
Abstract
The dual c-Met/vascular endothelial growth factor receptor 2 (VEGFR-2) TK inhibition is a good strategy to overcome therapeutic resistance to small molecules VEGFR-2 inhibitors. In this study, we designed 3-substituted quinazoline-2,4(1H,3H)-dione derivatives as dual c-Met/VEGFR-2 TK inhibitors. We introduced new synthetic methods for reported derivatives of 3-substituted quinazoline-2,4(1H,3H)-dione 2a–g, in addition to the preparation of some new derivatives namely, 3 and 4a–j. Three compounds namely, 2c, 4b, and 4e showed substantial amount of inhibition for both c-Met and VEGFR-2 TK (IC50 range 0.052–0.084 µM). Both compounds 4b, 4e showed HB with highly conserved residue Asp1222 in the HB region of c-Met TK. For VEGFR-2 TK, compound 4b showed HB with a highly conserved residue Asp1046 in the HB region. Compound 4e showed HB with Glu885 and Asp1046. Moreover, in silico prediction of pharmacokinetic and physicochemical parameters of target compounds was carried out using SwissADME website. The quinazoline-2,4(1H,3H)-dione derivatives are promising antiproliferative candidates that require further optimisation.
New 3-substituted quinazoline-2,4(1H,3H)-dione derivatives were synthesised and characterised.
Compounds 4b and 4e showed higher cytotoxic activity than cabozantinib against HCT-116 colorectal cell lines.
Both compounds 4b and 4e showed less toxicity to WI38 normal cell line compared to HCT 116 colon cancer cell line.
Compound 4b was superior to cabozantinib in VEGFR-2 inhibition while compound 2c was equipotent to cabozantinib.
Compounds 4b and 4e showed remarkable c-Met inhibitory activity.
Compounds 4b and 4e arrested cell cycle and induced significant levels of apoptosis.
In silico ADME prediction revealed high oral bioavailability and enhanced water solubility of target compounds as compared to cabozantinib.
Target compounds interacted with both c-Met and VEGFR-2 active site in similar way to cabozantinib.
Highlights
Introduction
Globally, cancer kills about 10 million people every year and its mortality rate will reach 16.3 million deaths in 2040Citation1. Conventional chemotherapeutic drugs can raise the survival rate of cancer patients but they have negative impact on patients’ quality of life because of their severe adverse effectsCitation2. With growing knowledge of chemistry and function of cellular molecular targets, tyrosine kinase (TK) inhibitors become the main arm of chemotherapeutic arsenalCitation3. Compared to conventional chemotherapy, TK inhibitors afford good anticancer activity with few and tolerable adverse effectsCitation2.
The activation of c-Met TK receptor is achieved by binding of HGF/SF, that binding promotes different cellular signalling pathways and results finally in cell proliferation, motility, migration, and survivalCitation4. These cellular activities are fundamental in normal physiological processes like wound healing but can lead to tumorigenesis when these pathways are deregulatedCitation5. c-Met TK was found to be overexpressed or mutated in human tumours and it was related to poor prognosisCitation4. Consequently, c-Met was considered as a potential molecular target for cancer therapyCitation6.
Another TK is a vascular endothelial growth factor receptor 2 (VEGFR-2), which is expressed in endothelial cells, mediated by VEGF, and fundamentally regulates the angiogenesis of tumoursCitation7,Citation8. Albeit VEGF/VEGFR pathway is overexpressed in cancer cells and it is considered as important drug target, many antiangiogenic drugs suffering from humble efficacyCitation9. Many neoplasms acquired therapeutic resistance against bevacizumab as well as small molecules inhibitors that target catalytic domain of VEGFR-2Citation10–13. There are several mechanisms beyond this resistance like expression of Tie2 by macrophageCitation9, tumour hypoxiaCitation14, upregulation of SDF,1Citation15 and overexpression of pERKCitation16. It is found that HGF/c-Met pathway drives this resistance and dual inhibition of c-Met/VEGFR-2 was found to be effective against tumours that are resistant to pan-VEGFR inhibitionCitation14,Citation17. Single-target VEGFR-2 inhibitors does increase hypoxia which leads to c-Met mediated invasion and metastasis of cancer cells. On the contrary, the combination of VEGFR-2 and c-Met inhibitors, like sunitinib and PF-04217903 remarkably lowers tumour invasion and metastasis.Citation18 Hence regarding drug resistance, multitarget TK inhibitors are more superior than single target inhibitorsCitation19.
Four structural requirements for dual c-Met/VEGFR-2 inhibition are heterocycle, linker, HB domain, and hydrophobic tail ()Citation19. Aza-heterocycle occupies adenine site in hinge region and forms from 0 to 3 HBs with highly conserved residues Met1160 and Cys919 in c-Met and VEGFR-2, respectively. Heterocycle ring can be surrogated by phenyl ring with HB groups like halogens and carboxamide ()Citation18,Citation20. Linker occupied the region adjacent to gatekeeper. It has a little role in protein binding, but it is important to fix other pharmacophoric elements in the corresponding binding sitesCitation8. It may be (hetero)aromatic or aliphatic chain ()Citation6,Citation7,Citation18,Citation20–22.
Figure 1. Pharmacophoric elements for dual inhibition of c-Met/VEGFR-2 TKs.
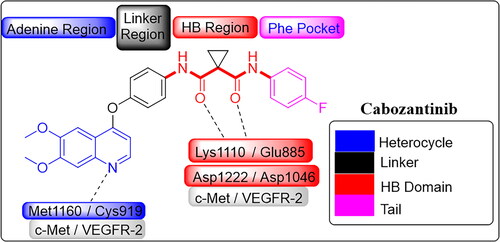
Figure 2. Four structural elements of reported dual c-Met/VEGFR-2 inhibitors.
HB domain can form HB with Asp1046 and Asp1222 of the DFG motif in c-Met and VEGFR-2, respectively. For c-Met inhibition, HB domain acts as HBA by forming two HB by two carbonyl oxygen with Asp1222 and Lys1110 while for VEGFR-2 inhibition, HB domain acts as dual HBA/HBD by forming two HB by carbonyl oxygen and amidic NH with Asp1046 and Glu885Citation8,Citation23. HB domain can be dicarboxamide, 3-sulfonylacrylamide, (thio)urea, N-acyl(thio)urea, carbamoylmethyl carboxylate, N-acylhydrazone, and semicarbazoneCitation3,Citation18,Citation22,Citation24–27. It also can be incorporated in nitrogenous five or six membered ring system with carboxamide substituent like pyrazole, imidazole, 1,2,3-triazole, pyridine, pyrimidine, pyridazine, quinoline, cinnoline, and naphthyridineCitation3,Citation4,Citation6,Citation7,Citation20,Citation21,Citation24,Citation25,Citation28–33. Hydrophobic tail occupies the phenyl pocket in the allosteric site of TK and it may be phenyl, (non)aromatic heterocyclic, or alicyclic ring ()Citation3,Citation19,Citation34.
Therefore, encouraged by the abovementioned rationale, we designed and synthesised a series of 3-substituted quinazoline-2,4(1H,3H)-dione analogues via a multi component reaction (). The antiproliferative activity of target compounds was performed against c-Met/VEGFR-2 overexpressing HCT-116 colon cancer cell line. In addition, cytotoxicity of selected compounds against WI38 normal cells was carried out to assess safety profile of new series of compounds. In vitro screening of inhibitory activity against both c-Met and VEGFR-2 was carried out using cabozantinib as a positive control. Cell cycle analysis and apoptosis assay were carried out. Furthermore, the molecular docking studies were performed to investigate the interactions of the target molecules with c-Met and VEGFR-2 TKs. Finally, in silico pharmacokinetic profile and druglikeness of the synthesised molecules were investigated by SwissADME.
Figure 3. General structures of target compounds.
Results and discussion
Chemistry
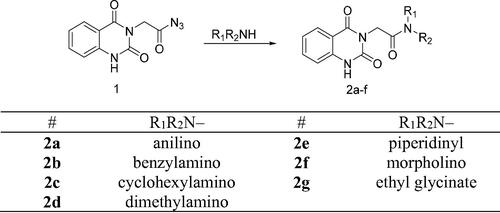
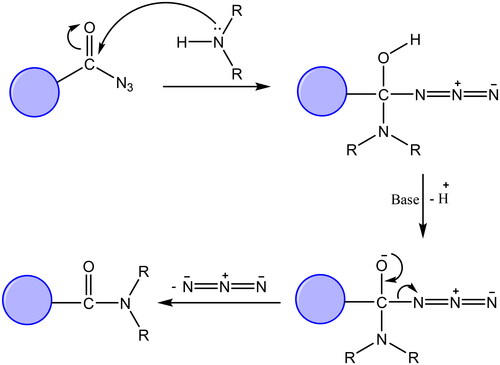
The synthetic protocol adopted in this study is illustrated in Schemes 1 and 2. The azide 1 was first prepared by Younes et al. by treatment of a suspension of 2-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)acetohydrazide in hydrochloric acid with a solution of sodium nitrite in water at 0–5 °CCitation35. Treatment of a cold solution of azide 1 with different primary and secondary amines afforded a new method to synthesise reported amides 2a–gCitation36–40. In these reactions, a low temperature (around 0 °C) is required to prevent decomposition of the acyl azide which releases nitrogen gas. The coupling step was performed under basic conditions. In our study, the amines were used as reactants as well as basic catalysts. For the synthesis of compound 2g, we used triethylamine as a catalyst. Another method was also used for the synthesis of compounds 2b, 2c, 2g in which it was observed that the coupling reaction can be performed under reflux as it is finished in a time less than that needed for decomposition of the azide. The reflux was carried out in dry benzene for compounds 2b and 2c, while dry pyridine was utilised as a solvent and basic catalyst for the synthesis of compound 2g ()Citation41.
Scheme 1. Synthesis of target compounds 2a–g.
Scheme 2. Synthesis of target compounds 3 and 4a–j; reagents and conditions: (a) N2H4·H2O, EtOH, reflux. (b) Ar-CHO, piperidine, EtOH, reflux. (c) Cyclohexanone, EtOH, reflux. (d) Isatin, AcOH, reflux. (e) Acetylacetone, EtOH, reflux. (f) CS2, pyridine, reflux. (g) PhNCS, EtOH, reflux. (h) NaOH.
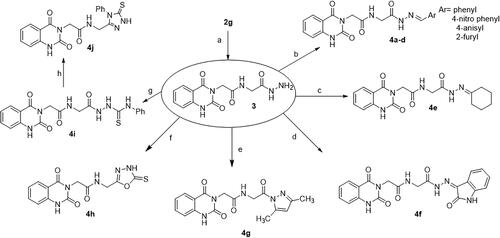
Figure 4. Suggested mechanism of reaction of acyl azide with primary and secondary amines.
The reaction of azide 1 with glycine ethyl ester hydrochloride gave 2g. Then, treatment of compound 2g with hydrazine hydrate yielded hydrazide 3. IR spectrum of 3 showed characteristic absorption bands for NH2 and NH’s at 3291 and 3175, and for C═O’s at 1704 and 1644. Besides, the 1H NMR spectrum revealed the disappearance of the triplet and quartet signals related to the ethyl group of the starting ester 2g, and the appearance of new characteristic singlet signals for NH and NH2 groups of the resulting hydrazide at δ 9.02 and 4.26 ppm, respectively. Compound 3 is a useful precursor in the synthesis of target compounds 4a–j.
Condensation of hydrazide 3 with various aromatic aldehydes namely, benzaldehyde, 4-nitrobenzaldehyde, anisaldehyde, and furfural afforded a series of hydrazone derivatives 4a–d, respectively in good yields. It is known that N-acylhydrazones may exist as E/Z geometrical isomers with respect to the imine C═N double bond, and cis/trans amide conformers due to rotation across the amide C(O)–NH single bond ()Citation42,Citation43. According to the literature, N-acylhydrazones derived from aryl and heteroaryl aldehydes tend to exist in the form of the less hindered E geometrical isomer both in the solid state and in DMSO solution; however, in less polar solvents, the Z isomer can be detected due to its stabilisation with intramolecular hydrogen bonds.Citation42–48
Figure 5. Isomers of N-acylhydrazones.
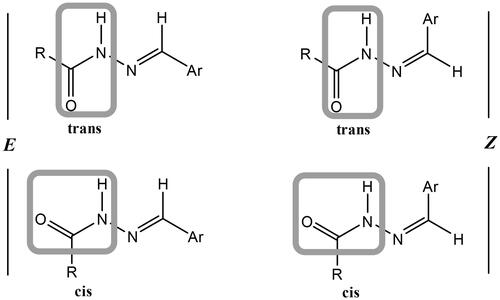
From the spectroscopic studies of the obtained hydrazones 4a–d, IR spectra showed characteristic stretching bands for NH’s in the range of 3249–3127 cm−1, for C═O’s in the range of 1736–1644 cm−1 and for C═N in the range of 1570–1518 cm−1. The 1H NMR spectra of these hydrazones exhibited two sets of signals for certain protons (NCH2C(O)NH, α-CH2, C(O)–NH–N, N═CH), which revealed their existence as an equilibrium mixture from cis/trans conformers of the E geometrical isomer. In addition to the abovementioned studies, this conclusion is supported by previous studies on similar hydrazones.Citation49,Citation50 It was found that the cis-conformer predominates in DMSO solution.Citation42 For α-CH2 protons, the smaller upfield signal is assigned to the trans-conformer while the larger downfield signal to the cis-conformer. On the contrary, for N═CH proton the larger upfield signal is attributed to the cis-conformer while the smaller downfield signal to the trans-conformer. These data are in accordance with literature results.Citation51 The signals of NCH2C(O)NH and C(O)–NH–N protons of cis- and trans-conformers were recognised from the integration of their signals relative to those of α-CH2 and N═CH protons (). The ratio of cis- to trans-conformers for every compound was calculated from the ratio of integrations of the paired peaks (see Supplementary Data). 13C NMR spectra also displayed duplicated signals for certain carbon atoms, which confirmed the presence of a mixture of conformational isomers.
Table 1. In vitro cytotoxicity of compounds 2a–g, 3, 4a–j, and cabozantinib against HCT116 cell line.
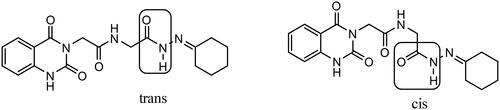
The reaction of carbohydrazide 3 with cyclohexanone afforded 4e. IR spectrum of 4e showed characteristic stretching bands for NH’s at 3256 and 3210 cm−1, for C═O’s at 1738 and 1647 cm−1 and for C═N at 1540 cm−1. In 1H NMR spectrum of hydrazone 4e in DMSO-d6, the NHCH2 appeared as two doublets with similar intensities at δ 3.84 and 4.15 ppm for CH2 protons and two triplets with similar intensities at δ 8.32 and 8.51 ppm for NH proton. Furthermore, C(O)–NH–N proton appeared as two singlets at δ 10.14 and 10.46 ppm also with similar intensities. We might conclude that the hydrazone 4e solution in DMSO is present in the form of cis/trans amide conformers in 1:1 ratio due to rotation across the amide C(O)–NH single bond ().
Figure 6. Cis/trans amide conformers of hydrazone 4e.
Refluxing of carbohydrazide 3 with isatin resulted in the formation of hydrazone 4f. The 1H NMR spectrum of this hydrazone in DMSO-d6 showed that some protons exhibit two sets of signals for each proton (NCH2C(O)NH, α CH2, C(O)–NH–N). The downfield shift of the two peaks related to the NH–N proton (δ 12.55 and 13.26 ppm) indicates the presence of intramolecular hydrogen bonding stabilising the Z geometrical isomer. Thus, it is more likely that compound 4f is found as an equilibrium mixture of cis/trans conformers of the Z geometrical isomer due to rotation across the amide CO–NH single bond, and the ratio of cis to trans conformers is 7:4, respectively.Citation52,Citation53
The compound 4g was prepared via the Knorr pyrazole synthesis by treatment of hydrazide 3 with acetylacetone as a β-diketone. The 1HNMR spectrum displayed singlet signals at δ 1.77, 1.96, and 6.40 ppm indicating the presence of two methyl groups and one methine group, beside the disappearance of NH and NH2 signals related to the hydrazide 3. 13C NMR spectrum was also used to confirm the structure where the two CH3 carbons appeared at 16.32 and 26.38 ppm.
The compound 4h was prepared by heating carbohydrazide 3 with carbon disulphide in pyridine. IR spectrum showed absorption bands at 3304 and 3214 attributed to NH’s, at 1714 and 1661 attributed to C═O’s, and at 1140 attributed to C═S. The 1H NMR spectrum showed the disappearance of NH2 signal of hydrazide 3 and the appearance of a new characteristic signal at δ 10.98 ppm related to NHCS proton of compound 4h.
The reaction of hydrazide 3 with phenyl isothiocyanate yielded 4i. IR spectrum showed bands for NH’s at 3444, 3285, and 3201 cm–,Citation1 for C═O’s at 1735, 1704, and 1668 cm–,Citation1 and for C═S at 1267 cm−1. The 1H NMR spectrum indicated a multiplet for aromatic protons (9H) at δ 7.06–7.95 ppm, besides the disappearance of NH2 signal of hydrazide 3 and the appearance of new characteristic signals at δ 9.31, 9.74, and 10.21 ppm related to the new NH groups.
Heating the thiosemicarbazide 4i in NaOH solution under gentle reflux leads to the formation of 4j. IR spectrum showed bands for NH’s at 3272 and 3200 cm−1, for C═O’s at 1718 and 1658 cm−1, and for C═S at 1210 cm−1. The 1H NMR spectrum showed the disappearance of the signals of NH groups of the thiosemicarbazide and the appearance of a new characteristic signal at δ 13.86 ppm related to the NH proton of the triazole ring.
Biology
In vitro antiproliferative activity against HCT-116
The target compounds were evaluated for their anticancer activity against HCT-116 colon cancer cell line in which both c-Met and VEGFR-2 are overexpressed.Citation54,Citation55 MTT assay was carried out to test the effect of different concentrations of the target compounds on HCT-116 cell line using DMSO as a negative control whereas cabozantinib was used as positive control. The half maximal inhibitory concentration (IC50) was calculated for each compound (). Most target compounds exhibited significant anticancer activities. Amongst all compounds, compound 4b exhibited promising inhibition activity with IC50 less than 1 µM. Eleven out of 18 compounds namely, 2a, 2b, 2c, 2f, 2g, 4b, 4d, 4e, 4g, 4h, and 4i displayed remarkable anticancer activity (IC50=0.734–16.02 µM) and they were more superior than cabozantinib. On the other hand, compound 4j (IC50=18.89 µM) showed comparable anticancer activities to cabozantinib. Compounds 2d, 2e, 3, 4a, 4c, and 4f showed noticeable anticancer activity (IC50=24.29–47.52 µM).
In vitro toxicity against normal cells
To scrutinise the safety of target compounds on normal cells, compounds 4b and 4e were selected to evaluate their cytotoxic activity against WI38 normal cell line (). Both compounds showed less toxicity to WI38 normal cell line compared to HCT 116 colon cancer cell line. Compounds 4b and 4e exhibited cytotoxic activity against HCT-116 colon cancer cells 25 and 7.5 times more than WI38 normal cells, respectively.
Table 2. In vitro cytotoxicity of compounds 4b and 4e against WI38 normal cell line.
In vitro activity against c-Met and VEGFR-2 tyrosine kinases
As our rationale is to target c-Met/VEGFR-2 TKs, herein, we selected six 3-substituted quinazoline-2,4(1H,3H)-dione derivatives for evaluation of their inhibitory activity against both c-Met and VEGFR-2 enzymes.Citation8 Cabozantinib was used as a positive control. The results in showed that all scrutinised compounds have dual c-Met/VEGFR-2 inhibitory activity in the nanomolar range (IC50 range 0.035–0.297 µM). Three compounds namely, 2c, 4b, and 4e showed substantial amount of inhibition for c-Met enzyme (IC50 range 0.063–0.084 µM). Interestingly, to agree with the c-Met inhibition results, compounds 2c, 4b, and 4e were found to be the most potent agents against VEGFR-2 enzyme (IC50 range 0.035–0.082 µM) (). This correlation between the inhibitory activity of target compounds against c-Met and VEGFR-2 TKs is in consonance with the shared pharmacophoric requirements for inhibition of the two TKs. Compared to cabozantinib, all target compounds showed less inhibitory activity against c-Met enzyme. On the other hand, compound 4b (IC50=0.035 µM) exhibited superior inhibition than cabozantinib against VEFGR-2. Additionally, compound 2c (IC50=0.052 µM) was equipotent to cabozantinib.
Figure 7. Correlation of the inhibitory activity of c-Met enzyme and VEGFR-2 enzyme.
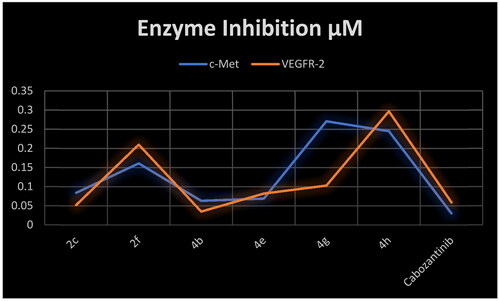
Table 3. Inhibitory activity of selected compounds against c-Met and VEGFR-2.
Apoptosis assay
Aiming to further study the mechanism of the target compounds, the cell cycle arrest experiment of 4b and 4e blocking the cell cycle of HCT-116 was carried out using Annexin V-FITC/PI staining ( and ).Citation56 HCT-116 colon cancer cells were treated with target compounds 4b, 4e, and cabozantinib at their IC50 concentrations. Compared to the control, the percentage of HCT-116 cells in the S process increased to 51.07% and 46.89% after treatment with 4b and 4e, respectively.
Figure 8. Cell cycle analysis in HCT-116 colon cancer cell line treated with compounds 4b and 4e.
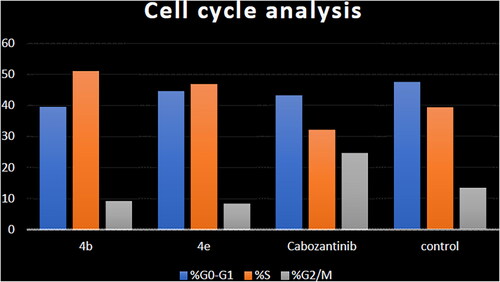
Table 4. Cell cycle analysis in HCT-116 colon cancer cell line treated with compounds 4b and 4e.
The mechanism of HCT-116 apoptosis induced by compounds 4b, 4e, and cabozantinib was further scrutinised by annexin V/PI staining ().Citation56 Compared to the control group, the number of both early and late apoptotic cells of target compounds 4b and 4e was increased. The number of early apoptotic cells of 4b and 4e (15.28 and 26.76%, respectively) was higher than that of cabozantinib. In addition, the number of late apoptotic and death cells of 4b (26.41%) was higher than that of cabozantinib. It can be concluded that the synthesised compounds 4b and 4e can induce apoptosis of HCT-116 cells more efficiently than cabozantinib ().
Figure 9. Apoptosis induction analysis using Annexin-V/propidium iodide (PI) staining assay.
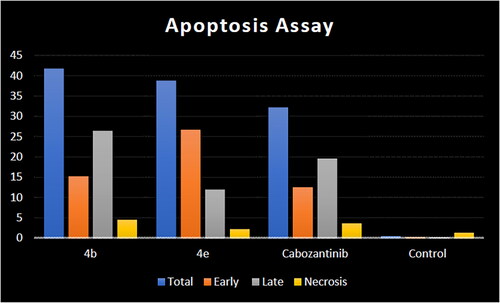
Figure 10. Representative cytograms of apoptotic HCT-116 cells induced by 4b and 4e compared to cabozantinib for 24 h.
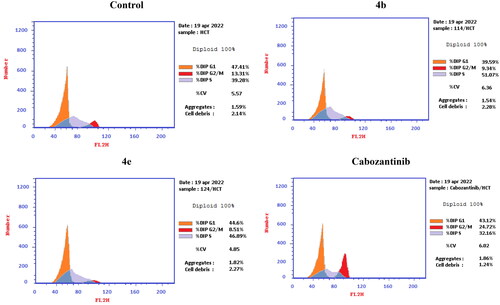
Figure 11. Representative cytograms of apoptotic HCT-116 cells induced by 4b and 4e compared to cabozantinib for 24 h.
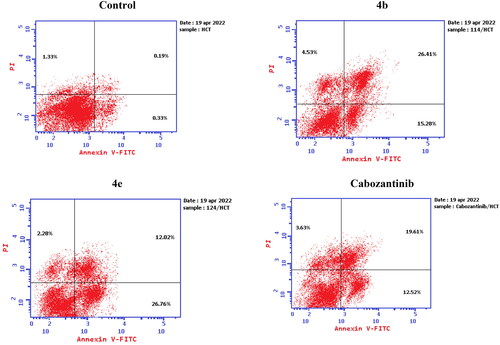
Table 5. Apoptosis induction analysis for compounds 4b, 4e, and cabozantinib.
In silico studies
Molecular modelling studies
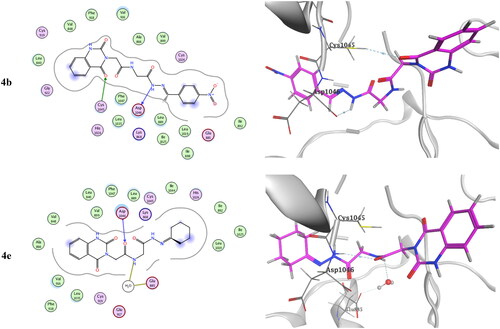
Molecular docking of the target compounds was carried out in the active site of both c-Met (PDB: 3lq8) and VEGFR-2 (PDB: 4asd) TKsCitation8,Citation19. represents docking styles of compounds 4b and 4e in the active site of c-Met TK. Compound 4b showed dual HB with highly conserved residue Asp1222 (2.64 and 2.84 Å) in the HB region as well as hydrophobic interactions with Met1131 and with Met1211 in the hinge region. Compound 4e showed HB with Asp1222 (2.88 Å) and Met1131 (3.42 Å) in addition to hydrophobic interactions with Met1131 and Met1211. On the other hand, docking poses of compounds 4b and 4e in the active site of VEGFR-2 TK are shown in . Compound 4b showed HB with Cys1045 (3.73 Å) and with a highly conserved residue Asp1046 (3.13 Å) in the HB region. Compound 4e showed HB with Glu885 (H2O 3.28 Å) and Asp1046 (3.20 Å).Citation8,Citation56
Figure 12. Docking pose of compound 4b and 4e with c-Met TK (PDB: 3lq8).
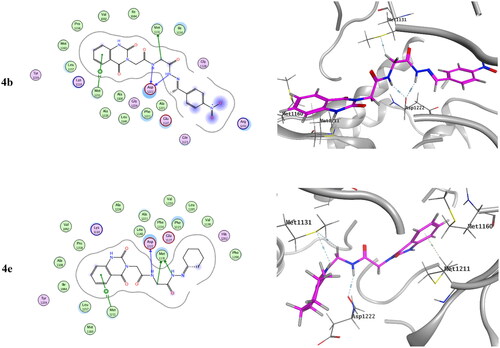
Figure 13. Docking pose of compound 4b and 4e with VEGFR-2 TK (PDB: 4asd).
In silico prediction of physicochemical and pharmacokinetic properties
Many drug candidates failed to reach the clinic because of their improper pharmacokinetics and physicochemical properties. Consequently, in silico prediction of pharmacokinetic and physicochemical parameters of target compounds was carried out using SwissADME website (see Supplementary Data)Citation8. SwissADME provides different reported filters to estimate drug likeness of tested compounds namely, Lipinski, Ghose, Veber, Egan, Muegge as well as bioavailability score. All target compounds showed zero or only one violation regarding all applied drug likeness filters. All target compounds have no Lipinski violation except compounds 4b and 4f which have only one violation just like cabozantinib. The Abbott Bioavailability Score of all tested compounds is equal to that of cabozantinib.
All target compounds have enhanced water solubility than cabozantinib. Like cabozantinib, most synthesised compounds were predicted to have high GI absorption (see Supplementary Data). Compared to cabozantinib, tested compounds were predicted not to be substrates for CYP2C19, CYP2C9, CYP2D6, and CYP3A4 that decrease the probability of drug–drug interactions.
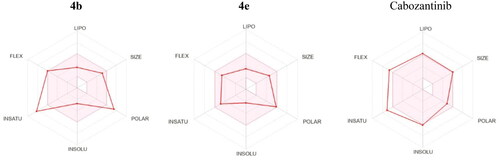
Another tool to assess the oral bioavailability of small molecules is the Bioavailability Radar (). It is a hexagon with specific physicochemical property at each vertex and the central pink region represents the optimum physicochemical space for good oral bioavailability. Compound 4e showed good fit to all six physicochemical parameters and it is predicted to have good oral bioavailability. Compound 4b exhibited deviation in both (Fraction Csp3 = 11) and (TPSA = 171.24 Å2) which expressed insaturation and polarity. In addition, cabozantinib showed deviation in both flexibility and insaturation as it has (10 rotatable bonds) and (Fraction Csp3 = 0.18).
Figure 14. Bioavailability Radar for 4b, 4e, and cabozantinib.
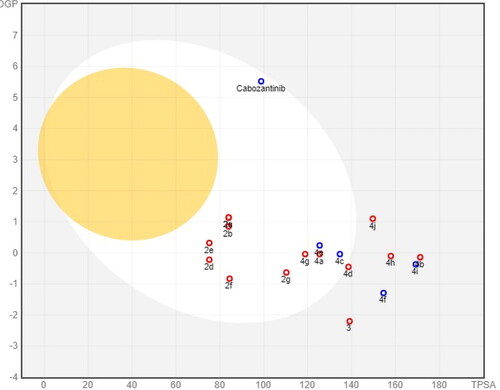
BOILED-Egg (The Brain Or IntestinaL EstimateD permeation) model is a robust method that carefully prognosticates both GI absorption and BBB accessibility by estimating both the lipophilicity (expressed in WLOGP) and polarity (expressed in TPSA) of target compounds (). Like cabozantinib, compounds 2a–g, 4a, 4c–e, and 4g showed a high GI absorption while compounds 3, 4b, 4f, and 4h–j showed low gastrointestinal absorption. The high GI absorption of some synthesised compounds is due to reasonable balance between their lipophilicity (WLOGP –0.83 to 1.14) and their polarity (TPSA 75.17–138.56 Å2). Moreover, all tested compounds as well as cabozantinib do not pass BBB confirming their good CNS safety profile.
Figure 15. BOILED-Egg model of target compounds and cabozantinib.
Conclusions
The current study was aimed at identifying novel drug-like molecules as anticancer drug candidates using in silico and in vitro techniques. Novel series of 3-substituted quinazoline-2,4(1H,3H)-dione derivatives of quinazolinone scaffold were synthesised and their chemical structures were confirmed by elemental and spectroscopic analyses. New synthetic methods were introduced for reported derivatives of 3-substituted quinazoline-2,4(1H,3H)-dione 2a–g, in addition to the preparation of some new derivatives namely, 3 and 4a–j. Compounds 2c, 4b, and 4e showed remarkable dual inhibition for both c-Met and VEGFR-2 TKs. Designed compounds exhibited binding mode with target proteins like that of cabozantinib. Compounds 4b and 4e showed HB with highly conserved residue Asp1222 in the HB region of c-Met TK. For VEGFR-2 TK, compound 4b showed HB with a highly conserved residue Asp1046 in the HB region. Compound 4e showed HB with Glu885 and Asp1046. Moreover, in silico prediction of pharmacokinetic and physicochemical parameters of target compounds was carried out using SwissADME website. The quinazoline-2,4(1H,3H)-dione derivatives are promising antiproliferative candidates that require further optimisation.
Experimental
Chemistry
All reactions were followed by thin layer chromatography (TLC). Melting points were measured using an electrothermal melting apparatus and are uncorrected. IR spectra were recorded on a Shimadzu FT-IR 8101 PC infra-red spectrophotometer (Kyoto, Japan). 1H and 13C NMR spectra were recorded on a BRUKER 400 MHz instrument (400.2 MHz for 1H and 100.64 MHz for 13C). Chemical shifts are expressed in parts per million (ppm) units using tetramethylsilane (TMS) as an internal reference. Mass spectra were recorded on a Shimadzu GC-MS QP-2010 Plus EX mass spectrometer (Kyoto, Japan) at 70 eV. Elemental analyses were carried out at the Microanalytical Centre of Cairo University, Giza, Egypt.
General procedure for synthesis of amides 2a–f
To a cold solution of azide 1 (0.49 g, 2 mmol) in ethyl acetate (15 ml), a solution of the corresponding amine derivative (2 mmol) in ethyl acetate (5 ml) was added dropwise with stirring. The reaction mixture was stirred at 0–5 °C for 6–14 h, then kept at room temperature overnight. The solid product was collected by filtration and crystallised from the appropriate solvent.
2-(2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-phenylacetamide (2a)
Crystallisation from acetic acid yielded 0.52 g (88%) of compound 2a as white crystals; m.p. over 300 °C (reported 294–296 °C)Citation36. FT-IR (KBr, υ, cm−1): 3204, 3136 (NH’s), 1737, 1636 (C═O’s). 1H NMR (DMSO-d6), δ, ppm: 4.72 (s, 2H, NCH2), 7.06 (t, 1H, ArH), 7.25 (m, 2H, ArHquinazoline), 7.32 (m, 2H, ArH), 7.57 (m, 2H, ArH), 7.71 (t, 1H, ArHquinazoline), 7.96 (d, 1H, ArHquinazoline), 10.31 (s, 1H, CONH), and 11.61 (s, 1H, quinazoline NH). MS m/z (%): 295 [M]+ (4.36%). Anal. Calcd. for C16H13N3O3: C, 65.08; H, 4.44; N, 14.23%. Found C, 65.16; H, 4.62; N, 14.12%.
N-Benzyl-2-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)acetamide (2b)
Crystallisation from acetic acid yielded 0.56 g (90.6%) of compound 2b as white crystals; m.p. 298–299 °C (reported 294–296 °C)Citation36. FT-IR (KBr, υ, cm−1): 3199 (NH’s), 2954, 2874 (aliphatic CH’s), 1735, 1640 (C═O’s). 1H NMR (DMSO-d6), δ, ppm: 4.31 (d, J= 6 Hz, 2H, NHCH2), 4.56 (s, 2H, NCH2), 7.20–7.35 (m, 7H, ArH), 7.69 (t, J= 7.6 Hz, 1H, ArHquinazoline), 7.96 (d, J= 7.6 Hz, 1H, ArHquinazoline), 8.66 (t, J= 6 Hz, 1H, NHCH2), and 11.54 (s, 1H, quinazoline NH). MS m/z (%): 309 [M]+ (9.03%) and 310 [M + 1]+ (2.06%). Anal. Calcd. for C17H15N3O3: C, 66.01; H, 4.89; N, 13.58%. Found C, 66.23; H, 5.05; N, 13.44%.
Another method for synthesis of compound 2b
A mixture of azide 1 (0.49 g, 2 mmol) and benzylamine (0.22 ml, 2 mmol) in dry benzene (15 ml) was refluxed for 4 h, then allowed to cool. The solid product was filtered off, dried, and crystallised from acetic acid to give 0.51 g (82.5%) of the target product 2b as white crystals.
N-Cyclohexyl-2-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)acetamide (2c)
Crystallisation from AcOH/H2O yielded 0.56 g (93%) of compound 2c as white crystals; m.p. over 300 °C (reported 308–310 °C)Citation36. FT-IR (KBr, υ, cm−1): 3252, 3202 (NH’s), 2929, 2855 (aliphatic CH’s), 1734, 1642 (C═O’s). 1H NMR (DMSO-d6), δ, ppm: 1.08–1.75 (m, 10H, cyclohexyl ring 5CH2), 3.52 (m, 1H, cyclohexyl ring CH), 4.46 (s, 2H, NCH2), 7.20–7.25 (m, 2H, ArH), 7.68 (t, J= 7.6 Hz, 1H, ArH), 7.94 (d, J= 7.6 Hz, 1H, ArH), 8.01 (d, J= 7.6 Hz, 1H, NH), and 11.48 (s, 1H, quinazoline NH). MS m/z (%): 301 [M]+ (6.80%). Anal. Calcd. for C16H19N3O3: C, 63.77; H, 6.36; N, 13.94%. Found C, 63.95; H, 6.48; N, 14.02%.
Another method for synthesis of compound 2c
A mixture of azide 1 (0.49 g, 2 mmol) and cyclohexylamine (0.23 ml, 2 mmol) in dry benzene (15 ml) was refluxed for 5 h, then allowed to cool. The solid product was filtered off, dried, and crystallised from AcOH/H2O to give 0.47 g (78%) of the target product as white crystals.
2-(2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N,N-dimethylacetamide (2d)
The solid product was collected by filtration and dried to give 0.21 g (42.5%) of the desired product; m.p. 218–220 °C. FT-IR (KBr, υ, cm−1): 3245 (NH), 1730, 1661 (C═O’s).1H NMR (DMSOd6), δ, ppm: 2.85, 3.08 (s, 6H, 2CH3), 4.73 (s, 2H, NCH2), 7.20–7.25 (m, 2H, ArH), 7.69 (t, J= 7.6 Hz, 1H, ArH), 7.93 (d, J= 6.8 Hz, 1H, ArH), and 11.54 (s, 1H, quinazoline NH). 13C NMR (DMSO-d6), δ, ppm: 35.56, 36.21, 41.94, 114.01, 115.66, 123.14, 127.86, 135.69, 139.87, 150.51, 162.22, and 166.35. MS m/z (%): 247 [M]+ (15.03%) and 248 [M + 1]+ (2.30%). Anal. Calcd. for C12H13N3O3: C, 58.29; H, 5.30; N, 17.00%. Found C, 58.11; H, 5.19; N, 16.91%Citation37.
3-(2-Oxo-2-(piperidin-1-yl)ethyl)quinazoline-2,4(1H,3H)-dione (2e)
Crystallisation from benzene yielded 0.39 g (68%) of compound 2e as white crystals; m.p. 204 °C. FT-IR (KBr, υ, cm−1): 3193 (NH), 2934, 2857 (aliphatic CH’s), 1721, 1652 (C═O’s).1H NMR (CDCl3), δ, ppm: 1.61–1.70 (m, 6H, CH2-3, 4 and 5), 3.53 and 3.62 (2t, 4H, CH2-2 and 6), 4.90 (s, 2H, NCH2), 6.97–7.04 (m, 2H, ArH), 7.35–7.40 (td, J= 7.6 Hz, 1H, ArH), 7.84–7.86 (dd, J= 8 Hz, 1H, ArH), and 10.38 (s, 1H, quinazoline NH). MS m/z (%): 287 [M]+ (27.44%), 288 [M + 1]+ (6.28%). Anal. Calcd. for C15H17N3O3: C, 62.71; H, 5.96; N, 14.63%. Found C, 62.87; H, 5.80; N, 14.56%Citation38.
3-(2-Morpholino-2-oxoethyl)quinazoline-2,4(1H,3H)-dione (2f)
Crystallisation from benzene/ethanol yielded 0.515 g (89%) of compound 2f as white crystals; m.p. 254–255 °C. FT-IR (KBr, υ, cm−1): ∼3430 (NH), 2907, 2874 (aliphatic CH’s), 1715, 1650 (C═O’s). 1H NMR (DMSO-d6), δ, ppm: 3.44 (t, 4H, morpholine ring 2 N-linked CH2), 3.64 (t, 4H, morpholine ring 2 O-linked CH2), 4.77 (s, 2H, NCH2), 7.21–7.25 (m, 2H, ArH), 7.69 (t, J= 7.6 Hz, 1H, ArH), 7.94 (d, J= 7.6 Hz, 1H, ArH), and 11.56 (s, 1H, quinazoline NH). MS m/z (%): 289 [M]+ (11.29%) and 290 [M + 1]+ (1.98%). Anal. Calcd. for C14H15N3O4: C, 58.13; H, 5.23; N, 14.53%. Found C, 58.03; H, 5.08; N, 14.45%Citation39.
Ethyl [2-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)acetyl]glycinate (2g)
Method A
A solution of glycine ethyl ester hydrochloride (0.14 g, 1 mmol) in ethyl acetate (10 ml) containing 0.2 ml of Et3N was added to a solution of azide 1 (0.245 g, 1 mmol) in ethyl acetate (10 ml). The mixture was stirred at 0–5 °C for 24 h, then kept at room temperature for another 24 h. The solvent was evaporated, and the residue was crystallised from benzene/ethanol to give the target product as white crystals; yield 0.15 g (49%); m.p. 228–230 °C (reported 223–225 °C)Citation40.
Method B
A mixture of azide 1 (0.245 g, 1 mmol) and glycine ethyl ester hydrochloride (0.14 g, 1 mmol) in dry pyridine (5 ml) was refluxed for 5 h. The reaction mixture was allowed to cool, then acidified with cold dilute hydrochloric acid (1:1). The resultant precipitate was collected by filtration, dried, and crystallised from benzene/ethanol to give the target product as white crystals; yield 0.235 g (77%). FT-IR (KBr, υ, cm−1): ∼3255, 3205 (NH’s), ∼2945, 2885 (aliphatic CH’s), 1740, 1645 (C═O’s). 1H NMR (DMSO-d6), δ, ppm: 1.19 (t, J= 7.2 Hz, 3H, OCH2CH3), 3.86 (d, J= 6 Hz, 2H, NHCH2), 4.10 (q, J= 7.2 Hz, 2H, OCH2CH3), 4.56 (s, 2H, NCH2), 7.20–7.24 (m, 2H, ArH), 7.68 (t, J= 7.2 Hz, 1H, ArH), 7.94 (d, J= 8 Hz, 1H, ArH), 8.62 (t, J = 6 Hz, 1H, CONHCH2), and 11.53 (s, 1H, quinazoline NH). 13C NMR (DMSO-d6), δ, ppm: 14.52, 41.13, 42.78, 60.93, 114.16, 115.66, 123.08, 127.88, 135.65, 139.96, 150.47, 162.25, 167.80, and 170.15. MS m/z (%): 305 [M]+ (6.86%) and 306 [M + 1]+ (1.30%). Anal. Calcd. for C14H15N3O5: C, 65.08; H, 4.95; N, 13.76%. Found C, 64.90; H, 5.09; N, 13.86%.
2-(2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-(2-hydrazineyl-2-oxo-ethyl)acetamide (3)
To a solution of ethyl [2-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)acetyl]glycinate 2g (0.305 g, 1 mmol) in absolute ethanol (10 ml), 0.5 ml of hydrazine hydrate was added. The reaction mixture was refluxed for 5 h, then allowed to cool. The solid product was collected by filtration, washed with boiling ethanol and dried to yield 0.217 g (74.6%) of compound 3 as white powder; m.p. 296–298 °C. FT-IR (KBr, υ, cm−1): 3291, 3175 (NH2, NH’s), 2933, 2894 cm−1 (aliphatic CH’s), 1704, 1644 (C═O’s). 1H NMR (DMSO-d6), δ, ppm: 3.70 (d, J= 6 Hz, 2H, NHCH2), 4.26 (s, 2H, NH2), 4.57 (s, 2H, NCH2), 7.20–7.25 (m, 2H, ArH), 7.69 (t, J= 7.8 Hz, 1H, ArH), 7.94 (d, J= 7.4 Hz, 1H, ArH), 8.49 (t, J= 6 Hz, 1H, CONHCH2), 9.02 (s, 1H, NHNH2), and 11.55 (s, 1H, quinazoline NH). 13C NMR (DMSO-d6), δ, ppm: 41.31, 43.00, 114.18, 115.66, 123.10, 127.89, 135.65, 139.93, 150.52, 162.33, 167.63, and 168.49. MS m/z (%): 291 [M]+ (4.95%). Anal. Calcd. for C12H13N5O4: C, 49.48; H, 4.50; N, 24.04%. Found C, 49.35; H, 4.39; N, 23.94%.
General procedure for synthesis of arylidenes 4a–d
To a solution of hydrazide 7 (0.291 g, 1 mmol) in absolute ethanol (15 ml), aldehyde (1 mmol) and few drops of piperidine were added. The reaction mixture was refluxed for 9–14 h (determined by TLC), then allowed to cool. The formed precipitate was filtered off and crystallised from the appropriate solvent.
(E)-N-[2-(2-Benzylidenehydrazineyl)-2-oxoethyl]-2-(2,4-dioxo-1,4-dihy-droquinazolin-3(2H)-yl)acetamide (4a)
Crystallisation from DMF/ethanol afforded compound 4a as white powder; yield 0.36 g (95%); m.p. 325 °C. FT-IR (KBr, υ, cm−1): 3252, 3202 (NH’s), 1735, 1690, 1644 (C═O’s), 1518 (C═N). 1H NMR (DMSO-d6), δ, ppm: 3.86, 4.30 (2d, 2H, NHCH2), 4.61 (s, 2H, NCH2), 7.21–7.25 (m, 2H, ArH), 7.42–7.44 (m, 3H, ArH), 7.68–7.69 (m, 3H, ArH), 7.95 (d, J= 7.6 Hz, 1H, ArH), 7.99, 8.22 (2s, 1H, N═CH), 8.45, 8.63 (2t, 1H, CONHCH2), 11.39, 11.52 (2s, 1H, NH–N), and 11.52 (s, 1H, quinazoline NH). 13C NMR (DMSO-d6), δ, ppm: 41.92, 42.89, 114.21, 115.66, 123.02, 127.23, 127.52, 127.87, 129.25, 130.30, 130.51, 134.54, 135.56, 139.97, 143.97, 147.37, 150.52, 162.26, 165.66, 167.54, 167.80, and 170.56. MS m/z (%): 379 [M]+ (22.86%). Anal. Calcd. for C19H17N5O4: C, 60.15; H, 4.52; N, 18.46%. Found C, 60.04; H, 4.45; N, 18.42%.
(E)-2-(2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-[2-(2-[4-nitrobenz-ylidene]hydrazineyl)-2-oxoethyl]acetamide (4b)
Crystallisation from DMF/H2O afforded compound 4b as yellowish white powder; yield 0.408 g (96%); m.p. over 300 °C. FT-IR (KBr, υ, cm−1): 3187, 3137 (NH’s), 1706, 1669 (C═O’s), 1563 (C═N). 1H NMR (DMSO-d6), δ, ppm: 3.91, 4.34 (2s, 2H, NHCH2), 4.60 (s, 2H, NCH2), 7.21 (m, 2H, ArH), 7.67 (t, 1H, ArH), 7.93 (m, 3H, ArH), 8.24–8.27 (m, 2H, ArH), 8.06, 8.29 (2s, 1H, N═CH), 8.50, 8.67 (2brs, CONHCH2), 11.52 (s, 1H, quinazoline NH), 11.73, 11.80 (2s, 1H, NH–N). 13C NMR (DMSO-d6), δ, ppm: 41.96, 42.91, 114.20, 115.66, 123.01, 124.43, 127.86, 128.18, 128.44, 130.03, 135.55, 139.96, 140.84, 141.56, 144.87, 148.18, 148.35, 150.51, 162.26, 166.17, 167.61, 167.86, and 170.99. MS m/z (%): 424 [M]+ (26.86%). Anal. Calcd. for C19H16N6O6: C, 53.78; H, 3.80; N, 19.80%. Found C, 65.67; H, 4.67; N, 14.64%.
(E)-2-(2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-[2-(2-[4-methoxy-benzylidene]hydrazineyl)-2-oxoethyl]acetamide (4c)
Crystallisation from ethanol afforded compound 4c as white powder; yield 0.393 g (96%); m.p. 290–292 °C. FT-IR (KBr, υ, cm−1): 3294, 3200 (NH’s), 1725, 1656 (C═O’s), 1570 (C═N). 1H NMR (DMSO-d6), δ, ppm: 3.80, 3.81 (2s, 3H, OCH3), 3.84, 4.27 (2d, 2H, NHCH2), 4.61 (s, 2H, NCH2), 6.98–7.03 (m, 2H, ArH), 7.21–7.26 (m, 2H, ArH), 7.60–7.72 (m, 3H, ArH), 7.96 (d, 1H, ArH), 7.94, 8.16 (2s, 1H, N═CH), 8.41, 8.60 (2t, CONHCH2), 11.22, 11.38 (2s, 1H, NH–N), and 11.52 (s, 1H, quinazoline NH). 13C NMR (DMSO-d6), δ, ppm: 41.84, 42.87, 43.02, 55.75, 114.17, 114.78, 115.66, 123.07, 123.12, 127.10, 127.15, 127.89, 128.83, 129.15, 135.62, 135.67, 139.96, 143.85, 147.19, 150.52, 150.61, 161.11, 161.31, 162.28, 162.39, 165.41, 167.53, 167.79, and 170.33. MS m/z (%): 409 [M]+ (13.29%). Anal. Calcd. for C20H19N5O5: C, 58.68; H, 4.68; N, 17.11%. Found C, 58.45; H, 4.53; N, 17.00%.
(E)-2-(2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-[2-(2-[furan-2-ylmethylene]hydrazineyl)-2-oxoethyl]acetamide (4d)
Crystallisation from DMF/ethanol afforded compound 4d as beige powder; yield 0.347 g (94%); m.p. over 300 °C. FT-IR (KBr, υ, cm−1): 3200, 3127 (NH’s), 1736, 1688, 1644 (C═O’s), and 1526 (C═N). 1H NMR (DMSO-d6), δ, ppm: 3.85, 4.23 (2d, 2H, NHCH2), 4.61 (s, 2H, NCH2), 6.61–6.64 (dd, 1H, furan ring HX), 6.88–6.93 (dd, 1H, furan ring HM), 7.21–7.26 (m, 2H, ArH), 7.69 (t, 1H, ArH), 7.81–7.84 (dd, 1H, furan ring HA), 7.95 (d, 1H, ArH), 7.88, 8.11 (2s, 1H, N═CH), 8.40, 8.61 (2t, CONHCH2), 11.32, 11.46 (s, 1H, NH–N), and 11.51 (s, 1H, quinazoline NH). MS m/z (%): 369 [M]+ (42.26%). Anal. Calcd. for C17H15N5O5: C, 55.28; H, 4.09; N, 18.96%. Found C, 55.37; H, 4.06; N, 18.91%.
N-[2-(2-Cyclohexylidenehydrazineyl)-2-oxoethyl]-2-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)acetamide (4e)
To a solution of compound 3 (0.58 g, 2 mmol) in ethanol (20 ml), cyclohexanone (0.21 ml, 2 mmol) was added, and the reaction mixture was refluxed for 10 h. After cooling, the solid product was filtered off and dried to afford pure compound 4e as a white powder without further purification. Yield 0.64 g (86%); m.p. 264–266 °C. FT-IR (KBr, υ, cm−1): 3256, 3210 (NH’s), 2942, 2860 (aliphatic CH’s), 1738,1647 (C═O’s), 1540 (C═N). 1H NMR (DMSO-d6), δ, ppm: 1.62–1.76 (m, 6H, 3CH2), 2.19–2.36 (m, 4H, 2CH2), 3.84, 4.15 (2d, 2H, NHCH2), 4.60 (s, 2H, NCH2), 7.21–7.30 (m, 2H, ArH), 7.69 (t, 1H, ArH), 7.94 (d, 1H, ArH), 8.32, 8.51 (2t, 1H, CONHCH2), 10.14, 10.46 (2s, 1H, NH), 11.51 (s, 1H, quinazoline NH). MS m/z (%): 371 [M]+ (23.14%). Anal. Calcd. for C18H21N5O4: C, 58.21; H, 5.70; N, 18.86%. Found C, 58.26; H, 5.72; N, 18.82%.
(Z)-2-[2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl]-N-[2-oxo-2-(2-(2-oxoindolin-3-ylidene)hydrazineyl)ethyl]acetamide (4f)
A mixture of hydrazide 3 (0.291 g, 1 mmol) and isatin (0.147 g, 1 mmol) in acetic acid (15) was heated under reflux for 8 h, then allowed to cool. The solid product was filtered off and recrystallised from acetic acid to give compound 4f as yellow crystals; yield 89.2%; m.p over 300 °C. FT-IR (KBr, υ, cm−1): 3328, 3228 (NH’s), 1724, 1695, 1650 (C═O’s). 1H NMR (DMSO-d6), δ, ppm: 4.01, 4.44 (2s, 2H, NHCH2), 4.64 (s, 2H, NCH2), 6.94 (d, J= 8 Hz, 1H, ArH), 7.09 (t, J= 8 Hz, 1H, ArH), 7.21–7.25 (m, 2H, ArHquinazoline), 7.38 (t, J= 8 Hz, 1H, ArH), 7.57 (d, J= 8 Hz, 1H, ArH), 7.69 (t, J= 8 Hz, 1H, ArHquinazoline), 7.95 (d, J= 8 Hz, 1H, ArHquinazoline), 8.60, 8.92 (2s, 1H, CONHCH2), 11.30 (s, 1H, indolinone NH), 11.51 (s, 1H,quinazoline NH), 12.55, 13.26 (2s, 1H, NH–N). MS m/z (%): 420.60 [M]+ (17.99%).
N-[2-(3,5-Dimethyl-1H-pyrazol-1-yl)-2-oxoethyl]-2-(2,4-dioxo-1,4-dihyd-roquinazolin-3(2H)-yl)acetamide (4g)
To a solution of compound 3 (0.58 g, 2 mmol) in ethanol (20 ml), acetyl acetone (0.2 ml, 2 mmol) was added in presence of few drops of piperidine, and the reaction mixture was refluxed for 16 h. After cooling, the solid product was filtered off and recrystallised from ethanol/benzene to afford compound 4g as white powder; yield 0.62 g (87.3%); m.p. 284–286 °C. FT-IR (KBr, υ, cm−1): 3241, 3199 (NH’s), 1732, 1646 (C═O’s). 1H NMR (DMSO-d6), δ, ppm: 1.77 (s, 3H, CH3), 1.96 (s, 3H, CH3), 4.10 (d, J= 4 Hz, 2H, NHCH2), 4.60 (s, 2H, NCH2), 6.40 (s, 1H, CH), 7.20–7.24 (m, 2H, ArH), 7.68 (t, J= 8 Hz, 1H, ArH), 7.94 (d, J= 8 Hz, 1H, ArH), 8.32 (t, J= 4 Hz, 1H, CONHCH2), and 11.49 (s, 1H, quinazoline NH). 13C NMR (DMSO-d6), δ, ppm: 16.32, 26.38, 42.19, 42.85, 52.37, 90.82, 114.13, 115.65, 123.08, 127.88, 135.61, 139.92, 150.50, 155.68, 162.26, 166.30, and 167.46. MS m/z (%): 355 [M]+ (23.14%). Anal. Calcd. for C17H17N5O4: C, 57.46; H, 4.82; N, 19.71%. Found C, 57.34; H, 4.74; N, 19.67%.
2-(2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-[(5-thioxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)methyl]acetamide (4h)
To a solution of compound 3 (0.58 g, 2 mmol) in pyridine (15 ml), 1 ml of carbon disulphide was added. The reaction mixture was refluxed for 10 h, then allowed to cool, acidified with cold dilute hydrochloric acid (1:1), and the solid formed was collected by filtration and dried to obtain the target product as grey powder without further purification; yield 0.24 g (36%); m.p. over 300 °C. FT-IR (KBr, υ, cm−1): 3304, 3214 (NH’s), 1714, 1661 (C═O’s), and 1140 (C═S). 1H NMR (DMSO-d6), δ, ppm: 3.86 (d, 2H, NHCH2), 4.59 (s, 2H, NCH2), 7.22–7.26 (m, 2H, ArH), 7.70 (t, 1H, ArH), 7.95 (d, 1H, ArH), 8.65 (t, 1H, CONHCH2), 10.98 (br s, 1H, NHCS), and 11.56 (s, 1H, quinazoline NH). 13C NMR (DMSO-d6), δ, ppm: 40.87, 42.89, 114.15, 115.71, 123.08, 127.86, 135.64, 139.97, 150.49, 162.29, 167.91, and 168.78. MS m/z (%): 333 [M]+ (20.79%). Anal. Calcd. for C13H11N5O4S: C, 46.84; H, 3.33; N, 21.01; S, 9.62%. Found C, 46.75; H, 3.28; N, 20.96; S, 9.66%.
2-(2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-[2-oxo-2-(2-[phenylcar-bamothioyl]hydrazineyl)ethyl]acetamide (4i)
To a solution of hydrazide 3 (0.58 g, 2 mmol) in absolute ethanol (20 ml), phenyl isothiocyanate (0.24 ml, 2 mmol) was added and the reaction mixture was refluxed for 15 h, then allowed to cool. The solid product was collected by filtration and crystallised from ethanol to afford compound 4i as white powder; yield 0.71 g (83.3%); m.p. 287–290 °C. FT-IR (KBr, υ, cm−1): 3444, 3285, 3201 (NH’s), 1735, 17.0, 1668 (C═O’s), and 1267 (C═S). 1H NMR (DMSO-d6), δ, ppm: 3.82 (d, 2H, NHCH2), 4.64 (s, 2H, NCH2), 7.06–7.95 (m, 9H, ArH), 8.75 (br s, 1H, CONHCH2), 9.31 (br s, 1H, NH), 9.74 (br s, 1H, NH), 10.21 (s, 1H, NH), and 11.56 (s, 1H, quinazoline NH). MS m/z (%): 426.56 [M]+ (60.16%). Anal. Calcd. for C19H18N6O4S: C, 53.51; H, 4.25; N, 19.71; S, 7.52%. Found C, 53.38; H, 4.18; N, 19.62; S, 7.61%.
2-(2,4-Dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-[(4-phenyl-5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl)methyl]acetamide (4j)
The thiosemicarbazide 4i (0.426 g, 1 mmol) was suspended in 2% solution of NaOH (15 ml) and heated under gentle reflux for 6 h. after cooling, the solution was neutralised with dil. HCl. The resulting precipitate was filtered off, washed with distilled water, and recrystallised from ethanol to afford compound 4j as white crystals. Yield 0.29 g (71%); m.p. over 300 °C. FT-IR (KBr, υ, cm−1): 3272, 3200 (NH’s), 1718, 1658 (C═O’s), and 1210 (C═S). 1H NMR (DMSO-d6), δ, ppm: 4.12 (d, J= 4 Hz, 2H, NHCH2), 4.42 (s, 2H, NCH2), 7.19–7.24 (m, 2H, ArHquinazoline), 7.40–7.57 (m, 5H, ArH), 7.68 (t, J= 8 Hz, 1H, ArHquinazoline), 7.94 (d, J= 8 Hz, 1H, ArHquinazoline), 8.61 (t, J= 4 Hz, 1H, CONHCH2), 11.50 (s, 1H, quinazoline NH), and 13.86 (s, 1H, NHCS). MS m/z (%): 408 [M]+ (39.11%). Anal. Calcd. for C19H16N6O3S: C, 55.87; H, 3.95; N, 20.58; S, 7.85%. Found C, 55.74; H, 3.78; N, 20.52; S, 7.93%.
Biological evaluation
In vitro antiproliferative activity against normal and cancer cells
To scrutinise the cytotoxicity of the target compounds on colorectal cancer and normal cells, MTT assay was carried out against HCT-116 and WI38 cell lines, respectively.
In vitro activity against c-Met and VEGFR-2
Both c-Met and VEGFR-2 assay of the target compounds 2c, 2f, 4b, 4e, 4g, and 4h was performed by a well-known methodology of BPS KDR Assay Kit Catalog # 40325 (BPS Bioscience, San Diego, CA) (see Supplementary Data)Citation8.
Cell cycle analysis and apoptosis assay
The effects of both 4b and 4e on progress of cell cycle and induction of apoptosis in HCT-166 cells were carried out using Annexin V-FITC Apoptosis Detection Kit (BioVision Research Products, Mountain View, CA) (see Supplementary Data)Citation8.
In silico studies
Molecular modelling study
Molecular modelling of compounds 4b and 4e and 2D and 3D visualisation processes were carried out within c-Met and VEFGR-2 TK active sites using MOE 2014.0901 software. The co-crystal structures of c-Met (PDB: 3lq8) and VEGFR-2 (PDB: 4asd) proteins were retrieved from the Protein Data Bank (RCSB). At first, 4b and 4e were prepared with the standard protocol of MOE 2014. After that the structures of the two proteins were prepared by using the MOE structure preparation protocol. Validation of the molecular docking study at the active sites was carried out by redocking of the original ligands into the active sites. The RMSD values of the most stable poses were 1.0648 (foretinib) and 0.9797 (sorafenib) in c-Met and VEGFR-2 active sites, respectively. Then, compounds 4b and 4e were docked into the active sites using the Alpha Triangle placement method. Refinement was carried out using Forcefield and scored by London dG scoring system.
In silico prediction of physicochemical and pharmacokinetic properties
The Swiss Institute of Bioinformatics provides a freely available tools for prediction of pharmacokinetics and physicochemical properties of small molecules via URL: http://www.swissadme.ch/index.php#. Different filters were used to compute druglikeness as follows: Pfizer Lipinski (MW ≤ 500, MLOGP ≤ 4.15, N or O ≤ 10, NH or OH ≤ 5); Ghose (160 ≤ MW ≤ 480, −0.4 ≤ WLOGP ≤ 5.6, 40 ≤ MR ≤ 130, 20 ≤ atoms ≤ 70); Veber (rotatable bonds ≤ 10, TPSA ≤ 140); Egan (WLOGP ≤ 5.88, TPSA ≤ 131.6); Muegge (200 ≤ MW ≤ 600, −2 ≤ XLOGP ≤ 5, TPSA ≤ 150, rings ≤ 7, C > 4, heteroatoms > 1, rotatable bonds > 15, HBA ≤ 10, HBD ≤ 5); Abbott Bioavailability Score (probability of F > 10% in rat).Citation57 iLOGCitation58. BOILED Egg is a plot of TPSA on the X-axis versus WLOGP on the Y-axis. The white oval is the suitable TPSA/WLOGP value for the highest probability of GI absorption, and the yolk circle is the suitable TPSA/WLOGP value for the highest probability to BBB accessibility. The optimal values of physicochemical properties involved in Bioavailability Radar are as follows: size (150 g/mol < MV > 500 g/mol), polarity (20 Å2<TPSA > 130 Å2), lipophilicity (–0.7 < XLOGP3> +5.0), insolubility (0 < Log S (ESOL)>6), insaturation (0.25 < Fraction Csp3 > 1.0), and flexibility (rotatable bonds < 9)Citation59.
Supplemental Material
Download PDF (2.8 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Global Cancer Observatory. Cancer today. Lyon: International Agency for Research on Cancer; 2022. https://gco.iarc.fr/
- Zhang J, Chen P, Duan Y, Xiong H, Li H, Zeng Y, Liang G, Tang Q, Wu D. Design, synthesis and biological evaluation of 7H-pyrrolo[2,3-d]pyrimidine derivatives containing 1,8-naphthyridine-4-one fragment. Eur J Med Chem. 2021;215:113273.
- Zhang J, Jiang X, Jiang Y, Guo M, Zhang S, Li J, He J, Liu J, Wang J, Ouyang L, et al. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. Eur J Med Chem. 2016;108:495–504.
- Xu H, Wang M, Wu F, Zhuo L, Huang W, She N. Discovery of N-substituted-3-phenyl-1,6-naphthyridinone derivatives bearing quinoline moiety as selective type II c-Met kinase inhibitors against VEGFR-2. Bioorg Med Chem. 2020;28(12):115555.
- Qiang H, Gu W, Huang DD, Shi W, Qiu Q, Dai Y, Huang W, Qian H. Design, synthesis and biological evaluation of 4-aminopyrimidine-5-cabaldehyde oximes as dual inhibitors of c-Met and VEGFR-2. Bioorg Med Chem. 2016;24(16):3353–3358.
- Li S, Zhao Y, Wang K, Gao Y, Han J, Cui B, Gong P. Discovery of novel 4-(2-fluorophenoxy) quinoline derivatives bearing 4-oxo-1, 4-dihydrocinnoline-3-carboxamide moiety as c-Met kinase inhibitors. Bioorg Med Chem. 2013;21(11):2843–2855.
- Gu W, Dai Y, Qiang H, Shi W, Liao C, Zhao F, Huang W, Qian H. Discovery of novel 2-substituted-4-(2-fluorophenoxy) pyridine derivatives possessing pyrazolone and triazole moieties as dual c-Met/VEGFR-2 receptor tyrosine kinase inhibitors. Bioorg Chem. 2017;72:116–122.
- Hassan A, Badr M, Abdelhamid D, Hassan HA, Abourehab MAS, Abuo‐Rahma GEDA. Design, synthesis, in vitro antiproliferative evaluation and in silico studies of new VEGFR-2 inhibitors based on 4-piperazinylquinolin-2(1H)-one scaffold. Bioorg Chem. 2022;120:105631.
- Dalton HJ, Pradeep S, McGuire M, Hailemichael Y, Ma S, Lyons Y, Armaiz-Pena GN, Previs RA, Hansen JM, Rupaimoole R, et al. Macrophages facilitate resistance to anti-VEGF therapy by altered VEGFR expression. Clin Cancer Res. 2017;23(22):7034–7046.
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342.
- Crawford Y, Ferrara N. Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol Sci. 2009;30(12):624–630.
- Poletto V, Rosti V, Biggiogera M, Guerra G, Moccia F, Porta C. The role of endothelial colony forming cells in kidney cancer’s pathogenesis, and in resistance to anti-VEGFR agents and mTOR inhibitors: a speculative review. Crit Rev Oncol Hematol. 2018;132:89–99.
- Goel HL, Pursell B, Shultz LD, Greiner DL, Brekken RA, Vander Kooi CW, Mercurio AM. P-Rex1 promotes resistance to VEGF/VEGFR-targeted therapy in prostate cancer. Cell Rep. 2016;14(9):2193–2208.
- Daudigeos-Dubus E, Le Dret L, Bawa O, Opolon P, Vievard A, Villa I, Bosq J, Vassal G, Geoerger B. Dual inhibition using cabozantinib overcomes HGF/MET signaling mediated resistance to pan-VEGFR inhibition in orthotopic and metastatic neuroblastoma tumors. Int J Oncol. 2017;50(1):203–211.
- Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19(4):512–526.
- Negri FV, Dal Bello B, Porta C, Campanini N, Rossi S, Tinelli C, Poggi G, Missale G, Fanello S, Salvagni S, et al. Expression of pERK and VEGFR-2 in advanced hepatocellular carcinoma and resistance to sorafenib treatment. Liver Int. 2015;35(8):2001–2008.
- Cascone T, Xu L, Lin HY, Liu W, Tran HT, Liu Y, Howells K, Haddad V, Hanrahan E, Nilsson MB, et al. The HGF/c-MET pathway is a driver and biomarker of VEGFR-inhibitor resistance and vascular remodeling in non-small cell lung cancer. Clin Cancer Res. 2017;23(18):5489–5501.
- Liang J-w, Li S-l, Wang S, Li W-q, Meng F-h. Synthesis and biological evaluation of novel (E)-N′-benzylidene hydrazides as novel c-Met inhibitors through fragment based virtual screening. J Enzyme Inhib Med Chem. 2020;35(1):468–477.
- Zhang Q, Zheng P, Zhu W. Research progress of small molecule VEGFR/c-Met inhibitors as anticancer agents (2016–present). Molecules. 2020;25(11):2666.
- Shi L, Wu T-T, Wang Z, Xue J-Y, Xu Y-G. Discovery of quinazolin-4-amines bearing benzimidazole fragments as dual inhibitors of c-Met and VEGFR-2. Bioorg Med Chem. 2014;22(17):4735–4744.
- Ibrahim HA, Awadallah FM, Refaat HM, Amin KM. Design, synthesis and molecular modeling study for some new 2-substituted benzimidazoles as dual inhibitors for VEGFR-2 and c-Met. Future Med Chem. 2018;10(5):493–509.
- Li C, Shan Y, Sun Y, Si R, Liang L, Pan X, Wang B, Zhang J. Discovery of novel anti-angiogenesis agents. Part 7: multitarget inhibitors of VEGFR-2, TIE-2 and EphB4. Eur J Med Chem. 2017;141:506–518.
- Zhang J, Dai J, Lan X, Zhao Y, Yang F, Zhang H, Tang S, Liang G, Wang X, Tang Q, et al. Synthesis, bioevaluation and molecular dynamics of pyrrolo-pyridine benzamide derivatives as potential antitumor agents in vitro and in vivo. Eur J Med Chem. 2022;233:114215.
- Li S, Huang Q, Liu Y, Zhang X, Liu S, He C, Gong P. Design, synthesis and antitumour activity of bisquinoline derivatives connected by 4-oxy-3-fluoroaniline moiety. Eur J Med Chem. 2013;64:62–73.
- Martorana A, Monica GL, Lauria A. Quinoline-based molecules targeting c-Met, EGF, and VEGF receptors and the proteins involved in related carcinogenic pathways. Molecules. 2020;25(18):4279.
- Nan X, Li H-J, Fang S-B, Li Q-Y, Wu Y-C. Structure-based discovery of novel 4-(2-fluorophenoxy)quinoline derivatives as c-Met inhibitors using isocyanide-involved multicomponent reactions. Eur J Med Chem. 2020;193:112241.
- Qi B, Tao H, Wu D, Bai J, Shi Y, Gong P. Synthesis and biological evaluation of 4‐phenoxy‐6,7‐disubstituted quinolines possessing semicarbazone scaffolds as selective c‐Met inhibitors. Arch Pharm Chem Life Sci. 2013;346(8):596–609.
- Liao W, Hu G, Guo Z, Sun D, Zhang L, Bu Y, Li Y, Liu Y, Gong P. Design and biological evaluation of novel 4-(2-fluorophenoxy) quinoline derivatives bearing an imidazolone moiety as c-Met kinase inhibitors. Bioorg Med Chem. 2015;23(15):4410–4422.
- Tang Q, Zhao Y, Du X, Chong L, Gong P, Guo C. Design, synthesis, and structure–activity relationships of novel 6,7-disubstituted-4-phenoxyquinoline derivatives as potential antitumor agents. Eur J Med Chem. 2013;69:77–89.
- Zhou S, Liao H, Liu M, Feng G, Fu B, Li R, Cheng M, Zhao Y, Gong P. Discovery and biological evaluation of novel 6,7-disubstituted-4-(2-fluorophenoxy)quinoline derivatives possessing 1,2,3-triazole-4-carboxamide moiety as c-Met kinase inhibitors. Bioorg Med Chem. 2014;22(22):6438–6452.
- Zhou S, Liao H, He C, Dou Y, Jiang M, Ren L, Zhao Y, Gong P. Design, synthesis and structure–activity relationships of novel 4-phenoxyquinoline derivatives containing pyridazinone moiety as potential antitumor agents. Eur J Med Chem. 2014;83:581–593.
- Liu M, Hou Y, Yin W, Zhou S, Qian P, Guo Z, Xu L, Zhao Y. Discovery of a novel 6,7-disubstituted-4-(2-fluorophenoxy) quinolines bearing 1,2,3-triazole-4-carboxamide moiety as potent c-Met kinase inhibitors. Eur J Med Chem. 2016;119:96–108.
- Wang M, Xu S, Lei H, Wang C, Xiao Z, Jia S, Zhi J, Zheng P, Zhu W. Design, synthesis and antitumor activity of novel sorafenib derivatives bearing pyrazole scaffold. Bioorg Med Chem. 2017;25(20):5754–5763.
- Zeidan MA, Mostafa AS, Gomaa RM, Abou-Zeid LA, El-Mesery M, El-Sayed MA-A, Selim KB. Design, synthesis and docking study of novel picolinamide derivatives as anticancer agents and VEGFR-2 inhibitors. Eur J Med Chem. 2019;168:315–329.
- Younes AMM, Taha MM, Abdel-Monsef A-BH. (2,4-Dioxo-1,4-dihydro-2H-quinazolin-3-yl)-acetic acid hydrazide: synthesis and reactions. J Asian Sci Res. 2013;3(8):800–809.
- El Kayal WM, Shtrygol SY, Zalevskyi SV, Shark AA, Tsyvunin VV, Kovalenko SM, Bunyatyan ND, Perekhoda LO, Severina HI, Georgiyants VA. Synthesis, in vivo and in silico anticonvulsant activity studies of new derivatives of 2-(2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)acetamide. Eur J Med Chem. 2019;180:134–142.
- National Center for Biotechnology Information. PubChem compound summary for CID 37143654; 2022. https://pubchem.ncbi.nlm.nih.gov/compound/37143654
- National Center for Biotechnology Information. PubChem compound summary for CID 4975215; 2022. https://pubchem.ncbi.nlm.nih.gov/compound/4975215
- National Center for Biotechnology Information. PubChem compound summary for CID 4906019; 2022. https://pubchem.ncbi.nlm.nih.gov/compound/4906019
- Rivero IA, Somanathan R, Hellberg LH. Synthesis of 3-dipeptidyl-2,4(1H,3H)-quinazolinediones as potential anti-hypertensive agents. Synth Commun. 1998;28(11):2077–2086.
- Mata A, Weigl U, Flögel O, Baur P, Hone CA, Kappe CO. Acyl azide generation and amide bond formation in continuous-flow for the synthesis of peptides. React Chem Eng. 2020;5(4):645–650.
- Palla G, Predieri G, Domiano P, Vignali C, Turner W. Conformational behaviour and E/Z isomerization of N-acyl and N-aroylhydrazones. Tetrahedron. 1986;42(13):3649–3654.
- Syakaev VV, Podyachev SN, Buzykin BI, Latypov SK, Habicher WD, Konovalov AI. NMR study of conformation and isomerization of aryl- and heteroarylaldehyde 4-tert-butylphenoxyacetylhydrazones. J Mol Struct. 2006;788(1–3):55–62.
- Korcz M, Sączewski F, Bednarski PJ, Kornicka A. Synthesis, structure, chemical stability, and in vitro cytotoxic properties of novel quinoline-3-carbaldehyde hydrazones bearing a 1,2,4-triazole or benzotriazole moiety. Molecules. 2018;23(6):1497.
- Lacerda RB, da Silva LL, de Lima CKF, Miguez E, Miranda ALP, Laufer SA, Barreiro EJ, Fraga CAM. Discovery of novel orally active anti-inflammatory N-phenylpyrazolyl-N-glycinyl-hydrazone derivatives that inhibit TNF-α production. PLOS One. 2012;7(10):e46925.
- Lopes AB, Miguez E, Kümmerle AE, Rumjanek VM, Fraga CAM, Barreiro EJ. Characterization of amide bond conformers for a novel heterocyclic template of N-acylhydrazone derivatives. Molecules. 2013;18(10):11683–11704.
- Uygun Y, Bayrak H, Özkan H. Synthesis and biological activities of methylenebis-4H-1,2,4-triazole derivatives. Turk J Chem. 2013;37(5):812–824.
- Huan LC, Phuong CV, Truc LC, Thanh VN, Pham-The H, Huong L-T-T, Thuan NT, Park EJ, Ji AY, Kang JS, et al. (E)-N′-arylidene-2-(4-oxoquinazolin-4(3H)-yl) acetohydrazides: synthesis and evaluation of antitumor cytotoxicity and caspase activation activity. J Enzyme Inhib Med Chem. 2019;34(1):465–478.
- Aboelmagd A, Salem EMS, Ali IAI, Gomaab MS. Synthesis of quinazolindionyl amino acid and hydrazone derivatives as possible antitumour agents. Org Chem. 2018;2018(3):20–35.
- Aboelmagd A, Salem EMS, Ali IAI, Gomaab MS. Synthesis of quinazolindionyl amino acid and dipeptide derivatives as possible antitumour agents. Org Chem. 2019;2019(5).
- Kuodis Z, Rutavičius A, Matijoška A, Eicher-Lorka O. Synthesis and isomerism of hydrazones of 2-(5-thioxo-4,5-dihydro-1,3,4-thiadiazol-2-ylthio) acetohydrazide. Open Chem. 2007;5(4):996–1006.
- Tumosienė I, Jonuškienė I, Kantminienė K, Mickevičius V, Petrikaitė V. Novel N-substituted amino acid hydrazone-isatin derivatives: synthesis, antioxidant activity, and anticancer activity in 2D and 3D models in vitro. Int J Mol Sci. 2021;22(15):7799.
- Sharma PK, Balwani S, Mathur D, Malhotra S, Singh BK, Prasad AK, Len C, Van der Eycken EV, Ghosh B, Richards NGJ, et al. Synthesis and anti-inflammatory activity evaluation of novel triazolyl-isatin hybrids. J Enzyme Inhib Med Chem. 2016;31(6):1520–1526.
- Huang D, Yang J, Zhang Q, Wang G, Zhang Z, Zhang Y, Li J. Structure-guided design and development of novel N-phenylpyrimidin-2-amine derivatives as potential c-Met inhibitors. Eur J Med Chem. 2021;223:113648.
- Seliem IA, Panda SS, Girgis AS, Tran QL, Said MF, Bekheit MS, Abdelnaser A, Nasr S, Fayad W, Soliman AAF, et al. Development of isatin-based Schiff bases targeting VEGFR-2 inhibition: synthesis, characterization, antiproliferative properties, and QSAR studies. ChemMedChem. 2022;17(13):e202200164.
- Hassan A, Badr M, Hassan HA, Abdelhamid D, Abuo‐Rahma GEDA. Novel 4-(piperazin-1-yl)quinolin-2(1H)-one bearing thiazoles with antiproliferative activity through VEGFR-2-TK inhibition. Bioorg Med Chem. 2021;40:116168.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717.
- Daina A, Michielin O, Zoete V. iLOGP: a simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J Chem Inf Model. 2014;54(12):3284–3301.
- Daina A, Zoete V. A boiled‐egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11(11):1117–1121.