
Abstract
The design of kinase inhibitors targeting the oncogenic kinase BCR-ABL constitutes a promising paradigm for treating chronic myeloid leukaemia (CML). Nevertheless, the efficacy of imatinib, the first FDA-approved targeted therapy for CML, is curbed by the emergence of resistance. Herein, we report the identification of the 2-methoxyphenyl ureidobenzothiazole AK-HW-90 (2b) as a potent pan-BCR-ABL inhibitor against imatinib-resistant mutants, particularly T315I. A concise array of six compounds 2a–f was designed based on our previously reported benzothiazole lead AKE-5l to improve its BCR-ABLT315I inhibitory activity. Replacing the 6-oxypicolinamide moiety of AKE-5l with o-methoxyphenyl and changing the propyl spacer with phenyl afforded 2a and AK-HW-90 (2b) with IC50 values of 2.0 and 0.65 nM against BCR-ABLT315I, respectively. AK-HW-90 showed superior anticancer potency to imatinib against multiple cancer cells (NCI), including leukaemia K-562. The obtained outcomes offer AK-HW-90 as a promising candidate for the treatment of CML and other types of cancer.
Graphical abstract
Introduction
Chronic myeloid leukaemia (CML) is a myeloproliferative neoplasm characterised by excessive production of immature white blood cells and constitutes approximately 15% of leukaemia cases in adultsCitation1. The fusion oncoprotein product of the Philadelphia chromosome (Ph), Break-point cluster region-Abelson (BCR-ABL), is the critical driver for the pathogenesis of CML and acute lymphoblastic leukaemia (Ph + ALL)Citation2. Therefore, the design of BCR-ABL kinase inhibitors offers a legitimate approach for the treatment of CML and ALL.
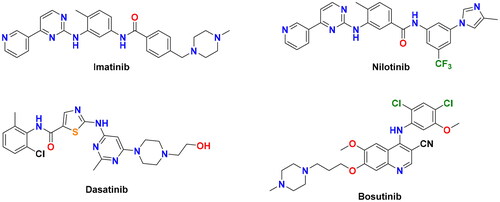
Imatinib (Gleevec®, ()), a diarylamide-4-(pyridin-3-yl)pyrimidine conjugate, is the first FDA-approved BCR-ABL inhibitor for the treatment of CML patientsCitation3. Despite its initial significant response in most of the CML patients, the clinical efficacy of imatinib was hampered in about 40% of patients due to dose intolerance and the emergence of drug resistanceCitation4. Point mutations within the BCR-ABL kinase domain represent the most recurrent mechanism of imatinib resistanceCitation5, where they hinder its proper binding with the targetCitation6. Among more than 100 different mutations detected in CML patients, the gatekeeper T315I mutation is the most refractory form, which emerges in more than 20% of CML patientsCitation7. Second-generation BCR-ABL inhibitors, nilotinib (Tasigna®), dasatinib (Sprycel®), and bosutinib (Bosulif®) (), have been approved to treat CML adult patients with acquired resistance to imatinibCitation8–10. However, these second-line drugs failed to inhibit several imatinib-resistant mutations, including the T315I formCitation11.
Figure 1. Chemical structures of the first and second-generation BCR-ABL inhibitors.
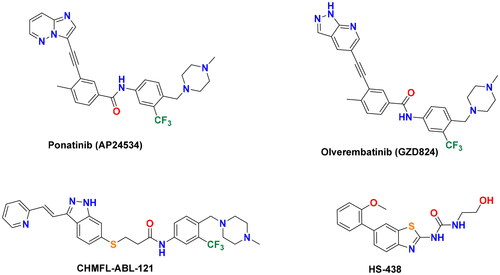
In 2012, the multikinase inhibitor ponatinib (Iclusig®, ) received FDA approval as the third-generation ABL inhibitor for the treatment of patients with CML and Ph + ALLCitation12. It demonstrated excellent potency against imatinib-clinically resistant ABL mutants, including T315I and other forms like Q252H and H396PCitation13,Citation14. However, the serious vascular/cardiotoxic effects associated with ponatinib restricted its use for these tumours harbouring T315I mutant. Ponatinib cardiotoxicity is thought to arise from its multikinase inhibitory mode and synchronous inhibition of kinases that are important for cardiovascular functionCitation15. Therefore, a number of structurally assorted BCR-ABLT315I inhibitors with improved selectivity/safety have been developed (). Olverembatinib (GZD824), a pyrazolopyridine-diarylamide conjugate, was developed from ponatinib as a potent BCR-ABLT315I inhibitor with a favourable safety profileCitation16. The indazole derivative CHMFL-ABL-121 is a type-II inhibitor derived from axitinib with an IC50 value of 0.2 nM against BCR-ABLT315ICitation17. The ureidobenzothiazole HS-438 was discovered by Park et al. as a pico-molar BCR-ABLT315I inhibitor, which potently inhibited the proliferation of imatinib-resistant Ba/F3T315I cellsCitation18.
Figure 2. Chemical structures of ponatinib and other potent BCR-ABLT315I inhibitors.
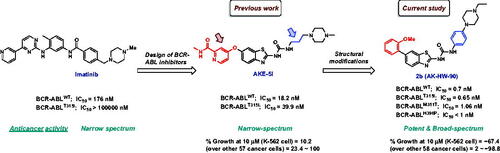
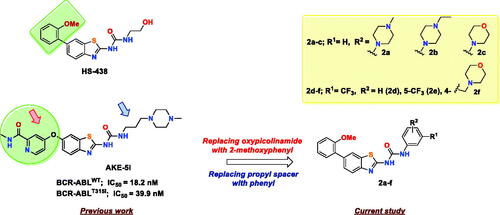
In a previous study, our group reported a series of benzothiazoles featuring picolinamide moiety as BCR-ABL inhibitorsCitation19. In this set, compound AKE-5l () was identified as the most potent BCR-ABL inhibitor with IC50 values of 18.2 and 39.9 nM against wild BCR-ABL and its T315I mutant, respectively. Moreover, AKE-5l showed selective anti-leukemic activity against only the K-562 cell line out of 58 tested cancer cells. In continuation of our ongoing efforts to identify new chemical entities as potent anticancer kinase inhibitors,Citation20–25 we herein aimed at further optimisation of AKE-5l activity. Based on the exceptional potency of HS-438 towards BCR-ABLT315I (IC50 = 0.064 nM), it was evident that installing 2-methoxyphenyl motif directly at C-6 of benzothiazole is optimal for activity than 6-oxypicolinamide. Therefore, in the current study, we replaced the picolinamide ether of AKE-5l with 2-methoxyphenyl moiety, while conserving the ureidobenzothiazole scaffold (). In addition, the aliphatic propyl spacer, which links ureidobenzothiazole and N-methylpiperazine in AKE-5l, was replaced by a phenyl ring to improve the anticancer spectrum while retaining the BCR-ABL inhibitory activity. The terminal phenyl was substituted with various hydrophilic and lipophilic moieties to underscore their impact on both kinase activity and cellular potency. These two major structural modifications generated a concise library of six 1–(6–(2-methoxyphenyl)benzo[d]thiazol-2-yl)-3-phenylurea derivatives 2a–f. All target compounds were evaluated against both BCR-ABLWT and BCR-ABLT315I, and a representative derivative 2b was further profiled against an array of imatinib-resistant BCR-ABL mutants.
Figure 3. Rational design of the target compounds.
Results and discussion
Experimental
The detailed experimental section is included in the supplemental material.
Chemistry
The synthetic outline for the preparation of the final compounds is illustrated in Scheme 1. The key starting material, 6-(2-methoxyphenyl)benzo[d]thiazol-2-amine (1), was obtained via Suzuki coupling of 2-amino-6-bromobenzothiazole with (2-methoxyphenyl)boronic acid using Pd(dppf)Cl2• CH2Cl2 as a catalyst under two different conditions. Initially, the reaction was conducted in 1,4-dioxane:H2O, (3:1, v/v) at 95 0C for 4 h using potassium carbonate as a base, and afforded compound 1 in 58% yield. Changing the solvent with 1,2-dimethoxyethane (DME):H2O (3:1, v/v), using sodium bicarbonate and running the reaction at 85 0C for 2 h furnished 1 in higher yield (87%). Treatment of benzothiazol-2-yl amine 1 with 1,1′-carbonyldiimidazole (CDI) in DMF afforded the corresponding isocyanate, which was coupled with the proper aniline at 100 0C to generate the target ureidobenzothiazoles.
Scheme 1. Reagents and reaction conditions: i) (2-Methoxyphenyl)boronic acid, Pd(dppf)Cl2• CH2Cl2, K2CO3, 1,4-dioxane:H2O (3:1, v:v), 95 0C, 4 h, 58%; ii) (2-Methoxyphenyl)boronic acid, Pd(dppf)Cl2• CH2Cl2, NaHCO3, DME:H2O (3:1, v:v), 85 0C, 2 h, 87% iii) a) CDI, DMF, rt, overnight; b) Appropriate substituted aniline, DMF, 100 0C, 3 h, 18–76% (over two steps).
In vitro kinase screening
All final compounds 2a–f were tested against the native BCR-ABLWT along with its clinically imatinib-resistant T315I mutant at Reaction Biology Corporation (RBC, Malvern, PA, USA)Citation26, using imatinib as a reference compound and the pan kinase inhibitor staurosporine as a positive control (). As disclosed from the data, ureidobenzothiazoles 2a–c, with hydrophilic cyclic amine at the 4-position of the terminal phenyl ring, showed excellent inhibitory potency with IC50 values spanning in the range of 0.701–2.16 nM and 0.651–8.11 nM against BCR-ABLWT and BCR-ABLT315I, respectively, being superior to the lead compound AKE-5l (BCR-ABLWT IC50 = 18.2 nM, BCR-ABLT315I IC50 = 39.9 nM). Such biochemical outcomes indicate that replacing the oxypicolinamide moiety of AKE-5l with o-methoxyphenyl, along with changing the propyl spacer into phenyl, resulted in improved BCR-ABL inhibitory activity. The existence of terminal nitrogen with small alkyl substituents, 2a and 2b, was found to be optimal for BCR-ABL inhibition. Compound 2b, with N-ethylpiperazine, exerted the best activity with sub-nanomolar IC50 values of 0.701 and 0.651 nM against BCR-ABLWT and BCR-ABLT315I, respectively. It showed 2–3 folds better activity than its methylpiperazine congener 2a (BCR-ABLWT IC50 = 1.50 nM, BCR-ABLT315I IC50 = 2.03 nM). Replacing the terminal nitrogen of piperazines 2a and 2b with oxygen 2c retained the BCR-ABLWT activity, however, it led to reduction in BCR-ABLT315I inhibition (BCR-ABLT315I IC50 = 8.11 nM). Insertion of m-trifluoromethyl group on the distal phenyl while removal of the hydrophilic cyclic amines 2d resulted in a dramatic drop in potency (BCR-ABLWT IC50 = 681 nM, BCR-ABLT315I IC50 = 1240 nM). Such decline in activity was much more pronounced upon the introduction of an additional trifluoromethyl group at the 5-position, as in 2e (BCR-ABLWT IC50 = 4090 nM, BCR-ABLT315I IC50 = 9560 nM). In contrast, incorporation of morpholine methylene at the 4-position of 2d afforded 2f, which restored inhibitory potency with IC50 values of 74.6 and 226 nM against BCR-ABLWT and BCR-ABLT315I, respectively. These findings point out the indispensable role of cyclic amines, particularly piperazine, in achieving sound BCR-ABL inhibition for this set of ureidobenzothiazoles. This importance might be attributed to the hydrophilic nature of cyclic amines, which offers a favourable orientation towards the solvent exposure region of BCR-ABL. Such a hypothesis could be emphasised by noticing the detrimental impact of the existence of m-trifluoromethyl group (2d and 2e) on the distal phenyl ring.
Table 1. In vitro inhibitory activity (IC50, nM) of the target compounds 2a–f, lead AKE-5l, and imatinib against BCR-ABLWT and BCR-ABLT315I kinases.
As an example of the piperazine ureidobenzothiazoles, compound 2b was further profiled against a panel of clinically imatinib-resistant BCR-ABL mutants, including the P-loop mutants E255K and Q252H, activation loop mutant H396P, catalytic segment mutant M351T, and mutant F317I in the ATP binding region (). The obtained findings confirmed the potent ability of 2b to inhibit the activity of the clinically relevant BCR-ABL with single-digit nanomolar IC50 values.
Table 2. In vitro enzymatic activity of 2b over a panel of clinically-resistant BCR-ABL mutants.
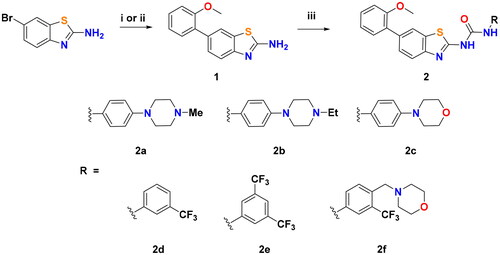
On the other hand, to gain certain insights about the kinase selectivity of this array of ureidobenzothiazoles, compounds 2a and 2b were tested against DDR1 (). Discoidin domain receptors (DDR1/2) are transmembrane receptor tyrosine kinases (RTKs), activated by fibrillar collagensCitation27. DDR1/2 and BCR-ABL share almost 61% sequence similarities in their ATP binding domains. Therefore, several BCR-ABL inhibitors like imatinib, nilotinib, and dasatinib possess potent nanomolar DDR inhibitory effects comparable to BCR-ABLCitation28. Interestingly, compounds 2a and 2b showed 130 and 284 folds selectivity for BCR-ABL (IC50 = 1.5 and 0.7 nM, respectively) over DDR1 (IC50 = 195 and 199 nM, respectively). This observation points out the merit of these ureidobenzothiazoles over the known BCR-ABL inhibitors in achieving exceptional selectivity towards BCR-ABL.
Figure 4. Dose-response curves of compounds 2a and 2b against DDR1.
In vitro cell-based evaluation of the antiproliferative activities
Assessment of the antiproliferative activity by MTT assay
Motivated by the promising results of cell-free assay, we examined the antiproliferative activity of target compounds against the BCR-ABL overexpressing human leukaemia K-562 cells, along with L132 normal cell by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay (). Consistent with biochemical kinase findings, the most active ureidobenzothiazoles 2a–c displayed good anticancer activity against K-562 cell with GI50 values of 3.39–4.52 µM. On the other hand, the m-trifluoromethylphenyl substituted members 2d–f exerted modest cellular potency (GI50 > 10 µM). These cellular outcomes define the impact of BCR-ABL inhibition on the proliferation of K-562 cells. Moreover, compounds 2a–c showed weak cytotoxic effects over L132 normal cell, as evidenced by high GI50 values (17.86–26.78 µM). In terms of selectivity index (SI), the top potent compounds 2a–c had reasonable SI values spanning in the range of 5.3–6.8, which indicate their selective antiproliferative activity towards cancer cell K-562.
Table 3. MTT antiproliferative activity (GI50, μM) of compounds 2a–f against leukaemia K-562 and L132 normal cella.
Broader assessment of anticancer activity by sulforhodamine B (SRB) assay
Single dose assay
To gain further insights about the anticancer profile of this new ureidobenzothiazole chemotypes, the structures of all final compounds were submitted to the National Cancer Institute (NCI, Developmental Therapeutics Program, Bethesda, MD, USA). Based on NCI selection criteria, compounds 2b (AK-HW-90), 2e, and 2f were chosen for broad screening against a library of 60 human cancer cell lines, representing nine types of blood and solid cancers. Employing the highly sensitive sulforhodamine B (SRB) assayCitation29, compounds 2b, 2e, and 2f were tested initially at a single dose of 10 μM, and their detailed activities on the percentages growth of the NCI-60 cell lines were illustrated in , and compared with that observed for the lead benzothiazole AKE-5l.
Figure 5. Growth percentages of the NCI-60 cell lines panel after treatment with 10 µM dose of compounds 2b (blue), 2e (red) 2f (green), and lead benzothiazole AKE-5l (purple).
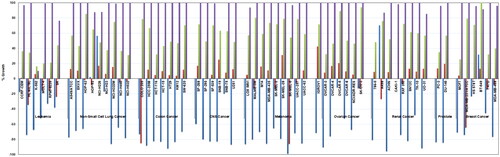
Interestingly, all three compounds 2b, 2e, and 2f showed superior anticancer potency (low % growth values) rather the lead ureidobenzothiazole AKE-5l. While AKE-5l showed narrow spectrum activity towards only K-562 cell line (% growth = 10.22), the o-methoxyphenyl benzothiazoles 2b, 2e, and 2f exerted broad spectrum anticancer effect against multiple cancer cells (). Among these compounds, the most potent BCR-ABL inhibitor 2b elicited the best cellular activity, where it manifested remarkable lethal effects (minus % growth values) over 55 cancer cells. Since both lead AKE-5l and 2b (AK-HW-90) share N-alkylpiperazine and ureidobenzothiazoles as common structural features, it was evident that replacing oxypicolinamide moiety and propyl spacer of AKE-5l with o-methoxyphenyl and phenyl ring in 2b, respectively, led to dramatic change in the anticancer property from narrow into expansive spectrum covering diverse cancer cells. Next in the activity rank is the 3,5-bis(trifluoromethyl)phenyl derivative 2e, which showed cytotoxic rather cytostatic activity against 21 cancer cells. In contrast to 2b and 2e, compound 2f evinced reasonable cytostatic growth inhibitory activity (% growth less than 40) towards 14 cancer cells. Despite of the modest BCR-ABL inhibitory potency of 2e and 2f, they showed good anticancer activity over NCI-60 cell lines, particularly 2e, which points out the existence of other underlying mechanism(s), beside kinase inhibition, that contribute to the anticancer property of this novel chemotype.
Five dose assay
The promising anticancer activities of compounds 2b and 2e fulfilled the NCI criteria, therefore, they were advanced to five-dose testing mode to determine their GI50 (the molar concentration causing 50% growth inhibition), TGI (the molar concentration producing 100% GI) and LC50 (the molar concentration achieving 50% lethality or tumour regression). The GI50 values, a measure of compound’s potency, of 2b and 2e along with imatinibCitation30 are presented in . Both compounds 2b and 2e showed high potency with one-digit micromolar GI50 values against all examined cell lines. Interestingly, 2b and 2e elicited superior anticancer potencies than imatinib over 55 and 54 cell lines, respectively. Regarding leukaemia, both 2b and 2e outperformed imatinib over five leukaemia cell lines; CCRF-CEM and MOLT-4 cells (ALL), HL-60 (TB) (AML), SR (ALK-positive anaplastic large cell lymphoma), and PRMI-8226 (multiple myeloma). For example, 2b and 2e showed equipotent activity against SR cell with GI50 value of ∼ 1.70 µM. Moreover, the growth of imatinib-insensitive lymphoma CCRF-CEM cell was potently suppressed by 2b (GI50 = 1.81 µM) and 2e (GI50 = 2.43 µM). The ethylpiperazine derivative 2b exhibited significant antileukemic activity over the BCR-ABL positive cell K-562 with GI50 value of 0.293 µM.
Table 4. GI50 values (µM) of 2b, 2e, and imatinib against NCI 60-cell line panela,b.
From another perspective, it was documented that imatinib could ameliorate the hypoxic circumstances in non-small cell lung cancer (NSCLC)Citation31, beside its ability to improve drug delivery and efficacy in NSCLC xenograftsCitation32. In this aspect, it is interesting to observe that both compounds 2b and 2e possess superior potency to imatinib over seven of the tested NSCL cancer cells. For instance, compounds 2b and 2e displayed GI50 values of 1.47 µM and 1.19 µM against HOP-92 cell line, respectively, with 9–11 folds better potency than imatinib (GI50 = 13.34 µM). The ethylpiperazine member 2b slightly surpassed the potency of its 3,5-bis-trifluoromethyl congener 2e over six cell lines of NSCL cancer panel. Compound 2b exerted equipotent anticancer effects (GI50 ∼ 1.76 µM) over NCI-H460 and NCI-H522 cell lines.
On the other hand, imatinib has shown beneficial clinical effects for the treatment of certain metastatic/inoperable gastrointestinal stromal tumours (GIST)Citation33. In this aspect, it is noteworthy reporting that both 2b and 2e elicited considerable higher anticancer activities than imatinib over all tested colon cancer cell lines. The growth of HT29, the most imatinib-responsive colorectal cancer cell, was strongly inhibited by 2b and 2e, with GI50 values of 1.31 µM and 2.15 µM, respectively. While imatinib exerted modest anticancer activity over COLO205, HCC-2998, HCT-116 and multidrug-resistant HCT-15 colon cancer cells (GI50 = 12.59–21.23 µM), 2b and 2e showed single-digit micromolar GI50 values spanning in the range of 1.07–2.65 µM. Such promising cellular findings recommend further investigations of 2b and 2e as potential candidates for colorectal cancer.
Since glioblastoma (GBM), the most aggressive form of brain tumoursCitation34, is resistant to the majority of tyrosine kinase inhibitors, including imatinibCitation35, it is important to point out that both 2b and 2e were remarkably superior to imatinib across all six examined brain cancer cell lines. Moreover, 2b and 2e exerted good anticancer effects against the temozolomide (TMZ)-resistant cell lines SF-295, SNB-75, SNB-19, and SF-268 gliomas. For instance, the ethyl piperazine member 2b suppressed the growth of the SNB-19 cell line with a GI50 value of 2.03 µM. Referring to melanoma, both 2b and 2e showed comparable potencies against all tested melanoma cell lines, surpassing imatinib, with GI50 values of 1.54–2.51 µM.
Moreover, imatinib efficacy in the treatment of ovarian cancers as a combination with docetaxel was reportedCitation36. In this line, it is interesting to find that compounds 2b and 2e have significant anticancer activity outperforming imatinib over all tested ovarian cancer cells, except OVCAR-5. Particularly, 2b showed GI50 values less than 2.0 µM over all cell lines. Of special interest, ureidobenzothiazoles 2b and 2e exerted appreciable potency towards the paclitaxel-resistant NCI/ADR-RES cell line, with GI50 values of 1.72 µM and 2.37 µM, respectively.
Beside the aforesaid-targeted cellular potencies of 2b and 2e, they elicited broad-spectrum activities over multiple cell lines originating from different cancer tissues. As an example, multidrug-resistant (MDR) UO-31 and RXF-393 renal cells showed high sensitivity to 2b (GI50 = 1.52 µM and 1.70 µM) and 2e (GI50 = 1.79 µM and 2.19 µM), respectively. Moreover, the top active member 2b exhibited pronounced anticancer potencies over the prostate cancer cell PC-3, MCF7, and triple-negative breast cancer cell MDA-MB-468 with GI50 values of 1.79, 1.31, and 2.17 µM, respectively.
Regarding the efficacy parameters (TGI and LC50 values) of ureidobenzothiazoles 2b and 2e (), it was found that compound 2b with hydrophilic ethylpiperazine motif possesses better efficacy than 2e against several cancer cells with TGI < 5.0 µM and LC50 < 10.0 µM. For example, 2b induced total growth inhibition against K-562 and HCT-15 cell lines, with TGI values of 1.34 and 2.47 µM, respectively.
Table 5. TGI and LC50 values (µM) of 2b and 2e over the most sensitive cell linesa,b.
2b
Molecular docking study
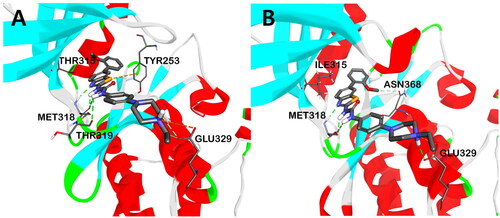
To get insights about the binding mode of this new set of ureidobenzothiazoles, molecular docking for the most potent candidate 2b (AK-HW-90) into the catalytic kinase domain of wild-type BCR-ABL (PDB code: 2GQG)Citation37 and its T315I mutant (PDB code: 2Z60)Citation38, in their DFG-in conformation, was conducted. As shown in , compound 2b showed tight binding within the ATP binding site of both BCR-ABLWT and BCR-ABLT315I kinases, as demonstrated by the formation of crucial three hydrogen bonds (HB) with hinge region residue Met318. The ring nitrogen of the benzothiazole scaffold was engaged in HB with amide hydrogen of Met318, while the two NH hydrogens of urea moiety formed two HBs with the amino carbonyl oxygen of Met318. In addition, attractive charge interaction was noticed between the terminal nitrogen of ethyl piperazine and the side chain carboxyl group of Glu329 at the solvent-accessible channel of the BCR-ABL kinase. Moreover, the methoxy group shared carbon-hydrogen bonds with the side chain hydroxyl group of Thr315 in BCR-ABLWT. However, in the case of BCR-ABLT315I, due to steric hindrance with Ile315, which has a larger residual volume than Thr315, such carbon-hydrogen interactions were lacking. Instead, π-alkyl interactions were observed between the methoxy group and the side chain phenyl group of the Tyr253 residue (Figure S1). Besides, in the case of BCR-ABLWT, π-sulphur interaction was noticed between the sulphur atom of benzothiazole and the phenyl group of the Tyr253 residue. The aforementioned binding interactions might justify the sub-nanomolar potency of 2b against both BCR-ABLWT and its T315 mutant.
Figure 6. The putative binding mode of compound 2b in the kinase domain of BCR-ABLWT (A) and BCR-ABLT315I (B). The hydrogen bond interactions are shown in green dotted lines.
In silico bioavailability prediction
The bioavailability of compound 2b was predicted by SwissADME online platformCitation39. The bioavailability radar panel evaluates the bioactive candidates in terms of their physicochemical parameters, such as lipophilicity, flexibility, polarity insolubility, size, and saturation. Interestingly, compound 2b was found to be in the pink region (optimal ranges) with favourable drug-like properties (Figure S2).
Conclusion
In the current investigation, we report the design and synthesis of new phenyl-ureidobenzothiazoles 2a–f in an effort to improve the BCR-ABL inhibitory activity of our previously reported lead AKE-5l. Two major structural modifications of AKE-5l were undertaken herein, replacing the 6-oxypicolinamide moiety and the propyl spacer of AKE-5l with o-methoxyphenyl and phenyl ring, respectively. Among the target compounds, both 2a and its ethylpiperazine congener AK-HW-90 (2b) stood as the most potent members with IC50 values of 2.0 and 0.65 nM against BCR-ABLT315I, respectively, surpassing the activity of lead AKE-5l (IC50 = 39.9 nM). Furthermore, AK-HW-90 (2b) potently inhibited the activity of a panel of imatinib-resistant mutants with single digit nanomolar IC50 values. On the cellular level, in contrast to the narrow spectrum anticancer activity of the lead AKE-5l, compound AK-HW-90 (2b) exerted considerable broad-spectrum anticancer activity across multiple cell lines including the CML K-562 cell. In addition, AK-HW-90 (2b) surpassed the activity of imatinib over more than 50 cancer cell lines with single digit micromolar GI50 values. Molecular docking study of AK-HW-90 (2b) was conducted to get insights about its postulated binding mode with BCR-ABLWT and BCR-ABLT315I. Considering the cell-free and cell-based assay results, compound AK-HW-90 (2b) may serve as a promising chemotherapeutic candidate for CML and other cancer types.
Supplemental Material
Download PDF (2.2 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am J Hematol. 2020;95(6):691–709.
- Faderl S, Garcia‐Manero G, Thomas DA, Kantarjian HM. Philadelphia chromosome‐positive acute lymphoblastic leukemia–current concepts and future perspectives. Rev Clin Exp Hematol. 2002;6(2):142–160.
- Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1(7):493–502.
- Pandrala M, Bruyneel AAN, Hnatiuk AP, Mercola M, Malhotra SV. Designing Novel BCR-ABL Inhibitors for Chronic Myeloid Leukemia with Improved Cardiac Safety. J Med Chem. 2022;65(16):10898–10919.
- Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaecla J, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108(1):28–37.
- O'Hare T, Eide CA, Deininger MWN. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110(7):2242–2249.
- Quintas-Cardama A, Cortes J. Therapeutic options against BCR-ABL1 T315I-positive chronic myelogenous leukemia. Clin Cancer Res. 2008;14(14):4392–4399.
- Le Coutre P, Ottmann OG, Giles F, Kim D-W, Cortes J, Gattermann N, Apperley JF, Larson RA, Abruzzese E, O'Brien SG, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111(4):1834–1839.
- Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305(5682):399–401.
- Boschelli DH, Ye F, Wang YD, Dutia M, Johnson SL, Wu B, Miller K, Powell DW, Yaczko D, Young M, et al. Optimization of 4-phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J Med Chem. 2001;44(2001):3965–3977.
- Vener C, Banzi R, Ambrogi F, Ferrero A, Saglio G, Pravettoni G, et al. First-line imatinib vs second-and third-generation TKIs for chronic-phase CML: a systematic review and meta-analysis. Blood Adv . 2020;4(12):2723–2735.
- FDA. https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/ponatinib-marketed-iclusig-informaton
- Huang WS, Metcalf CA, Sundaramoorthi R, Wang YH, Zou D, Thomas RM, et al. Discovery of 3-[2-(Imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a Potent, Orally Active Pan-Inhibitor of Breakpoint Cluster Region-Abelson (BCR-ABL) Kinase Including the T315I gatekeeper mutant. J Med Chem. 2010;53(12):4701–4719.
- O'Hare T, Shakespeare WC, Zhu XT, Eide CA, Rivera VM, Wang F, et al. AP24534, a Pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412.
- Moslehi JJ, Deininger M. Tyrosine kinase inhibitor–associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol. 2015;33(35):4210–4218.
- Jiang Q, Li ZR, Qin YZ, Li WM, Xu N, Liu BC, et al. Olverembatinib (HQP1351), a well-tolerated and effective tyrosine kinase inhibitor for patients with T315I-mutated chronic myeloid leukemia: results of an open-label, multicenter phase 1/2 trial. J Hematol Oncol. 2022;15(1):1–17.
- Liu XS, Wang BL, Chen C, Jiang ZR, Hu C, Wu H, et al. Discovery of (E)-N-(4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)-3-((3-(2-(pyridin-2-yl)vinyl)-1H-indazol-6-yl)thio)propanamide (CHMFL-ABL-121) as a highly potent ABL kinase inhibitor capable of overcoming a variety of ABL mutants including T315I for chronic myeloid leukemia. Eur J Med Chem. 2018;160:61–81.
- Hong S, Kim J, Yun SM, Lee H, Park Y, Hong SS, et al. Discovery of new benzothiazole-based inhibitors of breakpoint cluster region-abelson kinase including the T315I mutant. J Med Chem. 2013;56(9):3531–3545.
- El-Damasy AK, Cho NC, Kang SB, Pae AN, Keum G. ABL kinase inhibitory and antiproliferative activity of novel picolinamide based benzothiazoles. Bioorg Med Chem Lett. 2015;25(10):2162–2168.
- El-Damasy AK, Cho NC, Nam G, Pae AN, Keum G. Discovery of a nanomolar multikinase inhibitor (KST016366): A new benzothiazole derivative with remarkable broad-spectrum antiproliferative activity. ChemMedChem. 2016;11(15):1587–1595.
- Lee JH, El-Damasy AK, Seo SH, Gadhe CG, Pae AN, Jeong N, et al. Novel 5,6-disubstituted pyrrolo[2,3-d]pyrimidine derivatives as broad spectrum antiproliferative agents: Synthesis, cell based assays, kinase profile and molecular docking study. Bioorg Med Chem. 2018;26(21):5596–5611.
- Fang ZH, Han B, Jung KH, Lee JH, El-Damasy AK, Gadhe CG, et al. A novel tropomyosin-related kinase A inhibitor, KK5101 to treat pancreatic cancer. Cancer Lett. 2018;426:25–36.
- El-Damasy AK, Haque MM, Park JW, Shin SC, Lee JS, Kim EE, et al. 2-Anilinoquinoline based arylamides as broad spectrum anticancer agents with B-RAF(V600E)/C-RAF kinase inhibitory effects: Design, synthesis, in vitro cell-based and oncogenic kinase assessments. Eur J Med Chem. 2020;208:112756.
- El-Damasy AK, Jin H, Seo SH, Bang EK, Keum G. Design, synthesis, and biological evaluations of novel 3-amino-4-ethynyl indazole derivatives as Bcr-Abl kinase inhibitors with potent cellular antileukemic activity. Eur J Med Chem. 2020;207:112710.
- El-Damasy AK, Seo SH, Cho NC, Pae AN, Kim EE, Keum G. Design and synthesis of new 2-anilinoquinolines bearing N-methylpicolinamide moiety as potential antiproliferative agents. Chem Biol Drug Des. 2017;89(1):98–113.
- Reaction Biology Corporation. http://www.reactionbiology.com/webapps/site/Kinase_Assay_Protocol.aspx
- Vogel W. Discoidin domain receptors: structural relations and functional implications. Faseb J. 1999;13(9001):S77–S82.
- Day E, Waters B, Spiegel K, Alnadaf T, Manley PW, Buchdunger E, et al. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur J Pharm. 2008;599(1-3):44–53.
- Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116.
- https://dtp.cancer.gov/dtpstandard/dwindex/index.jsp
- Vlahovic G, Rabbani ZN, Herndon JE, Dewhirst MW, Vujaskovic Z. Treatment with Imatinib in NSCLC is associated with decrease of phosphorylated PDGFR-beta and VEGF expression, decrease in interstitial fluid pressure and improvement of oxygenation. Brit J Cancer. 2006;95(8):1013–1019.
- Vlahovic G, Ponce AM, Rabbani Z, Salahuddin FK, Zgonjanin L, Spasojevic I, et al. Treatment with imatinib improves drug delivery and efficacy in NSCLC xenografts. Brit J Cancer. 2007;97(6):735–740.
- Mazzocca A, Napolitano A, Silletta M, Ceruso MS, Santini D, Tonini G, et al. New frontiers in the medical management of gastrointestinal stromal tumours. Ther Adv Med Oncol. 2019;11:1–13.
- Shergalis A, Bankhead A, Luesakul U, Muangsin N, Neamati N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol Rev. 2018;70(3):412–445.
- Kim G, Ko YT. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch Pharmacal Res. 2020;43(4):385–394.
- Matei D, Emerson RE, Schilder J, Menning N, Baldridge LA, Johnson CS, et al. Imatinib mesylate in combination with docetaxel for the treatment of patients with advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis - A hoosier oncology group trial. Cancer. 2008;113(4):723–732.
- Tokarski JS, Newitt JA, Chang CYJ, Cheng JD, Wittekind M, Kiefer SE, et al. The structure of dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006;66(11):5790–5797.
- Zhou T, Parillon L, Li F, Wang Y, Keats J, Lamore S, Xu Q, Shakespeare W, Dalgarno D, Zhu X, et al. Crystal structure of the T315I mutant of Abl kinase. Chem Biol Drug Des. 2007;70(3):171–181.
- http://www.swissadme.ch/index.php