
Abstract
A small library of novel thiazolone-benzenesulphonamides has been prepared and evaluated for their ability to inhibit three human cytosolic carbonic anhydrases (hCA I, hCA II, and hCA VII) and three bacterial carbonic anhydrases (MscCAβ, StCA1, and StCA2). All investigated hCAs were inhibited by the prepared compounds 4a–4j in the low nanomolar range. These compounds were effective hCA I inhibitors (KIs of 31.5–637.3 nM) and excellent hCA II (KIs in the range of 1.3–13.7 nM) and hCA VII inhibitors (KIs in the range of 0.9–14.6 nM). The most active analog in the series, 4-((4-oxo-5-propyl-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide 4d, strongly inhibited bacterial MscCAβ, with KI of 73.6 nM, considerably better than AAZ (KI of 625 nM). The tested compounds displayed medium inhibitory potency against StCA1 (KIs of 69.2–163.3 nM) when compared to the standard drug (KI of 59 nM). However, StCA2 was poorly inhibited by the sulphonamides reported here, with KIs in the micromolar range between 275.2 and 4875.0 nM.
Introduction
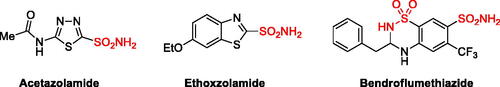
The carbonic anhydrases (CAs; EC 4.2.1.1) are a ubiquitous family of metalloenzymes that are found in both prokaryotes and eukaryotes and catalyse the reversible reaction between carbon dioxide and water with formation of bicarbonate and a protonCitation1. This simple reaction is fundamental to a variety of physiological and pathological processes in which cellular pH buffering plays an essential roleCitation2. In humans, 15 carbonic anhydrase isoforms (hCAs) have been identified till date, among which 3 have non-catalytic functions (VIII, X, and XI) and 12 are catalytically active (CA I–IV, VA–VB, VI–VII, IX, and XII–XIV)Citation3. Emerging scientific evidence indicates that CAs are involved in an array of disorders, including haemolytic anaemia (CA I), edoema (CA II), altitude sickness (CA II), glaucoma (CA II, IV), obesity (CA VA), neuropathic pain (CA VII), cancer (CA IX, XII), sterility (CA XIII), and many othersCitation4. They are therefore a target for drug discovery and since so far dozens of CA inhibitors have been FDA-approved for the treatment/management of various diseases (see )Citation5. On the other hand, a possible new approach for the treatment of infectious diseases which are caused by microorganisms such as bacteria, fungi or parasites is based on the inhibition of carbonic anhydrases families being present in these organismsCitation6.
Figure 1. Selected example of FDA-approved sulphonamide carbonic anhydrase inhibitors.
Sulphonamide moiety (R-SO2NH2) is a common substructure in marketed small-molecule drugsCitation5,Citation7 and a relevant “warhead” to target CAs, mainly because of it strong zinc-binding affinityCitation8. The large number of publications on the development of novel sulphonamide-based compounds and investigation of their inhibitory capacity against CAs for expanding the database of CAIs implies that the field is a hot topic in current drug discovery researchCitation9. However, a major challenge faced when designing hCA inhibitors for therapy is the lack of selectivity for a specific hCA isoform at least for the first and second generation compoundsCitation10.
For about two decades the tail approach was employed for developing isoform selective CA inhibitors. In this strategy, various moieties are attached to the terminal part of scaffolds of simple sulphonamides that may lead to interactions with the external part of the CA isoforms active site, which is the region at the entrance of the cavity that is more diverse than the active site residues between the 12 catalytically active human isoformsCitation11.
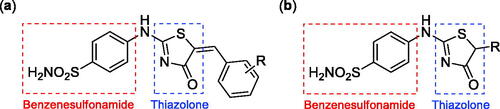
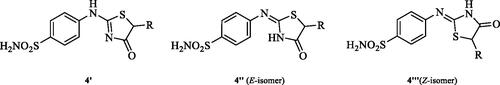
Very recently, the research group of Hassan has synthesised a small library of thiazolone-linked benzenesulphonamides () and reported their strong and selective hCA IX inhibitory activity over hCA II, the most active human CA isoformCitation12. Furthermore, it is reported that some of those compounds showed significant inhibitory effect against triple-negative breast cancer cell lines. In parallel with anticancer properties of thiazolone derivativesCitation13, compounds possessing this heterocyclic ring have also been reported to exhibit antibacterial and antifungal activitiesCitation14. Based on these literatures and in continuation of our interest in sulphonamide CAIsCitation15, herein, we present a novel, highly efficient and step-economic method for the synthesis of thiazol-2(5H)-one-containing sulphanilamides () and evaluate their capability to inhibit three bacterial and three human cytosolic carbonic anhydrases.
Figure 2. (a) General structure of thiazolone-benzenesulphonamides as selective hCA IX inhibitors developed by Hassan Citation12; (b) General structure of thiazolone-benzenesulphonamides discussed in the paper.
Experimental section
Chemistry
Reagents, starting materials and solvents were obtained from commercial sources and used as received. Thin-layer chromatography was performed on silica gel, spots were visualised with UV light (254 and 365 nm). NMR spectra were recorded on Bruker 500 spectrometer with chemical shifts values (δ) in ppm relative to TMS using the residual DMSO-d6 signal (1H 2.50; 13C 39.52). High-resolution mass spectra (HRMS) were recorded on a mass spectrometer with a Q-TOF micro mass analyser using the ESI technique.
General procedure for the synthesis of compounds 4a–j
To a stirred solution of 1-(4-aminophenyl)thiourea (300 mg, 1.3 mmol) in DMF (4 ml) appropriate α-bromo ester (1.0 equiv., 1.3 mmol) was added at room temperature and the mixture was stirred at 25–40 °C for 15–48 h. Water (20. mL) was added to the reaction mixture and precipitate formed was collected by filtration, washed with water (100 ml) and air dried to afford the corresponding thiazolone-benzenesulphonamides (26–92%) as white solids.
4-((4-Oxo-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4a)
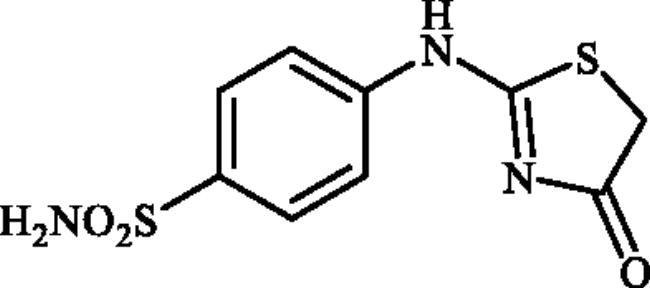 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromoacetate (0.143 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 15 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4a (322. mg, 92%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromoacetate (0.143 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 15 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4a (322. mg, 92%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 4.06 (2H, s), 7.15 (1H, s), 7.35 (2H, s), 7.86 (3H, s), 11.49 and 11.93 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 35.5, 39.4, 121.0, 122.4, 128.1, 140.6, 141.4, 151.9, 159.1, 175.3, 18.0.0, 189.3 ppm. HRMS (ESI) [M–H]−: m/z calcd for (C9H8N3O3S2) 270.0007. Found 270.0009.
4-((5-Methyl-4-oxo-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4b)
 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromopropanoate (0.169 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4b (311 mg, 84%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromopropanoate (0.169 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4b (311 mg, 84%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 1.54 (3H, s), 4.37 (1H, s), 7.14 (1H, s), 7.35 (2H, s), 7.85 (3H, s), 11.48 and 11.93 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 19.6, 45.0, 49.7, 121.1, 122.4, 128.1, 140.6, 142.3, 152.0, 157.2, 178.1, 178.5, 192.0 ppm. HRMS (ESI) [M–H]−: m/z calcd for (C10H10N3O3S2) 284.0164. Found 284.0166.
4-((5-Ethyl-4-oxo-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4c)
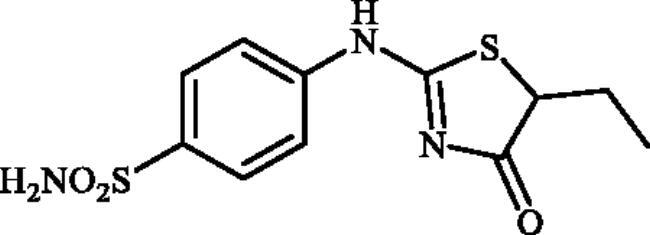 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromobutanoate (0.191 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4c (298 mg, 77%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromobutanoate (0.191 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4c (298 mg, 77%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 0.95 (3H, s), 1.85 (1H, s), 2.00 (1H, s), 4.41 (1H, s), 7.15 (1H, s), 7.35 (2H, s), 7.86 (3H, s), 11.51 and 11.94 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 11.4, 11.9, 26.4, 52.1, 57.1, 121.1, 12.4, 140.6, 142.3, 152.1, 157.3, 177.2, 178.7, 191.0 ppm. HRMS (ESI) [M–H]−: m/z calcd for (C11H12N3O3S2) 298.0320. Found 298.0327.
4-((4-Oxo-5-propyl-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4d)
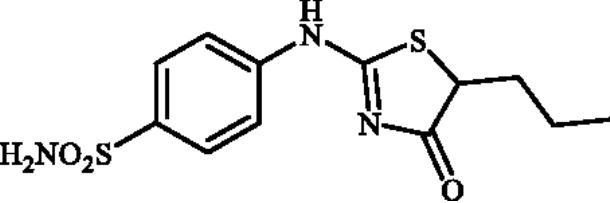 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromopentanoate (0.426 ml, 2.6 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 36 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4d (264 mg, 65%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromopentanoate (0.426 ml, 2.6 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 36 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4d (264 mg, 65%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 0.91 (3H, s), 1.33 (1H, s), 1.44 (1H, s), 1.77 (1H, s), 1.99 (1H, s), 4.41 (1H, s), 7.15 (1H, s), 7.34 (2H, s), 7.86 (3H, s), 11.49 and 11.95 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 14.4, 20.6, 21.4, 35.4, 35.6, 50.7, 55.7, 121.1, 122.4, 127.9, 128.1, 140.7, 142.4, 152.1, 157.3, 177.4, 178.7, 191.2 ppm. HRMS (ESI) [M–H]−: m/z calcd for (C12H14N3O3S2) 312.0477. Found 312.0478.
4-((5-Isopropyl-4-oxo-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4e)
 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromo-3-methylbutanoate (0.424 ml, 2.6 mmol) were dissolved in DMF (4 ml). After stirring at 40 °C for 36 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4e (106 mg, 26%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromo-3-methylbutanoate (0.424 ml, 2.6 mmol) were dissolved in DMF (4 ml). After stirring at 40 °C for 36 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4e (106 mg, 26%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 0.94 (5H, d, J = 9.2 Hz), 1.05 (1H, s), 2.42 (1H, s), 4.50 (1H, s), 7.16 (1H, s), 7.34 (2H, s), 7.86 (3H, s), 11.52 and 11.97 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 16.8, 20.8, 22.2, 30.4, 57.2, 62.6, 120.6, 121.9, 127.4, 127.6, 140.1, 141.8, 151.6, 156.7, 176.2, 178.3, 190.0 ppm. HRMS (ESI) [M–H]−: m/z calcd for (C12H14N3O3S2) 312.0477. Found 312.0475.
4-((5-Butyl-4-oxo-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4f)
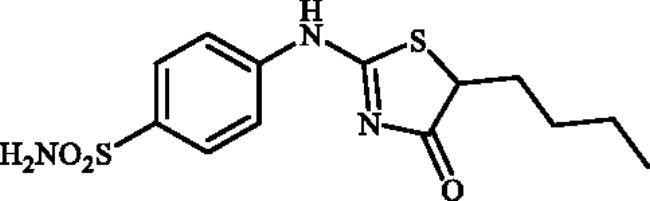 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromohexanoate (0.23.7 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 36 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4f (281 mg, 66%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromohexanoate (0.23.7 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 36 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4f (281 mg, 66%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 0.60 (3H, s), 1.03 (4H, s), 1.14 (1H, s), 1.50 (1H, s), 1.74 (1H, s), 4.13 (1H, s), 6.87 (1H, s), 7.06 (2H, s), 7.57 (3H, s), 11.21 and 11.67 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 14.7, 22.6, 29.3, 30.1, 32.98, 50.8, 55.9, 121.1, 122.4, 127.9, 128.1, 140.7, 142.3, 152.1, 157.2, 177.3, 178.7, 191.2 ppm. HRMS (ESI) [M–H]−: m/z calcd for (C13H16N3O3S2) 326.0633. Found 326.0634.
4-((5-Hexyl-4-oxo-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4g)
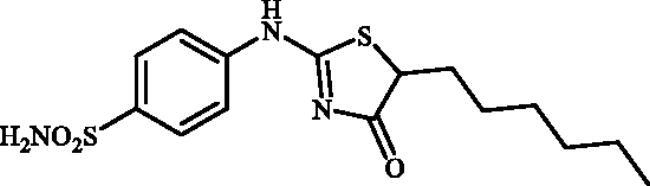 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromooctanoate (0.546 ml, 2.6 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 48 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4g (221 mg, 48%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromooctanoate (0.546 ml, 2.6 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 48 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4g (221 mg, 48%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 0.87 (3H, s), 1.27 (6H, s), 1.42 (2H, s), 1.77 (1H, s), 2.02 (1H, s), 7.14 (1H, s), 7.34 (2H, s), 7.85 (3H, s), 11.49 and 11.81 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 14.8, 22.9, 27.1, 27.8, 29.1, 31.9, 33.2, 50.8, 55.8, 121.0, 122.3, 127.9, 128.1, 140.6, 142.3, 152.0, 157.3, 177.3, 178.5, 191.1 ppm. HRMS (ESI) [M–H]−: m/z calcd for (C15H20N3O3S2) 354.0946. Found 354.0955.
4-((4-Oxo-5-phenyl-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4h)
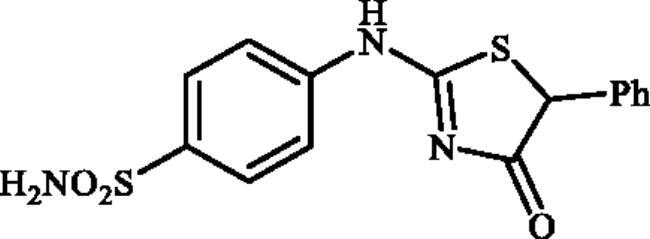 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromo-2-phenylacetate (0.546 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4 h (397 mg, 88%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromo-2-phenylacetate (0.546 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4 h (397 mg, 88%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 0.60 (3H, s), 5.65 (1H, d, J = 6.2 Hz), 7.25 (1H, s), 7.41 (7H, d, J = 6.2 Hz), 7.88–7.98 (3H, m), 11.71 and 12.23 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 53.9, 58.5, 121.3, 122.5, 127.9, 128.2, 129.1, 129.4, 129.9, 137.2, 137.7, 140.9, 142.3, 151.5, 157.5, 176.2, 178.7, 189.4 ppm. HRMS (ESI) [M–H]−: m/z calcd for (C15H12N3O3S2) 346.0320. Found 346.0322.
4-((5-(2-Bromoethyl)-4-oxo-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4i)
 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2,4-dibromobutanoate (0.205 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4i (396 mg, 81%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2,4-dibromobutanoate (0.205 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h water (20 ml) was added and the precipitated product was filtered, washed with water (100 ml), and air dried to afford 4i (396 mg, 81%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 2.32 (1H, s), 2.62 (1H, s), 3.67 (2H, s), 4.48 (1H, s), 7.16 (1H, s), 7.35 (2H, s), 7.86 (3H, s), 11.58 and 12.00 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 32.6, 33.5, 36.4, 36.9, 49.5, 54.3, 121.2, 122.4, 127.9, 128.1, 140.7, 142.3, 151.8, 157.3, 176.9, 178.7, 190.4 ppm. HRMS (ESI) [M–H]−: m/z calcd for (C11H11N3O3S2Br) 375.9425. Found 377.9416.
4-((5-Fluoro-4-oxo-4,5-dihydrothiazol-2-yl)amino)benzenesulphonamide (4j)
 1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromo-2-fluoroacetate (0.153 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h the reaction mixture was transferred to a separating funnel and mixture of water/dichloromethane (15:10 ml) was added and it was shaken vigorously for a few minutes. After 1 h, the precipitated product was filtered, washed with water (100 ml) and CH2Cl2 (20 ml), and air dried to afford 4j (247 mg, 66%) as a white powder.
1-(4-Aminophenyl)thiourea (300 mg, 1.3 mmol) and ethyl 2-bromo-2-fluoroacetate (0.153 ml, 1.3 mmol) were dissolved in DMF (4 ml). After stirring at room temperature for 20 h the reaction mixture was transferred to a separating funnel and mixture of water/dichloromethane (15:10 ml) was added and it was shaken vigorously for a few minutes. After 1 h, the precipitated product was filtered, washed with water (100 ml) and CH2Cl2 (20 ml), and air dried to afford 4j (247 mg, 66%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 6.76 (1H, d, J = 44.6 Hz), 7.18 (1H, d, J = 5.0 Hz), 7.40 (2H, d, J = 14.2 Hz), 7.88 (1H, d, J = 5.4 Hz), 7.92 (2H, s), 11.97 and 12.61 (1H, s and s) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 94.0 (d, J = 221.8 Hz), 97.6 (d, J = 223.5 Hz), 122.2 (d, J = 42.0 Hz), 128.1 (d, J = 33.8 Hz), 141.4, 141.6 (d, J = 9.8 Hz), 150.9, 153.9, 171.0, 176.1, 184.6 (d, J = 13.6 Hz) ppm. HRMS (ESI) [M–H]−: m/z calcd for (C9H7N3O3FS2) 287.9913. Found 287.9910.
CA inhibition assay
An applied photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation16. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer for α-CAs or 20 mM TRIS (pH 8.4) as buffer for β-CAs, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10 – 100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor, at least six traces of the initial 5 – 10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled – deionised water, and dilutions up to 0.1 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay in order to allow for the formation of the enzyme – inhibitor complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3 and the Cheng–Prusoff equation, as reported earlierCitation17–19, and represent the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in-house as reported earlierCitation20–22 and their concentrations in the assay system ranged between 10 and 14 nM.
Results and discussion
Chemistry
In Scheme 1, the synthesis of the target compounds reported here. The synthesis starts with the selective catalyst- and additive-free S-alkylation of easily accessible 4-thioureidobenzenesulphonamide 1 with commercially available α-bromo esters 2 under mild conditions. Following spontaneous intramolecular cyclisation in in situ generated ethyl 2-((N-(4-sulphamoylphenyl)carbamimidoyl)thio)acetate intermediate 3 into the desired products 4 in fair to excellent yields, ranging from 26% to 92%. The analytical and spectroscopic data (NMR and MS spectra) of the prepared compounds are in agreement with the proposed structures. Noteworthy, as observed in NMRs all compounds 4 exist as a mixture of tautomers ()Citation23. All synthesised inhibitors are novel and not known in literature.
Scheme 1. Reagents and conditions: (i) DMF, 25–40 °C, 15–48 h.
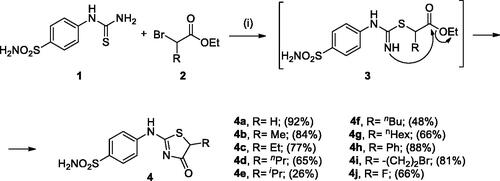
Figure 3. Representative tautomers of compounds 4.
Carbonic anhydrase inhibition
The newly synthesised target compounds 4a-4j were evaluated for their inhibitory activity towards three human (h) CA isoforms, the cytosolic hCA I, II, and VII, as well as three bacterial β-CAs from one gram-positive bacteria, Mammaliicoccus (Staphylococcus) sciuri, MscCAβ, and one gram-negative bacteria, Salmonella enterica (serovar Typhimurium), StCA1 and StCA2, by using a stopped-flow CO2 hydrase assayCitation16.
Some important information from the data of , where the inhibition data of these enzymes are presented, are listed below:
Table 1. Inhibition data of human CA isoforms hCA I, II, and VII and bacterial β-CA isoforms MscCA, from Mammaliicoccus (Staphylococcus) sciuri, and StCA1 and StCA2, from Salmonella enterica (serovar Typhimurium) with compounds 4a–j in comparison with the standard sulphonamide inhibitor AAZ by a stopped flow CO2 hydrase assay.
Ninety percent of the evaluated compounds exhibited superior potency against the slowest cytosolic human isoform hCA I with KIs in the range of 31.5–137.5 nM compared to the standard drug (acetazolamide; AAZ) whose KI value is 250 nM. The SAR analysis showed that the inhibitory activity was decreased with the increased chain length in alkyl-substituted compounds 4b–4g. Therefore, the best inhibitor was compound 4b (KI of 31.5 nM) with the smallest alkyl group (Me), whereas the poorest inhibitor was compound 4g (KI of 637.3 nM) with the longest alkyl group (n-Hex). However, the replacement of the aliphatic groups with a hydrogen atom did not increase the inhibitory activity as unsubstituted compound 4a showed almost equal inhibitory activity with Et-substituted one 4c. Additionally, no remarkable change was observed when the aliphatic groups were replaced with a benzene ring so that Ph-substituted compound 4h displayed almost the same inhibitory activity with iPr-substituted one 4e. However, the inhibitory activity was decreased with the insertion of a fluorine atom at the C5-position. In summary, the relative inhibitory rates of newly designed compounds against hCA I followed the order: Methyl (C1) substituted- > unsubstituted- ≈ medium aliphatic chain (C2-C4) substituted- ≈ benzene-substituted ≥ F-substituted- > long aliphatic chain (C6) substituted derivatives.
Against the fastest cytosolic human isoform hCA II, similar to hCA I, except long aliphatic chain (C6)-substituted thiazolone-benzenesulphonamide 4g with KI values of 13.7 nM, other compounds acted as more effective inhibitors (KIs in the range of 1.3–7.3 nM) compared to the standard drug (KI of 12.5 nM). Therefore, all substitution patterns present in these compounds lead to highly effective CAIs. It should be mentioned that the only aryl-substituted derivative 4h of the series exhibited the best inhibition for this isozyme which was 10-fold more effective than AAZ. Thus, synthesis and evaluation of novel aryl-substituted thiazolone-benzenesulphonamide may be useful for the development of effective and selective inhibitor of hCA II.
The brain-associated cytosolic isoform hCA VII which was recently validated as therapeutic target for the relief of neuropathic painCitation24 showed a rather similar inhibition profile with hCA I, with sulphonamides investigated here. Therefore, methyl-substituted compound 4b was the strongest inhibitor with a KI of 0.9 nM and nHex-substituted compound 4g, showed poorest inhibitory capacities with a KI of 14.6 nM. It is worth mentioning that 60% of the compounds investigated here (4a–d, 4i, 4j), showed even better inhibitory activities against hCA VII in comparison with the reference drug.
The bacterial β-CA from Mammaliicoccus (Staphylococcus) sciuri, MscCAβ, was effectively inhibited by some of the obtained sulphonamides, with the KI values in the range of 73.6 and 4026.3 nM. Indeed, Among the 10 investigated compounds, three exhibited superior inhibitory activity compared to AAZ which all are alkyl-substituted derivatives. Compound 4d showed the best inhibition against MscCA with a KI of 73.6 nM, which was 8-fold higher than that of AAZ (KI of 625 nM). Although this compound 4d displayed negligible selectivity for MscCA over StCA1 (MscCA/StCA1:1.4), its high selectivity index greater than 10 for MscCAversus StCA2 is remarkable.
One of the S. enterica (serovar Typhimurium) CA isoform, StCA1, was effectively inhibited by the sulphonamides reported here, with KIs in the range between 69.2 and 163.3 nM. The most promising compound, 4g, showed the most significant selectivity for this isozyme versus MscCAβ and StCA2, 48- and 58-fold higher selectivity, respectively. Therefore, this compound is an excellent candidate for future in vivo preclinical studies.
The second S. enterica (serovar Typhimurium) CA isoform, StCA2, was on the other hand poorly inhibited with targeted sulphonamides compared to MscCAβ and StCA1, and the KIs were in the range of 275.2–4875.0 nM. Among the tested compounds, unsubstituted derivative 4a demonstrated the most potent StCA2 inhibitory activity with a KI of 275.2 nM, although the activity was three-fold lower than the reference compound (KI of 84 nM).
Conclusion
A small library of C5-functionalized thiazol-2(5H)-one-containing sulphonamide derivatives 4a–4j has been prepared through a high atom- and step-economical reaction between easily accessible 4-thioureidobenzenesulphonamide and commercially available α-bromo esters under mild and additive-free conditions. These compounds were tested for their inhibition of three cytosolic human carbonic anhydrase isoforms (hCA I, hCA II, and hCA VII) as well as three bacterial enzymes belonging to the β-CA class (MscCAβ, StCA1, and StCA2). The results showed that majority of the examined compounds effectively inhibited hCAs, even better than the standard drug (AAZ). Noteworthy, in all cases these compounds showed significant selectivity for hCA II and hCA VII over hCA I. However, they did not show selectivity for the inhibition of hCA II versus hCA VII. The bacterial CAs were also inhibited by these compounds, but not as effectively as the hCAs. Importantly, compound 4g which showed the poorest capacity to inhibit human CAs, inhibited StCA1 almost in the same range of AAZ, while it has a 48- and 58-fold high selectivity for this isozyme versus MscCA and StCA2, respectively. The activity of this analog against StCA1 is especially noteworthy, and suggests that it is an excellent candidate for future in vivo preclinical studies.
Disclosure statement
No potential conflict of interest was reported by all author(s) except CTS. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J. 2016;473(14):2023–2032.
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem. 2007;15(13):4336–4350.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7(2):168–181.
- Alterio V, Di Fiore A, D'Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev. 2012;112(8):4421–4468.
- Scott KA, Njardarson JT. Analysis of US FDA-approved drugs containing sulfur atoms. Top Curr Chem. 2018;376(1):5.
- (a) Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat. 2013;23(6):693–704. (b) Capasso C, Supuran CT, An overview of the alpha-, beta-and gamma-carbonic anhydrases from bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem. 2015;30(2):325–332. (c) Supuran CT, Capasso C. An overview of the bacterial carbonic anhydrases. Metabolites. 2017;7(4):56.
- Smith DA, Jones RM. The sulfonamide group as a structural alert: a distorted story? Curr Opin Drug Discov Dev. 2008;11(1):72–79.
- (a) Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev. 2003;23(2):146–189. (b) Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem. 2014;6(10):1149–1165.
- (a) Grandane A, Tanc M, Zalubovskis R, Supuran CT. Synthesis of 6-tetrazolyl-substituted sulfocoumarins acting as highly potent and selective inhibitors of the tumor-associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem. 2014;22(5):1522–1528. (b) Grandane A, Tanc M, Zalubovskis R, Supuran CT. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett. 2014;24(5):1256–1260. (c) Angeli A, Carta F, Nocentini A, Winum JY, Zalubovskis R, Akdemir A, Onnis V, Eldehna WM, Capasso C, Simone G, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites. 2020;10:412. (d) Ivanova J, Carta F, Vullo D, Leitans J, Kazaks A, Tars K, Žalubovskis R, Supuran CT. N-Substituted and ring opened saccharin derivatives selectively inhibit transmembrane, tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem. 2017;25(13):3583–3589. (e) Grandāne A, Nocentini A, Domračeva I, Žalubovskis R, Supuran CT. Development of oxathiino [6, 5-b] pyridine 2, 2-dioxide derivatives as selective inhibitors of tumor-related carbonic anhydrases IX and XII. Eur J Med Chem. 2020;200:112300. (f) Nocentini A, Angeli A, Carta F, Winum JY, Zalubovskis R, Carradori S, Capasso C, Donald WA, Supuran CT. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem. 2021;36:561–580. (g) Grandane A, Nocentini A, Werner T, Zalubovskis R, Supuran CT. Benzoxepinones: a new isoform-selective class of tumor associated carbonic anhydrase inhibitors. Bioorg Med Chem. 2020;28(11):115496. (h) Podolski-Renić A, Dinić J, Stanković T, Jovanović M, Ramović A, Pustenko A, Žalubovskis R, Pešić M. Sulfocoumarins, specific carbonic anhydrase IX and XII inhibitors, interact with cancer multidrug resistant phenotype through pH regulation and reverse P-glycoprotein mediated resistance. Eur J Pharm Sci. 2019;138:105012.
- Galati S, Yonchev D, Rodríguez-Pérez R, Vogt M, Tuccinardi T, Bajorath J. Predicting isoform-selective carbonic anhydrase inhibitors via machine learning and rationalizing structural features important for selectivity. ACS Omega. 2021;6(5):4080–4089.
- Angeli A, Carta F, Nocentini A, Winum J-Y, Zalubovskis R, Akdemir A, Onnis V, Eldehna WM, Capasso C, Simone GD, et al. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites. 2020;10(10):412.
- Nemr MT, AboulMagd AM, Hassan HM, Hamed AA, Hamed MI, Elsaadi MT. Design, synthesis and mechanistic study of new benzenesulfonamide derivatives as anticancer and antimicrobial agents via carbonic anhydrase IX inhibition. RSC Adv. 2021;11(42):26241–26257.
- (a) Havrylyuk D, Zimenkovsky B, Vasylenko O, Zaprutko L, Gzella A, Lesyk R. Synthesis of novel thiazolone-based compounds containing pyrazoline moiety and evaluation of their anticancer activity. Eur J Med Chem. 2009;44(4):1396–1404. (b) Roaiah HM, Ghannam IA, Ali IH, El Kerdawy AM, Ali MM, Abbas SE, El‐Nakkady SS. Design, synthesis, and molecular docking of novel indole scaffold‐based VEGFR‐2 inhibitors as targeted anticancer agents. Arch Pharm. 2018;351(2):1700299.
- (a) Othman IM, Mahross MH, Gad-Elkareem MA, Rudrapal M, Gogoi N, Chetia D, Aouadi K, Snoussi M, Kadri A. Toward a treatment of antibacterial and antifungal infections: design, synthesis and in vitro activity of novel arylhydrazothiazolylsulfonamides analogues and their insight of DFT, docking and molecular dynamic simulations. J Mol Struct. 2021;1243(10):130862–1562. (b) Kalaria PN, Makawana JA, Satasia SP, Raval DK, Zhu HL. Design, synthesis and molecular docking of novel bipyrazolyl thiazolone scaffold as a new class of antibacterial agents. MedChemComm. 2014;5:1555.
- (a) Abdoli M, Angeli A, Bozdag M, Carta F, Kakanejadifard A, Saeidian H, Supuran CT. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of benzo [d] thiazole-5-and 6-sulfonamides. J Enzyme Inhib Med Chem. 2017;32(1):1071–1078. (b) Abdoli M, Bozdag M, Angeli A, Supuran C. Benzamide-4-sulfonamides are effective human carbonic anhydrase I, II, VII, and IX inhibitors. Metabolites. 2018;8(2):37. (c) Abdoli M, Giovannuzzi S, Supuran CT, Žalubovskis R. 4-(3-Alkyl/benzyl-guanidino) benzenesulfonamides as selective carbonic anhydrase VII inhibitors. J Enzyme Inhib Med Chem. 2022;37(1):1568–1576. (d) Abdoli M, Bonardi A, Supuran CT, Žalubovskis R. 4-Cyanamidobenzenesulfonamide derivatives: a novel class of human and bacterial carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2023;38(1):156–165. (e) Ivanova J, Abdoli M, Nocentini A, Žalubovskis R, Supuran CT. 1, 2, 3-Benzoxathiazine-2, 2-dioxides–effective inhibitors of human carbonic anhydrases. J Enzyme Inhib Med Chem. 2023;38(1):225–238. (f) Abdoli M, De Luca V, Capasso C, Supuran CT, Žalubovskis R. Benzenesulfonamides incorporating hydantoin moieties effectively inhibit eukaryotic and human carbonic anhydrases. Int J Mol Sci. 2022;23(22):14115.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- (a) Nishimori I, Minakuchi T, Morimoto K, Sano S, Onishi S, Takeuchi H, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: DNA cloning and inhibition studies of the alpha-carbonic anhydrase from Helicobacter pylori, a new target for developing sulfonamide and sulfamate gastric drugs. J Med Chem. 2006;49(6):2117–2126. (b) Supuran CT, Clare BW. Carbonic anhydrase inhibitors. Part 57. Quantum chemical QSAR of a group of 1,3,4-thiadiazole and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur J Med Chem. 1999;34:41–50. (c) Gieling RG, Babur M, Mamnani L, Burrows N, Telfer BA, Carta F, Winum JY, Scozzafava A, Supuran CT, Williams KJ. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J Med Chem. 2012;55(11):5591–600.
- (a) Abbate F, Winum JY, Potter BV, Casini A, Montero JL, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with EMATE, a dual inhibitor of carbonic anhydrases and steroid sulfatase. Bioorg Med Chem Lett. 2004;14(1):231–234. (b) Gülçin İ, Scozzafava A, Supuran CT, Akıncıoğlu H, Koksal Z, Turkan F, Alwasel S. The effect of caffeic acid phenethyl ester (CAPE) on metabolic enzymes including acetylcholinesterase, butyrylcholinesterase, glutathione S-transferase, lactoperoxidase, and carbonic anhydrase isoenzymes I, II, IX, and XII. J Enzyme Inhib Med Chem. 2016;31(6):1095–1101.
- (a) Zimmerman SA, Ferry JG, Supuran CT. Inhibition of the archaeal beta-class (Cab) and gamma-class (Cam) carbonic anhydrases. Curr Top Med Chem. 2007;7(9):901–908. (b) Supuran CT, Barboiu M, Luca C, Pop E, Brewster ME, Dinculescu A. Carbonic anhydrase activators. Part 14. Synthesis of mono- and bis- pyridinium salt derivatives of 2-amino-5-(2-aminoethyl)- and 2-amino-5-(3-aminopropyl)-1,3,4-thiadiazole, and their interaction with isozyme II. Eur J Med Chem. 1996; 31:597–606. (c) Aspatwar A, Barker H, Aisala H, Zueva K, Kuuslahti M, Tolvanen M, Primmer CR, Lumme J, Bonardi A, Tripathi A, et al. Cloning, purification, kinetic and anion inhibition studies of a recombinant β-carbonic anhydrase from the Atlantic salmon parasite platyhelminth Gyrodactylus salaris. J Enzyme Inhib Med Chem. 2022;37:1577–1586.
- (a) Nishimori I, Minakuchi T, Vullo D, Scozzafava A, Supuran CT. Inhibition studies of the β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium with sulfonamides and sulfamates. Bioorg Med Chem. 2011;19(16):5023–5030. (b) Vullo D, Nishimori I, Minakuchi T, Scozzafava A, Supuran CT. Inhibition studies with anions and small molecules of two novel β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium. Bioorg Med Chem Lett. 2011;21(12):3591–3595. (c) Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol. 2011;2:34.
- (a) Urbanski LJ, Vullo D, Parkkila S, Supuran CT. An anion and small molecule inhibition study of the β-carbonic anhydrase from Staphylococcus aureus. J Enzyme Inhib Med Chem. 2021;36(1):1088–1092. (b) Urbanski LJ, Bua S, Angeli A, Kuuslahti M, Hytönen VP, Supuran CT, Parkkila S. Sulphonamide inhibition profile of Staphylococcus aureus β-carbonic anhydrase. J Enzyme Inhib Med Chem. 2020;35(1):1834–1839. (c) Angeli A, Urbański LJ, Capasso C, Parkkila S, Supuran CT. Activation studies with amino acids and amines of a β-carbonic anhydrase from Mammaliicoccus (Staphylococcus) sciuri previously annotated as Staphylococcus aureus (SauBCA) carbonic anhydrase. J Enzyme Inhib Med Chem. 2022;37:2786–2792.
- De Luca V, Giovannuzzi S, Supuran CT, Capasso C. May sulfonamide inhibitors of carbonic anhydrases from Mammaliicoccus sciuri prevent antimicrobial resistance due to gene transfer to other harmful staphylococci? IJMS. 2022;23(22):13827.
- Mahboobi S, Sellmer A, Höcher H, Eichhorn E, Bär T, Schmidt M, Maier T, Stadlwieser JF, Beckers TL. [4-(Imidazol-1-yl)thiazol-2-yl]phenylamines. A novel class of highly potent colchicine site binding tubulin inhibitors: synthesis and cytotoxic activity on selected human cancer cell lines. J Med Chem. 2006;49(19):5769–5776.
- Supuran CT. Carbonic anhydrase inhibition and the management of neuropathic pain. Expert Rev Neurother. 2016;16(8):961–968.