
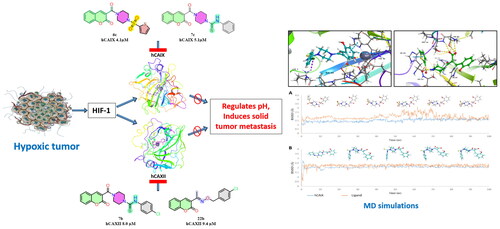
Abstract
The Carbonic anhydrase isoforms IX and XII play a significant role in regulating the intracellular and extracellular pH in hypoxic tumours abetting the metastasis of solid tumours. Selective and potent inhibitors targeting carbonic anhydrase IX and XII reduce the activity of these isoforms in hypoxic tumours, representing an antitumor and antimetastatic mechanism. Coumarin-based derivatives are selective inhibitors of CA isoforms IX and XII. In this study, we report the design and synthesis of new 3-substituted coumarin derivatives with different functional moieties and their inhibitory activity against various carbonic anhydrase isoforms. We found that the tertiary sulphonamide derivative 6c showed selective inhibition against CA IX with IC50 of 4.1 µM. Similarly, the carbothioamides 7c, 7b and oxime ether derivative 20a exhibited good inhibition against CA IX and CA XII. Additionally, the binding mode was predicted and validated using molecular docking and dynamic simulations.
Graphical Abstract
Introduction
Carbonic anhydrases (CAs; EC 4.2.1.1) are a superfamily of metalloenzymes with Zn2+ ion as the active site core, catalyses the reversible hydration of carbon dioxide into bicarbonate ions and protons, controlling the acid-base balance in the blood and tissues. Carbonic anhydrases are ubiquitous in all organisms. Eight families, namely α, β, γ, δ, ζ, η, θ and ι, were reported, of which the sixteen different isoforms of α-CAs were isolated from mammals and classified as cytosolic (CA I, CA II, CA III, CA VII, CA XIII), membrane-bound (CA IV, CA IX, CA XII, CA XIV and CA XV), mitochondrial (CA VA, CA VB) and CA VI secreted in body fluids like colostrum and salivaCitation1–3.
Hypoxia in tumour cells remains a significant barrier to the effective treatment of most malignanciesCitation4. Hypoxic cancer cells are known to express HIF-1, which in turn stimulates downstream signalling leading to the overexpression of glucose/proton transporters, bicarbonate and monocarboxylate transporters, as well as tumour-associated isoforms hCA IX and hCA XII, on the surfaces of the hypoxic tumour cells. These enzymes regulate the pH of the hypoxic tumour cells by converting CO2 into bicarbonate and protons, where the bicarbonate buffers the internal pH and the protons form an acidic extracellular pH, thereby inducing the metastasis of the tumourCitation5–7. In addition, CA IX and XII are scarcely expressed in normal cells; hence, selective inhibition of these isoforms, when combined with chemotherapy, can result in an effective, innovative treatment for solid tumoursCitation4.
Carbonic anhydrase inhibitors are classified into three classes based on their binding in the active site: (a) Zn2+ binders (sulphonamides) (b) CAIs anchoring to the zinc-coordinated water/hydroxide ion (phenols, polyamines) and (c) compounds blocking the active site’s entrance (coumarins and lacosamide)Citation8. The coumarins, in particular, inhibit the CAs in a prodrug approach where the lactone ring of the coumarin is hydrolysed by the enzyme, with the obtained 2-hydroxycinnamic acids blocking the entry of the substrates into the catalytic siteCitation9,Citation10. Many studies involving the carboxylic acid groups as the zinc binding groups reveal that the carboxylic acid groups are more selective towards the hCA IX and hCA XIICitation11,Citation12.
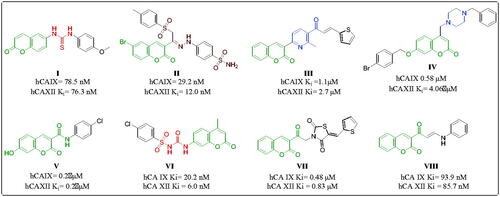
Coumarin is a neo-flavonoid found in secondary metabolites of plants, and its natural and synthetic derivatives are known to possess various therapeutic effectsCitation13–16. The various substituted positions of the coumarin ring led to compounds with potent hCA IX and XII inhibitorsCitation10. Thacker et al. reported coumarin-linked thiourea derivative (I) () selectively inhibiting the hCA IX and hCA XII in nanomolar concentrationsCitation17. Abdelrahman et al. reported the compound (II) () with selective nanomolar inhibition of isoforms hCA IX and hCA XIICitation18. Tawfik et al. reported compound (III) () with micromolar inhibition of cancer-associated isoformsCitation19. Chandra et al. reported piperazine-linked coumarin derivative (IV) () with submicromolar inhibition of hCA IXCitation20. Thacker and team reported coumarin-3-carboxamide derivative (V) () with submicromolar inhibition of hCA IX and hCA XIICitation21. Bozdag et al. reported coumarin arylsulfonylureido derivative (VI) () with selective nanomolar inhibition of the tumour-associated isoforms of carbonic anhydraseCitation22. Eldehna et al. reported the 3-substituted thiazolidinedione derivatives VII with submicromolar carbonic anhydrase inhibition selectively against hCA IX and hCA XII with a Ki value of 0.48 and 0.83, respectivelyCitation23. Ibrahim et al. reported the 3-substituted coumarin enaminone derivative VIII selectively inhibiting the hCA IX and hCA XII with a Ki value of 93.9 nM and 85.7 nM respectivelyCitation24.
Figure 1. Carbonic anhydrase activity of previously reported coumarin derivatives.
Based on the above literature survey, which revealed the potential of coumarin and its derivatives selectively inhibiting hCA IX and hCA XII, we designed several new coumarin derivatives incorporating various functional moieties such as sulphonamides, carbothioamides and oxime ethers with piperazine as a linker. This work focuses on designing and synthesising new coumarin derivatives and evaluating their inhibitory activity against different isoforms of carbonic anhydrase. Further, the binding pattern of these inhibitors at the active site is predicted using molecular docking and simulations studies.
Materials and methods
Chemistry
All the reagents and solvents were procured from commercial suppliers and were used without further purification. Thin layer chromatography (TLC) was performed on MERCK pre-coated silica gel 60-F254 (0.5 mm) aluminium plates. Visualisation of the TLC plates was done under UV light 254 nm and 280 nm. 1H and 13C NMR spectra were recorded on Bruker Avance III AV500 MHz using DMSO-d6 as a solvent with tetramethyl silane (TMS) as the internal standard. Chemical shifts (δ) for 1H and 13C are reported in parts per million(ppm) downfield from TMS. Spin multiplicities are described as s(singlet), brs (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Coupling constant (J) values are reported in hertz (Hz). HR-MS were determined with Agilent QTOF mass spectrometer 6540. All the purifications were performed using silica gel (230–400) on Biotage isolera one flash chromatography. The reactions are carried under nitrogen positive pressure using freshly distilled anhydrous solvents. All the structures and IUPAC names of the compounds were generated using ChemDraw professional 20.1.
Synthesis of ethyl 2-oxo-2H-chromene-3-carboxylateCitation25 (2)
An equimolar mixture of 2-hydroxy benzaldehyde (1 equiv.) and diethyl malonate (1.1 equiv.) in ethanol was refluxed in the presence of piperidine (0.1 equiv.) and a catalytic amount of acetic acid for 4–6 h at 70–80 °C. The reaction progress was monitored by TLC. After the completion of the reaction, the solvent was removed under reduced pressure and the reaction mixture was poured slowly into the beaker containing crushed ice and the contents were stirred well. The resultant white solid was filtered and recrystallized from ethanol to give a white crystalline solid with a yield of 82%. m.p. 130–133 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.77 (s, 1H), 7.93 (dd, J = 7.7, 1.5 Hz, 1H), 7.77–7.73 (m, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.44–7.40 (m, 1H), 4.31 (q, J = 7.1 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H).
Synthesis of 2-oxo-2H-chromene-3-carboxylic acidCitation25 (3)
To the stirring solution of ethyl 2-oxo-2H-chromene-3-carboxylate (1 equiv.) in methanol, a solution of sodium hydroxide (2 equiv.) in water was added dropwise. The reaction mixture was stirred at room temperature for 2 h. After the completion of the reaction confirmed by TLC, the solvent was removed under reduced pressure and acidified with hydrochloric acid to give 3 as a white solid, which was then filtered and recrystallized from methanol to give white crystalline solid with a yield of 74%. m.p. 146–148 °C. 1H NMR (500 MHz, DMSO-d6) δ 13.25 (s, 1H), 8.76 (s, 1H), 7.92 (dd, J = 7.8, 1.5 Hz, 1H), 7.77–7.71 (m, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.43–7.39 (m, 1H).
Synthesis of tert-butyl 4–(2-oxo-2H-chromene-3-carbonyl) piperazine-1-carboxylateCitation26 (4)
To an oven-dried round-bottomed flask equipped with a magnetic stir bar was added 2-oxo-2H-chromene-3-carboxylic acid (1 equiv.) and N-Boc piperazine (1 equiv.), HOBt (0.5 equiv.) and EDC.HCl (2 equiv.) in DMF maintaining inert conditions, and DIPEA (3 equiv.) was added slowly to the reaction mixture and stirred for 8–12 h at room temperature. After the completion of the reaction confirmed by TLC, the reaction mixture was poured into a beaker with crushed ice, and the resultant solid was filtered, dried and used for the subsequent reaction without any purification with a 61% yield. m.p. 140–143 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.78 (dd, J = 7.7, 1.4 Hz, 1H), 7.71–7.67 (m, 1H), 7.47 (d, J = 8.3 Hz, 1H), 7.44–7.39 (m, 1H), 3.62–3.57 (m, 2H), 3.43–3.36 (m, 4H), 3.32–3.30 (m, 2H), 1.42 (s, 9H).
Synthesis of 3-(piperazine-1-carbonyl)-2H-chromene-2-oneCitation26 (5)
To the stirring solution of tert-butyl 4–(2-oxo-2H-chromene-3-carbonyl)piperazine-1-carboxylate (1 equiv.) in DCM, TFA (4 equiv.) was added dropwise at 0–5 °C. The reaction mixture was allowed to stir for 1 h. TLC confirmed the completion of the reaction, and the reaction mixture was neutralised with sodium bicarbonate solution and extracted with dichloromethane. The organic layer was dried over sodium sulphate and concentrated to get the product as a pale buff solid with a 69% yield. m.p. 152–155 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.19 (s, 1H), 7.78 (d, J = 7.4 Hz, 1H), 7.68 (t, J = 7.5 Hz, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.41 (t, J = 7.4 Hz, 1H), 3.53 (s, 2H), 3.29 (s, 2H), 2.73 (s, 2H), 2.65 (s, 2H).
General procedure for synthesis of 3–(4-(sulphonyl)piperazine-1-carbonyl)-2H-chromen-2-one derivatives(6a–e)
To the solution of 3-(piperazine-1-carbonyl)-2H-chromen-2-one (1 equiv.) and various substituted sulphonyl chlorides (1.2 equiv.) in DCM, DIPEA was added and stirred at room temperature for 2–3 h. After the completion of the reaction, the solvent was evaporated under reduced pressure, and the crude residue was subjected to column chromatography with 6:4 ethyl acetate and hexane to yield the title compounds (75–81%).
3–(4-((4-Nitrophenyl)sulphonyl)piperazine-1-carbonyl)-2H-chromen-2-one (6a)
Pale-yellow solid; yield 81%; m.p. 222–224 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.47 (d, J = 8.9 Hz, 2H), 8.16 (s, 1H), 8.04 (d, J = 8.9 Hz, 2H), 7.73 (dd, J = 7.7, 1.5 Hz, 1H), 7.71–7.65 (m, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 3.75–3.69 (m, 2H), 3.54–3.48 (m, 2H), 3.13–3.05 (m, 2H), 3.02–2.96 (m, 2H).13C NMR (125 MHz, DMSO-d6) δ 163.4, 158.1, 153.9, 150.7, 143.3, 141.2, 133.3, 129.6, 129.5, 125.3, 125.3, 124.5, 118.7, 116.7, 46.3, 46.1, 46.0, 41.0. HRMS (ESI): m/z calculated for [M + H]+ C20H18N3O7S 444.4375; found 444.4379.
N-(4-((4–(2-oxo-2H-chromene-3-carbonyl)piperazin-yl)sulphonyl)phenyl)acetamide (6b)
Off-white solid; yield 70%; m.p. 194–196 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.42 (s, 1H), 8.18 (s, 1H), 7.85 (d, J = 8.9 Hz, 2H), 7.73 (dd, J = 7.7, 1.8 Hz, 1H), 7.71–7.65 (m, 3H), 7.44 (d, J = 8.4 Hz, 1H), 7.39 (t, J = 7.6 Hz, 1H), 3.74–3.66 (m, 2H), 3.53–3.44 (m, 2H), 3.01–2.93 (m, 2H), 2.90–2.83 (m, 2H) 2.11 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 169.7, 163.4, 158.1, 153.9, 144.2, 143.2, 133.3, 129.4, 129.3, 128.6, 125.3, 124.6, 119.2, 118.8, 116.8, 46.5, 46.2, 46.1, 41.0, 24.7. HRMS (ESI): m/z calculated for [M + H]+ C22H22N3O6S 456.4925; found 456.4950.
3–(4-(Thiophen-2-ylsulfonyl)piperazine-1-carbonyl)-2H-chromen-2-one (6c)
Pale-brown solid; yield 76%; m.p. 175–177 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.20 (s, 1H), 8.11 (d, J = 4.4 Hz, 1H), 7.75 (d, J = 7.5 Hz, 1H), 7.71–7.66 (m, 2H), 7.45 (d, J = 8.3 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.33 (t, J = 4.4 Hz, 1H), 3.77–3.72 (m, 2H), 3.56–3.51 (m, 2H), 3.06–3.00 (m, 2H), 2.96–2.91 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 163.4, 158.1, 153.9, 143.3, 134.9, 134.8, 133.9, 133.3, 129.5, 129.0, 125.4, 124.7, 118.8, 116.8, 46.5, 46.2, 45.9, 40.9. HRMS (ESI): m/z calculated for [M + H]+ C18H17N2O5S2 405.0573; found 405.0582.
3–(4-((5-Bromothiophen-2-yl)sulphonyl)piperazine-1-carbonyl)-2H-chromen-2-one (6d)
Pale-brown solid; yield 78%; m.p. 191–193 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.76 (d, J = 7.2 Hz, 1H), 7.69 (t, J = 7.1 Hz, 1H), 7.55 (d, J = 3.3 Hz, 1H), 7.52 (d, J = 3.3 Hz, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.41 (t, J = 7.0 Hz, 1H), 3.81–3.72 (m, 2H), 3.58–3.51 (m, 2H), 3.11–3.04 (m, 2H), 3.01–2.94 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 163.4, 158.1, 153.9, 143.3, 136.2, 134.6, 133.3, 132.8, 129.6, 125.4, 124.5, 120.4, 118.8, 116.8, 46.5, 46.1, 45.9, 40.9. HRMS (ESI): m/z calculated for [M + H]+ C18H16BrN2O5S2 482.9679; found 482.9655.
3–(4-(Ethylsulfonyl)piperazine-1-carbonyl)-2H-chromen-2-one (6e)
Pale-buff solid; yield 65%; m.p. 234–236 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.24 (s, 1H), 7.78 (d, J = 7.7. Hz, 1H), 7.70 (t, J = 7.8 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 3.73–3.67 (m, 2H), 3.52–3.47 (m, 2H), 3.29–3.25 (m, 2H), 3.20–3.16 (m, 2H), 3.11 (q, J = 7.3 Hz, 2H), 1.23 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 163.5, 158.2, 154.0, 143.1, 133.3, 129.5, 125.4, 124.8, 118.8, 116.8, 46.8, 45.9, 45.5, 43.3, 41.7, 7.8. HRMS (ESI): m/z calculated for [M + H]+ C16H19N2O5S 351.1009; found 351.1014.
General procedure for synthesis of carbothioamide derivatives of coumarin(7a–c)
In an oven-dried RBF containing a solution of 3-(piperazine-1-carbonyl)-2H-chromen-2-one (1 mmol.) in ACN, substituted phenylisothiocyanates (1.2 mmol.) were added and refluxed at 80–90 °C for 2–3 h. After the completion of the reaction, confirmed by TLC, the solvent was removed under reduced pressure, and the crude mixture was subjected to column chromatography with 6:4 ethyl acetate and hexane to afford the titled compounds with a yield of 65–75%.
N-(2-chlorophenyl)-4–(2-oxo-2H-chromene-3-carbonyl)piperazine-1-carbothioamide (7a)
White solid; yield 65%; m.p. 213–215 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.39 (s, 1H), 8.27 (s, 1H), 7.80 (d, J = 7.4 Hz, 1H), 7.70 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.4 Hz, 2H), 7.43 (t, J = 7.3 Hz, 1H), 7.37–7.26 (m, 3H), 4.09–4.02 (m, 2H), 3.97–3.91 (m, 2H), 3.77–3.70 (m, 2H), 3.60–3.54 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 182.7, 163.8, 158.2, 154.1, 143.2, 1387, 133.3, 132.4, 131.5, 129.8, 129.5, 128.3, 127.7, 125.3, 124.9, 118.9, 116.8, 48.6, 47.8, 46.0, 41.7. HRMS (ESI): m/z calculated for [M + H]+ C21H19ClN3O3S 428.0830; found 428.0836.
N-(4-chlorophenyl)-4–(2-oxo-2H-chromene-3-carbonyl)piperazine-1-carbothioamide (7b)
Off-white solid; yield 73%; m.p. 224–226 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.49 (s, 1H), 8.26 (s, 1H), 7.80 (dd, J = 7.7, 1.3 Hz, 1H), 7.70 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 8.3 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 7.39–7.32 (m, 4H), 4.05–4.00 (m, 2H), 3.94–3.88 (m, 2H), 3.75–3.69 (m, 2H), 3.59–3.54 (m, 2H).13C NMR (125 MHz, DMSO-d6) δ 182.2, 163.7, 158.2, 154.1, 143.2, 140.4, 133.3, 129.5, 128.8, 128.4, 127.3, 125.3, 124.9, 118.9, 116.8, 487, 47.9, 45.9, 41.7. HRMS (ESI): m/z calculated for [M + H]+ C21H19ClN3O3S 428.0830; found 428.0838.
4–(2-Oxo-2H-chromene-3-carbonyl)-N-phenylpiperazine-1-carbothioamide (7c)
Off-white solid; yield 71%; m.p. 231–233 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.45 (s, 1H), 8.26 (s, 1H), 7.80 (dd, J = 7.7, 1.1 Hz, 1H), 7.70 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 8.3 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.33–7.29 (m, J = 4.4 Hz, 4H), 7.15–7.10 (m, 1H), 4.06–3.99 (m, 2H), 3.94–3.88 (m, 2H), 3.76–3.69 (m, 2H), 3.59–3.53 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 182.4, 163.7, 158.2, 154.1, 143.2, 141.4, 133.3, 129.5, 128.5, 125.7, 125.3, 124.9, 124.9, 118.9, 116.8, 48.6, 47.8, 46.0, 41.8. HRMS (ESI): m/z calculated for [M + H]+ C21H20N3O3S 394.1220; found 394.1250.
Synthesis of 7-hydroxy-2-oxo-2H-chromene-3-carboxylic acid (10)
A mixture of 2,4-dihydroxy benzaldehyde (1 equiv.) and diethyl malonate (1.1 equiv.) in ethanol was refluxed at 70–80 °C in the presence of a catalytic amount of piperidine and acetic acid for 4–6 h. After completion of the reaction, the solvent was removed under reduced pressure, and the concentrate was poured slowly into crushed ice to yield ethyl 7-hydroxy-2-oxo-2H-chromene-3-carboxylate as a yellow solid which was filtered and used as such for the subsequent reaction. To the stirring solution of ethyl 7-hydroxy-2-oxo-2H-chromene-3-carboxylate (1 equiv.) in methanol, a solution of sodium hydroxide (3 equiv.) in water was added dropwise over 15 min. The reaction mixture was stirred at room temperature for 2 h. After the completion of the reaction, confirmed by TLC, the solvent was removed under reduced pressure and acidified with hydrochloric acid, which produced an off-white solid. The solid was filtered and dried with a yield of 70%.
Synthesis of 4-hydroxy-2-oxo-2H-chromene-3-carboxylic acidCitation27 (15)
To an oven-dried RBF, equimolar amounts of salicylic acid (1 equiv.) and malonic acid (1 equiv.) were added with trifluoroacetic acid as solvent and the contents were refluxed at 90–100 °C for 8 h. After the completion of the reaction, confirmed by TLC, the solvent was removed under reduced pressure, and the crushed ice was added to the action mixture to give a white solid, filtered and dried with a yield of 88%.
Synthesis of hydroxy-3–(4-(sulphonyl) piperazine-1-carbonyl)-2H-chromen-2-one derivatives (12a–c, 16a–c)
To an oven-dried RBF equipped with a magnetic stir bar was added suitable hydroxy 2-oxo-2H-chromene-3-carboxylic acid (1 equiv.) and substituted sulphonyl piperazine (1 equiv.), HOBt (0.5 equiv.) and EDC.HCl (2 equiv.) in DMF maintaining inert conditions, and DIPEA (3 equiv.) was added slowly to the reaction mixture and was allowed to stir at room temperature for 8–12 h. After the completion of the reaction confirmed by TLC, the reaction mixture was poured into a beaker with crushed ice, and the resultant solid was filtered and purified by column chromatography with 6:4 ethyl acetate and hexane as the solvent for elution to afford the title compound with a yield of 60–75%.
3–(4-((4-Fluorophenyl)sulphonyl)piperazine-1-carbonyl)-7-hydroxy-2H-chromen-2-one (12a)
Off-white solid; yield 70%; m.p. 154–156 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.78 (s, 1H), 8.13 (s, 1H), 7.61 (d, J = 8.6 Hz, 1H), 7.08 (t, J = 8.9 Hz, 2H), 7.01–6.97 (m, 2H), 6.85 (dd, J = 8.5, 2.3 Hz, 1H), 6.77 (d, J = 2.2 Hz, 1H), 3.77–3.71 (m, 2H), 3.54–3.47 (m, 2H), 3.17–3.11 (m, 2H), 3.09–3.03 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 163.84, 162.65, 158.51, 157.76, 156.15, 155.88, 148.13, 143.77, 130.94, 120.14, 118.38, 118.32, 115.93, 115.76, 114.14, 111.27, 102.54, 50.09, 49.62, 46.78, 41.75. HRMS (ESI): m/z calculated for [M + H]+ C20H18FN2O6S 433.0864; found 433.0873.
7-Hydroxy-3–(4-(phenylsulfonyl)piperazine-1-carbonyl)-2H-chromen-2-one (12b)
Off-white solid; yield 71%; m.p. 190–192 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.79 (s, 1H), 8.05 (s, 1H), 7.83–7.74 (m, 3H), 7.69 (t, J = 7.5 Hz, 2H), 7.56 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 8.3 Hz, 1H), 6.74 (s, 1H), 3.74–3.64 (m, 2H), 3.47–3.42 (m, 2H), 3.00–2.95 (m, 2H), 2.92–2.86 (m, 2H).13C NMR (125 MHz, DMSO-d6) δ 163.9, 162.7, 158.5, 156.1, 144.2, 135.4, 133.9, 130.9, 130.0, 127.9, 119.6, 114.2, 111.2, 102.5, 46.5, 46.3, 46.2, 41.1. HRMS (ESI): m/z calculated for [M + H]+ C20H19N2O6S 415.0958; found 415.0966.
3–(4-((5-Bromothiophen-2-yl)sulphonyl)piperazine-1-carbonyl)-7-hydroxy-2H-chromen-2-one (12c)
Pale-brown solid; yield 68%; m.p. 217–219 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.81 (s, 1H), 8.09 (s, 1H), 7.58 (d, J = 7.5 Hz, 1H), 7.52 (d, J = 16.9 Hz, 2H), 6.84 (d, J = 6.4 Hz, 1H), 6.75 (s, 1H), 3.79–3.67 (m, 2H), 3.56 − 3.45 (m, 2H) 3.10–3.02 (m, 2H), 3.01–2.95 (m, 2H). 13C NMR (125 MHz, DMSO-d6) δ 163.9, 162.8, 158.5, 156.2, 144.2, 136.3, 134.5, 132.7, 131.0, 120.38, 119.5, 114.2, 111.2, 102.5, 46.5, 46.1, 40.9. HRMS (ESI): m/z calculated for [M + H]+ C18H16BrN2O6S2 498.9628; found 498.9637.
3–(4-((4-Fluorophenyl)sulphonyl)piperazine-1-carbonyl)-4-hydroxy-2H-chromen-2-one (16a)
Pale-green solid; yield 72%; m.p. 170–172 °C. 1H NMR (500 MHz, DMSO-d6) 1H NMR (500 MHz, DMSO) δ 9.81 (s, 1H), 7.23 (t, J = 7.7 Hz, 1H), 7.14 (dd, J = 7.4, 1.2 Hz, 1H), 7.06 (t, J = 8.8 Hz, 2H), 6.97 (d, J = 4.6 Hz, 1H), 6.95 (d, J = 4.6 Hz, 1H), 6.88 (d, J = 8.2 Hz, 1H), 6.85 (t, J = 7.4 Hz, 1H), 3.84–3.61 (m, 2H), 3.44–3.34 (m, 2H), 3.13–3.01 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ 167.48, 157.69, 155.81, 153.73, 148.20, 148.19, 130.67, 128.62, 124.26, 119.60, 118.27, 118.21, 116.11, 115.91, 115.73, 49.88. HRMS (ESI): m/z calculated for [M + H]+ C20H18FN2O6S 433.0864; found 433.0877.
4-Hydroxy-3–(4-(phenylsulfonyl)piperazine-1-carbonyl)-2H-chromen-2-one (16b)
Pale-green solid; yield 73%; m.p. 224–226 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.76 (s, 1H), 7.74 (d, J = 7.7 Hz, 3H), 7.67 (t, J = 7.6 Hz, 2H), 7.20 (t, J = 7.1 Hz, 1H), 7.05 (d, J = 7.3 Hz, 1H), 6.83–6.77 (m, 2H), 3.76–3.59 (m, 2H), 3.30–3.20 (m, 2H), 2.98–2.86 (m, 4H). 13C NMR (125 MHz, DMSO-d6) δ 167.5, 153.6, 135.4, 135.4, 133.8, 130.8, 129.9, 128.7, 127.9, 123.7, 119.6, 116.1, 116.1, 46.5. HRMS (ESI): m/z calculated for [M + H]+ C20H19N2O6S 415.0958; found 415.0963.
3–(4-((5-Bromothiophen-2-yl)sulphonyl)piperazine-1-carbonyl)-4-hydroxy-2H-chromene-2-one (16c)
Pale-brown solid; yield 70%; m.p. 254–256 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.82 (s, 1H), 7.53 (d, J = 4.0 Hz, 1H), 7.49 (d, J = 4.0 Hz, 1H), 7.22 (t, J = 7.8 Hz, 2H), 7.11 (dd, J = 7.5, 1.5 Hz, 1H), 6.88–6.81 (m, 2H), 3.74 (s, 2H), 3.01 (s, 6H). 13C NMR (125 MHz, DMSO-d6) δ 167.5, 153.6, 136.3, 134.5, 132.6, 130.8, 128.8, 123.6, 120.3, 119.6, 116.1, 46.4. HRMS (ESI): m/z calculated for [M + H]+ C18H16BrN2O6S2 498.9628; found 498.9635.
Synthesis of 3-acetyl-2H-chromene-2-oneCitation2 (18)
A mixture of 2-hydroxy benzaldehyde (1 equiv.) and ethyl acetoacetate (1 equiv.) in ethanol was stirred at room temperature in the presence of a catalytic amount of piperidine for 4–6 h. Reaction progress was monitored by TLC. After the completion of the reaction, the solvent was removed under reduced pressure, and the reaction mixture was added to crushed ice and the precipitate was filtered and recrystallized from ethanol with a yield of 70%.
General procedure for the synthesis of ketooxime ether derivatives of coumarin (20a, b)
To the solution of 3-acetyl coumarin(1 equiv.), O-benzyl hydroxylamine saltsCitation28 (1.5 equiv.) in DMF under nitrogen was added a catalytic amount of acetic acid. The reaction mixture was stirred at room temperature for 2–3 h. After completion of the reaction, the reaction mixture was poured into crushed iceCitation28. Further, the obtained solid was filtered and subjected to column chromatography with ethyl acetate and hexane 1:9 to get the titled molecules with a 70–73% yield.
(E)-3–(1-(((4-fluorobenzyl)oxy)imino)ethyl)-2H-chromen-2-one (20a)
Pale-green solid; yield 70%; m.p. 123–125 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.12 (s, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.66 (t, J = 7.9 Hz, 1H), 7.46 (td, J = 9.5, 6.6 Hz, 3H), 7.39 (t, J = 7.6 Hz, 1H), 7.22 (t, J = 8.9 Hz, 2H), 5.20 (s, 2H), 2.15 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 163.3, 161.3, 159.4, 154.2, 153.9, 142.0, 134.4, 133.0, 130.7, 130.6, 129.7, 125.3, 124.6, 119.0, 116.5, 115.6, 115.6, 75.2, 14.8. HRMS (ESI): m/z calculated for [M + H]+ C18H15FNO3 312.1030; found 312.1036.
(E)-3–(1-(((4-chlorobenzyl)oxy)imino)ethyl)-2H-chromen-2-one (20b)
Pale-green solid; yield 73%; m.p. 128–130 °C. 1H NMR (500 MHz, DMSO-d6) δ 8.11 (s, 1H), 7.83 (dd, J = 7.8, 1.5 Hz, 1H), 7.68 − 7.63 (m, 1H), 7.45 (d, J = 1.8 Hz, 4H), 7.43 (s, 1H), 7.41–7.37 (m, 1H), 5.21 (s, 2H), 2.16 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 159.4, 154.4, 153.9, 142.0, 137.2, 133.0, 132.9, 130.2, 129.7, 128.9, 125.3, 124.6, 119.0, 116.5, 75.0, 14.9. HRMS (ESI): m/z calculated for [M + H]+ C18H15ClNO3 328.0735; found 328.0742.
CA inhibitory assay
An Applied Photophysics stopped-flow instrument was used for assaying the CA-catalysed CO2 hydration activityCitation29. Phenol red (at a concentration of 0.2 mM) was used as an indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as a buffer and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM to determine the kinetic parameters and inhibition constants. Enzyme concentrations in the assay system were in the range of 4–13 nM. For each inhibitor, at least six traces of the initial 5–10% of the reaction were used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water, and dilutions up to 0.01 nM were done thereafter with the assay buffer. The inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng-Prusoff equation, as reported earlierCitation30,Citation31, representing the mean from at least three different determinations. All CA isoforms were recombinant ones obtained in house as reported earlierCitation32,Citation33.
In silico studies
The crystal structure of CA IX (pdb 5DVX)Citation34 and XII (pdb 1JD0)Citation35 were prepared using the Protein Preparation WizardCitation36 and minimised using the OPLS3e force field. The 3D ligand structures were prepared using LigprepCitation37 at pH 7.4 ± 0.5. To correctly estimate the binding mode of the synthesised compounds in the catalytic pocket of hCA IX, the grid was prepared using the native coumarin coordinates co-crystallized in hCA II. The prepared structure of hCA IX (pdb: 5DVX) was superimposed against the structure of hCA II (pdb: 3F8E). The co-crystallized ligand (2-amino-2-hydroxymethyl-propene-1,3-diol) from the hCA IX isoform was removed, and the hydrolysed coumarin of the hCA II isoform, together with its Zn2+ coordinated water molecule was inserted inside the prepared structure of the hCA IX and the docking grid was prepared by taking the centroid of the co-crystallized hydrolysed coumarin ligand with default settings. The grid for the hCA XII was generated by taking the central coordinates of co-crystallized acetazolamide and keeping the default settings. Ligands were docked using extra precision mode (XP) of GlideCitation38 and the best pose of each molecule was refined with PrimeCitation39 with a VSGB solvation model considering the target flexible within 3 Å around the ligand. The best poses of 6c and 7c in complex with CA IX and 6d and 7b in complex with CA XII were taken to perform a 100 ns MD simulation using DesmondCitation40 using the OPLS 2005 force field. The orthorhombic box system was built using TIP4PEW water molecules as solvent system extended 15 Å away from any protein atom and neutralised by adding chlorine and sodium ions. The simulation protocol comprised a starting relaxation step followed by a final production phase of 100 ns. The relaxation step is divided into five stages having: (a) a stage of 100 ps at 10 K retaining the harmonic restraints on the solute heavy atoms (force constant of 50.0 kcal mol–1 Å–2) using the NPT ensemble with Brownian dynamics; (b) a stage of 12 ps at 10 K with harmonic restraints on the solute heavy atoms (force constant of 50.0 kcal mol–1 Å–2), using the NVT ensemble and Berendsen thermostat; (c) a stage of 12 ps at 10 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (d) a stage of 12 ps at 300 K and 1 atm, retaining the harmonic restraints and using the NPT ensemble and Berendsen thermostat and barostat; (e) a final 24 ps stage at 300 K and 1 atm without harmonic restraints, using the NPT Berendsen thermostat and barostat. Then the system enters the final production phase of the MD simulation run using a canonical NPT Berendsen ensemble at temperature 300 K. During the MD simulation, a time step of 2 fs was used while constraining the bond lengths of hydrogen atoms with the MSHAKE algorithm. The atomic coordinates of the system were saved every 100 ps along the MD trajectory. Protein and ligand RMSD values, ligand torsions evolution and occupancy of intermolecular hydrogen bonds and hydrophobic contacts were computed along the production phase of the MD simulation with the Simulation Interaction Diagram tools implemented in MaestroCitation18.
Results and discussion
Chemistry
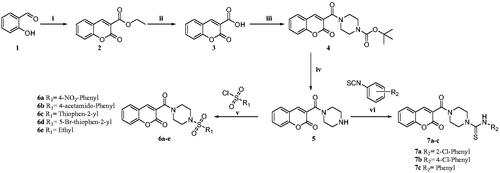
The synthetic route followed for the new coumarin-sulphonamide derivatives is summarised in Scheme 1. Salicylaldehyde (1), on reaction with diethyl malonate in the presence of a catalytic amount of piperidine and acetic acid, gave ethyl 2-oxo-2H-chromene-3-carboxylate (2), which on hydrolysis with NaOH afforded 2-oxo-2H-chromene-3-carboxylic acid (3). The acid (3) was then coupled with N-Boc piperazine in the presence of EDC.HCl, HOBt and DIPEA to afford tert-butyl 4–(2-oxo-2H-chromene-3-carbonyl) piperazine-1-carboxylate (4). The Boc deprotection with TFA in DCM afforded 3-(piperazine-1-carbonyl)-2H-chromen-2-one (5), which on reaction with various benzene sulphonyl chlorides using triethylamine as base and DCM as solvent afforded the titled sulphonamide derivatives (6a–e). The reaction of intermediate 5 with substituted phenylisothiocyanates in acetonitrile yielded the carbothioamide derivatives (7a–c) (Scheme 1).
Scheme 1. Reagents and conditions: (i) diethyl malonate, Piperidine, EtOH, 70–80 °C, 4–6 h, 82% (ii) NaOH, H2O, MeOH, rt., 2 h, 74% (iii) N-Boc piperazine, EDC.HCl, HOBt, DIPEA, DMF, rt, 8-12 h, 61% (iv) TFA, DCM, 0–5 °C to rt., 1 h, 69% (v) substituted sulphonyl chlorides, DIPEA, DCM, rt., 2–3 h, 75–81%. (vi) substituted phenylisothiocyanates, ACN, reflux, 2–3 h, 65–75%.
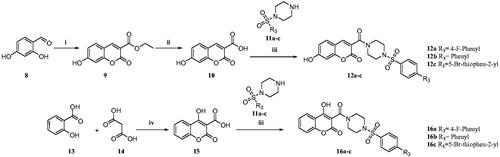
In another modification, 7-hydroxy-3-carboxy coumarin (10) was prepared using 2,4-dihydroxy benzaldehyde as the starting material. The 4-hydroxy-3-carboxy coumarin (15) was prepared by heating salicylic acid with malonic acid in the presence of trifluoroacetic acid. These acid intermediates 10 and 15 were coupled with substituted sulphonyl piperazines to afford compounds 12a–c and 16a–c (Scheme 2).
Scheme 2. Reagents and conditions: (i) diethyl malonate, piperidine, glacial acetic acid (Cat.), reflux, 80 °C, 4–6 h, 79% (ii) NaOH, H2O, MeOH, rt.,2 h, 70% (iii) substituted sulphonyl piperazines, EDC.HCl, HOBt, DIPEA, DMF, rt,8–12 h, 60–75% (iv) TFA, 90–100 °C 8 h, 88% Citation27.
Similarly, 3-acetyl coumarin (18) intermediate was prepared from salicylaldehyde and ethyl acetoacetate. The intermediate 18 was further treated with substituted O-benzyl hydroxylamine. HCl to yield compounds 20a, b (Scheme 3).
Scheme 3. Reagents and conditions: (i) ethyl acetoacetate, piperidine, EtOH, rt, 4–6 h, 70% (ii) cat. AcOH, DMF, rt, 2–3 h, 70–73%.
CA inhibition
The synthesised compounds were tested for their CA inhibitory potential against the isoforms hCA I, II, IX, and XII, and the results are given in . The CA inhibition was evaluated using the stopped-flow carbon dioxide hydrase assayCitation29, and acetazolamide (AAZ) was used as a reference CAI. The concentration-response curves of the most active molecules compared with the control acetazolamide against the CA isoforms IX and XII are shown in Figures S1 and S2 of the supporting information.
Table 1. Inhibition data for hCA isozymes I, II, IX and XII for synthesised compounds using AAZ as a standard CAI (Ki in µM).a,b
The synthesised coumarin derivatives showed selective CA inhibitory activity against hCA IX and hCA XII (associated with tumour) with Ki values ranging from 4 to 100 µM.
While the sulphonamide derivatives 6c exhibited hCA IX inhibition with Ki of 4.1 µM, compound 6a showed inhibition against hCA IX and hCA XII with Ki of 7.1 and 9.1 µM, respectively. The compound 6d inhibited both hCA IX and hCA XII with Ki of 5.56 and 9.8 µM, respectively. The compounds 12b and 16b exhibited selective inhibition of hCA XII with Ki of 9.3 and 9.5 µM, respectively.
The Structure-Activity Relationship studies indicate that the sulphonamides with phenyl and thiophen-2-yl substitutions with EWG, like NO2 (6d) and Br (6a), were found to be active against both the isoforms. In contrast, aliphatic ethyl (6e) substitutions on piperazine led to a decrease in activity. The hydroxyl substitutions on the coumarin ring (12a–c and 16a–c) resulted in a loss of activity against hCA IX.
The carbothioamide derivatives (7a–c) inhibited hCA IX and hCA XII, of which 7c showed inhibition against hCA IX with Ki of 5.1 µM, and 7b inhibited both the isoforms hCA IX and hCA XII with Ki of 9.2 and 8.0 µM respectively.
The unsubstituted carbothioamide derivative 7c inhibited hCA IX with better Ki than hCA XII, and the 4-Cl derivative 7b inhibited hCA XII better than hCA IX. The 2-Cl (7a) substitution resulted in a 5–7-fold decrease in the hCA IX and hCA XII inhibition.
The coumarin oxime ether derivatives inhibited both the isoforms, hCA IX and hCA XII with a Ki of 8.9–62.1 µM, where 4-Cl-phenyl substituted compound 20b was equally active against both hCA IX and hCA XII with a Ki of 8.9 and 9.4 µM, the 4-F-phenyl substituted 20a exhibited a 6-fold decrease in activity against the hCAIX isoform.
In silico studies
In an attempt to predict the binding mode of the synthesised compounds with hCA IX (PDB ID: 5DVX) and hCA XII (PDB ID: 1JD0), molecular docking and dynamic simulation studies were performed. The α-carbonic anhydrases are found to cleave the lactone ring of the coumarins to form a cinnamic acid derivativeCitation9,Citation10,Citation30. The predicted hydrolysed structures of the synthesised compounds were drawn accordingly, and the in silico studies were performed using protocols adopted from the literatureCitation18,Citation41.
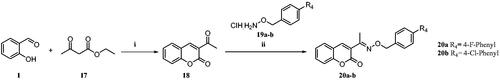
Owing to the flexibility of the hydrolysed forms of the synthesised molecules exhibiting better binding affinities towards the CA active site residues when compared to the closed forms, the compounds 6c and 7c showed potent inhibition against the hCA IX (PDB ID: 5DVX). The carboxyl group of compound 6c showed key H-bonding interactions with Thr332 and Thr333, and the thiophene ring showed a π–π stacking interaction with His200. Similarly, compound 7c showed two hydrogen bonding interactions with Gln224, Thr332 and Thr333 and the hydrophobic interactions stabilised the binding mode in the active pocket of hCA IX as shown in .
Figure 2. (A) Docking pose of compound 6c (green) (B) docking pose of compound 7c (grey) in the active site of hCA IX (PDB ID: 5DVX) Zn2+ is represented as grey sphere.
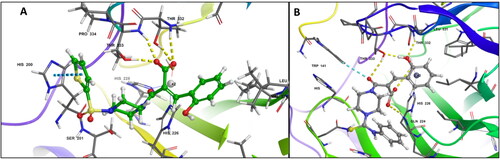
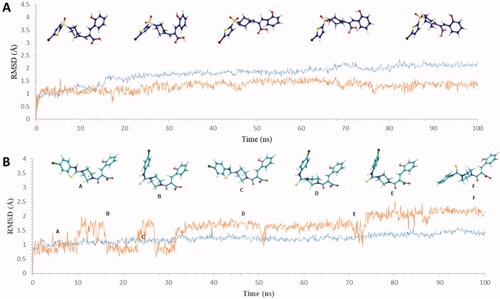
Further, the molecular dynamics simulation for 100 ns showed the molecules to be stable in the binding pocket throughout the simulation. The plots of the ligand (red) and protein (blue) in show the changes in RMSD and the conformation of the ligands during the 100 ns simulation in the binding site of hCA IX (PDB ID: 5DVX). The compound 6c attained a stable conformation in the binding pocket at 20 ns, and minor changes in the conformation at 50 ns resulted in the fluctuation in the RMSD below 2.5 Å retaining the key interactions of Thr332 and Thr333. Likewise, compound 7c stabilised into the binding site at 20 ns and with minor fluctuations in the RMSD throughout the simulation, retaining the interactions with Gln224.
Figure 3. Graph showing analysis of ligand RMSD for MD simulation time of 100 ns on hCA IX (PDB ID: 5DVX) (A) change in conformations of 6c. (B) change in conformations of 7c.
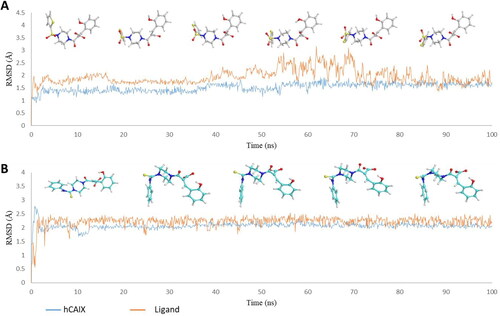
The compounds 6d and 7b, active against hCA XII, showed important interactions with the binding site residues of hCA XII (PDB ID: 1JD0). Interestingly, compound 6d showed metal coordination through the carboxylate anion and Zn2+, hydrogen bonding interactions with Thr199, Thr200, Thr91, and a halogen bond with Lys67. Additionally, the π–π stacking interaction with His94 stabilised the ligand in the hCA XII binding pocket. Similarly, compound 7b coordinated with Zn2+ in addition to the hydrogen bonding interactions with Thr199, Thr200, the π–π stacking interaction with His94 and His96, and a halogen bonding interaction with Lys4 stabilised 7c as shown in . It should be mentioned that the hydrolysed coumarins were not seen bound to the metal ion by X-ray crystallography, as mentioned in the introductionCitation10. Thus, the obtained computational data reported here must be validated by crystallographic experiments.
Figure 4. (A) Docking pose of compound 6d (purple) (B) docking pose of compound 7b (teal) in the active site of hCA IX (PDB ID: 1JD0) Zn2+ is represented as grey sphere.
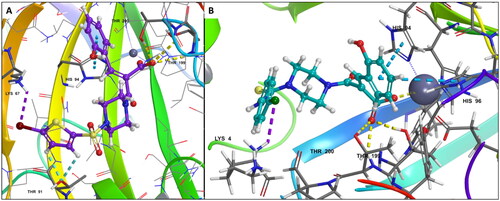
Moreover, it is evident that the compounds showed stable interactions throughout the 100 ns simulation. The plots in showed the compound 6d settled in a stable conformation from 40 ns and with minor fluctuations in the RMSD around 1–1.5 Å. Whereas the compound 7b stabilised in the pocket at 10 ns later, the changes in the conformations from 20 to 35 ns leading to the final stabilisation of the ligand at 40–60 ns with minor changes in RMSD around 2.5 Å with stable key interactions with the metal ion and the residues accounting for the potency of the compounds against hCA XII.
Figure 5. Graph showing analysis of ligand RMSD for MD simulation time of 100 ns on hCA XII (PDB ID: 1JD0) (A) change in conformations of 6d. (B) change in conformations of 7b.
Conclusion
In summary, this work describes the design and synthesis of 16 new coumarin derivatives consisting of 11 coumarin-linked sulphonamides (6a–e, 12a–c, 16a–c), 3 coumarin-linked carbothioamides (7a–c) and 2 coumarin oxime ether derivatives (20a, 20b) as potential carbonic anhydrase inhibitors. The synthesised compounds selectively inhibited the hCA IX and hCA XII with Ki ranging from 4 to 93 µM. Compounds 6c and 7c exhibited selectivity towards hCA IX with a Ki of 4.1 and 5.1 µM, respectively. At the same time, compounds 6d and 7b inhibited hCA XII with a Ki of 9.8 and 8.0 µM, respectively. The oxime ether 20a selectively inhibited hCA XII with a Ki of 9.2 µM. Further, the in-silico studies revealed the key interacting residues of the synthesised molecules in the binding pocket and the molecular dynamic simulations confirmed the binding mode and the stability of the interactions of the ligands in the binding pockets of the enzymes hCA IX and hCA XII which are overexpressed in the hypoxic cancer cells suggesting the compounds 6c, 7c and 7b can further be explored to develop into effectiive inhibitors of hCA IX and hCA XII.
Supplemental Material
Download PDF (2.4 MB)Acknowledgements
M.G, K.B, and K.K.C is thankful to Department of Pharmaceuticals, Ministry of Chemicals & Fertilizers, Govt. of India, for the award of NIPER fellowship G.P thanks DBT for the fellowship.
Disclosure statement
No potential competing interest was reported by all authors except CTS. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Zengin Kurt B, Sonmez F, Ozturk D, Akdemir A, Angeli A, Supuran CT. Synthesis of coumarin-sulfonamide derivatives and determination of their cytotoxicity, carbonic anhydrase inhibitory and molecular docking studies. Eur J Med Chem. 2019;183:111702.
- Thacker PS, Sridhar Goud N, Argulwar OS, Soman J, Angeli A, Alvala M, Arifuddin M, Supuran CT. Synthesis and biological evaluation of some coumarin hybrids as selective carbonic anhydrase IX and XII inhibitors. Bioorg Chem. 2020;104:104272.
- Aimene Y, Eychenne R, Rodriguez F, Mallet-Ladeira S, Saffon-Merceron N, Winum J-Y, Nocentini A, Supuran CT, Benoist E, Seridi A. Synthesis, crystal structure, inhibitory activity and molecular docking of coumarins/sulfonamides containing triazolyl pyridine moiety as potent selective carbonic anhydrase IX and XII inhibitors. Crystals. 2021;11(9):1076.
- McDonald PC, Chafe SC, Supuran CT, Dedhar S. Cancer therapeutic targeting of hypoxia induced carbonic anhydrase IX: from bench to bedside. Cancers. 2022;14(14):3297.
- Mahon BP, Pinard MA, McKenna R. Targeting carbonic anhydrase IX activity and expression. Molecules. 2015;20(2):2323–2348.
- Bonardi A, Bua S, Combs J, Lomelino C, Andring J, Osman SM, Toti A, Di Cesare Mannelli L, Gratteri P, Ghelardini C, et al. The three-tails approach as a new strategy to improve selectivity of action of sulphonamide inhibitors against tumour-associated carbonic anhydrase IX and XII. J Enzyme Inhib Med Chem. 2022;37(1):930–939.
- Neri D, Supuran CT. Interfering with PH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 2011;10(10):767–777.
- Alterio V, Di Fiore A, D'Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev. 2012;112(8):4421–4468.
- Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem. 2016;31(3):345–360.
- Maresca A, Temperini C, Vu H, Pham NB, Poulsen S-A, Scozzafava A, Quinn RJ, Supuran CT. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc. 2009;131(8):3057–3062.
- Abdel-Aziz AA-M, El-Azab AS, Ceruso M, Supuran CT. Carbonic anhydrase inhibitory activity of sulfonamides and carboxylic acids incorporating cyclic imide scaffolds. Bioorg Med Chem Lett. 2014;24(22):5185–5189.
- Nocentini A, Lucidi A, Perut F, Massa A, Tomaselli D, Gratteri P, Baldini N, Rotili D, Mai A, Supuran CT. α,γ-Diketocarboxylic acids and their esters act as carbonic anhydrase IX and XII selective inhibitors. ACS Med Chem Lett. 2019;10(4):661–665.
- Giovannuzzi S, Hewitt CS, Nocentini A, Capasso C, Flaherty DP, Supuran CT. Coumarins effectively inhibit bacterial α-carbonic anhydrases. J Enzyme Inhib Med Chem. 2022;37(1):333–338.
- Lipeeva AV, Zakharov DO, Burova LG, Frolova TS, Baev DS, Shirokikh IV, Evstropov AN, Sinitsyna OI, Tolsikova TG, Shults EE. Design, synthesis and antibacterial activity of coumarin-1,2,3-triazole hybrids obtained from natural furocoumarin peucedanin. Molecules. 2019;24(11):2126.
- Chougala BM, Samundeeswari S, Holiyachi M, Shastri LA, Dodamani S, Jalalpure S, Dixit SR, Joshi SD, Sunagar VA. Synthesis, characterization and molecular docking studies of substituted 4-coumarinylpyrano[2,3-c]pyrazole derivatives as potent antibacterial and anti-inflammatory agents. Eur J Med Chem. 2017;125:101–116.
- Fayed EA, Sabour R, Harras MF, Mehany ABM. Design, synthesis, biological evaluation and molecular modeling of new coumarin derivatives as potent anticancer agents. Med Chem Res. 2019;28(8):1284–1297.
- Thacker PS, Srikanth D, Angeli A, Singh P, Chinchilli KK, Arifuddin M, Supuran CT. Coumarin‐thiourea hybrids show potent carbonic anhydrase IX and XIII inhibitory action. ChemMedChem. 2021;16(8):1252–1256.
- Abdelrahman MA, Ibrahim HS, Nocentini A, Eldehna WM, Bonardi A, Abdel-Aziz HA, Gratteri P, Abou-Seri SM, Supuran CT. Novel 3-substituted coumarins as selective human carbonic anhydrase IX and XII inhibitors: synthesis, biological and molecular dynamics analysis. Eur J Med Chem. 2021;209:112897.
- Tawfik HO, Shaldam MA, Nocentini A, Salem R, Almahli H, Al-Rashood ST, Supuran CT, Eldehna WM. Novel 3-(6-methylpyridin-2-yl)coumarin-based chalcones as selective inhibitors of cancer-related carbonic anhydrases IX and XII endowed with anti-proliferative activity. J Enzyme Inhib Med Chem. 2022;37(1):1043–1052.
- Chandra KM, Goud NS, Arifuddin M, Alvala M, Alvala R, Angeli A, Supuran CT. Synthesis and biological evaluation of novel 4,7-disubstituted coumarins as selective tumor-associated carbonic anhydrase IX and XII inhibitors. Bioorg Med Chem Lett. 2021;39:127877.
- Thacker PS, Alvala M, Arifuddin M, Angeli A, Supuran CT. Design, synthesis and biological evaluation of coumarin-3-carboxamides as selective carbonic anhydrase IX and XII inhibitors. Bioorg Chem. 2019;86:386–392.
- Bozdag M, Ferraroni M, Carta F, Vullo D, Lucarini L, Orlandini E, Rossello A, Nuti E, Scozzafava A, Masini E, et al. Structural insights on carbonic anhydrase inhibitory action, isoform selectivity, and potency of sulfonamides and coumarins incorporating arylsulfonylureido groups. J Med Chem. 2014;57(21):9152–9167.
- Eldehna WM, Taghour MS, Al-Warhi T, Nocentini A, Elbadawi MM, Mahdy HA, Abdelrahman MA, Alotaibi OJ, Aljaeed N, Elimam DM, et al. Discovery of 2,4-thiazolidinedione-tethered coumarins as novel selective inhibitors for carbonic anhydrase IX and XII isoforms. J Enzyme Inhib Med Chem. 2022;37(1):531–541.
- Ibrahim HS, Abdelrahman MA, Nocentini A, Bua S, Abdel-Aziz HA, Supuran CT, Abou-Seri SM, Eldehna WM. Insights into the effect of elaborating coumarin-based aryl enaminones with sulfonamide or carboxylic acid functionality on carbonic anhydrase inhibitory potency and selectivity. Bioorg Chem. 2022;126:105888.
- Mesiti F, Gaspar A, Chavarria D, Maruca A, Rocca R, Gil Martins E, Barreiro S, Silva R, Fernandes C, Gul S, et al. Mapping chromone-3-phenylcarboxamide pharmacophore: Quid Est Veritas? J Med Chem. 2021; 64 (15):11169–11182.
- Zhang J, Jiang C-S. Synthesis and evaluation of coumarin/piperazine hybrids as acetylcholinesterase inhibitors. Med Chem Res. 2018;27(6):1717–1727.
- Woods LL, Johnson D. Coumarins from 2-hydroxyaryl acids and malonic acid. J Org Chem. 1965;30(12):4343–4344.
- Akunuri R, Veerareddy V, Kaul G, Akhir A, Unnissa T, Parupalli R, Madhavi YV, Chopra S, Nanduri S. Synthesis and antibacterial evaluation of (E)-1-(1H-indol-3-yl) ethanone O-benzyl oxime derivatives against MRSA and VRSA strains. Bioorg Chem. 2021;116:105288.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase: I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem. 2010;53(1):335–344.
- Carta F, Temperini C, Innocenti A, Scozzafava A, Kaila K, Supuran CT. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem. 2010;53(15):5511–5522.
- Hilvo M, Baranauskiene L, Salzano AM, Scaloni A, Matulis D, Innocenti A, Scozzafava A, Monti SM, Fiore AD, Simone GD, et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem. 2008;283(41):27799–27809.
- Di Fiore A, Truppo E, Supuran CT, Alterio V, Dathan N, Bootorabi F, Parkkila S, Monti SM, Simone GD. Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorg Med Chem Lett. 2010;20(17):5023–5026.
- Mahon BP, Bhatt A, Socorro L, Driscoll JM, Okoh C, Lomelino CL, Mboge MY, Kurian JJ, Tu C, Agbandje-McKenna M, et al. The structure of carbonic anhydrase IX Is adapted for low-pH catalysis. Biochemistry. 2016;55(33):4642–4653.
- Whittington DA, Waheed A, Ulmasov B, Shah GN, Grubb JH, Sly WS, Christianson DW. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci USA. 2001;98(17):9545–9550.
- Protein Preparation Wizard. New York (NY): Schrödinger, LLC; 2021.
- LigPrep. New York (NY): Schrödinger, LLC; 2021.
- Glide. New York (NY): Schrödinger, LLC; 2021.
- Prime. New York (NY): Schrödinger, LLC; 2021.
- Schrödinger Release 2021-2: Desmond Molecular Dynamics System. New York (NY): D. E. Shaw Research; 2021.
- Pustenko A, Nocentini A, Gratteri P, Bonardi A, Vozny I, Žalubovskis R, Supuran CT. The antibiotic furagin and its derivatives are isoform-selective human carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2020;35(1):1011–1020.