIA1 inhibitors. The PDIA1 a-domain, PDIA1a (residues 18–137) consisting of a thioredoxin fold was overexpressed in E. Coli to verify the activity of the aromatic sulphonamides of Az-COOH towards the catalytic CGHC motif. The PDIA1a activity was also compared with full-length PDIA1 (purchased from MyBioSource) in an insulin turbidity assay. Both proteins showed similar affinity towards our compounds.
The 15N- and 15N,13C-labelled versions of PDIA1a were expressed to elucidate the binding mechanism of the Az-COOH derivatives by means of NMR. The chemical shifts obtained for the PDIA1a-apo form from 2D 1H-15N-HSQC were compared with the chemical shifts of known NMR structure of PDIA1a, PDB ID: 1MEKCitation21. Both structures had similar chemical shifts except for the CGHC active site, indicating that cysteines C53 and C56 did not form disulphide bonds. As the protein was purified in an excess of DTT, the reduced PDIA1a form was obtained. The first three N-terminal residues (S17, D18, and A19) as well as the residues D24, H25, H55, and A118 were not observed in the 2D 1H-15N-HSQC spectra (residue numbering by full-length P4HB sequence, uniport P07237, Figure S1)Citation22 most probably due to increased flexibility.
Two compounds, C-3389 and C-3399, with different activities towards PDIA1 and PDIA3 (selective PDIA1 and non-selective PDIA3 and PDIA1 inhibitor, respectively, see and ) were chosen for further analysis. As the Az-CONH2 and its activated analogues easily undergo ring opening and self-alkylation reactionsCitation6, the stability of the compounds in the aqueous solutions was tested prior to the experiments with the protein. Two buffers were used: 10 mM AcOH-NaOH pH 5.1, 50 mM NaCl and 10 mM Tris–HCl pH 7.6, 50 mM NaCl. The impact of the DTT was tested as well. The ring opening occurred faster at pH 7.4 in the absence of DTT for both compounds. 49%, and 16% of C-3389 and C-3399, respectively, were degraded in 3.5 h. Similar degradation levels were reached in 39 h at pH 5.1. The addition of DTT slowed down the degradation by a factor of two at the higher pH and increased degradation by 10% at the lower pH. Thus, 10 mM AcOH-NaOH pH 5.1, 50 mM NaCl buffer without DTT was chosen for the primary 2D 1H-15N-HSQC experiments.
The DTT was removed from the protein solution prior to ligand binding by overnight dialysis at 4 °C. The oxidation state of the apo-PDIA1a was monitored by NMR at 25 °C in the following 48 h by acquiring 1H-15N HSQC spectra. The experimental data showed no changes in the chemical shifts of the CGHC motif, indicating that the protein was maintained in its reduced state without DTT.
Next, PDIA1a was titrated with the previously mentioned ligands to ensure complete protein saturation. The 2D 1H-15N-HSQC spectrum of apo-PDIA1a was compared with the same spectra of protein–ligand complexes at different ratios. Both compounds C-3399 () and C-3389 () showed binding to PDIA1a. In both cases, minimal chemical shift perturbations and intensity changes were observed at a 1:0.5 ratio, whereas several signals were weakened or doubled at a 1:1 ratio. Complete protein saturation was achieved only at 1:2 ratios.
Figure 2. 2D 1H-15N HSQC spectra of the reduced form of uniformly labelled 15 N-PDIA1a apo-protein (blue) and protein–ligand complex with C-3399 at 1:1 ratio (red) and 1:2 ratio (green) in 10 mM AcOH-NaOH pH 5.1, 50 mM NaCl.
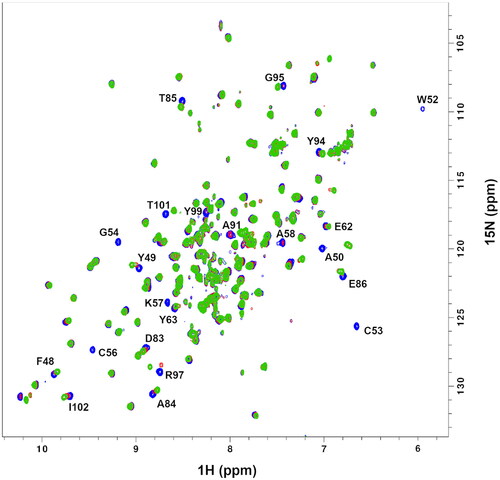
Figure 3. 2D 1H-15N HSQC spectra of the reduced form of uniformly labelled 15 N-PDIA1a apo-protein (blue) and protein–ligand complex with C-3389 at 1:1 ratio (red) and 1:2 ratio (green) in 10 mM AcOH-NaOH pH 5.1, 50 mM NaCl.
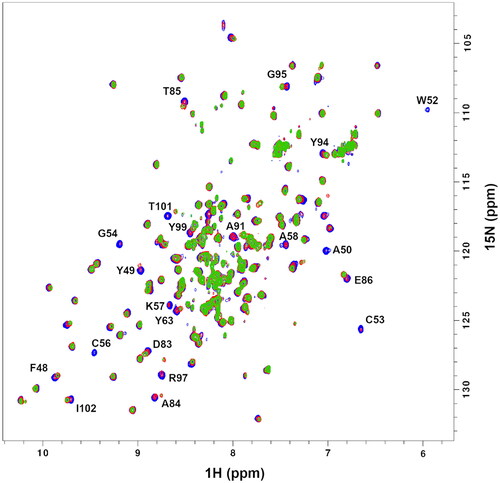
The alignment of the 2D 1H-15N HSQC spectra of apo-protein with protein-C-3399 () and protein–C-3389 () complexes at 1:1 and 1:2 ratios allowed to identify 22 residue cross-peaks, 7 of which disappeared, and 15 were shifted due to ligand binding event.
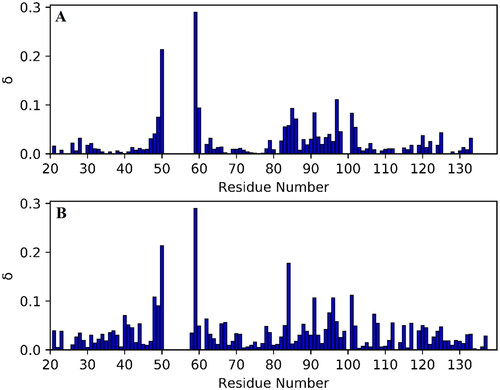
Chemical shift perturbation plots for C-3399 and C-3389 were calculated using EquationEquation (1)(1)
(1) . Faded residues (W52, C53, G54, C56, K57, A58, and Y99) were excluded from the comparison. As one can see, both compounds C-3399 () and C-3389 () were bound in a similar manner, causing significant changes in the chemical shifts of the active site residues. C-3389 caused larger chemical shift differences for F48, A84, and V96 than C-3399. Thus, it was proposed that such a perturbation was caused by binding of the n-hexyl substituent of the compounds (see ).
Figure 4. Chemical shift perturbation, δ, plots for PDIA1a complex with covalently bound C-3399 (A) and C-3389 (B). Disappeared residues (W52, C53, G54, C56, K57, A58, and Y99) were excluded from the comparison.
In order to test whether the ligands were bound covalently, overnight dialysis at 4 °C to the same buffer was performed. The spectra of the protein–ligand complexes did not change, revealing that a covalent bond was formed. The addition of the excess of DTT also did not change the spectra in 24 h. Thus, the newly formed covalent bond is rather strong and could not be reduced by reducing agents under experimental conditions.
Afterwards, the reaction between apo-protein and C-3399 and C-3389 was performed in the presence of 2 mM DTT. The reaction occurred in the same manner as it was observed without DTT. However, complete protein saturation was achieved at a slightly higher protein-ligand ratio of 1:2.5, suggesting that DTT might slow down binding or degrade the ligand prior its binding to the protein.
An additional 3D CBCA(CO)NH and 3D [1H,1H]-NOESY-15N-HSQC spectra were acquired for the PDIA1a–C-3399 complex to identify which cysteine (C53 or C56) was involved in the covalent bond formation. The analysis of the CB chemical shifts indicated that the reaction with activated derivatives of Az-COOH was not selective because both cysteines of the catalytic centre formed corresponding reaction products and could explain why a twofold excess of the ligand was necessary to fully saturate the protein.
Antiproliferative activity
The antiproliferative effects of PDIA1 inhibitors (Scaffolds I–III) were tested in four cancer cell lines: HT-1080, CaCo-2, MCF-7, and MDA-MB-231. We found that depending on the structure of the compounds and the cell line used in the experiment, the cytotoxicity of these compounds fluctuated over a large range – from IC50 = 0.13 ± 0.02 µM to IC50 > 500 µM ().
Table 4. The in vitro antiproliferative effect of benzene sulphonamides of aziridine-2-carboxylic acid derivatives (Scaffold I) towards panel of cancer cells.
Table 5. Inhibiting activity of 1-naphthalene sulphonamides of aziridine-2-carboxylic acid derivatives (Scaffold II) towards panel of cancer cell lines in vitro.
Table 6. Inhibiting activity of 2-naphthalene sulphonamides of aziridine-2-carboxylic acid derivatives (Scaffold III) towards panel of cancer cells in vitro.
When comparing the average IC50 values of the ten most active compounds across each of the cell lines being studied, the most sensitive to Az-COOH derivatives was the cell line HT-1080 with an average IC50 = 3.9 µM, followed by a line of MDA-MB-231 with a median IC50 = 5.73 µM. The sensitivities of the cell line MCF-7 with median IC50 = 7.18 µM and CaCo-2 with IC50 = 7.48 µM were remarkably similar to each to other.
Among the tested compounds, the most active compound on HT-1080 was C-3546 (IC50 = 0.13 ± 0.02 µM), whereas against CaCo-2 cells, it was C-3336 that had the highest activity (IC50 = 0.5 ± 0.1 µM). In two other lines – MCF-7 and MDA-MB-231 – the Az-COOH derivatives tested seemed to be less potent. Still, the most active in cell line MCF-7 was the compound C-3256 with IC50 = 2.8 ± 0.2 µM, and against MDA-MB-231 was the compound C-3376 with IC50 = 2.3 ± 0.2 µM.
In most cases (except C-3546, Scaffold I), the naphthalen-1- and naphthalen-2-sulphonic acid amides of the Az-COOH derivatives (Scaffolds II and III) were more cytotoxic against HT-1080 compared to the corresponding benzosulphonamides (Scaffold I) with IC50 values from 4.6 ± 0.4 µM (C-3336) to 100 ± 5 µM (C-3326).
The cytostatic activity of the Az-COOMe derivatives of Scaffold I towards HT-1080 cells can be improved by p-substitution of the hydrogen atom with electron-donating alkyl groups (t-Bu > dodecyl > octyl > propyl > heptyl > i-Pr > cyclohexyl > hexyl > Bu > Et) or by some other electron donors, such as Me2N or BuO as well as by lipophilic substituents (F, NO2, and H-bound donating/accepting MeCONH group). In contrast, some electronegative substituents (Cl, CF3) in the p-position of the benzene ring decreased the anti-cancer activity of the tested compounds. This finding correlates to some extent with observations that p-alkyl groups increased the PDIA1 inhibiting activity of Az-COOH derivatives of Scaffold I.
Similarly, for HT-1080, the non-substituted benzosulphonamides of Az-COOMe (C-3251) also had relatively weak activity against CaCo-2 cells (IC50 = 110 ± 8 µM). Comparing the sensitivity of the HT-1080 and CaCo-2 cell lines against the compounds of Scaffold I, the latter cell line was found to be more resistant.
In CaCo-2 cells, the cytotoxicity of benzosulphonamides (Scaffold I, Subtype Az-COOMe) with substituents in the p-position decreased in the following order: Et > t-Bu > MeO > F > i-Pr > Me > octyl > Cl > NO2 > propyl > Me2N > cyclohexyl > dodecyl > CH3CONH > H > heptyl > pentyl > BuO > Ph > hexyl > Bu > CF3, which did not correlate well with changes of the PDIA1 inhibiting activity for the same compounds, potentially reflecting the low level of PDIA1 expressed by this cell lineCitation11.
The cytotoxicity against CaCo-2 of p-substituted benzosulphonamides (Scaffold I, Subtype Az-CONH2) decreased as follows: t-Bu > CF3 > NO2 > Me > H > NH2, which correlates to some extent with changes of their lipophilicity.
For benzosulphonamides of Scaffold I, Subtype Az-CONHMe, the effect of p-substituents on the cytotoxicity of compounds was even more pronounced: t-Bu (C-3593, IC50 = 25 ± 4 µM) > C6H11 (C-3380, IC50 = 157 ± 25 µM).
The cytotoxicity against CaCo-2 cells of naphthalene-1-sulphonamides (Scaffold II, subtype Az-COOMe) decreased in the following order: 4-Me > 5-CHO, 6-NMe2 > 6-NMe2 > 4-NMe2 > 5-NMe2. Among the Az-CONMe2 derivatives of Scaffold II, the 6-NMe2 derivative was more active compared to 4-NMe2. For the sulphonamides of Az-COOMe (Scaffold III), the cytotoxicity decreased in the following range: 5-CHO,6-NMe2 > H > 6-Me > 8-NMe2 > 5-Cl,6-NHMe.
The cytotoxicity of Az-COOH derivatives towards the MCF-7 cell line in vitro varied from IC50 = 2.8 ± 3.2 µM (for C-3256) to IC50 = 485 ± 32 µM (for C-3308). In general, the cytostatic activity of benzosulphonamides (Scaffold I, subtype Az-COOMe) towards the MCF-7 cell line in vitro decreased in the following order:
4-CF3 > 3-CF3 > 2,4-di-F > 4-octyl > 4-Et > 4-Me > 4-t-Bu > 4-i-Pr = 4-Cl > 4-C12H23 > 4-Bu > 4-H > 4-Pr > 4-Me2N > 2,4,6-tri-Me = 4-MeO > 4-BuO = 4-NO2 > 4-heptyl > 4-pentyl > 4-hexyl > 4-cyclohexyl > 3-Me > 4-CH3CONH > 2-Me.
In most cases, the Az-CONMe2 derivatives of Scaffolds I–III were more cytotoxic against cell line MCF-7 when compared to the corresponding Az-COOMe derivatives. This observation is most likely explained by an increased hydrolytic stability of dimethyl amides as compared with methyl ester.
Further, we evaluated the antiproliferative activity of PDIA1 inhibitors against the triple-negative breast cancer cell line MDA-MB-231. The cytotoxicity of Az-COOMe benzosulphonamides (Scaffold I) towards MDA-MB-231 cell line in vitro varied from IC50 = 5.6 ± 0.6 µM (C-3297, 2-F, 4-Me) to IC50 = 380 ± 21 µM (C-3320, 2-Me) and decreased in the following order:
2-F,4-Me > 4-t-Bu > 4-CF3 > 4-heptyl > 3-CF3 > 4-hexyl > 2,4,6-trimethyl > 4-BuO > 4-Bu > 4-pentyl > 4-iso-propyl > 4-F > 4-Pr > H > 4-octyl > 4-NMe2 > 4-Cl > 2-F,4-Me > 4-MeO > 4-Et > 4-cyclohexyl > 2-Me > 4-CH3CONH.
We have found that the cytotoxicity of p-alkylbenzene sulphonamides (Scaffold I, Subtype Az-COOMe) against MDA-MB-231 correlated to some extent with PDIA1 inhibitory activity () and may reflect the fact that these cells do have relatively higher PDIA1 expression levels among all of the PDI isoforms expressed by these cellsCitation11.
Cytotoxicity of aromatic sulphonamides towards normal embryonic murine cells BALB/3T3 and estimation of basal cytotoxicity
We tested the cytotoxicity of all synthesised compounds towards Balb/c 3T3 (Mouse Swiss Albino embryo fibroblast) cells. According to the data presented in , aromatic sulphonamides depending on structure possessed quite different toxicities towards the normal cell line, starting from highly toxic compounds (C-3272, C-3273, IC50 < 1 µM) and ending with practically non-toxic derivatives (C-3251, IC50 = 3307 ± 947 µM; C-3294, IC50 = 1248 ± 191 µM; C-3315, IC50 = 2800 ± 200 µM;). In general, most active PDIA1 inhibitors are slightly more toxic compared to less potent compounds.
Table 7. Cytotoxicity of aromatic sulphonamides (Scaffolds I–III) towards mouse Swiss Albino embryo fibroblast cells (Balb/c 3T3).
Nevertheless, the ratio of PDIA1 inhibiting activity/cytotoxicity on 3T3 cells was 500:1 for C-3389, 300:1 for C-3380, 275:1 for C-3369 and 126:1 for C-3287, which means that these compounds should have substantial therapeutic safety intervals if used as PDIA1 inhibitors in vivo. Interestingly, the medium active (IC50 = 6 µM) non-toxic PDIA1 inhibitor compound C-3251 did have an even better safety interval (1:551).
Data from the in vitro tests were used for estimating the starting dose for acute oral systemic toxicity tests in rodents. The predicted oral toxicity for rodents of the compounds tested was also in a broad scope, but for many of the compounds, the calculated LD50 values were low.
In vivo acute toxicity of selected PDI inhibitors
The acute toxicity for the selected compounds C-3380, C-3389, C-3390, and C-3399 was determined using a fixed-dose procedureCitation27, and these compounds were classified to category 5/unclassified for GHS. After a single 300 and 2000 mg/kg b.w. administration to individual animals (sighting study), and after a single dose of 2000 mg/kg b.w. to four animals (main study), no symptoms of toxicity were observed. The animals survived the entire period of the study. During 14 days of observation, body weight gains were noted. At the doses tested, none of the compounds caused alteration of behaviour or led to symptoms related to the central nervous system, autonomic, circulatory and respiratory during the period tested. A necropsy of the animals at the dose of 2000 mg/kg b.w. showed macroscopic aspects of normal organs. In acute toxicity, all compounds exhibited a significant margin of safety, demonstrated by the absence of behavioural and systemic toxicity at the tested dose of 2000 mg/kg b.w., and there were no adverse effects related to the animals.
Influence of PDIA1 inhibitors on clonogenic potential, cell cycle, and cell death of human colon cancer cell lines
To confirm the anti-cancer effect of a selected PDIA1 inhibitor, C-3251 was chosen (low cytotoxicity, potent inhibition of PDIA1, see ). For that purpose, HT-29 and CaCo-2 colon cancer cells were used. In a 72-h antiproliferative assay, the IC50 of the tested and control compounds was as follows: CaCo-2 cells: C-3251 − 3.37 ± 0.2 µg/mL; CCF642 − 0.41 ± 0.1 µg/mL; LOC14 − 9.12 ± 3.4 µg/mL. HT-29 cells: C-3251 − 30.22 ± 4.73 µg/mL; CCF642 − 0.41 ± 0.05 µg/mL; LOC14 − 12.64 ± 8.4 µg/mL. Concentrations close to the IC50 were used in further studies.
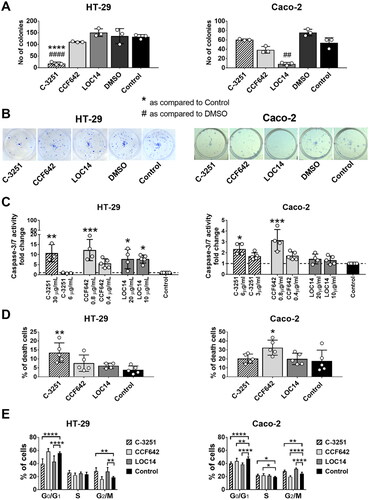
As shown in , C-3251 considerably diminished the number of colonies, and the effect was more pronounced as compared to reference PDI inhibitors, such as CCF642 or LOC14, on HT-29 cells (). Furthermore, C-3251 activated caspase-3/7 in both colon cancer cell lines and increased the percentage of dead cells in the HT-29 cell line (). C-3251 arrested the cell cycle in G2/M phase in HT-29 cells or in the S phase in CaCo-2 cells and diminished the percentage of cells in G0/G1 stage in both colon cancer cell lines, as shown in .
Figure 5. The influence of PDIA1 inhibitor C-3251 on the clonogenic potential (A, B), cell death (C, D) and cell cycle (E) of human colon cancer HT-29 and CaCo-2 cells. Dashed line designated control level. Data represent the means ± SD of least three independent experiments. Statistical analysis was performed using Kruskal–Wallis followed by Dunn’s (A, C: CaCo-2), one-way ANOVA followed by Sidak’s (A, C: HT-29; D) or two-way ANOVA followed by Dunnett’s (E) multiple comparisons test (*p = 0.05, **, ##p = 0.01, ***p = 0.001, ****, ####p = 0.0001).
Anti-cancer activity of selected PDIA1 inhibitors in vivo
The anti-cancer activity of some PDIA1-inhibitors with various potency to inhibit PDIA1 (C-3281, C-3329, and C-3342) was tested in the Lewis lung carcinoma (LLC) murine model in vivo ( and , Tables S1–S13). We selected these compounds based on their PDIA1 inhibition activity. C-3281 displayed one of the highest PDIA1 inhibiting activities (IC50 = 0.14 µM, ), C-3329 was a modest PDIA1 inhibitor with IC50 = 1.1 µM ), and C-3342 was the weakest PDIA1 inhibitor (IC50 = 33.4 µM, ). We also estimated the cytotoxicity of these compounds against normal murine fibroblast 3T3 cells in vitro and calculated their basal toxicity. We found that the strongest inhibitor of PDIA1 (C-3281) was also the most potent anti-cancer agent among those compounds tested in vivo, slowing tumour growth by 65–71% in dosages of 5–15 mg i.p. once a day for 16 days. Of note, this compound possessed the lowest cytotoxicity towards normal 3T3 cells (IC50 = 120 ± 25 µM), with a calculated basal toxicity of approximately 530 ± 45 mg/kg (). C-3329 demonstrated relatively weaker anti-cancer activity but still inhibited tumour growth by 58% at a high dosage (i.p., 100 mg/kg for 16 days). This compound had a calculated basal toxicity of 324 ± 26 mg/kg with IC50 = 44 ± 6 µM for 3T3 cells in vitro. The weakest PDIA1 inhibitor C-3342 was the least active anti-cancer agent among the three tested in vivo. It was cytotoxic on the 3T3 cell line in vitro with IC50 = 93 ± 22 µM, with the calculated basal toxicity being 399 ± 42 mg/kg. Therefore, all three compounds (C-3281, C-3329, and C-3342) could be categorised as essentially non-toxic and with anti-cancer effects that seemed related to the inhibition of PDIA1. Good toleration of these compounds after 16 days long administration in vivo to animals was also reflected in the undisturbed weight gain of the treated animals in all test groups that, in general, did not differ significantly from the control animals, except for the group treated with C-3281 (5 mg/kg for 16 days) (Table S8).
Figure 6. Cytostatic activity of C-3281 (i.p., 5 mg/kg for 16 days and 15 mg/kg for 16 days), C-3329 (i.p., 100 mg/kg for 16 days) and C-3342 (i.p. 170 mg/kg for 16 days) on Lewis lung carcinoma model in mice.
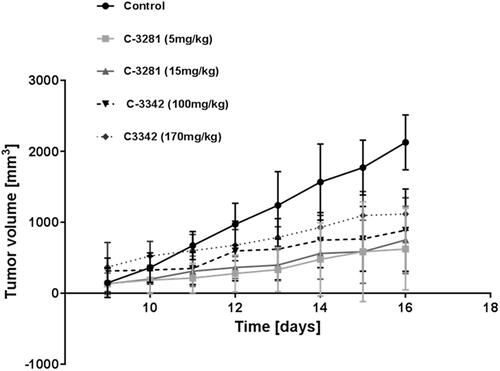
Figure 7. Inhibition of tumour growth in Lewis lung carcinoma control group (1) and Lewis lung carcinoma mice groups (2–5) treated with C-3281 (i.p., 5 mg/kg) for 16 days (2), C-3281 (i.p., 15 mg/kg, for 16 days) (3), C-3342 (i.p., 170 mg/kg for 16 days) (4) and C-3329 (i.p., 100 mg/kg for 16 days) (5).
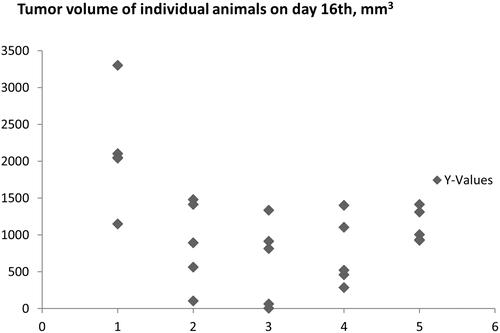
Interestingly, C-3281, the most potent among these three inhibitors of PDIA1, demonstrated the lowest cytotoxicity towards HT-1080, CaCo-2, MCF-7, and MDA-MB-231 in in vitro assays, as compared with C-3329 and C-3342 (). These results suggest that the systematic in vivo effects of PDIA1 inhibitors may involve a possible metabolic transformation of these compounds into more active PDIA inhibitors in vivo.
Anti-thrombotic effects of PDIA1 inhibitors in vivo
To confirm the anti-thrombotic effects of selected PDI inhibitors C-3389 and C-3257, these compounds were investigated in a rat model of arterial thrombosis under in vivo conditions compared to rutin and isoquercetin, both of which have been previously reported as PDIA1 inhibitors with anti-thrombotic propertiesCitation38. The C-3389 and C-3257 compounds were chosen based on their lower IC values for PDIA1 and PDIA3 (in relation to rutin and isoquercetin); for C-3389, IC50 was 0.03 or 23.3 µM, and for C-3257, it was 0.08 or 48.5 µM for PDIA1 and PDIA3, respectively; in turn, for rutin, it was 4 ± 0.3 and > 200 µM PDIA1 and PDIA3, respectively.
As shown in reference PDI inhibitors, rutin significantly inhibited arterial thrombus formation, whereas isoquercetin only tended to reduce thrombosis as assessed by thrombus weight in rats treated intravenously with 3% ethanol (Vehicle), isoquercetin or rutin, administered 10 min prior to the induction of arterial thrombosis. Interestingly, in identical experimental settings, the anti-thrombotic effects of C-3389 or C-3257 administered to rats at an equivalent dose of rutin were comparable to the effects observed for rutin, despite the fact that rutin displayed a significantly higher IC50 value against PDIA1. Furthermore, it is important to add that C-3257 decreased arterial thrombus formation in rats in a dose-dependent manner, with a pronounced effect at a dose of 0.05 μmol/kg ().
Figure 8. Anti-thrombotic effects of reference PDIA1 inhibitors (A) and C-3257 (B) in arterial thrombosis in rats under in vivo conditions. Data are means with SEM; n = 5–11, $, &, * or ** indicate p = 0.068, p = 0.054, p < 0.05 and p < 0.01 respectively vs. corresponding vehicle control. Statistical analysis was performed using Student’s t-test or Mann-Whitney test.
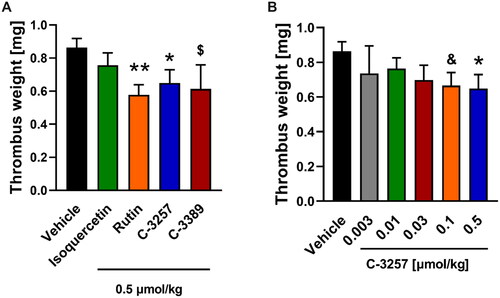
Clearly, our results show that C-3389 and C-3257 compounds represent a novel pharmacological tool to inhibit thrombosis. However, further studies are required to elucidate in detail the mechanism of anti-thrombotic action of these compounds.
Summing up, presently, most available PDI inhibitors are non-selective sulphydryl-reactive compounds that act broadly on thiol isomerasesCitation39–41. In the present work, for the first time, we demonstrated that aromatic N-sulphonamides of Az-COOH derivatives are potent and relatively selective PDIA1 inhibitors; these compounds also have additional activity towards PDIA3 with low or moderate cytotoxic activity in vitro towards a panel of cell lines and very low acute toxicity in vivo.
Binding experiments with the use of a protein NMR spectroscopy confirmed the covalent binding of novel PDI inhibitors to the catalytic active sites of PDIA1a via alkylation of cysteine thiol groups. Among the 82 compounds tested, there was a correlation between increased potency for the inhibition of PDIA1 activity of the tested compounds with improved anti-cancer activity in in vitro assays in multiple cancer cell lines. Anti-cancer effects were also confirmed in a 3D model of colon cancer and in a murine in vivo model of cancer. We also demonstrated that novel PDI inhibitors display anti-thrombotic effects in vivo.
Aromatic N-sulphonamides of Az-COOH derivatives described here were designed as targeted covalent inhibitors (TCIs) of PDIA1 and PDIA3, and thus belong to the same class of inhibitors as number of existing drugs such as for example clopidogrel or omeprazoleCitation42,Citation43. However, further studies are required to better understand metabolism of our PDIA1/PDIA3 inhibitors in cellular environment, and precise mechanisms of the inhibition of the catalytic domains of PDIA1/PDIA3.
Altogether, aromatic N-sulphonamides of Az-COOH derivatives described here represent promising candidates for the development of novel anti-cancer and anti-thrombotic agents, that might also have a vasoprotective action, encompassing compounds with relatively selective PDIA1 or non-selective PDIA1 and PDIA3 blocking mechanisms of action.
Supplemental Material
Download PDF (515.8 KB)Acknowledgements
Authors acknowledge Latvian National Fundamental and Applied Research Grant No. lzp-2018/1–0143 “Isoform-selective PDI inhibitors: design, synthesis and SAR” and The National Centre for Research and Development (STRATEGMED1/233226 /11/NCBR/2015) for financial support.
Disclosure statement
Zelencova-Gopejenko D., Andrianov V., Domracheva I., Kanepe-Lapsa I., Milczarek M., Stojak M., Przyborowski K., Kramkowski K., Wietrzyk J., Chlopicki S. .and Kalvins I. have filed a patent on aromatic sulphonamides derivatives that inhibits PDIA1 (WO/2021/141506) and PDIA3 (WO/2021/141507). Zelencova D., Andrianov V., Domracheva I., Kanepe-Lapsa I., Chlopicki S., .and Kalvins I. are co-authors of patent application WO/2021/141508. Fedak FA. and Walczak M. declare no conflict of interest. The authors are originators of data and are responsible for the content and writing of this article.
Additional information
Funding
References
- Hemminki K, Ludlum DB. Covalent modification of DNA by antineoplastic agent. J Natl Cancer Inst. 1984;73(5):1021–1028.
- Akhtar R, Naqvi SAR, Zahoor AF, Saleem S. Nucleophilic ring opening reactions of aziridines. Mol Divers. 2018;22(2):447–501.
- Gaitonde M. A spectrophotometric method for the direct determination of cysteine in the presence of other naturally occurring amino acids. Biochem J. 1967;104(2):627–633.
- Morodor L, Musiol HJ, Scharf R. Aziridine-2-carboxylic acid A reactive amino acid unit for a new class of cysteine proteinase inhibitors. FEBS Lett. 1992;299(1):51–53.
- Leite I, Andrianov V, Zelencova-Gopejenko D, Loza E, Kazhoka-Lapsa I, Domracheva I, Stoyak M, Chlopicki S, Kalvins I. Aziridine-2-carboxylic acid derivatives and its open-ring isomers as a novel PDIA1 inhibitors. Chem Heterocycl Comp. 2021;57(11):1086–1106.
- Aleksis R, Jaudzems K, Ivanova J, Žalubovskis R, Kalvinsh I, Liepinsh E. Reactivity of aziridine-2-carboxamide (leakadine) with nucleophiles in aqueous solutions. Chem Heterocycl Comp. 2014;49(11):1589–1598.
- Ali Khan H, Mutus B. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front Chem. 2014;2:70.
- Kozlov G, Määttänen P, Thomas DY, Gehring K. A structural overview of the PDI family of proteins. FEBS J. 2010;277(19):3924–3936.
- Xu S, Sankar S, Neamati N. Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today. 2014;19(3):222–240.
- Benham AM. The protein disulfide isomerase family: key players in health and disease. Antioxid Redox Signal. 2012;16(8):781–789.
- Stojak M, Milczarek M, Kurpinska A, Suraj-Prazmowska J, Kaczara P, Wojnar-Lason K, Banach J, Stachowicz-Suhs M, Rossowska J, Kalviņš I, et al. Protein disulphide isomerase A1 is involved in the regulation of breast cancer cell adhesion and transmigration via lung microvascular endothelial cells. Cancers (Basel). 2020;12(10):2850.
- Wu Y, Essex DW. Vascular thiol isomerases in thrombosis: the yin and yang. J Thromb Haemost. 2020;18(11):2790–2800.
- Keller R. The computer aided resonance assignment tutorial. Goldau, Switzerland: Cantina Verlag; 2004.
- Williamson MP. Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16.
- Nadir UK, Singh A. Synthesis of functionalized N ‐arylsulfonyl aziridines from α,β‐unsaturated esters, amides, ketones, and nitriles using N, N ‐dichloroarylsulfonamides as nitrogen source. Synth Commun. 2004;34(7):1337–1347.
- Lapinsky DJ, Bergmeier SC. A Suzuki cross-coupling route to substituted aziridines. Tetrahedron Lett. 2001;42(49):8583–8586.
- Smith AB, Kim DS. A general, convergent strategy for the construction of indolizidine alkaloids: total syntheses of (−)-indolizidine 223AB and alkaloid (−)-205B. J Org Chem. 2006;71(7):2547–2557.
- Baldwin JE, Spivey AC, Schofield CJ, Sweeney JB. Amino acid synthesis via ring opening of N-sulphonyl aziridine-2-carboxylate esters with organometallic reagents. Tetrahedron. 1993;49(28):6309–6330.
- Smith AM, Chan J, Oksenberg D, Urfer R, Wexler DS, Ow A, Gao L, McAlorum A, Huang SG. A high-throughput turbidometric assay for screening inhibitors of protein disulfide isomerase activity. J Biomol Screen. 2004;9(7):614–620.
- Watanabe MM, Laurindo FRM, Fernandes DC. Methods of measuring protein disulfide isomerase activity: a critical overview. Front Chem. 2014;2:73.
- Kemmink J, Darby NJ, Creighton TE, Dijkstra K, Scheek RM. Nuclear magnetic resonance characterization of the N-terminal thioredoxin-like domain of protein disulfide isomerase. Protein Sci. 1995;4(12):2587–2593.
- Kemmink J, Darby NJ, Dijkstra K, Nilges M, Creighton TE. Structure determination of the N-terminal thioredoxin-like domain of protein disulfide isomerase using multidimensional heteronuclear 13C/15N NMR spectroscopy. Biochemistry. 1996;35(24):7684–7691.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63.
- Stokes WS, Casati S, Strickland J, Paris M. Neutral red uptake cytotoxicity tests for estimating starting doses for acute oral toxicity tests. Curr Protoc Toxicol. 2008;36:20.4.1–20.4.20.
- ICCVAM. Test method valuation report (TMER): in vitro cytotoxicity test methods for estimating starting doses for acute oral systemic toxicity tests. Research Triangle Park (NC): NIH Publication; 2006.
- ICCVAM. 2006a, Background review document: in vitro basal cytotoxicity test methods for estimating acute oral systemic toxicity. Research Triangle Park (NC): National Institute for Environmental Health Sciences.
- OECD, Guidelines 420. Acute oral toxicity − fixed dose procedure. OECD guidelines for testing of chemicals. Paris, France: OECD; 2001. p. 1.
- Wietrzyk J, Chodyński M, Fitak H, Wojdat E, Kutner A, Opolski A. Antitumor properties of diastereomeric and geometric analogs of vitamin D3. Anticancer Drugs. 2007;18(4):447–457.
- Pawlak A, Ziolo E, Fiedorowicz A, Fidyt K, Strzadala L, Kalas W. Long-lasting reduction in clonogenic potential of colorectal cancer cells by sequential treatments with 5-azanucleosides and topoisomerase inhibitors. BMC Cancer. 2016;16(1):893.
- Milczarek M, Psurski M, Kutner A, Wietrzyk J. Vitamin D analogs enhance the anticancer activity of 5-fluorouracil in an in vivomouse colon cancer model. BMC Cancer. 2013;13(1):294.
- Kramkowski K, Leszczynska A, Mogielnicki A, Chlopicki S, Fedorowicz A, Grochal E, Mann B, Brzoska T, Urano T, Motterlini R, et al. Antithrombotic properties of water-soluble carbon monoxide-releasing molecules. Arterioscler Thromb Vasc Biol. 2012;32(9):2149–2157.
- Vuori K, Myllylä R, Pihlajaniemi T, Kivirikko KI. Expression and site-directed mutagenesis of human protein disulfide isomerase in Escherichia coli. This multifunctional polypeptide has two independently acting catalytic sites for the isomerase activity. J Biol Chem. 1992;267(11):7211–7214.
- Gilfix BM, Blank DW, Rosenblatt DS. Novel reductant for determination of total plasma homocysteine. Clin Chem. 1997;43(4):687–688.
- Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, Zandi E, Petasis NA, Neamati N. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc Natl Acad Sci USA. 2012;109(40):16348–16353.
- Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L, Bowley SR, Joshi SS, Dilks JR, Furie B, et al. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest. 2012;122(6):2104–2113.
- Rancy PC, Thorpe C. Oxidative protein folding in vitro: a study of the cooperation between quiescin-sulfhydryl oxidase and protein disulfide isomerase. Biochemistry. 2008;47(46):12047–12056.
- Santos GB, Gonzalez-Perilli L, Mastrogiovanni M, Aicardo A, Cerdeira CD, Trostchansky A, Brigagão MRPL. Nitroxide 4-hydroxy-2,2′,6,6′-tetramethylpiperidine 1-oxyl (Tempol) inhibits the reductase activity of protein disulfide isomerase via covalent binding to the Cys 400 residue on CXXC redox motif at the a′active site. Chem Biol Interact. 2017;272:117–124.
- Stainer AR, Sasikumar P, Bye AP, Unsworth AJ, Holbrook LM, Tindall M, Lovegrove JA, Gibbins JM. The metabolites of the dietary flavonoid quercetin possess potent antithrombotic activity, and interact with aspirin to enhance antiplatelet effects. TH Open. 2019;3(3):e244–e258.
- Karala AR, Ruddock LW. Bacitracin is not a specific inhibitor of protein disulfide isomerase. FEBS J. 2010;277(11):2454–2462.
- Khan MMG, Simizu S, Lai NS, Kawatani M, Shimizu T, Osada H. Discovery of a small molecule PDI inhibitor that inhibits reduction of HIV-1 envelope glycoprotein gp120. ACS Chem Biol. 2011;6(3):245–251.
- Lovat PE, Corazzari M, Armstrong JL, Martin S, Pagliarini V, Hill D, Brown AM, Piacentini M, Birch-Machin MA, Redfern CPF. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008; 68(13):5363–5369.
- Potashman MH, Duggan ME. Covalent modifiers: an orthogonal approach to drug design. J Med Chem. 2009;52(5):1231–1246.
- Boike L, Henning NJ, Nomura DK. Advances in covalent drug discovery. Nat Rev Drug Discov. 2022;21(12):881–898.