
Abstract
Topoisomerase II (TOP-2) is a promising molecular target for cancer therapy. Numerous antibiotics could interact with biologically relevant macromolecules and provoke antitumor potential. Herein, molecular docking studies were used to investigate the binding interactions of 138 antibiotics against the human topoisomerase II-DNA complex. Followed by the MD simulations for 200 ns and MM-GBSA calculations. On the other hand, the antitumor activities of the most promising candidates were investigated against three cancer cell lines using doxorubicin (DOX) as a reference drug. Notably, spiramycin (SP) and clarithromycin (CL) showed promising anticancer potentials on the MCF-7 cell line. Moreover, azithromycin (AZ) and CL exhibited good anticancer potentials against the HCT-116 cell line. Finally, the TOP-2 enzyme inhibition assay was carried out to confirm the proposed rationale. Briefly, potent TOP-2 inhibitory potentials were recorded for erythromycin (ER) and roxithromycin (RO). Additionally, a SAR study opened eyes to promising anticancer pharmacophores encountered by these antibiotics.
Molecular docking studies of 139 antibiotics against the topoisomerase II-DNA complex.
SP, RO, AZ, CL, and ER were the most promising and commercially available candidates.
Molecular dynamics simulations for 200 ns for the most promising five complexes.
MM-GBSA calculations for the frontier five complexes.
SP and CL showed promising anticancer potentials on the MCF-7 cell line, besides, AZ and CL exhibited good anticancer potentials against the HCT-116 cell line.
Potent TOP-2 inhibitory potentials were recorded for ER and RO.
Highlights
Graphical Abstract
Introduction
Numerous patients worldwide suffer from variable types of cancer and the prevalence of cancer is increasingCitation1. For instance, according to GLOBOCAN 2020, the global number of cancer cases was 19 292 789 while death cases attributed to cancer were 9 958 133Citation2,Citation3. Numerous factors contribute to this, such as increased pollution and exposure to ultraviolet radiationCitation4. Additionally, change in population age structure and the rise of bad lifestyle habits such as smoking, alcohol consumption, being overweight, obesity, physical inactivity, and malnutrition all contribute to the increased global prevalence of cancerCitation5,Citation6. Therefore, the development of cancer therapy is an ongoing research quest. In the past, cancer therapy relied on surgeries and radiotherapy. Currently, molecular cancer cell targets are the main approach for the development of an anticancer agentCitation7,Citation8.
Topoisomerase II (TOP-2) is a promising molecular target for cancer therapy. It is a nuclear enzyme that is essential for cell survival. TOP-2 is involved in various stages of DNA replication as well as chromosome assembly and segregation. It acts by breaking the double-stranded DNA and promoting the relaxation of over-coiled strandsCitation9,Citation10. Therefore, it is responsible for the topological changes in double-stranded DNA that are required prior to replication and transcription. TOP-2 exerts its function via introducing a protein-bridged cut in DNA strandsCitation11. A higher level of TOP-2 expression and a prolonged intracellular half-life is observed in malignant cells over normal ones making the enzyme a potential target for anticancer therapy. Drug candidates that target TOP-2 can be classified as TOP-2 poisons and TOP-2 catalytic inhibitors. The TOP-2 poisons exert their function by increasing the covalent binding between the TOP-2 and DNA. This group includes most of the clinically relevant compounds e.g. doxorubicin (DOX). The TOP-2 catalytic inhibitors act by halting the activity of TOP-2 which is crucial for tumour survivalCitation12,Citation13.
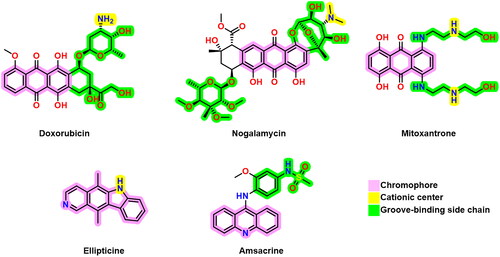
DNA intercalators and TOP-2 poisons usually exhibit three pharmacophoric features; a polyaromatic planar structure (chromophore) to be placed between two neighbouring DNA nucleobasesCitation14, a cationic centre that interacts with the negatively charged phosphate group on DNA nucleobases, and a groove-binding site which stays in the DNA minor grooveCitation15. Some reported DNA intercalators and TOP-2 poisons are depicted in Citation16–18.
Figure 1. Some previously reported topoisomerase II inhibitors and DNA intercalators display their pharmacophoric characteristics.
Immense research is taking place on the development of novel TOP-2 inhibitors. The journey for the development of a new drug candidate from drug discovery to FDA approval is an economically exhausting process with a low degree of success and uncertaintyCitation19,Citation20. Repurposing drug candidates that are already approved by the FDA can surpass such time and cost resource consumption; meanwhile, reducing the incidence of developing a new agent with significant side effects that may impact the health of patientsCitation21,Citation22. Repurposing FDA-approved drugs may also reduce the chemical consumption that is exerted in the development process which is a global quest for climate change. In addition, the utilisation of some in-silico approaches, such as molecular docking and molecular dynamics have a crucial role in the drug repurposing process by affording deep insights about the affinity of the repurposed compounds to the new specified targets and putting eyes on the possible capability of these compounds to be dedicated for new therapeutic uses along with their primary therapeutic usesCitation23–25.
In this respect, antibiotics are fruitful candidates for drug repurposing. Numerous antibiotics may have protein and nucleic acid targets that exhibit similarities with those of the eukaryotic cells. For example; antibiotics that involve complexing with DNA or the associated proteins such as anthracycline and ionophores. This interaction with biologically relevant macromolecules provokes an antitumor potential of antibiotics. Those classes are either clinically approved or investigated for their antitumor activityCitation26–30. In addition to providing a new therapeutic candidate, the repurposing of antibiotics may find a new role for drug molecules that lost their clinical relevance due to the development of bacterial resistanceCitation31.
Consequently, the rationale of this study is based on screening a reasonable number of available FDA-approved antibiotics that may interact with nucleic acid synthesis or replication. Also, they may share the same previously described pharmacophores commonly retained by most TOP-2 inhibitors. Therefore, 138 antibiotics belonging to aminoglycosides, lincosamides, macrolides, oxazolidinones, peptidyl transferases, streptogramins, sulphonamides, tetracyclines, quinolones, and other classes were in silico tested for their activity against TOP-2 using molecular docking. Then, some of the most promising and commercially available antibiotics were subjected to molecular dynamics simulations for 200 ns to get deep insights regarding their exact stability within the receptor pocket of Top-2. In addition, the MM-GBSA calculations for the frontier members were calculated. Additionally, the antitumor activities of the most promising candidates were investigated against three cancer cell lines (breast, liver, and colorectal) using DOX as a reference drug. Finally, the TOP-2 enzyme inhibition assay for the outstanding promising antibiotics compared to DOX as an FDA-approved TOP-2 inhibitor and DNA intercalator was carried out to confirm the proposed rationale.
Materials and methods
SP, RO, AZ, CL, and ER (as ethyl succinate) were provided from NODQCAR at a purity > 99.87%. Human mammary tumours, hepatocellular carcinoma (HepG2), and colorectal cancer (HCT-116) cell lines were obtained from VACSERA; Egypt. RPMI 1640 doped with 10% FBS was provided from VACSERA; Egypt. The inhibitory potential against TOP-2 was assessed by the Topo GEN assay kit (TOPOGEN, CO, USA).
In silico studies
Docking studies
The molecular docking studies were used to screen the binding interactions of the investigated antibiotics utilising the MOE 2019 suiteCitation32.
Preparation of the investigated compounds
PerkinElmer ChemOffice Suite 2017 toll was employed for drawing the screened compounds which were then made available for the docking process as previously described in the default procedureCitation33,Citation34. By importing them into one database and saving them as an MDB file, the screened compounds and the co-crystallised inhibitor (EVP) were ready for the docking process towards the human topoisomerase II-DNA complex.
Preparation of human topoisomerase II-DNA complex target
Using the protein data bank, the human topoisomerase II-DNA complex X-ray structure was downloaded (PDB code: 3QX3, https://www.rcsb.org/structure/3QX3). The target receptor was protonated, broken bonds were corrected, and the entire protein was energetically minimised to prepare for the molecular docking process as discussed previously in detailCitation35,Citation36.
Molecular docking against the prepared target complex
The general docking process was started after the file for the prepared active site was loaded. The docking process site was (Ligand atoms), the scoring methodology was (London dG) and the placement methodology was (triangle matcher). Rigid receptor docking was utilised as the refinement methodology and the scoring methodology was GBVI/WSA dG with the selection of the best 10 poses for each tested compound from 30 different poses. Subsequently, the investigated ligands’ MDB file was uploaded to the program and the calculations of the running docking were automatically executed. Following completion, the obtained poses were examined, and the best ones with the highest scores, acceptable RMSD values, and better ligand–enzyme interactions were chosen and saved for further investigationCitation37,Citation38.
Molecular dynamics simulations (MDs)
The MD simulationsCitation39,Citation40 for the six studied complexes (SP-3QX3, RO-3QX3, AZ-3QX3, CL-3QX3, ER-3QX3, and EVP-3QX3) were performed using the Desmond package (Schrödinger LLC)Citation41. Moreover, the thermal_mmgbsa.py python script of Schrodinger was applied to calculate the Molecular Mechanics Generalised Born Surface Area (MM-GBSA) energiesCitation42 for the aforementioned six complexes. The detailed methodologies were depicted in the Supplementary Information (SI 1 and SI 2).
Biological evaluations
The antiproliferative activities of the most promising antibiotics against human breast, liver, and colorectal cancer cell lines
To determine the cytotoxic concentration of the tested antibiotics. Antibiotics were transferred into flat-bottomed cell culture microtiter plates (96 well plates, TPP-Swiss) at a concentration of 179.2, 133.5, 241.9, 227.3, and 205.5 µM for SP, RO, AZ, CL, and ER, respectively. An eight-stage four-fold dilution of the antibiotic with the culture media was performed. Cells were seeded (2 × 105 cell/ml; 50 µL) in the wells and incubated for 24 h at 37 °C in a humidified and 5% CO2 atmosphere.
The viability of cells was determined using the tetrazolium-salt-based biochemical reaction, MTT assayCitation43, where the cell’s treatment medium was decanted and washed thoroughly using a 200 µL buffering system (PBS) twice for 3 min each. This was followed by the addition of MTT (50 µL), and cells were incubated with the dye for another 4 h at 37 °C. The dye was discarded, and acidified isopropanol (0.4%, 50 µL) was dispensed to each well to dissolve the formazan crystals. The purple formazan colour was measured at a wavelength of 570 nm and a reference wavelength of 630 nm using an EL-x 800 microplate reader. Results were compared to a positive control (cells and solvent). DOX was used as standard (920 µM). Results were expressed as the concentration of antibiotics that inhibited the growth of cells by using the following equation: Viability % = OD test materials x 100/OD of cell control, then, the IC50 was calculated.
Topoisomerase II inhibitory assay
The drugs which showed good activity in silico were tested in vitro to estimate their inhibitory potential against TOP-2 using the TopGEN plasmid-based kit according to the user manual protocol TG1001Citation10. Results were reported as IC50 and compared to DOX.
Results and discussion
In silico findings
Molecular docking studies
The molecular docking studies were used to screen the binding interactions of the investigated antibiotics utilising the MOE 2019 suiteCitation32,Citation42,Citation44. Molecular docking studies were employed to unveil the suggested binding interactionsCitation45,Citation46 of these investigated compounds against the human topoisomerase II-DNA complex (PDB ID: 3QX3), with the native co-crystallised ligand (EVP) as a reference control.
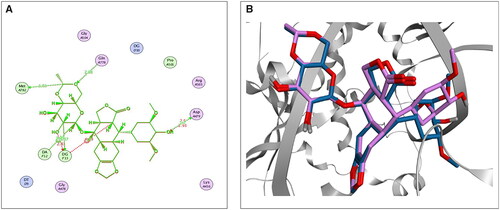
First, the accuracy and suitability of the used force field and the docking protocols were validated via re-docking the co-crystallised EVP inhibitor against the human topoisomerase II-DNA complex target receptor. Consequently, a low RMSD value (1.42 Å) was obtained, enclosing the validity of the Amber10: EHT force fieldCitation20,Citation47–49; also, a similar binding mode was observed, as depicted in . The 2D superimposition of the native and re-docked co-crystallised EVP is shown in .
Figure 2. The 2D (A) and 3D (B) illustrations display the superimposition of the native co-crystallised EVP and redocked one (Mauve and Light Blue, respectively, in 3D picture) at the topoisomerase II-DNA complex (PDB entry: 3QX3) with RMSD value 1.42 Å.
Molecular docking was conducted for screening the investigated compounds utilising the co-crystallised ligand (EVP) as a reference standard. Hence, deep insights into the binding interactions of the screened compounds against the DNA–topoisomerase II complex could be attained. The binding scores of the screened antibiotics towards the human topoisomerase II-DNA complex (PDB ID: 3QX3) compared to the co-crystallised inhibitor (EVP) are represented in the Supplementary Data (Table SI 1).
The binding scores, RMSD values, as well as the details of the binding interactions with their corresponding bond types for the most promising and commercially available five compounds (spiramycin (SP), roxithromycin (RO), azithromycin (AZ), clarithromycin (CL), and erythromycin (ER)), besides, the co-crystallised ligand (EVP), are depicted in .
Table 1. The binding free energy scores, RMSD values, and binding interactions of the most promising compounds (SP, RO, AZ, CL, and ER) along with the co-crystallised ligand (EVP) into DNA–topoisomerase II complex.
It is worth noting that the nucleobases; Ade12, Gua13, Cyt8, Cyt11, and Thy9, and amino acids; ASP479, ARG503, GLN778, and MET782 could be regarded as the key binding sites of DNA–topoisomerase II complexCitation50.
Analysing the binding interactions of the re-docked native inhibitor (EVP) revealed that EVP displayed a binding score of −10.51 kcal/mol with an RMSD value of 0.93 Å. Furthermore, it is worth noting that the dioxane nucleus of EVP could form H-bonds with the key amino acids GLN778 and MET782 at distances of 2.94 and 3.72 Å, respectively. Besides, the pyran nucleus of EVP could interact with key nucleobases forming an H-bond with DG13 and an H-pi bond with DA12 at distances of 3.37 and 3.75 Å, respectively. Finally, the terminal –OH of the cyclohexyl moiety of EVP could form H-bond with the key amino acid ASP479 at a distance of 2.70 Å, as shown in .
Figure 3. 2D (A) and 3D (B) illustrations revealing the binding interactions of the re-docked co-crystallised ligand (EVP) at the topoisomerase II-DNA complex active site, where red dashed lines stand for H-bonds, and black ones stand for H-pi bonds.
Compounds AZ, CL, and ER showed a docking score of −11.72, −10.99, and −10.83 kcal/mol, respectively, which is higher than that attained by the co-crystallised inhibitor.
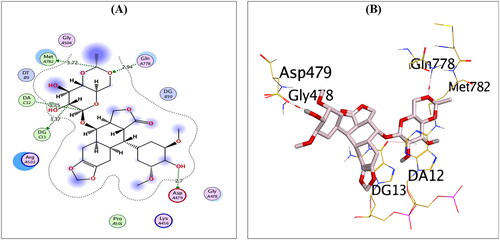
Compound AZ could compose H-bonds with the key amino acids ASP479, GLU477, and ARG503 at distances 2.93, 3.07, and 3.29 Å, respectively. Additionally, it could interact with the key nucleobase DT9 forming H-bond at a distance of 2.95 Å, as shown in .
Figure 4. The 2D and 3D binding interactions of compounds AZ (A,B), CL (C,D), and ER (E,F) reveal their binding interactions at the topoisomerase II-DNA complex active site.
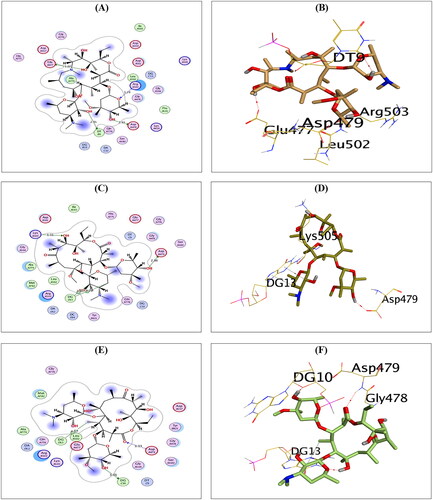
Compound CL, on the other hand, could compose H-bonds with the amino acids ASP479 and LYS505 at distances of 2.86 and 3.31 Å, respectively. In addition, it could interact with the key nucleobase DG13 forming two H-pi bonds at distances of 3.87 and 4.25 Å, as shown in . Finally, compound ER could compose H-bond with the key amino acid ASP479 at a distance of 3.33 Å. Moreover, it could interact with the nucleobases DG10 and DG13 forming one H-bond (3.01 Å) and one H-pi bond (4.57 Å), as shown in .
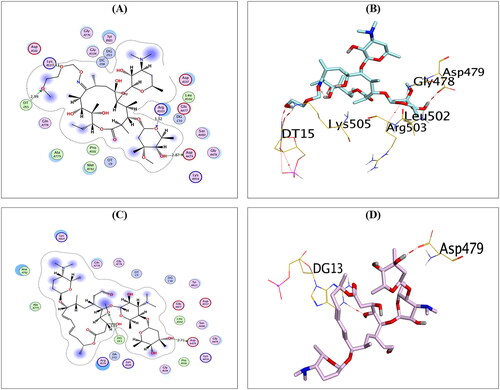
Furthermore, compounds RO and SP exhibited the best binding scores among selected compounds. Hence, compounds RO and SP revealed binding scores of −11.97 and −12.07 kcal/mol, which is superior to that of the co-crystallised inhibitor. Compound RO could compose H-bonds with the two key amino acids ASP479 and ARG503 and the amino acid LYS505 at distances 2.87, 3.32, and 3.11 Å, respectively. In addition, compound RO could interact with the nucleobase DT15 forming H-bond at a distance of 2.99 Å, as shown in . On the other hand, compound SP could compose H-bonds with the key amino acid ASP479 at a distance of 2.71 Å and the key nucleobase DG13 at a distance of 3.03 Å, as shown in .
Figure 5. The 2D and 3D binding interactions of compounds RO (A,B), and SP (C,D) reveal their binding interactions at the topoisomerase II-DNA complex active site (PDB: 3QX3).
Molecular dynamics simulations (MDs)
MD simulations were performed for 200 ns to monitor the exact behaviour of the superior and commercially available antibiotic members (SP, RO, AZ, CL, and ER) within the binding pocket of the topoisomerase II-DNA complex. This was done to clarify and deeply understand the protein–ligand interactions of the studied compoundsCitation37,Citation51,Citation52. Moreover, the EVP co-crystallised inhibitor was used as a reference.
RMSD analysis
In order to investigate the degree of deviation for the studied complexes regarding the initial position of their heavy atoms in a quantitative manner, the RMSD of the protein Cα atoms was recorded during the 200 ns of the simulation. It is an important parameter to judge the stability within the protein conformational changes all over the simulation time.
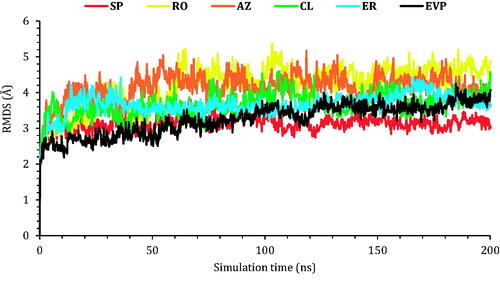
The protein RMSD values of the six studied complexes are described in . The protein RMSD for SP showed the most stable behaviour during the simulation time with an RMSD value of less than 3.00 Å. On the other hand, EVP, CL, and ER RMSD values were less than 4.00 Å indicating stable unchanged conformations for both proteins of CL and ER compared to that of the co-crystallised (EVP) inhibitor. Finally, the RMSD for RO and AZ proteins deviated at 60 and 40 ns, respectively, to 5.00 Å indicating slight conformational changes of the target TOP-2 protein.
Figure 6. The RMSD of the six complexes (SP, RO, AZ, CL, and ER) within the topoisomerase II-DNA complex compared to the co-crystallised (EVP) inhibitor as a function of simulation time (200 ns).
Next, the RMSD of the frontier ligands concerning their initial position inside the active site of the TOP-2 was reported in . Among other compounds, AZ was the most stable inside the active site; it held its position with RMDS of 1.00 Å from the beginning of the simulation till around 150 ns, where it moved by 2.00 with respect to its recent position and held it towards the end of the simulation. The co-crystallised ligand (EVP) and compound RO showed similar behaviours. The RMSD of both compounds was around 3.00 Å concerning their original site. Compound SP moved by 5.00 Å concerning its initial position, while compound ER moved by 6.50 Å, where both stayed steady towards the end of the simulation. Finally, compound CL showed the highest RMSD, and could not hold a fixed position; the compounds moved by almost 7.00 Å concerning its initial position. Snapshots of the ligands–proteins during a different simulation time are presented in Figures SI 1–SI 6 (SI).
Figure 7. The RMSD of the ligands (SP, RO, AZ, CL, and ER) within the topoisomerase II-DNA complex compared to the co-crystallised (EVP) inhibitor as a function of simulation time (200 ns).
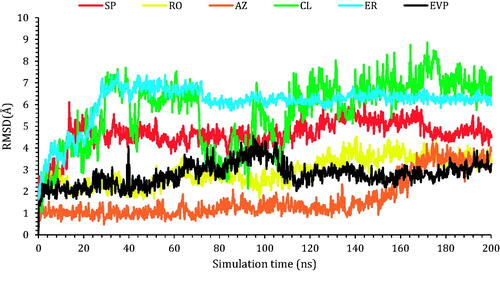
Figure SI 7 (SI) reports the RMSF (root mean square fluctuation). Where, most proteins fluctuated within the range of 3–4 Å, with C- and N- terminals fluctuating at around 8 Å. Such change is acceptable within such a large system.
Histograms analysis
The protein–ligand interaction fractions for the six studied complexes (SP-3QX3, RO-3QX3, AZ-3QX3, CL-3QX3, ER-3QX3, and EVP-3QX3) are described in as histogram presentations.
Figure 8. Histograms indicating the fractions of binding between the protein’s amino acids and its ligand for (A) SP-3QX3, (B) RO-3QX3, (C) AZ-3QX3, (D) CL-3QX3, (E) ER-3QX3, and (F) EVP-3QX3 complexes.
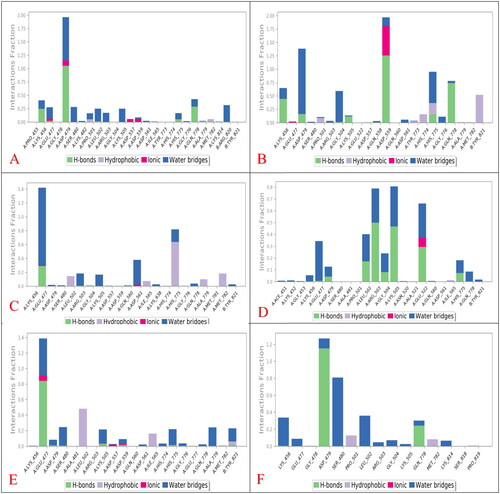
The histogram of the SP-3QX3 complex () showed that ASP479 contributed 195% of the interactions as the main amino acid. The interaction types were H-bonds (105%), water bridges (80%), and ionic interactions (10%).
However, the RO-3QX3 histogram () showed that ASP559 was the principal amino acid in the interactions with RO with 195% as follows (H-bonds (125%), water bridges (60%), and ionic interactions (10%)). Also, ASP479 contributed 140% in the form of water bridges (120%) and H-bonds (20%).
On the other hand, the AZ-3QX3 histogram () represented that GLU477 residue interaction reached 140% as water bridges (105%), H-bonds (30%), and ionic bonds (5%). Besides, HIS775 interactions were noticed to be 80% (hydrophobic (65%) and water bridges (15%)).
Moreover, the histogram of the CL-3QX3 complex () represented that LYS505, ARG503, and GLU522 were the most bound amino acids to CL with 80%, 78%, and 66% interaction fractions, respectively. The type of interactions for LYS505 and ARG503 were H-bonds (48 and 50%) and water bridges (32 and 28%), respectively. Besides, GLU522 interactions were through H-bonds (30%), water bridges (28%), and ionic bonds (8%) as well.
Furthermore, the ER-3QX3 complex histogram () clarified that GLU477 was the superior contributing amino acid to ER interactions with 140% through H-bonds (80%), water bridges (50%), and ionic bonds (10%).
Finally, regarding the histogram of the EVP-3QX3 complex (), ASP479 was noted to be the main amino acid included in the interactions with the co-crystallised EVP inhibitor (125%). It formed mainly H-bonds (115%) and a few water bridges (10%) to EVP. Also, the contributions of SER480 reached 80% through the formation of water bridges only to EVP.
Therefore, only SP (195%) and RO (140%) members showed a closely similar binding interaction to ASP479 amino acid of the binding pocket of the topoisomerase II-DNA complex compared to that of the co-crystallised EVP inhibitor (125%).
Heat maps analysis
The heat map analysis was considered to monitor the total number of interactions for each amino acid residue regarding the simulation time (200 ns). The heat maps for the six studied complexes (SP-3QX3, RO-3QX3, AZ-3QX3, CL-3QX3, ER-3QX3, and EVP-3QX3) are represented in and SI 8–SI 11, respectively.
Figure 9. (A) Heat map describing the protein–ligand interactions regarding the simulation time of 200 ns for SP-3QX3. (B) Heat map describing the protein–ligand interactions regarding the simulation time of 200 ns for RO-3QX3.


The heat map of the SP-3QX3 complex () showed that ASP479 could form more than one type of interaction with SP all over the simulation time (200 ns). It showed the most apparent interactions from (0–10 and 20–130 ns) of the simulation time.
On the other hand, the RO-3QX3 heat map () clarified that ASP559 interactions with RO were of high intensity all over the time of the simulation (200 ns). This was followed by Asp479 interactions, divided by the simulation time (200 ns), showing the most intense interactions from 10 to 30 ns.
Besides, the AZ-3QX3 heat map (Figure SI 8) showed that GLU447 amino acid interactions with AZ were more intense at (40–110 and 120–200 ns) of the simulation time. However, the HIS775 interactions were clearer from 50 to 190 ns of the simulation time.
However, the heat map of the CL-3QX3 complex (Figure SI 9) notified that LYS505 interactions were almost from 0–30 and 75–200 ns. ARG503 interactions on the other hand were distributed equally all over the simulation time (200 ns). Also, GLU522 interactions with CL were more explicit from 140 ns until the end of the simulation.
Moreover, the heat map of the ER-3QX3 complex (Figure SI 10) represented the equal distribution of GLU477 interactions to ER from the beginning to the end of the simulation time (200 ns).
Finally, the EVP-3QX3 heat map (Figure SI 11) clarified that the interactions of ASP479 to the co-crystallised EVP inhibitor were of high intensity from the start to the end of the simulation time (200 ns). Besides, SER480 contributions were mostly from 35 to 190 ns of the simulation time.
MM-GBSA calculations
The average MM-GBSA binding energy was calculated using the thermal_mmgbsa.py python script of SchrodingerCitation41,Citation53 to calculate Van der Waals, Hydrogen-bonding, Generalised Born electrostatic solvation, Coulomb, Lipophilic, and Covalent binding energies. The calculated energies for compounds SP, RO, AZ, CL, ER, and EVP at the topoisomerase II-DNA complex are represented in .
Table 2. Prime MM-GBSA energies for compounds SP, RO, AZ, CL, ER, and EVP at the binding pocket of the topoisomerase II-DNA complex.
Based on the results in , we can observe that the ΔG Binding (−73.77 kcal/mol) and VdW (−74.17 kcal/mol) energies of ER were superior to other members including the co-crystallised EVP inhibitor. Also, the Coulomb (−184.81 kcal/mol) and Solv_GB (199.54 kcal/mol) energies for AZ were higher than those of the studied candidates. The Lipo energy of AZ (−15.16 kcal/mol) was very close to that of EVP (−17.81 kcal/mol). On the other hand, SP showed the highest Covalent energy (3.26 kcal/mol) within the binding pocket of the topoisomerase II-DNA complex. However, RO formed the best H-bond energy (−2.08 kcal/mol) towards the binding pocket of the topoisomerase II-DNA complex compared to the other examined drugs.
Biological evaluations
The antiproliferative activities of the most promising antibiotics against human breast, liver, and colorectal cancer cell lines
The cytotoxicity (IC50) assay was tested against three cancer cell lines namely breast cancer cell line (MCF-7), hepatocellular carcinoma (HepG-2), and human colorectal carcinoma (HCT-116) utilising the MTT assay. In addition, the IC50 was compared to that of DOX as a reference drug. Results showed a comparable IC50 to that attained by DOX ().
Figure 10. Inhibitory potential (IC50) of the most promising and commercially available antibiotics (SP, RO, AZ, CL, and ER) against MCF-7, HepG2, and HCT-116.
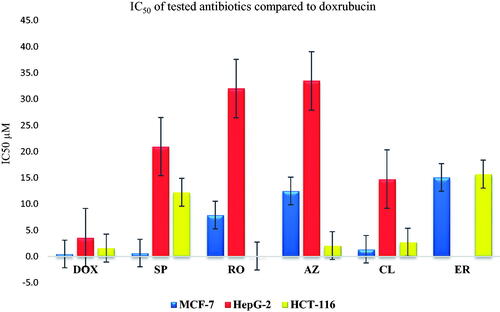
SP showed a promising anticancer potential on the MCF-7 cell line (IC50 0.67 ± 0.43 µM). This was followed by a weaker activity towards both HepG-2 (20.94 ± 1.33 µM) and HCT-116 (12.23 ± 3.27), respectively. On the other hand, RO exhibited an antiproliferative activity of 7.9 ± 0.79, 32 ± 0.18, and 0.097 ± 0.024 µM as IC50 on MCF-7, HepG-2, and HCT-116, respectively, representing moderate potential on MCF-7 and HCT-116, with a strong potential against HCT-116Citation5Citation2,Citation55. However, AZ exhibited a good anticancer potential against the HCT-116 cell line (2.08 ± 0.05 µM) compared to 1.6 ± 0.05 µM for DOX. Furthermore, moderate and weak anticancer activities were recorded for AZ with average IC50 of 7.9 ± 0.79 and 32 ± 0.18 µM for MCF7 and HepG-2, respectively, compared to 0.49 ± 0.1 (MCF7) and 3.6 ± 0.07 (HepG-2) µM for DOX. These data are supported by previous reports, indicating AZ’s close potential for Chronic Myeloid LeukaemiaCitation56. On the other hand, CL exhibited a promising antiproliferative pattern on different cell lines where 1.39 ± 0.15, 14.77 ± 0.15, and 2.74 ± 0.42 µM were recorded as the IC50 in MCF-7, HepG-2, and HCT-116 cell lines, respectively, representing good activity on MCF-7 and HCT-116, besides a moderate antiproliferative potential towards the HepG-2 type. These results are similar to previous reports, which highlight the potential of CL as an anticancer agentCitation54,Citation57–60. Moreover, ER showed weak to moderate inhibitory potential with IC50 of 15.07 ± 2.7 and 15.69 ± 1.32 µM against MCF-7 and HCT-116 cell lines, respectivelyCitation61.
Topoisomerase II inhibitory activity
The potential of the tested drugs to inhibit the ability of TOP-2 to decatenate an intertangled mass of DNA was assessed. In addition, the selected macrolides were further investigated to determine their inhibitory potential on the TOP-2 enzyme using DOX (an anthracycline antibiotic with potent inhibition towards TOP-2) as a positive control. The acquired results agreed with the cytotoxicity assay, and the obtained data indicated that the tested antibiotics have a promising inhibitory potential against the TOP-2 enzyme relative to DOX.
The recorded IC50 for DOX (positive control) was 6.37 ± 0.38 µM. In addition, potent TOP-2 inhibitory activity was recorded for ER and RO with IC50 of 11.58 ± 0.70 and 13.07 ± 0.77 µM, respectively. On the other hand, AZ showed moderate activity with an IC50 of 36.09 ± 2.11 µM. Finally, CL and SP exhibited moderate IC50 of 25.58 ± 1.08 and 21.23 ± 1.19 µM, respectively ().
Figure 11. (A) The measured IC50 of tested antibiotics (SP, RO, AZ, CL, and ER) on TOP-2 compared to DOX. (B) Gel electrophoresis image representing the variable decatenation potential of DNA by TOP-2 as inhibited by DOX and the tested antibiotics (SP, RO, AZ, CL, and ER). The N Lane represents no TOP-2 activity while the P lane is the uninhibited enzyme.
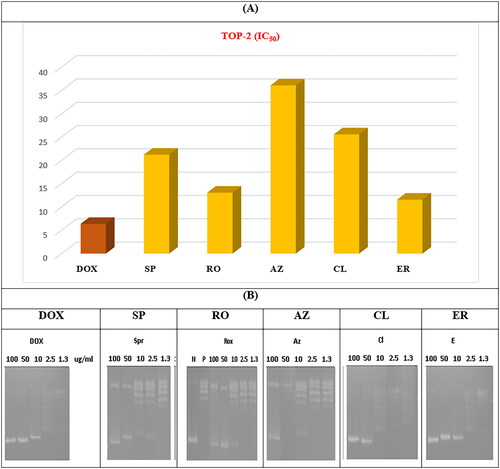
The order of IC50 of different antibiotics was slightly different in cytotoxicity and enzyme assay, especially for ER. Variations may explain the tested antibiotics’ cellular/nuclear permeability in different cell lines.
Structure-activity relationship (SAR)
Based on the molecular docking binding scores for the tested antibiotics (Table SI 1), a SAR study was conducted opening eyes to promising anticancer pharmacophores encountered by the screened antibiotics. Interestingly, the following outcomes were attained ():
Figure 12. SAR study for the affinity of the examined 138 antibiotics as topoisomerase II inhibitors and DNA intercalators.
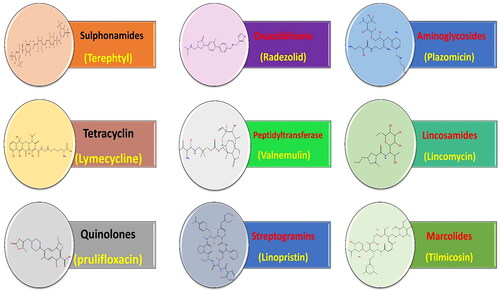
Among the screened antibiotics, it was disclosed that macrolides and streptogramins exhibited the most promising binding scores and, thus, higher anticancer potential.
Moreover, among screened aminoglycosides, it was revealed that plazomicin displayed the best binding affinity to TOP-2, whereas, among screened lincosamides, it was unravelled that lincomycin showed the best binding score.
Besides, regarding screened macrolides, it was shown that all compounds experienced promising binding scores that are better than that attained by the co-crystallised ligand assuring their stability at the target receptor. In particular, it was disclosed that tilmicosin showed the best binding score among the investigated macrolides.
Furthermore, considering screened oxazolidinone, it was disclosed that radezolid showed the best binding affinity to the target receptor, whereas, among screened peptidyltransferase, it was revealed that valnemulin has the best docking score.
In addition, regarding screened streptogramins, it was shown that linopristin, pristinamycin IA, quinupristin, and virginiamycin S1 antibiotics exhibited promising docking scores that are better than that attained by the co-crystallised ligand. In particular, it was disclosed that linopristin showed the best docking score among the screened streptogramins.
Additionally, regarding sulphonamides, tetracyclines, and quinolones, it was unveiled that the compounds with the best binding score in their corresponding class were terephtyl, lymecycline, and prulifloxacin, respectively.
It is worth noting that, among screened antibiotics, sulphonamides and quinolones exhibited the lowest binding scores, and thus, less anticancer potential could be attained.
Conclusions
This research aims to identify a potential TOP-2 inhibitor that is considered a promising molecular target for cancer therapy. Herein, the molecular docking studies of 139 antibiotics belonging to different classes revealed that SP, RO, AZ, CL, and ER were among the most promising and commercially available members. Afterward, the MD simulations for 200 ns clarified that AZ was the most stable inside the active site of the topoisomerase II-DNA complex, followed by the co-crystallised ligand (EVP) and RO, which showed similar behaviours according to the RMSD analysis. Also, the histogram analysis showed that only SP and RO members showed a closely similar binding interaction to ASP479 crucial amino acid of the TOP-2 receptor, compared to that of the co-crystallised EVP inhibitor. Moreover, based on the MM-GBSA calculations, it was observed that the ΔG Binding energy of ER (-73.77 kcal/mol) was superior to the other MD-studied compounds, including the co-crystallised EVP inhibitor. On the other hand, the MTT assay against MCF-7, HepG-2, and HCT-116 cancer cell lines clarified that SP and CL showed promising anticancer potentials on the MCF-7 cell line with IC50 values of 0.67 ± 0.43 and 1.39 ± 0.15 µM, respectively, compared to that of DOX (0.49 ± 0.1 µM). Besides, AZ and CL exhibited good anticancer potentials against the HCT-116 cell line (2.08 ± 0.05 and 2.74 ± 0.42 µM, respectively) compared to 1.6 ± 0.05 µM for DOX. Furthermore, the TOP-2 enzyme inhibition assay was carried out to confirm the proposed rationale compared to DOX as a TOP-2 inhibitor and DNA intercalator. Notably, potent TOP-2 inhibitory potentials were recorded for ER and RO with IC50 values of 11.58 ± 0.51 and 13.07 ± 0.65 µM, respectively, compared to DOX (6.37 ± 0.21 µM). Additionally, the SAR study revealed that macrolides and streptogramins exhibited the most promising binding scores among the tested antibiotics and thus expected better anticancer potentials. Finally, it is recommended to evaluate all the superior antibiotics (either examined or not) using more advanced preclinical and clinical studies to get a more accurate conclusion regarding their antitumor activities.
Author contributions
Conceptualisation: Ahmed A. Al-Karmalawy; Data curation and formal analysis: Faten Farouk, Ayman Abo Elmaaty, Radwan Alnajjar, and Ahmed A. Al‐Karmalawy; Methodology: Faten Farouk, Ayman Abo Elmaaty, Haytham O. Tawfik, Radwan Alnajjar, Mohamed A. Saleh, and Ahmed A. Al‐Karmalawy; Project administration and supervision: Ahmed A. Al-Karmalawy; Resources, software, and validation: Faten Farouk, Ayman Abo Elmaaty, and Radwan Alnajjar; Writing – review & editing: Faten Farouk, Ayman Abo Elmaaty, Ahmed Elkamhawy, Haytham O. Tawfik, Radwan Alnajjar, Mohammed A. S. Abourehab, Wagdy M. Eldehna, and Ahmed A. Al‐Karmalawy. All authors approved the final version of the manuscript.
Supplemental Material
Download PDF (1.7 MB)Acknowledgements
The authors would like to thank the Deanship of scientific research at Umm Al-Qura University for supporting this work by grant code (23UQU4290565DSR022), [email protected]. Also, the authors acknowledge the HPC, the University of Cape Town, for using supercomputing facilities.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Roderburg C, Loosen SH, Kunstein A, Mohr R, Jördens MS, Luedde M, Kostev K, Luedde T. Cancer patients have an increased incidence of dementia: a retrospective cohort study of 185,736 outpatients in Germany. Cancers. 2021;13(9):2027.
- Xia C, Dong X, Li H, Cao M, Sun D, He S, Yang F, Yan X, Zhang S, Li N, et al. Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin Med J. 2022;135(5):584–590.
- Mansour KA, Elbermawi A, Al-Karmalawy AA, Lahloub M-F, El-Neketi M. Cytotoxic effects of extracts obtained from plants of the Oleaceae family: bio-guided isolation and molecular docking of new secoiridoids from Jasminum humile. Pharm Biol. 2022;60(1):1374–1383.
- Häder D-P, Williamson CE, Wängberg S-Å, Rautio M, Rose KC, Gao K, Helbling EW, Sinha RP, Worrest R. Effects of UV radiation on aquatic ecosystems and interactions with other environmental factors. Photochem Photobiol Sci. 2015;14(1):108–126.
- Maddams J, Utley M, Møller H. Projections of cancer prevalence in the United Kingdom, 2010–2040. Br J Cancer. 2012;107(7):1195–1202.
- Attia EZ, Khalifa BA, Shaban GM, Amin MN, Akil L, Khadra I, Karmalawy AAA, Alnajjar R, Zaki MYW, Aly OM, et al. Potential topoisomerases inhibitors from Asergillus terreus using virtual screening. S Afr J Bot. 2022;149:632–645.
- Al-Karmalawy AA, Nafie MS, Shaldam MA, Elmaaty AA, Antar SA, El-Hamaky AA, Saleh MA, Elkamhawy A, Tawfik HO. Ligand-based design on the dog-bone-shaped BIBR1532 pharmacophoric features and synthesis of novel analogues as promising telomerase inhibitors with in vitro and in vivo evaluations. J Med Chem. 2023;66(1):777–792.
- Dong H, Yin H, Zhao C, Cao J, Xu W, Zhang Y. Design, synthesis and biological evaluation of novel osimertinib-based HDAC and EGFR dual inhibitors. Molecules. 2019;24(13):2407.
- Hande KR. Topoisomerase II inhibitors. Updat Cancer Therapeut. 2008;3(1):13–26.
- Hammoud MM, Nageeb AS, Morsi MA, Gomaa EA, Elmaaty AA, Al-Karmalawy AA. Design, synthesis, biological evaluation, and SAR studies of novel cyclopentaquinoline derivatives as DNA intercalators, topoisomerase II inhibitors, and apoptotic inducers. New J Chem. 2022;46(23):11422–11436.
- Villman K, Ståhl E, Liljegren G, Tidefelt U, Karlsson MG. Topoisomerase II- expression in different cell cycle phases in fresh human breast carcinomas. Mod Pathol. 2002;15(5):486–491.
- Baldwin E, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 2005;5(4):363–372.
- Khalifa MM, Al-Karmalawy AA, Elkaeed EB, Nafie MS, Tantawy MA, Eissa IH, Mahdy HA. Topo II inhibition and DNA intercalation by new phthalazine-based derivatives as potent anticancer agents: design, synthesis, anti-proliferative, docking, and in vivo studies. J Enzyme Inhib Med Chem. 2022;37(1):299–314.
- Romer DR. Synthesis of 2, 3‐dichloroquinoxalines via Vilsmeier reagent chlorination. J Heterocyclic Chem. 2009;46(2):317–319.
- Ducray R, Ballard P, Barlaam BC, Hickinson MD, Kettle JG, Ogilvie DJ, Trigwell CB, Novel 3-alkoxy-1H-pyrazolo [3, D 4. ] pyrimidines as EGFR and erbB2 receptor tyrosine kinase inhibitors. Bioorg Med Chem Lett. 2008;18(3):959–962.
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229.
- Gallego J, Ortiz AR, de Pascual-Teresa B, Gago F. Structure–affinity relationships for the binding of actinomycin D to DNA. J Comput Aided Mol Des. 1997;11(2):114–128.
- Bailey SA, Graves DE, Rill R. Binding of actinomycin D to the T (G) nT motif of double-stranded DNA: determination of the guanine requirement in nonclassical, non-GpC binding sites. Biochemistry. 1994;33(38):11493–11500.
- Hamed MIA, Darwish KM, Soltane R, Chrouda A, Mostafa A, Abo Shama NM, Elhady SS, Abulkhair HS, Khodir AE, Elmaaty AA, et al. β-Blockers bearing hydroxyethylamine and hydroxyethylene as potential SARS-CoV-2 Mpro inhibitors: rational based design, in silico, in vitro, and SAR studies for lead optimization. RSC Adv. 2021;11(56):35536–35558.
- Khattab M, Al-Karmalawy AA. Computational repurposing of benzimidazole anthelmintic drugs as potential colchicine binding site inhibitors. Future Med Chem. 2021;13(19):1623–1638.
- Reed MD. The rescue and repurposing of pharmaceuticals: augmenting the drug development paradigm. Pediatric Pharmacy Advocacy Group. 2016;21:4–6.
- Elmaaty AA, Eldehna WM, Khattab M, Kutkat O, Alnajjar R, El-Taweel AN, Al-Rashood ST, Abourehab MAS, Binjubair FA, Saleh MA, et al. Anticoagulants as potential SARS-CoV-2 Mpro inhibitors for COVID-19 patients: in vitro, molecular docking, molecular dynamics, DFT, and SAR studies. IJMS. 2022;23(20):12235.
- Shazia P, Alnoman RB, Bayazeed AA, Alqahtani AM. Computational insights into the drug repurposing and synergism of FDA-approved influenza drugs binding with SARS-CoV-2 protease against COVID-19. Am J Microbiol Res. 2020;8(3):93–102.
- Al-Karmalawy AA, Soltane R, Abo Elmaaty A, Tantawy MA, Antar SA, Yahya G, Chrouda A, Pashameah RA, Mustafa M, Abu Mraheil M, et al. Coronavirus disease (COVID-19) control between drug repurposing and vaccination: A comprehensive overview. Vaccines. 2021;9(11):1317.
- Abbas HA, Elsherbini AM, Shaldam MA. Repurposing metformin as a quorum sensing inhibitor in Pseudomonas aeruginosa. Afr H Sci. 2017;17(3):808–819.
- Dinić J, Efferth T, García-Sosa AT, Grahovac J, Padrón JM, Pajeva I, Rizzolio F, Saponara S, Spengler G, Tsakovska I. Repurposing old drugs to fight multidrug resistant cancers. Drug Resist Updat. 2020;52:100713.
- Gao Y, Shang Q, Li W, Guo W, Stojadinovic A, Mannion C, Man Y-g, Chen T. Antibiotics for cancer treatment: a double-edged sword. J Cancer. 2020;11(17):5135–5149.
- Petrelli F, Ghidini M, Ghidini A, Perego G, Cabiddu M, Khakoo S, Oggionni E, Abeni C, Hahne JC, Tomasello G, et al. Use of antibiotics and risk of cancer: a systematic review and meta-analysis of observational studies. Cancers. 2019;11(8):1174.
- Kingwell K. Antibiotics switch to anticancer target. Nat Rev Drug Discov. 2021;20(9):666.
- Paul M, Gafter-Gvili A, Fraser A, Leibovici L. The anti-cancer effects of quinolone antibiotics? Eur J Clin Microbiol Infect Dis. 2007;26(11):825–831.
- Qabaja A, Alshalalfa M, Alanazi E, Alhajj R. Prediction of novel drug indications using network driven biological data prioritization and integration. J Cheminform. 2014;6(1):1–14.
- Inc, C. Molecular operating environment (MOE). Chemical Computing Group Inc.; 2016. p. 1010. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Inc%2C+C.+Molecular+operating+environment+%28MOE%29.+Chemical+Computing+Group+Inc.%3B&btnG=.
- Elshal M, Eid N, El-Sayed I, El-Sayed W, Al-Karmalawy AA. Concanavalin-A shows synergistic cytotoxicity with tamoxifen via inducing apoptosis in estrogen receptor-positive breast cancer: in vitro and molecular docking studies. Pharm Sci. 2021;28(1):76–85.
- Ma C, Taghour MS, Belal A, Mehany AB, Mostafa N, Nabeeh A, Eissa IH, Al-Karmalawy AA. Design and synthesis of new quinoxaline derivatives as potential histone deacetylase inhibitors targeting hepatocellular carcinoma: in silico, in vitro, and SAR studies. Front Chem. 2021;9. https://doi.org/10.3389/fchem.2021.725135.
- Taher RF, Al-Karmalawy AA, Abd El Maksoud AI, Khalil H, Hassan A, El-Khrisy E-DA, El-Kashak W. Two new flavonoids and anticancer activity of Hymenosporum flavum: in vitro and molecular docking studies. J Herbmed Pharmacol. 2021;10(4):443–458.
- Al-Karmalawy AA, Farid MM, Mostafa A, Ragheb AYH, Mahmoud S, Shehata M, Shama NMA, GabAllah M, Mostafa-Hedeab G, Marzouk MM. Naturally available flavonoid aglycones as potential antiviral drug candidates against SARS-CoV-2. Molecules. 2021;26(21):6559.
- Elebeedy D, Badawy I, Elmaaty AA, Saleh MM, Kandeil A, Ghanem A, Kutkat O, Alnajjar R, Abd El Maksoud AI, Al-Karmalawy AA. In vitro and computational insights revealing the potential inhibitory effect of Tanshinone IIA against influenza A virus. Comput Biol Med. 2022;141:105149.
- Elmaaty AA, Darwish KM, Chrouda A, Boseila AA, Tantawy MA, Elhady SS, Shaik AB, Mustafa M, Al-Karmalawy AA. In silico and in vitro studies for benzimidazole anthelmintics repurposing as VEGFR-2 antagonists: novel mebendazole-loaded mixed micelles with enhanced dissolution and anticancer activity. ACS Omega. 2022;7(1):875–899.
- Zaki AA, Ashour A, Elhady SS, Darwish KM, Al-Karmalawy AA. Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies. J Tradit Complement Med. 2022;12(1):16–34.
- Elmaaty AA, Darwish KM, Khattab M, Elhady SS, Salah M, Hamed MIA, Al‐Karmalawy AA, Saleh MM. In a search for potential drug candidates for combating COVID-19: computational study revealed salvianolic acid B as a potential therapeutic targeting 3CLpro and spike proteins. J Biomol Struct Dyn. 2022;40(19):1–28.
- Tools M-DI. Schrödinger: New York (NY): Schrödinger; 2017.
- Elagawany M, Elmaaty AA, Mostafa A, Abo Shama NM, Santali EY, Elgendy B, Al-Karmalawy AA. Ligand-based design, synthesis, computational insights, and in vitro studies of novel N-(5-Nitrothiazol-2-yl)-carboxamido derivatives as potent inhibitors of SARS-CoV-2 main protease. J Enzyme Inhib Med Chem. 2022;37(1):2112–2132.
- Betancur-Galvis LA, Morales G, Forero J, Roldan J. Cytotoxic and antiviral activities of Colombian medicinal plant extracts of the Euphorbia genus. Mem Inst Oswaldo Cruz. 2002;97(4):541–546.
- Kutkat O, Moatasim Y, Al‐Karmalawy AA, Abulkhair HS, Gomaa MR, El-Taweel AN, Abo Shama NM, GabAllah M, Mahmoud DB, Kayali G, et al. Robust antiviral activity of commonly prescribed antidepressants against emerging coronaviruses: in vitro and in silico drug repurposing studies. Sci Rep. 2022;12(1):12920.
- Mahmoud A, Mostafa A, Al-Karmalawy AA, Zidan A, Abulkhair HS, Mahmoud SH, Shehata M, Elhefnawi MM, Ali MA. Telaprevir is a potential drug for repurposing against SARS-CoV-2: computational and in vitro studies. Heliyon. 2021;7(9):e07962.
- El-Azab MF, Al-Karmalawy AA, Antar SA, Hanna PA, Tawfik KM, Hazem RM. A novel role of Nano selenium and sildenafil on streptozotocin-induced diabetic nephropathy in rats by modulation of inflammatory, oxidative, and apoptotic pathways. Life Sci. 2022;303:120691.
- El-Demerdash A, Al-Karmalawy AA, Abdel-Aziz TM, Elhady SS, Darwish KM, Hassan AHE. Investigating the structure–activity relationship of marine natural polyketides as promising SARS-CoV-2 main protease inhibitors. RSC Adv. 2021;11(50):31339–31363.
- Elebeedy D, Elkhatib WF, Kandeil A, Ghanem A, Kutkat O, Alnajjar R, Saleh MA, Abd El Maksoud AI, Badawy I, Al-Karmalawy AA. Anti-SARS-CoV-2 activities of tanshinone IIA, carnosic acid, rosmarinic acid, salvianolic acid, baicalein, and glycyrrhetinic acid between computational and in vitro insights. RSC Adv. 2021;11(47):29267–29286.
- Elmaaty AA, Alnajjar R, Hamed MI, Khattab M, Khalifa MM, Al-Karmalawy AA. Revisiting activity of some glucocorticoids as a potential inhibitor of SARS-CoV-2 main protease: theoretical study. RSC Adv. 2021;11(17):10027–10042.
- El-Naggar AM, Abou-El-Regal MM, El-Metwally SA, Sherbiny FF, Eissa IH. Synthesis, characterization and molecular docking studies of thiouracil derivatives as potent thymidylate synthase inhibitors and potential anticancer agents. Mol Divers. 2017;21(4):967–983.
- El-Masry RM, Al-Karmalawy AA, Alnajjar R, Mahmoud SH, Mostafa A, Kadry HH, Abou-Seri SM, Taher AT. Newly synthesized series of oxoindole–oxadiazole conjugates as potential anti-SARS-CoV-2 agents: in silico and in vitro studies. New J Chem. 2022;46(11):5078–5090.
- El Gizawy HA, Boshra SA, Mostafa A, Mahmoud SH, Ismail MI, Alsfouk AA, Taher AT, Al-Karmalawy AA. Pimenta dioica (L.) Merr. bioactive constituents exert anti-SARS-CoV-2 and anti-inflammatory activities: molecular docking and dynamics, in vitro, and in vivo studies. Molecules. 2021;26(19):5844.
- Alnajjar R, Mostafa A, Kandeil A, Al-Karmalawy AA. Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease. Heliyon. 2020;6(12):e05641.
- Oyama T, Maruyama I, Aikou T. Roxithromycin inhibits constitutive activation of nuclear factor KB by diminishing oxidative stress in a rat model of hepatocellular carcinoma. Clin Cancer Res. 2005;11(15):5645–5650.
- Chlebda E, Merwid-L1d A, Szumny D, Trocha M. g, Fereniec-Go³Êbiewska L, Gebarowska E, Kowalski P, Szel1g A. Antitumor effect of macrolides–erythromycin and roxithromycin in B16 melanoma-transplanted mice. Pharmacol Rep. 2007;59:269–274.
- Ozkan T, Hekmatshoar Y, Karabay AZ, Koc A, Altinok Gunes B, Karadag Gurel A, Sunguroglu A. Assessment of azithromycin as an anticancer agent for treatment of imatinib sensitive and resistant CML cells. Leuk Res. 2021;102:106523.
- Wang L, Kitaichi K, Hui CS, Takagi K, Takagi K, Sakai M, Yokogawa K, Miyamoto K-i, Hasegawa T. Reversal of anticancer drug resistance by macrolide antibiotics in vitro and in vivo. Clin Exp Pharmacol Physiol. 2000;27(8):587–593.
- Ishimatsu Y, Mukae H, Sakamoto N. Activity of clarithromycin in mucosa-associated lymphoid tissue-type lymphomas: response. Chest. 2011;139(3):725–726.
- Ferreri AJ. Activity of clarithromycin in mucosa-associated lymphoid tissue-type lymphomas: antiproliferative drug or simple antibiotic? Chest. 2011;139(3):724–725.
- Ishii A, Kumasaka M, Nagashima Y, Nakajima Y, Kuramochi K, Sugawara F, Narukawa M, Kamakura T. A eukaryotic molecular target candidate of roxithromycin: fungal differentiation as a sensitive drug target analysis system. Biosci Biotechnol Biochem. 2013;77(7):1539–1547.
- Teixeira DS, Louzeiro GC, Figueiredo M, Cherubini K, Salum FG. Erythromycin: an alternative for the management of oral mucositis? Medicina Oral, Patologia Oral y Cirugia Bucal. 2022;27(5):25439–25439.