
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
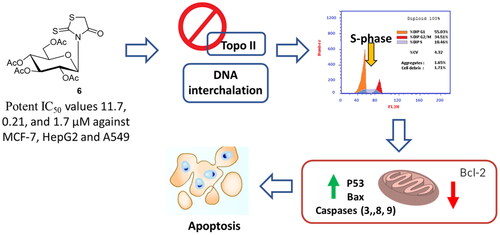
In the present study, 5-arylidene rhodanine derivatives 3a–f, N-glucosylation rhodanine 6, S-glucosylation rhodanine 7, N-glucoside rhodanine 8 and S-glucosylation 5-arylidene rhodanines 13a–c were synthesised and screened for cytotoxicity against a panel of cancer cells with investigating the effective molecular target and mechanistic cell death. The anomers were separated by flash column chromatography and their configurations were assigned by NMR spectroscopy. The stable structures of the compounds under study were modelled on a molecular level, and DFT calculations were carried out at the B3LYP/6-31 + G (d,p) level to examine their electronic and geometric features. A good correlation between the quantum chemical descriptors and experimental observations was found. Interestingly, compound 6 induced potent cytotoxicity against MCF-7, HepG2 and A549 cells, with IC50 values of 11.7, 0.21, and 1.7 µM, compared to Dox 7.67, 8.28, and 6.62 µM, respectively. For the molecular target, compound 6 exhibited topoisomerase II inhibition and DNA intercalation with IC50 values of 6.9 and 19.6 µM, respectively compared to Dox (IC50 = 9.65 and 31.27 µM). Additionally, compound 6 treatmnet significantly activated apoptotic cell death in HepG2 cells by 80.7-fold, it induced total apoptosis by 34.73% (23.07% for early apoptosis, 11.66% for late apoptosis) compared to the untreated control group (0.43%) arresting the cell population at the S-phase by 49.6% compared to control 39.15%. Finally, compound 6 upregulated the apoptosis-related genes, while it inhibted the Bcl-2 expression. Hence, glucosylated rhodanines may serve as a promising drug candidates against cancer with promising topoisomerase II and DNA intercalation.
Graphical Abstract
Introduction
Worldwide, cancer is a major contributor to mortality rates and a major bottleneck to extending the human lifespan. Estimates for 2020 indicate that there would be 19.3 million new cases of cancer and about 10.0 million deaths from cancer worldwide. With an expected 1.8 million fatalities (18%), lung cancer continued to be the largest cause of cancer-related mortality. This was followed by colorectal (9.4%), liver (8.3%), stomach (7.5%), and female breast (6.9%) cancersCitation1. With the continuous failure of current medicines on one side and the development of drug resistance on the other, cancer offers one of the greatest challenges to medical scienceCitation2. Topoisomerases are nuclear enzymes that temporarily break DNA strands to facilitate DNA topological manipulationCitation3,Citation4. Topoisomerase I intentionally tear off individual molecules. In order to unwind tightly wound DNA, topoisomerase II creates double-strand breaks and shuttles the strands through a nick in the strandCitation5. Topoisomerases are enzymes that are present in all eukaryotic cells and are crucial to their survivalCitation6. One of the most effective ways to combat cancer is by inhibiting human DNA topoisomerase II. Drugs that inhibit topoisomerase II have shown great promise in the clinic, however, drug resistance in cancer cells can reduce their usefulnessCitation7. Combination therapy and multitarget medications have been proposed in numerous research to increase the efficacy of anticancer medicationCitation8. Inhibitors of topoisomerase have been used extensively in chemotherapy. Etoposide, doxorubicin, daunorubicin, mitoxantrone, teniposide, and amsacrine are some of the most widely utilised topoisomerase II inhibitorsCitation9. Topo II inhibitors were initially quite effective in treating the disease, but further treatments proved unsuccessfulCitation10. As a result, new medications targeting topoisomerase II are needed to eliminate these limitationsCitation11.
2-Thioxo-4-thiazolidinone and its derivatives have been the subject of organic synthesis research for well over half a century due to their usefulness in a wide variety of chemical and biological processes. From a biological point of view, derivatives of 2-thioxo-4-thiazolidinone have been demonstrated to possess biological activities, including antibacterialCitation12–15, antifungalCitation16,Citation17, anticonvulsantCitation18, anticancerCitation19, antihuman immunodeficiency virus type 1 (HIV-1)Citation20–22, antimicrobialCitation23, antidiabeticCitation24, antituberculoticCitation25–27, antiparasiticCitation28, hypnoticCitation29 and anthelmintic activitiesCitation30. Nucleic acid synthesis also involves the use of 2-thioxo-4-thiazolidinones, which function as analogues of purine basesCitation31. Over the past decade, thiazole nucleoside analogues have emerged as powerful and increasingly essential antimetabolite agentsCitation32,Citation33. N-(beta-D-glucopyranosyl)-5-(4-nitrobenzylidene)-2-thioxo-4-thiazolidinone demonstrated antiviral efficacy by inhibiting viral RNA synthesis, and although N-glucosyl derivatives of 2-thioxo-4-thiazolidinones are not widely studied, they have shown promise as antiviral agentsCitation34. Previous studies of glycosyl derivatives of structurally related heterocyclic systems were reportedCitation35–50. While bearing in mind the biological relevance of 2-thioxo-4-thiazolidinones, we have continued our work on the synthesis of new nucleosides as possible antiviral and anticancer medicinesCitation51–58. Herein, we detail the design, synthesis, anticancer screening, and spectroscopy of a panel of N- and S-glucosylated analogues with thioxo-4-thiazolidinone bases. New synthesis methods have enabled the first preparation of N- and S-glucosyl derivatives of 2-thioxo-4-thiazolidinones. Metwally et al.Citation24 were able to synthesise N-glycosyl derivatives of 2-thioxo-4-thiazolidinones without resorting to the S-glycosylation of 2-thioxo-4-thiazolidinone. Conversely, they were unable to synthesise 2-thioxo-4-thiazolidine N-glycosyl derivatives that would have been required for this process. Based on the results of these analyses, we investigated alternative synthetic routes leading to 2-thioxo-4-thiazolidinone nucleosides for use as antiviral and anticancer medicines. Nitrogen glycosylated compounds and their sulphur counterparts with 2-thioxo-4-thiazolidinone bases are synthesised and analysed for conformational stability and anticancer activity in this study. To the best of our knowledge, this represents the first time that S-glycosides of 2-thioxo-4-thiazolidinone have been prepared using novel synthetic approaches.
Computational chemistry has come a long way in the past few decades, and it is now commonly used alongside experimental methods to study organic and biological structures and reactions. Structures, characteristics of molecules, processes, and selectivity of reactions can all be better understood with the use of computationsCitation59. Density functional theory (DFT) is widely used to calculate many different types of molecular properties, including but not limited to molecular structures, vibrational frequencies, chemical shifts, non-linear optical (NLO) effects, natural bond orbital (NBO) analysis, molecular electrostatic potential, frontier molecular orbitals, and thermodynamic propertiesCitation60–68. Herein, we detail the design, synthesis, anticancer screening, and spectroscopic analysis of a series of nitrogen glucosylated carrying 2-thioxo-4-thiazolidinone bases. The purpose of this work is to use density functional theory to analyse how alterations to molecular and electronic structure affect the biological activity of the substances under research, and to try to locate a strong correlation between theoretical data and actual observations.
Results and discussion
Chemistry
2-Thioxo-4-thiazolidinone (1) was reacted with aromatic aldehydes (2a–f) in ethanol and morpholine as secondary amine at room temperature to give 5-((Z)-arylidene)-2-thioxo-4-thiazolidinones (3a–f) (Scheme 1)Citation69–71. The compositions of 5-((Z)-arylidene)-2-thioxo-4-thiazolidinones (3a–f) were confirmed on the basis of elemental analysis and spectroscopic data (IR, 1H NMR, 13C NMR and MS). The infra-red absorption spectrum of compound 3a was characterised by the presence of signals for the NH and CO groups at 3190 and 1726 cm−1, respectively. The 1H NMR spectrum (DMSO-d6) of 5-((Z)-benzylidene)-2-thioxo-4-thiazolidinone (3a) showed a single piece at δ 7.62 ppm assigned to the vinyl proton, indicating a Z configuration of the outer cyclic double bond. This is consistent with the 1H NMR spectrum (DMSO-d6) of 5-((Z)-(2-pyridylmethylene)-2-thioxo-4-thiazolidinone showing its vinyl proton at δ 7.70 ppmCitation72. The 13C NMR spectrum (DMSO-d6) for 5-((Z)-benzylidene)-2-thioxo-4-thiazolidinone (3a) showed signals at δ 125.90, 169.76 and 196.70 ppm assigned to vinyl, carbonyl and thiocarbonyl carbons, indicating respectively the presence of a Z configuration of the outer cyclic double bond. This is in agreement with the 13C NMR spectrum (DMSO-d6) of 5-((Z)-(2-pyridylmethylene)-2-thioxo-4-thiazolidinone showing vinyl, carbonyl and thiocarbonyl carbons respectively at δ 124.66, 170.05 and 202.66 ppmCitation72 (Scheme 1).

The silylation of the nucleoside base 1 was accomplished with bis(trimethylsilyl)acetamide (BSA) in anhydrous MeCN at 70–80 °C, and furnished the trimethylsilylated derivative 4. This derivative 4 was condensed, devised by Vorbrüggen et al.Citation73, with 1,2,3,4,6-penta-O-acetyl-α-D-glucopyranose (5) in the presence of trimethylsilyl trifluoromethyanesulfonate (TMSOTf) as catalyst at 70–80 °C for 60 min. The nucleosides 6 and 7 were isolated by silica gel column chromatography in 6% and 30% yields, respectively. The structures of 6 and 7 were established and confirmed by elemental analyses and spectral data (IR, 1H NMR, 13C NMR and MS). The absence of signal for NH and the presence of signal for the thiocarbonyl group at vmax 1225 cm−1 characterised the IR absorption spectrum of compound 6. While the IR absorption spectrum of compound 7 was characterised by the absence of signal for thiocarbonyl group. The 1H NMR spectrum of compound 6 showed the anomeric proton as a doublet at δH 6.82 ppm (J = 9.30 Hz) indicating the presence of the β-D-glucopyranose moietyCitation35–39. The 13C NMR (CDCl3) spectrum of compound 6 showed a singlet at δC 172.1 and 201.5 ppm assigned to the carbonyl at C-4 and the thiocarbonyl group at C-2, respectively. These data are also in agreement with the 13C NMR (CDCl3) spectrum of 3-(4-morpholinomethyl)-2-thioxo-4-thiazolidinone (10) (Scheme 2). The later was prepared from the reaction of 1 with morpholine and formaldehyde in EtOH at room temperature, since the carbonyl at C-4 appears at δC 175.1 ppm and the thiocarbonyl group at C-2 appears at δC 203.2 ppm indicating the presence of N-glycosylation. The 1H NMR spectrum of compound 7 showed the anomeric proton as a doublet at δH 5.82 ppm (J = 10.4 Hz) indicating the presence of the β-D-glucopyranose moietyCitation35–39. The 13C NMR (CDCl3) spectrum of compound 7 showed a singlet at δc 186.5 and 198.83 ppm assigned to the carbonyl at C-4 and the thiocarbonyl group at C-2, respectively. These data are also in agreement with the 13C NMR (CDCl3) spectrum of hitherto known 2-methylmercapto-4-thiazolidinone (11)Citation70 (Scheme 2) since the methylmercapto at C-2 appears at δC 15.9 ppm, the carbonyl at C-4 appears at δC 187.1 ppm and the thiocarbonyl group at C-2 appears at δC 202.5 ppm indicating the presence of S-glycosylation. Treatment of the protected nucleoside 6 with conc. HCl/MeOH (3.5%) at 50 °C for 2 h afforded in both cases the corresponding deprotected nucleoside 8 indicating N-glycosylation. In the same manner, treatment of 7 with conc. HCl/MeOH (3.5%) at 50 °C for 2 h furnished hitherto known 2,4-thazolidinedione (9)Citation74 indicating S-glycosylation. This type of cleavage explains why we did not successful in the preparation of the corresponding deprotected nucleoside of 7 (Scheme 3).


Correspondingly, the protected nucleoside 7 was condensed with the appropriate aromatic aldehydes (12a–c) in ethanol in the presence of morpholine as secondary amine catalyst to afford the corresponding 5-((Z)-arylidene-2-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinones (13a–c) in good yields. Treatment of the protected nucleoside 13b with a solution of sodium methoxide in methanol furnished 5-((Z)-2,4-dichlorobenzylidene)-2,4-thiazolidindione (14). This type of cleavage explains why we did not successful in the preparation of the corresponding deprotected nucleosides of 13a–c. The structures of 13a–c and 14 were established and confirmed by elemental analyses and spectral data (IR, 1H NMR, 13C NMR and MS). The absence of signals for the NH and CS groups characterises the IR absorption spectra of 13a. The 1H NMR (CDCl3) spectrum of 5-((Z)-benzylidene-2-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinones (13a) showed a single piece at δH 7.91 ppm assigned to the vinyl proton, indicating a Z configuration of the cyclic double bond. This is consistent with the 1H NMR (CDCl3) spectrum of 5-((Z)-benzylidene)-2-allylmercapto-4-thiazolidinone showing a vinyl proton at δ H 7.84 ppmCitation70. While the abnormal proton appears as a doublet at δ H 5.98 ppm (J = 10.4 Hz), indicating the presence of the β-D-glucopyranose moietyCitation35–39. It showed a 13C NMR spectrum (CDCl3) for 5-((Z)-benzylidene-2-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinones (13a) singlet at C 178.8 and 189.2 ppm assigned to a carbonyl at C-4 and a CS group at C-2, respectively. These data are also consistent with the 13C NMR (CDCl3) spectrum of 5-((Z)-benzylidene)-2-allylmercapto-4-thiazolidinoneCitation70, since the carbonyl at C-4 appears at 179.8 ppm. The CS group is shown in C-2 at −191.8 ppm, indicating a binding to S-glycosylation (Scheme 4).

Computational method
DFT (density functional theory) was utilised to optimise the molecule structures of the substances under study by utilising the Beck’s three parameter exchange functional and the Lee-Yang-Parr non local correlation functional (B3LYP)Citation75–77 with 6–31 + G(d,p) basis set which is implemented in Gaussian 09 program packageCitation78. The DFT/B3LYP combination was used to determine molecular characteristics relevant to molecular reactivityCitation79. Highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), global hardness and softness, electronegativity, electron affinity, and ionisation potential are examples of molecular characteristicsCitation80–82.
Quantum chemical study
Quantum chemistry approaches and molecular modelling techniques can describe a wide variety of molecular characteristics that characterise the reactivity, shape, and binding properties of an entire molecule, a molecule fragment, or a substituent. Some substances’ biological activity was shown to be affected by changing structural factors, and these changes were explored using quantum chemical simulations. According to the results of the computations, the geometrical structures of the organic molecules under study are not planar.
Our calculations began with a comparison of the Z- and E-form stabilities of the compounds under study; we found that, for all molecules under study, the Z-form is more stable than the E-form by a factor of 0.003–0.062 au, which is in good agreement with experimental results. Thus, we used density functional theory (DFT) computations to determine the Z-form stable structures of all molecules.
We started our calculations to make a comparison between the stability of the investigated compounds in Z- and E-forms and the calculations showed that they are more stable in the Z-form than E-form by 0.003–0.062 au, for all investigated molecules which is in a good agreement with the experimental observations. So, we performed DFT calculations on the stable structures of Z-form for all molecules.
Compounds 3a–f, 10 and 14
The optimised molecular structures with minimum energies obtained from the calculations of the investigated compounds are shown in . The calculations were done to investigate the effect of substituents on the biological activity of the thioxothiazoline-4-one 3a compound. In general, it was shown from the quantum chemical parameters obtained from the calculations that the presence of substituents on thioxothiazoline-4-one increase the reactivity of the inhibitors 3b–f.
Figure 1. The optimised molecular structures of the investigated inhibitors obtained from the calculations.


The interaction between the HOMO and LUMO levels of the reacting species determines chemical reactivity, according to the frontier molecular orbital theory, or FMO. The EHOMO and ELUMO symbols on a molecule represent its capacity to give electrons to an applicable acceptor with vacant molecular orbitals and its capacity to accept electrons, respectively. The ability of the molecule to receive electrons increases with the value of ELUMO. Comparison the reactivity between compound 3a and 3d, the insertion of methoxy group substituent increase the energy of the HOMO of 3d inhibitor by 0.017 au, which means that this compound could react as nucleophile (electron donor), . Also, many theoretical models for describing the structure and conformation barriers in molecular systems make use of the HOMO-LUMO energy gap, E, which is an important stability index. For a given molecule, the lower its value of E, the greater the likelihood that it possesses inhibitory efficiency. The calculations showed that inhibitor 3d has a smaller ΔE (0.132 au) than that of 3a (0.146 au), , which means increasing the biological activity for 3d compound which agree well with experimental observations, . The dipole moment, D, the first derivative of the energy with respect to an applied electric field, was used to discuss and rationalise the structure. There is a good correlation between D and inhibition efficiency. This was shown from increasing the dipole moment. The molecule with higher efficiency, compound 3d, has higher dipole moment (5.229 D) than that of compound 3a (3.738 D) ().
Figure 2. The calculated electronic transition between HOMO-LUMO for the highest and lowest biologically active 3a and 14 inhibitors.

Table 1. The calculated quantum chemical parameters obtained from DFT/B3LYP/6-31 + G (d) of the investigated compounds.
Absolute hardness, η, and softness, σ are important properties that measure both the stability and reactivity of a molecule. The energy gap between a hard molecule and a soft molecule is considerable for the former and small for the latter. Because they are more likely to donate electrons to an acceptor, soft molecules are more reactive than their rigid counterparts. A biological system’s inhibitor serves as a Lewis base, whereas the enzyme plays the role of a Lewis acid. The increasing the softness and chemical potential of 3d inhibitor (15.383 au−1, and −0.153 au) respectively could be responsible for increasing its biological activity more than that 3a (13.158 au−1 and −0.163 au) respectively, , which agrees well with the experimental observations.
It could be observed from the calculations that the insertion of hydroxyl and methoxy groups on the oxothizlidine-4-one moiety, increases highly the reactivity of 3e molecule with respect to 3a inhibitor. This was shown by increasing the energy of HOMO, dipole, softness and decreasing the ΔE, and electronegativity (−0.219 au, 0.130 au, 4.616 D, 15.384 au−1 and 0.154 au) respectively, .
To compare the effect of dioxolane, 3f, and dioxane, 3c, substituents on the biological activity of inhibitor 3a, the calculations showed that inhibitors 3f and 3c have higher activity than that of unsubstituted 3a inhibitor. Moreover, the calculations showed that the dioxolane substituent has higher reactivity than that of dioxane substituent. This was shown from the decreasing the energy of the LUMO (−0.087 au) which means that 3f inhibitor has more electron accepting ability from enzyme than that of 3c compound (-0.084 au), . Also, 3f has a lower energy gap, ΔE (0.131 au), than that of 3c (0.134) which could be responsible for increasing the reactivity of 3f more that of 3c. Meanwhile, increasing the softness and chemical potential of compound 3f (15.152 au−1 and −0.153 au, respectively) could increase its reactivity with respect to 3c with dioxane substituent. This is in a good agreement with the experimental data.
Meanwhile, one can see that the presence of dichloro-substituent on thiazlolidine-4-one moiety is slightly increased the reactivity of compound 3b with respect to compound 3a. This could be attributed to increasing the electron affinity, softness and electronegativity of 3b inhibitor (0.099 au, 13.889 au−1 and 0.172 au, respectively) more than that of 3a inhibitor (0.090 au, 13.158 au−1 and 0.163 au, respectively). Also, the lowering of ΔE value, which is a function of reactivity, for 3b (0.144 au) more than that of 3a (0.146 au) could be responsible for increasing the its reactivity towards the enzyme, , which is in agreement with experimental observation.
Experimental observations showed that the replacement of sulphur atom in thiazolidine-2,4-dione moiety by oxygen atom is highly decreased the biological activity of inhibitor 14 with respect to inhibitor 3a. This was confirmed from the calculations which showed the effect of substituent on the calculated quantum chemical parameters. The calculations showed the highly increasing the energy gap between HOMO-LUMO of inhibitor 14 (0.161 au) which means increasing its stability (less reactive) more than that of 3a (0.146 au), . Also, the decreasing the energy of HOMO, dipole moment, electron affinity and softness (−0.248 au, 1.350 D, 0.087 au, and 12.500 au−1, respectively) and increasing the electronegativity (0.167) could be responsible for the decreasing the reactivity of inhibitor 14 and accordingly decreasing its biological activity which is in a good agreement with the experimental observation.
Surprisingly, the morpholinomethyl-2-thioxothiazolidin-4-one, inhibitor 10, was found experimentally to be biological inactive which was confirmed from the calculated quantum parameters. The calculations showed that compound 10 has high energy gap, ΔE (0.142 au), low electron affinity (0.065 au) which probably responsible for biological inactive which agrees well with experimental data.
From the above, we could conclude the importance of computational chemistry to explain the reactivity of the investigated molecules and accordingly the biological activity.
Compounds 6 and 7
Quantum chemical calculations were performed to investigate the effect of structural parameters on the biological activity of the investigated compounds 6 and 7. It was shown from the calculations that the geometrical structures of the investigated organic compounds are not planner. The optimised molecular structures with minimum energies obtained from the calculations of the investigated compounds are shown in .
Figure 3. The optimised molecular structures, charge density distributions (HOMO and LUMO) for the investigated compounds 6 and 7.

We started our calculations to make a comparison between the stability of the investigated compounds 6 and 7 and the calculations showed that compound 6 is more stable than compound 7 by about 0.035 au. which is in a good agreement with the experimental observations.
Also, the experimental data showed that the presence of sugar moiety at the nitrogen atom of thioxothiazoline-4-one moiety, compound 6, showed a higher biological activity than that at sulphur atom of the same moiety, compound 7.
The calculations showed that the efficient inhibitor 6 has a lower energy for LUMO (−0.069 au) which increase its ability to accept charges from the enzyme and accordingly increase its biological activity, which is in a good agreement with the experimental observations. It was shown from the calculations that the inhibitor 6 has the smaller HOMO–LUMO gap (0.166 au) compared with the that of molecule 7 (0.181 au), . Accordingly, it could be expected that the molecule 6 has more inclination to interact with reactivity higher than that of molecule 7 which agrees well with the experimental data.
The dipole moment, D, was used to discuss and rationalise the structure. There is a good correlation between D and inhibition efficiency. The molecule with higher efficiency, compound 6, has higher dipole moment (11.057 D) than that of compound 7 (5.089 D) ().
Absolute hardness, η, and softness, σ are important indices which measure both the stability and reactivity of a molecule. Accordingly, it was concluded from the calculations that inhibitor 6 with higher σ value (12.048 au−1), has higher ability inhibition efficiency compared to inhibitor 7 (11.050 au−1), , which is in a good agreement with the experimental data. Also, the calculations showed that inhibitor 6 has higher χ (0.152 au), , which leads to increase its donation ability to the enzyme and accordingly enhance its inhibition efficiency. It is concluded from the above discussion that the quantum chemical parameters confirm that the inhibitor with sugar substituents on nitrogen atom of thioxothiazoline-4-one moiety has higher inhibition efficiency than that on sulphur atom which agrees well with the experimental observations.
Additionally, the HOMO and LUMO levels—two widely used quantum chemical parameters—have an impact on how a molecule interacts with other molecules. displays the charge density distribution for the investigated compounds at the HOMO and LUMO level. The studied compounds revealed that the HOMO levels, which could react with the biological target as a nucleophile (hydrogen bond acceptor), are primarily localised on the thioxothiazolidine-4-one moiety with the lone pairs of sulphur, oxygen, and nitrogen atoms, indicating that these moieties are the preferred sites for the attacking electrophile at the enzyme. The LUMO, which could be reacted as an electrophile (hydrogen bond donor) with the biological target is also completely localised on localised on the thioxothiazoline-4-one moiety as antibonding n-π*character of C-S, C-N and C-O groups which means that these moieties could be reacted as electrophile (electron acceptor). Surprisingly, the calculations showed that there is no contribution at all for sugar moiety which means that the insertion of sugar moiety will not effect on the biological activity.
Compounds 13a–c
The DFT molecular modelling calculations were performed to study the effect of inserion the sugar moiety on the sulphur atom of the thiooxothiazolin-4-one on the inhibition efficiency of the investigated compounds for enzyme. The molecular structures of the investigated compounds 13a, 13b and 13c obtained from the calculations are shown in . By comparing between the inhibition of 3a, 3b and 3c compounds with that of 13a, 13b and 13c compounds, the calculations showed that the presence of sugar moiety decreases the biological activity which is in a good agreement with the experimental observations. Surprisingly, the calculations showed that inhibitors 3a, 3 b, and 3c have the lower energies of LUMO (−0.090, −0.099, and −0.094 au, respectively) than that of 13a, 13b and 13c (−0.077, −0.096, and −0.071 au, respectively), which means that these compounds could react as electrophiles (electron acceptor) with enzyme, . Also, the decreasing the energy gap, ΔE, between HOMO-LUMO for compounds 3a, 3b, and 3c (0.146, 0.144, and 0.134 au, respectively), means that those inhibitors are probably more favourable for the reactivity towards the enzyme. The increasing of the chemical potential, electronegativity and mean increasing the reactivity of the molecule and accordingly increase the biological activity, which agrees well with the experimental observations ().
Figure 4. The calculated molecular structures of the investigated compounds 13a, 13b, and 13c.

Absolute hardness, η, and softness, σ are important indices that measure both the stability and reactivity of a molecule. Accordingly, it was concluded that inhibitors (3a, 3b, and 3c) with higher σ value (13.158, 13.884, and 14.925 au−1, respectively) have higher ability inhibition efficiency compared to inhibitors 13a, 13b, and 13c (13.158, 13.333, and 14.885 au−1, respectively), , which is in a good agreement with the experimental data. Also, the calculations showed that inhibitors 3a, 3b, and 3c have higher χ (0.153, 0.172 and 0.151 au, respectively), which leads to increase its donation ability to the enzyme and accordingly enhance its inhibition efficiency. It was concluded from the above discussion that the quantum chemical parameters confirm that the inhibitor with sugar substituents on nitrogen atom of thioxothiazoline-4-one moiety has higher inhibition efficiency than that on sulphur atom which agrees well with the experimental observations.
Biological activities
The synthesised compounds were screened for their cytotoxicity against breast, liver and lung cancer cells using the MTT assay. As seen in , most exhibited good cytotoxicity against MCF-7, hepG2 and A549 cells with promising IC50 values. Interestingly, compounds 6, 7 and 13a showed potent cytotoxicity with IC50 values of (11.7, 0.21, 1.7 µM), (12.4, 0.76, 0.31) and (3.1, 17.2, 6.1), respectively compared to Dox (7.67, 8.28, 6.62 µM). While some compounds were found to be not active with IC50 values higher than 50. Hence, these compounds 6, 7 and 13a were investigated for molecular target and apoptosis activity.
Table 2. Cytotoxic IC50 values of the tested compounds against MCF-7, HepG2 and A549 cell lines using the MTT assay.
Topo II and DNA intercalation assay
Compounds 6, 7 and 13a with the highest cytotoxic activity against HepG2 cells were tested against the Topo II and DNA intercalation activities to highlight their mechanistic study. As seen in , the tested compounds exhibited promising dual Topo II and DNA intercalation activities, interestingly, compound 6 had IC50 values of 6.9 and 19.6 µM, respectively compared to Dox (IC50 = 9.65 and 31.27 µM). Additionally, compounds 13a and 7 exhibited promising inhibition activity with IC50 values of 8.3 and 9.1 µM against Topo II and 22.6 and 29.6 µM against DNA intercalation. Hence, compound 6 was further investigated for apoptotic cell death in HepG2 cells.
Table 3. IC50 values of Topoisomerase II inhibition and DNA intercalation of the tested compounds.
Apoptotic investigation of compound 6 against HepG2
Flow cytometric examination of Annexin V/PI staining was used to examine apoptotic cell death in untreated and treated HepG2 cells to determine the apoptotic activity of compound 6 (IC50 = 0.21 M, 48 h). showed compound 6 significantly activated apoptotic cell death in HepG2 cells by 80.7-fold, it induced total apoptosis by 34.73% (23.07% for early apoptosis, 11.66% for late apoptosis) compared to the untreated control group (0.43%).
Figure 5. (A) Cryptographs of annexin-V/Propidium Iodide staining of untreated and 6-treated HepG2 cells with the IC50 values, 48 h, “Q1-UL (necrosis, AV-/PI+), Q1-UR (late apoptotic cells, AV+/PI+), Q1-LL (normal cells, AV-/PI-), Q1-LR (early apoptotic cells, AV+/PI-)”, (B) Percentage of cell population at each cell cycle G0-G1, S, G2/M, and Pre-G1 using DNA content-flow cytometry aided cell cycle analysis.

Afterward, DNA flow cytometry was used to determine the cell population in each cell phase following treatment with a cytotoxic agent. As seen in , compound 6 treatment significantly increased the cell population at the S-phase by 49.6% compared to control 39.15%, while cells in G1 weren’t significantly increased, and cells in G2/M were decreased. Consequently, compound 6 induced apoptsis in HepG2 cells arresting the cell proliferation at S-phase.
RT-PCR of apoptosis-inducing agents against HepG2
For validating the apoptotic cell death in HepG2 cells upon treatment with compound 6, gene expression level using RT-PCR was investigated for the apoptosis-mediaged genes of P53, Bax, caspase-3,8,9 and Bcl-2 in both untreated and treated HepG2 cells. As seen in , compound 6, upregulated the apoptosis-related genes by 6.16, 5.7, 8.3, 3.29, 6.14-fold change, while it inhibted the Bcl-2 expression by 0.36-fold. Hence, these findings indicated the apoptosis-mediated cell death of compound 6 treatment through intrinsic and extrinsic pathways.
Table 4. Fold of change of apoptosis-related genes in untreated and treated HepG2 cells.
Molecular docking
Molecular docking study was conducted to highlight the mechanism of binding of the promising compound 6 against topoisomerase II enzymes, as they are important molecular drug targets and inhibitors of these enzymes are widely used as effective anticancer agents. Molecular docking using AutoDock vina software was validated by calculating the RMSD value to be 1.87 Å, and this was seen by overlying the co-crystallised ligand structuers in the self-docking. As seen in , binding disposition and ligand receptor interacion of compound 6 inside the protein active site with active amino acids of Arg 503, Gln 778, and Ala 779. Compound 6 was properly docked with binding energy of −11.31 Kcal/mol, and it formed a good binding interactions with Arg 503 as key amino acid.
Figure 6. Binding disposition and ligand-receptor interactions of the co-crystallised ligand Etoposide (A); (Green-colored) and the docked compound 6 (B) (Cyan-colored) inside the topoisomeasre II (PDB = 3QX3) with the highlighted key amino acids.

Conclusion
We have carried out the successful synthesis of hitherto unreported 3-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinone (6), 2–(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinone (7), 3-(β-D-glucopyranosyl)-2-thioxo-4-thiazolidinones (8) and their corresponding 5-(Z)-arylidene derivatives (13a–c). The conformational analyses of their most stable configurations were established by NMR spectroscopy. The antiviral and other antitumor activities of the new prepared compounds are being investigated and will be reported in due course. The nucleoside base 1 can be used as a starting material for the synthesis of other carbohydrate derivatives such as deoxy, amino and azido nucleosides. To examine the stable structure of the compounds, DFT calculations using the B3LYP/6-31 + G (d,p) level were used to infer the electronic and geometric structures. The calculated quantum chemical parameters and the experimental findings exhibited a strong correlation. Interestingly, compounds 6 exhibited promising cytotoxicity against MCF-7, HepG2 and A549 cells with IC50 values of 11.7, 0.21, and 1.7 µM, compared to Dox 7.67, 8.28, and 6.62 µM, respectively. For the molecular target, compound 6 exhibited topoisomerase II inhibition and DNA intercalation with IC50 values of 6.9 and 19.6 µM, respectively compared to Dox. Additionally, compound 6 treatmnet significantly activated apoptotic cell death in HepG2 cells by 80.7-fold, arresting the cell cycle at the S-phase by 49.6% Hence, glucosylated Rhodanines may serve as a promising drug candidates against cancer through apoptosis with promising topoisomerase II and DNA interchelation.
Experimental section
Chemistry and analysis
Melting points have been identified on the Büchi apparatus and have not been corrected. TLC was performed on a silica gel of aluminium 60 F254 (Merck) sheet, and detected by short UV light. Infra-red spectra of potassium bromide pellets were obtained using a Pye Unicam 1000 spectrometer. 1H NMR and 13C NMR spectra were measured on a 300 MHz Bruker Advance DPX spectrophotometer in DMSO-d6 or CDCl3 using TMS as an internal standard. Chemical shifts are given in ppm and J in Hz. Analytical data were acquired with a C, H, N Elemental Carlo Erba 1106 analyser. Mass spectra were recorded by EI on a Varian MAT 112 spectrometer and FAB on a Kratos MS spectrometer.
General procedures for the synthesizing 3a–3f
The nucleoside bases 3a–f, were prepared form the direct condensation of 1 with the appropriate aromatic aldehydes (5a–f) according to our previous reported procedure with using morpholine instead of piperidine in the previous methodCitation70 as the following: To a mixture of 2-thioxo-4-thiazolidinone (1) (1.33 g, 10 mmol), anhydrous morpholine (0.87 g, 10 mmol) and anhydrous EtOH (30 ml), appropriate aromatic aldehydes (2a–f) were added (11 mmol). The mixture was stirred until the starting material was consumed (12 h; TLC). The reaction mixture was diluted with water and neutralised with dilute hydrochloric acid. The yellow solid that was separated by filtration was collected and recrystallized from ethanol to give products 3a–f in quantitative yields.
5-((Z)-benzylidene)-2-thioxo-4-thiazolidinone (3a)
Yield 2.10 g (95%); mp 208–210 °C (lit.Citation69 yield 90%, mp 204–206 °C); IR (KBr): v 3190 (NH), 1726 (C=O) cm−1; 1H NMR (300 MHz, DMSO-d6): δ 7.49–7.59 (m, 5H, Ar-H), 7.62 (s, 1H, =CH), 13.85 (s, 1H, NH); 13C NMR (300 MHz, DMSO-d6): δ 125.90 (=CH), 129.84, 130.90, 131.14, 132.05, 133.36 (C-5, C-Ar), 169.76 (C-4), 196.07 (C-2).
5-((Z)-2,4-dichlorobenzylidene)-2-thioxo-4-thiazolidinone (3b)
Yield 2.84 g (98%); mp 230–232 °C (lit.Citation66 yield 90%, mp 228–230 °C); IR (KBr): v 3190 (NH), 1723 (C=O) cm−1; 1H NMR (300 MHz, DMSO-d6): δ 7.43–7.76 (m, 4H, =CH, Ar-H), 13.86 (s, 1H, NH); 13C NMR (300 MHz, DMSO-d6): δ 124.56 (=CH), 125.37, 128.33, 129.66, 129.88, 129.89, 130.23, 135.66 (C-5, C-Ar), 169.06 (C-4), 195.07 (C-2); MS, m/z 289 (M+).
5-((Z)-3,4-ethylenedioxybenzylidene)-2-thioxo-4-thiazolidinone (3c)
Yield 2.60 g (93%); mp 208–210 °C (lit.Citation70 yield 94%, mp 207– 208 °C); IR (KBr): v 3196 (NH), 1736 (C=O) cm−1; 1H NMR (300 MHz, DMSO-d6): δ 4.26 (m, 4H, 2 x OCH2), 6.94 (m, 3H, Ar-H), 7.38 (s, 1H, =CH) 13.65 (s, 1H, NH); 13C NMR (300 MHz, DMSO-d6) d 64.14, 64.71 (2 x OCH2), 118.19, 119.35, 122.99, 124.57, 125.69, 126.40, 131.79, 143.89, 146.15 (C-Ar, =CH, C-5), 169.52 (C-4), 195.46 (C-2); MS, m/z 279 (M+).
5-((Z)-4-methoxybenzylidene)-2-thioxo-4-thiazolidinone (3d)
Yield 2.44 g (97%); mp 257–259 °C (lit.Citation71 yield 90%, mp 254– 256 °C); IR (KBr): v 3194 (NH), 1728 (C=O) cm−1; 1H NMR (300 MHz, DMSO-d6): δ 7.13 (d, J = 8.76 Hz, 2H, 2′-H, 6′-H), 7.59 (d, J = 8.86 Hz, 2H, 3′-H, 5′-H), 7.63 (s, 1H, =CH), 13.76 (s, 1H, NH); 13C NMR (300 MHz, DMSO-d6): δ 125.90 (=CH), 129.84, 130.90, 131.14, 132.05, 133.36 (C-5, C-Ar), 169.76 (C-4), 196.07 (C-2).
5-((Z)-(2-hydroxy-3-methoxybenzylidene)-2-thioxo-4-thiazolidinone (3e)
Yield 2.40 g (90%), mp 199–201 °C (lit.Citation70 yield 86%, mp 197–198 °C); IR (KBr): v 3189 (NH), 1726 (C=O) cm−1; 1H NMR (300 MHz, DMSO-d6): δ 3.86 (s, 3H, OMe), 6.86–7.15 (m, 3H, Ar-H), 7.94 (s, 1H, =CH), 9.89 (s, 1H, OH), 13.77 (s, 1H, NH); 13C NMR (300 MHz, DMSO-d6): δ 56.23 (OMe), 114.43, 119.99, 120.50, 124.70, 127.19, 147.19, 148.26 (=CH, C-5, C-Ar), 170.10 (C-4), 196.43 (C-2); MS, m/z 267 (M+).
5-((Z)-3,4-methylenedioxybenzylidene)-2-thioxo-4-thiazolidinone (3f)
Yield 2.45 g (92%); mp 211–212 °C (lit.Citation61 yield 71%, mp 278– 279 °C); IR (KBr): v 3194 (NH), 1732 (C=O) cm−1; 1H NMR (300 MHz, DMSO-d6): δ 6.13 (s, 2H, OCH2O), 7.12 (m, 3H, Ar-H), 7.56 (s, 1H, =CH) 13.67 (s, 1H, NH).
General procedures for the synthesizing 6 and 7
2-Thioxo-4-thiazolidinone (1) (665 mg, 5 mmol) was suspended in anhydrous acetonitrile (25 ml) and BSA (1.25 ml, 5 mmol) was added, and the reaction mixture was heated at 70–80 °C for 30 min. 1,2,3,4,6-Penta-O-acetyl-α-D-glucopyranose (5) (1.87 g, 5 mmol) dissolved in anhydrous acetonitrile (25 ml) was added to the reaction mixture via a cannula. Finally, TMSOTf (1.00 ml, 5 mmol) was added, and the reaction mixture was heated at 70–80 °C for 60 min. Saturated NaHCO3 was added to quench the reaction and the resulting mixture was extracted with CH2Cl2. The collected organic fractions were washed with saturated NaCl solution, dried with MgSO4, filtered, and evaporated to dryness. The resulting solid was purified using flash chromatography (eluent 10–50%, ether/petroether ether, 40–60 °C) to give 6 and 7, respectively.
3-(2',3',4',6'-Tetra-O-acetyl-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinone (6)
This compound was separated as white foams; yield 0.14 g (6%); IR (KBr): v 1755 (CO), (CO), 1746 1238 (CS) cm−1; 1H NMR (300 MHz, CDCl3): δ 1.98, 2.04, 2.06, 2.10 (4 s, 12H, 4Ac), 3.88 (m, 3H, 5-H, H-5′), 4.23 (m, 2H, 6′-H, 6”-H), 5.25 (dd, J = 9.8, 9.8 Hz, 1H, 4′-H), 5.35 (dd, J = 9.4, 9.4 Hz, 1H, 2′-H), 5.92 (dd, J = 9.3, 9.8 Hz, 1H, 3′-H), 5.82 (d, J = 9.3 Hz, 1H, 1′-H); 13C NMR (300 MHz, CDCl3): δ 20.30, 20.40, 20.60, 20.74 (4Ac), 33.62 (C-5), 61.40 (C-6′), 67.50 (C-2′), 67.87 (C-3′), 72.82 (C-4′), 74.73 (C-5′), 82.43 (C-1′), 169.40, 169.90, 170.08, 170.67 (4Ac), 172.10 (C-4), 201.54 (C-2); EI ms: m/z = 463 (M+). Anal. Calcd. for C17H21NO10S2 (463.48): C, 44.05; H, 4.57; N, 3.02. Found: C, 44.24; H, 4.69; N, 2.83.
2–(2',3',4',6'-Tetra-O-acetyl-β-D-glucopyranosylmercapto)-4-thiazolidinone (7)
This compound was separated as white foams; yield 0.70 g (30%); IR (KBr): v 1752 (CO) cm−1; 1H NMR (300 MHz, CDCl3): δ 2.02, 2.05, 2.07, 2.08 (4 s, 12H, 4Ac), 3.98 (m, 1H, 5′-H), 4.07 (s, 2H, H-5), 4.15 (dd, J = 2.1, 12.6 Hz, 6′-H), 4.31 (dd, J = 4.6, 12.6 Hz, 1H, 6”-H), 5.11 (dd, J = 9.6, 9.6 Hz, 1H, 4′-H), 5.35 (dd, J = 9.3, 10.2 Hz, 1H, 2′-H), 5.33 (dd, J = 9.3, 9.8 Hz, 1H, 3′-H), 5.82 (d, J = 10.4 Hz, 1H, 1′-H); 13C NMR (300 MHz, CDCl3): δ 20.06, 20.16, 20.31, 20.40 (4Ac), 39.14 (C-5), 61.13 (C-6′), 67.23 (C-2′), 69.74 (C-3′), 73.04 (C-4′), 75.94 (C-5′), 82.24 (C-1′), 169.01, 169.03, 169.45, 170.12 (4Ac), 186.53 (C-4), 198.83 (C-2); EI ms: m/z = 463 (M+). Anal. Calcd. for C17H21NO10S2 (463.48): C, 44.05; H, 4.57; N, 3.02. Found: C, 44.16; H, 4.78; N, 2.76.
General procedures for the synthesizing 3-(-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinone (8)
The protected nucleoside 6 (463 mg, 1 mmol) was suspended in MeOH (15 ml) and concentrated HCl (0.5 ml) was added. The reaction mixture was stirred at 50 °C for 2 h, and the mixture was cooled to room temperature. A resin used for ion exchange (Amberlite IR-120, HO—form) that had been rinsed with MeOH was then added to the resultant solution. The reaction mixture was stirred for 5 min before being filtered and evaporated under vacuum, and the residue was purified by flash chromatography (eluent 0–5%, CHCl3/MeOH) to get 254 mg (86%) of 8. This compound was separated as pale yellow foams; IR (KBr): v 3383 (OH), 1733 (CO), 1228 (CS) cm−1; 1H NMR (300 MHz, CDCl3): δ 3.13–3.66 (m, 6H, 5′-H, 6′-H, 4′-H, 3′-H, 2′-H), 4.60 (d, J = 4.6 Hz, 1H, 6′-OH), 4.78 (t, J = 5.2 Hz, 1H, 4′-OH), 5.20 (d, J = 5.5 Hz, 1H, 3′-OH), 5.32 (d, J = 6.0 Hz, 1H, 2′-OH), 5.67 (d, J = 10.1 Hz, 1H, 1′-H); 13C NMR (DMSO-d6): δ 33.40 (C-5), 61.74 (C-6′), 68.32 (C-2′), 70.54 (C-3′), 77.99 (C-4′), 81.08 (C-5′), 86.07 (C-1′), 175.14 (C-4), 206.55 (C-2); EI ms: m/z = 295 (M+). Anal. Calcd. for C9H13NO6S2 (295.33): C, 36.60; H, 4.44; N, 4.74. Found: C, 36.68; H, 4.57; N, 4.62.
General procedures for the synthesizing 2,4-thazolidinedione (9)
The protected nucleoside 7 (463 mg, 1 mmol) was suspended in MeOH (15 ml) and concentrated HCl (0.5 ml) was added. The reaction mixture was stirred at 50 °C for 2 h, and the mixture was cooled to room temperature. After washing with MeOH, ion exchange resin (Amberlite IR-120, HO—form) was added to the resultant solution. After 5 min of stirring, the mixture was filtered, evaporated in vacuo, and the residue was purified by flash chromatography (eluent 0–5%, CHCl3/MeOH) to get 167 mg (94%) of 9. This compound has mp 125–127 °C (lit.Citation74 yield 57%, mp 124–126 °C); IR (KBr): v 3198, CO 1756, CO 1710 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 4.13 (s, 2H, CH2), 12.22 (s, 1H, NH); 13C NMR (300 MHz, DMSO-d6): δ 37.50 (C-5), 167.35 (C-2), 170.92 (C-4); EI ms: m/z = 177 (M+). Anal. Calcd. for C3H3NO2S (177.12): 30.77; H, 2.58; N, 11.96. Found: 30.89; H, 2.72; N, 11.63.
General procedures for the synthesizing 3-morpholinomethyl-2-thioxo-4-thiazolidinone (10)
A mixture of 2-thioxo-4-thiazolidinone (1) (665 mg, 5 mmol) and morpholine (87 mg, 1 mmol) in anhydrous ethanol (5 ml) and aqueous formaldehyde (0.1 ml) was stirred for 12 h at room temperature until the starting material was consumed (TLC). The separated solid was collected by filtration and recrystallized from ethanol to give 190 mg (82%) of 10 as a yellow solid. This compound has mp 104–106 °C; IR (KBr): v 1732 (CO), 1228 (CS) cm−1; 1H NMR (300 MHZ, CDCl3): δ 2.73 (t, J = 4.7 Hz, 4H, 2′-H, 6′-H), 3.64 (t, J = 4.7 Hz, 4H, 3′-H, 5′-H), 4.01 (s, 2H, 5-H), 4.92 (s, 2H, NCH2N); 13C NMR (300 MHZ, CDCl3): δ 35.26 (C-5), 51.70 (C-2′, C-6′), 65.29 (NCH2N), 66.78 (C-3′, C-5′), 175.06 (C-4), 203.18 (C-2); EI ms: m/z = 232 (M+). Anal. Calcd. for C8H12N2O2S2 (232.32): C, 41.45; H, 5.28; N, 11.87. Found: C, 41.36; H, 5.21; N, 12.06.
General procedures for the synthesizing 2-methylmercapto-4-thiazolidinone (11)
2-Thioxo-4-thiazolidinone (1) (1.33 g, 10 mmol) was dissolved in aqueous NaOH (2%, 25 ml) at room temperature. To this solution was added methyl iodide (1.56 g, 11 mmol), and the reaction mixture was stirred overnight at room temperature. The reaction mixture was diluted with CH2Cl2, washed with cold saturated aqueous NaHCO3 and water, and dried over anhydrous Na2SO4. CH2Cl2 was evaporated to dryness and the remaining methanol crystallised to yield 1.26 g (86%), melting point 72–74 °C (lit.Citation83 168 °C–170 °C).
General procedures for the synthesizing 13a-13c
To a mixture of the protected nucleoside 7 (0.46 g, 1 mmol), anhydrous morpholine (0.09 g, 1 mmol) and anhydrous ethanol (10 ml) was added the appropriate aromatic aldehydes (12a–c) (1 mmol). The mixture was stirred until the starting material was consumed (12 h; TLC). The reaction mixture was neutralised with HCl/MeOH. After stirring for 5 min, the solution was filtered and evaporated in vacuo and the residue was purified by flash chromatography (eluent 10–50%, ether/petroleum ether, 40–60 °C) to give the products 13a–c as yellow solids.
5-((Z)-benzylidene)-2–(2',3',4',6'-tetra-O-acetyl-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinone (13a)
This compound has mp 166–168 °C; yield 0.49 (90%) IR (KBr): v CO 1750 cm−1; 1H NMR (300 MHZ, CDCl3): δ 2.03, 2.06, 2.07, 2.11 (4 s, 12H, 4Ac), 3.98–4.34 (m, 3H, 5′-H, 6′-H, 6”-H), 5.14–5.40 (m, 3H, 4′-H, 2′-H, 3′-H), 5.98 (d, J = 10.4 Hz, 1H, 1′-H), 7.45–7.53 (m, 5H, Ar-H), 7.91 (s, 1H, =CH); 13C NMR (300 MHZ, CDCl3): δ 20.21, 20.33 (4Ac), 61.26 (C-6′), 67.36 (C-2′), 68.92 (C-3′), 73.21 (C-4′), 76.12 (C-5′), 81.76 (C-1′), 125.10 (=CH), 125.63 (C-5), 129.05, 130.28, 130.84, 132.93, 137.03 (C-Ar), 169.10, 169.14, 169.50, 170.20 (4Ac), 178.82 (C-4), 189.22 (C-2); EI ms: m/z = 551 (M+). Anal. Calcd. for C24H25NO10S2 (551.59): C, 52.26; H, 4.57; N, 2.54. Found: C, 52.36; H, 4.67; N, 2.42.
5-((Z)-2,4-dichlorobenzylidene)-2–(2',3',4',6'-tetra-O-acetyl-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinone (13 b)
This compound has mp 167–169 °C; IR (KBr): v CO 1752 cm−1; 1H NMR (300 MHZ, CDCl3): δ 2.02, 2.05, 2.07, 2.11 (4 s, 12H, 4Ac), 3.96–4.36 (m, 3H, 5′-H, 6′-H, 6”-H), 5.13–5.42 (m, 3H, 4′-H, 2′-H, 3′-H), 5.98 (d, J = 10.4 Hz, 1H, 1′-H), 7.45–7.68 (m, 3H, Ar-H), 7.93 (s, 1H, =CH); EI ms: m/z = 620 (M+). Anal. Calcd. for C24H23Cl2NO10S2 (620.48): C, 46.46; H, 3.74; N, 2.26. Found: C, 46.54; H, 3.86; N, 2.20.
5-((Z)-2,4-ethylenedioxybenzylidene)-2–(2',3',4',6'-tetra-O-acetyl-β-D-glucopyranosyl)-2-thioxo-4-thiazolidinone (13c)
This compound was separated as yellow foams; IR (KBr): v CO 1750 cm−1; 1H NMR (300 MHZ, CDCl3): δ 2.02, 2.04, 2.07, 2.09 (4 s, 12H, 4Ac), 3.76 (m, 1H, 5′-H), 4.07–4.30 (m, 6H, 6′-H, 6”-H, 2CH2), 4.88 (dd, J = 9.7, 9.7 Hz, 1H, 4′-H), 5.06 (dd, J = 9.3, 9.3 Hz, 2′-H), 5.54 (dd, J = 9.2, 9.2 Hz, 3′-H), 5.97 (d, J = 10.3 Hz, 1H, 1′-H), 6.93–7.07 (m, 3H, Ar-H), 7.80 (s, 1H, =CH); 13C NMR (300 MHZ, CDCl3): δ 20.33, 20.47, 20.50, 20.52 (4Ac), 61.82 (C-6′), 63.93, 64.50 (2 OCH2), 67.44 (C-2′, C-3′), 72.33 (C-4′), 73.35 (C-5′), 81.82 (C-1′), 118.08, 119.12, 122.83, 125.00, 126.46, 137.50, 143.84, 146.53 (C-Ar, =CH, C-5), 169.56, 170.06, 170.10, 170.56 (4Ac), 179.40 (C-4), 188.86 (C-2); EI ms: m/z = 609 (M+). Anal. Calcd. for C26H27NO12S2 (609.62.60): C, 51.22; H, 4.46; N, 2.30. Found: C, 50.94; H, 4.62; N, 2.17.
General procedures for the synthesizing 5-((Z)-2,4-dichlorobenzylidene)-2,4-thiazolidindione (14)
Method A
To a stirred suspension of protected nucleoside 13b (620 mg, 1 mmol) in anhydrous MeOH (15 ml) a portion of NaOMe (0.06 g, 1.1 mmol) in anhydrous MeOH (15 ml) was added at 0 °C. Then the reaction mixture was stirred for 2 h at room temperature. An ion exchange resin (Amberlite IR-120, H+-form) was added to the resulting solution, which was pre-washed with MeOH. After stirring for 5 min, the solution was filtered and evaporated in vacuo and the residue was purified by flash chromatography (0–5% elution, CHCl3/MeOH) to provide 214 mg (78%) of 14 as a pale yellow solid.
Method B
Protected nucleoside 13b (620 mg, 1 mmol) was suspended in MeOH (15 ml) and concentrated HCl (0.5 ml) was added. The reaction mixture was heated for 2 h at 50 °C. An ion exchange resin (Amberlite IR-120, OH--form) was added to the resulting solution, which was pre-washed with MeOH. After stirring for 5 min, the solution was filtered and evaporated in vacuo and the residue was purified by flash chromatography (0–5% elution, CHCl3/MeOH) to provide 255 mg (93%) of 14 as a pale yellow solid.
This compound has mp 206–208 °C; IR (KBr): v NH 3198, CO 1758, CO 1709 cm−1; 1H NMR (300 MHZ, DMSO-d6): δ 7.50–7.80 (m, 4H, Ar-H, =CH), 12.82 (s, 1H, NH); 13C NMR (300 MHZ, DMSO-d6): δ 125.35, 125.42, 127.76, 128.22, 129.82, 129.89, 129.94, 135.29, 135.37 (=CH, C-5, C-Ar), 166.79 (C-4), 167.27 (C-2); EI ms: m/z = 274 (M+). Anal. Calcd. for C10H5Cl2NO2S (274.12): C, 43.81; H, 1.84; N, 5.11. Found: C, 44.02; H, 2.00; N, 4.94.
Biology
Cytotoxicity against cancer cells
Using RPMI-1640 medium L-Glutamine (Lonza Verviers SPRL, Belgium, cat#12-604F), we successfully cultured liver (HepG2), breast (MCF-7), and lung (A549) cells bought from the National Research Institute, Egypt. Each culture was supplemented with 10% foetal bovine serum (FBS, Sigma-Aldrich, MO, USA) and 1% penicillin-streptomycin (Lonza, Belgium). Using a 96-well plate, cells were seeded at a density of 5,000 cells per well in triplicate. The chemicals were added to the cells on day 2 at doses of (0.01, 0.1, 1, 10, and 100 M). To determine cell viability, MTT solution was used after 48 h (Promega, USA)Citation84. Using GraphPad Prism 7, as was previously reported in, we computed the survival rate in comparison to the control and obtained the IC50 valuesCitation85.
Topo II and DNA intercalation assay
Topoisomerase II (TopoGEN, Inc., Columbus) and DNA intercalator (methyl green (20 mg) and Calfthymus DNA (10 mg) were tested for their capacity to block the enzyme’s activity. Following equation was used to determine compound inhibition percentages: IC50 was determined using GraphPad prism7 using inhibition curves at five different concentrations of each compoundCitation86.
Investigation of apoptosis
Annexin V/PI staining and cell cycle analysis
After incubation overnight, 6-well culture plates were populated with HepG2 cells (3–5 105 cells/well). After that, compound 6 was used to treat the cells for 48 h at concentrations that were found to be IC50 for them. Cells and medium supernatants were then collected and washed in ice-cold PBS. The next step was suspending the cells in 100 µL of annexin binding buffer solution "25 mM CaCl2, 1.4 M NaCl, and 0.1 M Hepes/NaOH, pH 7.4” and incubation with “Annexin V-FITC solution (1:100) and propidium iodide (PI)” at a concentration equals 10 µg/mL in the dark for 30 min. The Cytoflex FACS system was then used to acquire the stained cells. cytExpert was used for the statistical analysisCitation87–89.
Real time-polymerase chain reaction for the selected genes
The gene expression of P53, Bax, Caspasses-3,8,9 as proapoptotic genes and Bcl-2 as an anti-apoptotic gene were evaluated to investigate the apoptotic pathway. The IC50 values for compound 6 were then used to treat HepG2 cells for 48 h. Afterwards, the standard operating procedure called for an RT-PCR reaction to be performed. The Ct values were then used to determine the relative expression of each gene in each sample, normalised to the β-actin as house-keeping geneCitation90–92.
In silico studies
Chimaera-UCSF and AutoDock Vina were used for molecular modelling research on a Linux-based system. Binding sites within proteins were identified by measuring the dimensions of grid boxes surrounding the co-crystallised ligands; this entire process was performed utilising Maestro to build and optimise the structures of both proteins and compounds. After standard procedures, the Topo II (PDB= 3qx3) protein structures were docked against the chemicals under study using AutoDock VinaCitation84,Citation93–96. Protein and ligand structures were optimised with Vina to make them more energetically favourable. The results of the molecular docking were interpreted by the binding activities in terms of the binding energy and the interactions between the ligand and the receptor. Chimaera was then used to complete the visualisation.
The Supporting Information is available free of charge at 13C NMR, 1H NMR, and IR spectra of the new synthesised compounds (PDF).
Supplemental Material
Download PDF (281.7 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A, Bray F. Cancer statistics for the year 2020: an overview. Int J Cancer. 2021;149(4):778–789.
- Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S. Drug resistance in cancer: an overview. Cancers. 2014; 6(3):1769–1792.
- Zhuo S-T, Li C-Y, Hu M-H, Chen S-B, Yao P-F, Huang S-L, Ou T-M, Tan J-H, An L-K, Li D, et al. Synthesis and biological evaluation of benzo[a]phenazine derivatives as a dual inhibitor of topoisomerase I and II. Org Biomol Chem. 2013;11(24):3989–4005.
- Bailly C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem Rev. 2012;112(7):3611–3640.
- Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 2013;8(1):82–95.
- Pommier Y, Sun Y, Huang SN, Nitis JL. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol. 2016;17(11):703–721.
- Capranico G, Marinello J, Chillemi G. Type I DNA Topoisomerases. J Med Chem. 2017;60(6):2169–2192.
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9(5):338–350.
- Christodoulou MS, Zarate M, Ricci F, Damia G, Pieraccini S, Dapiaggi F, Sironi M, Lo Presti L, García-Argáez AN, Dalla Via L, et al. 4-(1,2-Diarylbut-1-en-1-yl)isobutyranilide derivatives as inhibitors of topoisomerase II. Eur J Med Chem. 2016; 118:79–89.
- Christodoulou MS, Calogero F, Baumann M, García-Argáez AN, Pieraccini S, Sironi M, Dapiaggi F, Bucci R, Broggini G, Gazzola S, et al. Boehmeriasin A as new lead compound for the inhibition of topoisomerases and SIRT2. Eur J Med Chem. 2015; 92:766–775.
- Yao BL, Mai YW, Chen SB, Hua HT, Yao PF, Ou TM, Tan JH, Wang HG, Li D, Huang SL, et al. Design, synthesis and biological evaluation of novel 7-alkylamino substituted benzo[a]phenazin derivatives as dual topoisomerase I/II inhibitors. Eur J Med Chem. 2015; 92:540–553.
- Kucukguzel SG, Oruc EE, Rollas S, Sahin F, Ozbek A. Synthesis, characterisation and biological activity of novel 4-thiazolidinones, 1,3,4-oxadiazoles and some related compounds. Eur J Med Chem. 2002;37(3):197–206.
- Gualtieri M, Bastide L, Villain-Guillot P, Michaux-Charachon S, Latouche J, Leonetti J-P. In vitro activity of a new antibacterial rhodanine derivative against Staphylococcus epidermidis biofilms. J Antimicrob Chemother. 2006;58(4):778–783.
- Sim MM, Ng SB, Buss AD, Crasta SC, Goh KL, Lee SK. Benzylidene Rhodanines as Novel Inhibitors of UDP-N-Acetylmuramate/l-Alanine Ligase. Bioorg Med Chem Lett. 2002;12(4):697–699.
- Petrikaite V, Tarasevicius E, Pavilonis A. New ethacridine derivatives as the potential antifungal and antibacterial preparations. Medicina (Kaunas)). 2007;43(8):657–663.
- Capan G, Ulusoy N, Ergenc N, Kiraz M. New 6-phenylimidazo[2,1-b]thiazole derivatives: synthesis and antifungal activity. Monatsh Chem. 1999;130:1399–1407.
- Sortino M, Delgado P, Juárez S, Quiroga J, Abonía R, Insuasty B, Nogueras M, Rodero L, Garibotto FM, Enriz RD, et al. Synthesis and antifungal activity of (Z)-5-arylidenerhodanines. Bioorg Med Chem. 2007;15(1):484–494.
- Ergenc N, Capan G. Synthesis and anticonvulsant activity of new 4- thiazolidone and 4-thiazoline derivatives. Farmaco. 1994;49:133–135.
- Bhatt JJ, Shah BR, Shah HP, Trivedi PB, Undavia NK, Desai NC. Synthesis of anti‐HIV, anticancer and antitubercular 4‐oxo‐ thiazolidines (III), 2‐imino‐4‐oxo‐thiazolidines (VI) and their 5‐ arylidine derivatives. Indian J Chem, Sect B: Org Chem Incl Med Chem. 1994;33(2):189–192.
- Barreca ML, Chimirri A, Luca LD, Monforte A, Monforte P, Rao A, Zappala M, Balzarini J, De Clercq E, Pannecouque C, et al. Discovery of 2,3-diaryl-1,3-thiazolidin-4-ones as potent anti-hiv-1 agents. Bioorg Med Chem Lett. 2001;11(13):1793–1796.
- Ozkirimli S, Kazan F, Tunali Y. Synthesis, antibacterial and antifungal activities of 3-(1,2,4-triazol-3-yl)-4-thiazolidinones. J Enzyme Inhib Med Chem. 2009;24(2):447–452.
- Chandrappa S, Benaka Prasad SB, Vinaya K, Ananda Kumar CS, Thimmegowda NR, Rangappa KS. Synthesis and in vitro antiproliferative activity against human cancer cell lines of novel 5-(4-methyl-benzylidene)-thiazolidine-2,4-diones. Invest New Drugs. 2008;26(5):437–444.
- Metwally NH, Abdalla MA, Mosselhi MAN, El-Desoky EA. Synthesis and antimicrobial activity of some new N-glycosides of 2-thioxo-4-thiazolidinone derivatives. Carbohydr Res. 2010;345(9):1135–1141.
- Murugan R, Anbazhagan S, Sriman Narayanan S. Synthesis and in vivo antidiabetic activity of novel dispiropyrrolidines through [3 + 2] cycloaddition reactions with thiazolidinedione and rhodanine derivatives. Eur J Med Chem. 2009;44(8):3272–3279.
- Bukowski L, Janowiec M, Zwolska-Kwiek Z, Andrzejczyk Z. Some reactions of 2-cyanomethylimidazo[4,5-b]pyridine with isothiocyanates. Antituberculotic activity of the obtained compounds. Pharmazie. 1998;53(6):373–376.
- Chandrappa S, Kavitha CV, Shahabuddin MS, Vinaya K, Ananda Kumar CS, Ranganatha SR, Raghavan SC, Rangappa KS. Synthesis of 2-(5-((5-(4-chlorophenyl)furan-2-yl)methylene)-4-oxo-2-thioxothiazolidin-3-yl)acetic acid derivatives and evaluation of their cytotoxicity and induction of apoptosis in human leukemia cells. Bioorg Med Chem. 2009;17(6):2576–2584.
- Brooke EW, Davies SG, Mulvaney AW, Okada M, Pompeo F, Sim E, Vickers RJ, Westwood IM. Synthesis and in vitro evaluation of novel small molecule inhibitors of bacterial arylamine N-acetyltransferases (NATs). Bioorg Med Chem Lett. 2003;13(15):2527–2530.
- Rauter AP, Padilha M, Figueiredo JA, Ismael MI, Justino J, Ferreira H, Ferreira MJ, Rajendran C, Wilkins R, Vaz PD, et al. Bioactive Pseudo‐C‐nucleosides Containing Thiazole, Thiazolidinone, and Tetrazole Rings. J. Carbohydr. Chem. 2005;24(3):275–296.
- Ergenc N, Capan G, Gunay NS, Ozkirimli S, Gungor M, Ozbey S, Kendi E. Synthesis and hypnotic activity of new 4-thiazolidinone and 2-thioxo-4,5-imidazolidinedione derivatives. Arch Pharm Pharm Med Chem. 1999;332(10):343–347.
- Verma A, Saraf SK. 4-Thiazolidinone – A biologically active scaffold. Eur J Med Chem. 2008;43(5):897–905.
- Hardy RW, Marcotrigiano J, Blight KJ, Majors JK, Rice CM. Hepatitis C Virus RNA synthesis in a cell-free system isolated from replicon-containing hepatoma cells. J Virol. 2003;77(3):2029–2037.
- Mallick SK, Martin AR, Lingard RG. Synthesis and antimicrobial evaluation of some 5-(5-nitrofurylidene)rhodanines, 5-(5-nitrofurylidene)thiazolidine-2,4-diones, and their vinylogs. J Med Chem. 1971;14(6):528–532.
- Kumar R, Lown JW. Synthesis and antitumor cytotoxicity evaluation of novel thiazole-containing glycosylated polyamides. Eur J Org Chem. 2003;2003(24):4842–4851.
- Foye WO, Tovivich P. N-glucopyranosyl-5-aralkylidenerhodanines: synthesis and antibacterial and antiviral activities. J Pharm Sci. 1977;66(11):1607–1611.
- El-Barbary AA, Khodair AI, Pedersen EB, Nielsen C. S-glucosylated hydantoins as new antiviral agents. J Med Chem. 1994;37(1):73–77.
- Khodair AI, el-Subbagh HI, el-Emam AA. Synthesis of certain 5-substituted 2-thiohydantoin derivatives as potential cytotoxic and antiviral agents. Boll Chim Farm. 1997;136(8):561–567.
- Al-Obaid AM, el-Subbagh HI, Khodair AI, Elmazar MM. 5-Substituted 2-thiohydantoin analogs as a novel class of antitumor agents. Anticancer Drugs. 1996;7(8):873–880.
- Khodair AI. Glycosylation of 2-thiohydantoin derivatives. synthesis of some novel S-alkylated and S-glucosylatedhydantoins. Carbohydr Res. 2001;331(4):445–453.
- Khodair AI. Synthesis of 2-thiohydantoins and their S-glucosylated derivatives as potential antiviral and antitumor agents. Nucleosides Nucleotides Nucleic Acids. 2001;20(9):1735–1750.
- El-Barbary AA, Khodair AI, Pedersen EB, Nielsen C. Synthesis and antiviral evaluation of hydantoin analogues of azt. Arch Pharm. 1994;327(10):653–655.
- Khodair AI, El-Ashry EH, Al-Masoudi NAL. Thiohydantoin nucleosides. Synthesis approaches, Monatsh Fur Chem. 2004;135:1061–1079.
- Khodair AI, Attia AM, Gendy EA, Elshaier YAMM, El-Magd MA. Design, synthesis and cytotoxicity evaluation of some novel of S-glycoside of 2-thioxopyridine and N-glycoside of 2-oxopyridine derivatives as antibreast cancer. J Heterocyclic Chem. 2019;56(6):1733–1747.
- Khodair AI, Alsafi MA, Nafie MS. Synthesis, molecular modeling and anti-cancer evaluation of a series of quinazoline derivatives. Carbohydr Res. 2019; 486:107832.
- Khodair AI, Elsafi MA, Al‐Issa SA. Simple and efficient synthesis of novel 3-substituded-2-thioxo-2,3-dihydro-1H-benzo[g]quinazolin-4-ones and their reactions with alkyl halides and α-glycopyranosyl bromides. J Heterocyclic Chem. 2019;56(9):2358–2368.
- Attia AM, Khodair AI, Gendy EA, El-Magd MA, Elshaier YAMM. New 2-oxopyridine/2-thiopyridine derivatives tethered to a benzotriazole with cytotoxicity on MCF7 cell lines and with antiviral activities. LDDD. 2020;17(2):124–137.
- Khodair AI, El-Barbary AA, Imam DR, Kheder NA, Elmalki F, Ben Hadda T. Synthesis, DFT, antiviral, and molecular docking studies of some novel 1,2,4-triazine nucleosides as potential bioactive compounds. Carbohydr Res. 2021; 500:108246.
- Khodair AI, Bakare SB, Awad MK, Al-Issa SA, Nafie MS. Design, synthesis and computational explorations of novel 2-thiohydantoin nucleosides with cytotoxic activities. J Heterocyclic Chem. 2022;59(4):664–685.
- Nafie MS, Khodair AI, Hassan HAY, Abd El-Fadeal NM, Bogari HA, Elhady SS, Ahmed SA. Evaluation of 2-thioxoimadazolidin-4-one derivatives as potent anti-cancer agents through apoptosis induction and antioxidant activation: in vitro and in vivo approaches. Molecules. 2021;27(1):83.
- Elsayed GH, Fahim AM, Khodair AI. Synthesis, anti-cancer activity, gene expression and docking stimulation of 2-thioxoimidazolidin-4-one derivatives. J Mol Struct. 2022;1265:133401.
- El-Barbary AA, Imam DR, El–Tahawy MMT, El-Hallouty SM, Kheder NA, Khodair AI. Unexpected synthesis, characterization and comutaional details of novel triazine-pyrrole hybrid nucleosides. J Mol Struct. 2023;1272:134182.
- Zsolnai T. Antimicrobial effect of potential isothiocyanate-formers. 5. Arzneimittelforschung. 1969;19(4):558–572.
- Werbel LM, Headen N, Elslager EF. 3-Phenylrhodanines as potential antimalarial agents. J Med Chem. 1968;11(2):364–365.
- Brown FC, Bradsher CK, Bond SM, Grantham RJ. Mildew-Preventing Activity of Rhodanine Derivatives. Ind Eng Chem. 1954;46(7):1508–1512.
- Takematsu T, Furushima M, Hasegawa Y, Morioka M, Tsuchiyama T. Herbicide containing 2-mercapto-4-keto-5-substituted thiazoline derivatives, JP 47013812. 1972. Google Scholar.
- Mackie A, Stewart GM. In vitro testing of chemical compounds against vinegar eelworm (Turbatrix aceti) attempted correlation of anthelmintic effect and chemical constitution. Arch Int Pharmacodyn Ther. 1955;102(4):476–486.
- Moers FG, Goossens JWM, Langhout JPM. Rhodanine complexes of copper(I), palladium(II) and platinum(II). J. Inorg. Nucl. Chem. 1973;35(3):855–859.
- Brockman RW, Sidwell RW, Arnett G, Shaddix S. Heterocyclic thiosemicarbazones: correlation between structure, inhibition of ribonucleotide reductase, and inhibition of DNA VIRUSES. Proc Soc Exp Biol Med. 1970;133(2):609–614.
- Foye WO, Lange WE, Feldmann EG. Comparative activities and toxicities II.*: sugar and amino acid derivatives of bis-(4-aminophenyl)-sulfone. J Am Pharm Assoc. 1958;47(11):831–833.
- Houk KN, Liu F. Holy grails for computational organic chemistry and biochemistry. Acc Chem Res. 2017;50(3):539–543.
- Kabanda MM, Murulana LC, Ozcan M, Karadag F Quantum chemical studies on the corrosion inhibition of mild steel by some triazoles and benzimidazole derivatives in acidic medium. Int. J. Electrochem. Sci. 2012; 7:5035–5056.
- Udhayakala P, Jayanthi A, Rajendiran TV. Adsorption and quantum chemical studies on the inhibition potentials of some formazan derivatives. Der Pharma Chem. 2011;3:528–539.
- Awad MK, Masoud MS, Shaker MA, Ali AE, El-Tahawy MMT. MP2 and DFT theoretical studies of the geometry, vibrational and electronic absorption spectra of 2-aminopyrimidine. Res Chem Intermed. 2013;39(6):2741–2761.
- Atlam FM, Awad MK, El-Bastawissy EA. Computational simulation of the effect of quantum chemical parameters on the molecular docking of HMG-CoA reductase drugs. J Mol Struct. 2014;1075:311–326.
- Khodair AI, Bakare SB, Awad MK, Nafie MS. Design, synthesis, DFT, molecular modelling studies and biological evaluation of novel 3-substituted (E)-5-(arylidene)-1-methyl-2-thioxoimidazolidin-4-ones with potent cytotoxic activities against breast MCF-7, liver HepG2, and lung A549. J Mol Struct. 2021;1229:129805.
- Elbadawi MM, Khodair AI, Awad MK, Kassab SE, Elsaady MT, KRA, Abdellatif Design, synthesis and biological evaluation of novel thiohydantoin derivatives as antiproliferative agents: a combined experimental and theoretical assessments. J. Mol. Struct. 2022;1249:131574.
- Khodair AI, Awad MK, Gesson J-P, Elshaier YAMM. New N-ribosides and N-mannosides of rhodanine derivatives with anticancer activity on leukemia cell line: Design, synthesis, DFT and molecular modelling studies. Carbohydr Res. 2020;487:107894.
- Awad MK, Abdel-Aal MF, Atlam FM, Hekal HA. Molecular docking, molecular modeling, vibrational and biological studies of some new heterocyclic α-aminophosphonates. Spectrochim Acta A Mol Biomol Spectrosc. 2019;206:78–88.
- El-Borai MA, Awad MK, Rizk HF, Atlam FM. Design, synthesis and docking study of novel imidazolyl pyrazolopyridine derivatives as antitumor agents targeting MCF7 cell line. COS. 2018;15(2):275–285.
- Khodair AI, Gesson J-P. A new approach for the N- and S-galactosylation of 5-arylidene-2-thioxo-4-thiazolidinones. Carbohydr. Res. 2011; 346:2831–2337.
- Khodair AI. Convenient synthesis of 2-arylidene-5H-thiazolo[2,3-b]quinazoline-3,5[2H]-diones and their benzoquinazoline derivatives. J. Heterocycl. Chem. 2002;39(6):1153–1160.
- Zhou J-F, Zhu F-X, Song Y-Z, Zhu Y-L. Synthesis of 5-arylalkylidenerhodanines catalyzed by tetrabutylammonium bromine in water under microwave irradiation. ARKIVOC. 2007;2006(14):175–180.
- Chowdhry MM, Mingos DMP, White AJP, Williams DJ. Syntheses and characterization of 5-substituted hydantoins and thiazolines—implications for crystal engineering of hydrogen bonded assemblies. Crystal structures of 5-(2-pyridylmethylene)-hydantoin, 5-(2-pyridylmethylene)-2-thiohydantoin, 5-(2-pyridylmethylene)thiazolidine-2,4-dione, 5-(2-pyridylmethylene)rhodanine and 5-(2-pyridylmethylene)pseudothiohydantoin. J Chem Soc Perkin Trans 1. 2000;(20):3495–3504.
- Vorbruggen H, Krolikiewicz K, Bennua B. Nucleoside syntheses, XXII1) Nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalysts. Chem Ber. 1981;114(4):1234–1255.
- Walter W, Randau G. Über die oxydationsprodukte von thiocarbonsäureamiden, VIII oxydationsreaktionen an N-acyl-thioamiden. Justus Liebigs Ann Chem. 1965;681(1):55–63.
- Becke AD. Density-functional thermochemistry II: the effect of the Perdew-Wang generalized-gradient correlation correction. J. Chem. Phys. 1992;97(12):9173–9177.
- Becke AD. A new mixing of Hartree–Fock and local density-functional theories. J Chem Phys. 1993;98(2):1372–1377.
- Lee C, Yang W, Parr RG. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter. 1988;37(2):785–789.
- Frisch MJ. Gaussian 09, revision B.01, Pittsburgh: Gaussian, Inc.; 2009. https://gaussian.com/g09citation/
- Senet PL. Chemical harnesses of atoms and molecules from frontier orbitals. Chem Phys Lett. 1997;275(5-6):527–532.
- Geerlings P, De Proft F, Langenaeker W. De Proft, F. Theory, conceptual density functional. Chem Rev. 2003;103(5):1793–1873.
- Pu SL. Materials for nonlinear optics-chemical perspectives, chapter 22: observing high second harmonic generation and control of molecular alignment in one dimension. ACS Symp. Ser. 1991;455:331–342.
- Scrocco E, Tomasi J. Electronic molecular structure, reactivity and intermolecular forces: an euristic interpretation by means of electrostatic molecular potentials. Adv Quant Chem. 1978;11:115–193.
- Girard ML, Dreux C. Possibilités réactionnelles et structurales de dérivés de la thiazolidine. I.-addition, substitution, Hydrolyse. Bull Soc Chim Fr. 1968;3461(2):34.
- Nafie MS, Tantawy MA, Elmgeed GA. Screening of different drug design tools to predict the mode of action of steroidal derivatives as anti-cancer agents. Steroids. 2019;152:108485.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1-2):55–63.
- Tantawy ES, Amer AM, Mohamed EK, Abd Alla MM, Nafie MS. Synthesis, characterization of some pyrazine derivatives as anti-cancer agents: In vitro and in Silico approaches. J Mol Struct. 2020;1210:128013.
- Nafie MS, Amer AM, Mohamed AK, Tantawy ES. Discovery of novel pyrazolo[3,4-b]pyridine scaffold-based derivatives as potential PIM-1 kinase inhibitors in breast cancer MCF-7 cells. Bioorg Med Chem. 2020;28(24):115828.
- Khalifa MM, Al-Karmalawy AA, Elkaeed EB, Nafie MS, Tantawy MA, Eissa IH, Mahdy HA. Topo II inhibition and DNA intercalation by new phthalazine-based derivatives as potent anticancer agents: design, synthesis, anti-proliferative, docking, and in vivo studies. J Enzyme Inhib Med Chem. 2022;37(1):299–314.
- Boraei ATA, Eltamany EH, Ali IAI, Gebriel SM, Nafie MS. Synthesis of new substituted pyridine derivatives as potent anti-liver cancer agents through apoptosis induction: In vitro, in vivo, and in silico integrated approaches. Bioorg Chem. 2021;111:104877.
- Dawood KM, Raslan MA, Abbas AA, Mohamed BE, Abdellattif MH, Nafie MS, Hassan MK. Novel bis-thiazole derivatives: synthesis and potential cytotoxic activity through apoptosis with molecular docking approaches. Front Chem. 2021;9:694870.
- Gad EM, Nafie MS, Eltamany EH, Hammad MSAG, Barakat A, Boraei ATA. Discovery of new apoptosis-inducing agents for breast cancer based on ethyl 2-amino-4,5,6,7-tetra hydrobenzo[b]thiophene-3-carboxylate: synthesis, in vitro, and in vivo activity evaluation. Molecules. 2020;25(11):2523.
- Tantawy MA, Shaheen S, Kattan S, Alelwani SW, Barnawi W, Elmgeed IO, Nafie GA, Cytotoxicity MS. In silico predictions and molecular studies for androstane heterocycle compounds revealed potential antitumor agent against lung cancer cells. J. Biomol. Struct. Dyn. 2020;40(10):4352–4365.
- ElZahabi HSA, Nafie MS, Osman D, Elghazawy NH, Soliman DH, EL-Helby AAH, Arafa RK. Design, synthesis and evaluation of new quinazolin-4-one derivatives as apoptotic enhancers and autophagy inhibitors with potent antitumor activity. Eur J Med Chem. 2021;222:113609.
- Eltamany EE, Elhady SS, Ahmed HA, Badr JM, Noor AO, Ahmed SA, Nafie MS. Chemical profiling, antioxidant, cytotoxic activities and molecular docking simulation of carrichtera annua DC. (Cruciferae). Antioxidants. 2020;9(12):1286.
- Kishk SM, Kishk RM, Yassen ASA, Nafie MS, Nemr NA, ElMasry G, Al-Rejaie S, Simons C. Molecular insights into human transmembrane protease serine-2 (tmps2) inhibitors against sars-cov2: homology modelling, molecular dynamics, and docking studies. Molecules. 2020;25(21):5007.
- Nafie MS, Boraei ATA. Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer. Bioorg Chem. 2022;122:105708.