
Abstract
Prions are infectious protein particles known to cause prion diseases. The biochemical entity of the pathogen is the misfolded prion protein (PrPSc) that forms insoluble amyloids to impair brain function. PrPSc interacts with the non-pathogenic, cellular prion protein (PrPC) and facilitates conversion into a nascent misfolded isoform. Several small molecules have been reported to inhibit the aggregation of PrPSc but no pharmacological intervention was well established thus far. We, here, report that acylthiosemicarbazides inhibit the prion aggregation. Compounds 7x and 7y showed almost perfect inhibition (EC50 = 5 µM) in prion aggregation formation assay. The activity was further confirmed by atomic force microscopy, semi-denaturing detergent agarose gel electrophoresis and real-time quaking induced conversion assay (EC50 = 0.9 and 2.8 µM, respectively). These compounds also disaggregated pre-existing aggregates in vitro and one of them decreased the level of PrPSc in cultured cells with permanent prion infection, suggesting their potential as a treatment platform. In conclusion, hydroxy-2-naphthoylthiosemicarbazides can be an excellent scaffold for the discovery of anti-prion therapeutics.
Introduction
Prions are the infectious protein that cause prion diseases, including bovine spongiform encephalopathy, scrapie, and Creutzfeldt–Jakob disease (CJD)Citation1,Citation2. The clinical signs of prion diseases are related to impaired brain function, such as cognitive dysfunction, cerebral ataxia and motor dysfunctionCitation1,Citation3. The neuropathological features of prion diseases include spongiform degeneration and gliosis in accociation with the accumulation of PrPsc in the brainCitation4. Recent studies at the cellular and molecular levels report that spongiosis and neurodegeneration are caused by prion-induced chronic endoplasmic reticulum (ER) stress leading to the depletion of an intracellular lipid molecule and impaired lysosomal trafficking in brain cellsCitation5,Citation6.
The ultimate pathogens, prions, are believed to be an aggregate of disease-causing prion protein (PrPSc), which is the misfolded isoform of normal cellular prion protein (PrPC). In the propagation mechanism of prion diseases, PrPSc induces the conformational change of normal PrPC to become a nascent misfolded PrPSc moleculeCitation7,Citation8, demonstrating its infectious nature. Therefore, PrPSc acts as a catalyst or template in this transformation to propagate the misfolded conformer. PrPSc is capable of forming aggregates that further afford amyloids or fibril structuresCitation9. Recent studies of PrP fibrils using cryo-electron microscopy demonstrated that PrPSc molecules adopt parallel in register intermolecular β-sheet amyloid structuresCitation10,Citation11.
Since PrP conversion and aggregation occur autocatalytically without involvement of a signalling pathway or enzymatic systemCitation9,Citation12, PrPSc molecule itself has been a direct drug target to interfere disease-causing aggregatesCitation13–16. Similarly, PrPC also has been a target to block PrP conversionCitation17–25. In addition, an auxiliary factor that assists in the PrP conversion was targeted to block the conversionCitation26–31. Regardless of targets, several small molecules have been reported to inhibit PrPSc propagation ()Citation32. Some of them were identified through library screening, medicinal chemistry, or in silico approaches. For example, GN8 and its analogues (R1–R2=cycloheptyl) were discovered by a compound screening and subsequent medicinal chemistry effortsCitation21,Citation22. On the other hand, many compounds that provided intriguing anti-prion activity were known drugs or literature compounds that have irrelevant indications. They include chlorpromazine (antipsychotic)Citation23, quinacrine (antimalarial)Citation33, doxycycline (antibiotic)Citation15, tegobuvir (antihepatitis C)Citation24, and SGI-1027 (DNA methyltransferase inhibitor)Citation20,Citation25. None of them, however, have shown robust in vivo and clinical activity thus far, suggesting tremendous medical unmet needs for the discovery of new anti-prion agents.
Figure 1. Representative compounds with anti-prion aggregation activity.
In search of compounds to intervene misfolding pathologies, we have synthesised a series of acylthiosemicarbazide compounds some of which were found to inhibit ER stress as chemical chaperonesCitation34. ER stress is triggered by the abnormal accumulation of misfolded protein in ER. Inhibitors of ER stress relieve misfolded protein load either by decreasing the formation of misfolded proteins or by removing them by ER stress-associated degradation or apoptosisCitation35–38. Because of the misfolding nature of PrPSc, it would be worth investigating the cross-activity of the compounds. In this study, the scope of the analogues was expanded and their anti-prion activity of was measured by using prion aggregate formation assay (PAFA)Citation39,Citation40 that we have fine-tuned for the purpose of screening. Several compounds showed significant inhibition against prion aggregation. Some of them showed the activity to disassemble pre-existing aggregates. These activities were further confirmed by atomic force microscopy (AFM), semi-denaturing detergent agarose gel electrophoresis (SDD-AGE), and real-time quacking-induced conversion (RT-QuIC) assay with different prion strains. Also, the activity of compounds was evaluated in cultured cells with chronic prion infection.
Materials and methods
Synthesis of compounds
Synthetic details and spectroscopic data of new compounds are provided in the supplementary material.
Preparation of recombinant PrP
The recPrP preparation was conducted according to the method described elsewhereCitation41. Briefly, the cDNA fragments encoding His-tagged mouse or bank vole PrP (23–231) was cloned into plasmid pET100/D-TOPO (Invitrogen, Carlsbad, CA, USA). The recombinant plasmid was used for transformation of E. coli BL21 Star (DE3) (Invitrogen). Expression of recPrP was induced using 1 mM isopropyl β-D-1-thiogalactopyranoside (iNtRON, Seongnam-si, Gyeonggi-do, Korea). After incubation for 16 h, the bacterial cells were lysed by sonication in lysis buffer [10% sucrose (Sigma-Aldrich, St. Louis, MO, USA)], 0.1 M Tris (VWR Life Science, West Chester, PA, USA), 50 mM ethylenediaminetetraacetic acid (EDTA, pH 8.0) (Invitrogen), 0.2 M sodium chloride (NaCl) (pH 7.9) (Sigma-Aldrich), 1 mM phenylmethylsulfonylfluoride (MP Biomedicals, Santa Ana, CA, USA)]. The inclusion bodies derived from cell lysate were solubilised and refolded using solubilisation buffer [8 M urea (Samchun, Seoul, Korea), 10 mM glycine (Sigma-Aldrich), pH 10.6] and refolding buffer [0.6 M urea, 10 mM glycine, pH 10.6], respectively. The his-tagged mouse and bank vole recPrP was purified by sequential chromatography steps including immobilised metal affinity, cation exchange, and reverse phase chromatography. The purified mouse and bank vole recPrP was desalted and dissolved in 20 mM Tris, 150 mM NaCl, pH 8.0. Finally, recPrP was stored at −80 °C after adding glycerol (Sigma-Aldrich) and dimethyl sulfoxide (Sigma-Aldrich) 10% and 2%, respectively, to maintain stability.
PrP aggregate formation assay
PAFA was conducted according to the method described elsewhereCitation39. Ten µg mouse recPrP was denatured to unfolded using 6 M guanidine hydrochloride (GdnHCl) (Sigma-Aldrich) for 5 min at room temperature and then mixed in 200 µl reaction buffer [4M GdnHCl (Sigma-Aldrich) and 10 µM ThT (Sigma-Aldrich) in phosphate buffered saline (PBS, pH7.4) (Welgene, Gyeongsan-si, Gyeongsangbuk-do, Korea)] together with compounds (1–100 µM) and vehicle, DMSO. The PAFA reaction mixture was prepared in a 96-well black flat bottom polystyrene not treated microplate (Corning, Corning, NY, USA), sealed with a microplate adhesive film (USA Scientific, Ocala, FL, USA) and incubated at 37 °C for 60 h with shaking at 335 rpm in Infinite M200 Pro Fluorescence reader (Tecan, Maennedorf, Zurich, Switzerland). The fibrils formation was measured every 1 h by top reading of the fluorescence intensity using a 444 nm excitation and 485 nm emission filter.
Atomic force microscopy
AFM was conducted according to the modified method described elsewhereCitation42. Ten-mm-diameter highest grade V1 AFM mica discs (Ted Pella, Redding, CA, USA) was coated with 100 µl of 1% 3-aminopropyltriethoxysilane (Sigma-Aldrich) in 1 mM acetic acid (Sigma-Aldrich) for 20 min for fibrils absorption. After removing the solution, the mica was rinsed three times with 100 µl of distilled water, dried for 30 min using compressed N2 gas, and stored in a cryo box for 1 h. PAFA products were dropped onto the coated mica and incubated for 30 min until the fibrils were absorbed. After removing the solution, the mica was rinsed, dried, and stored in a cryo box as described above until imaging. Fibrils were imaged using XE-100 AFM (Park Systems, Suwon-si, Gyeonggi-do, Korea). To minimise damage to fibrils, imaging was performed in a non-contact mode using a point probe plus non-contact/tapping mode high resonance frequency reflex coating (PPP-NCHR) (Park Systems). During measurement, the constant scan rate was maintained at 0.5 Hz. The scan size was 10 µm × 10 µm to 1.5 µm × 1.5 µm, and the pixel was 256 × 256. AFM scanning was performed using XEP software (Park Systems), and the scanned image was edited using XEI software (Park Systems).
Semi-denaturing detergent-agarose gel electrophoresis and western blotting
SDD-AGE was conducted according to the method described elsewhere with minor modificationCitation43. The medium for SDD-AGE was prepared with 1.5% agarose (Lonza, Basel, Switzerland) and 0.1% sodium dodecyl sulphate (SDS) (Sigma-Aldrich) in TAE (1 M Tris, 5.71% acetic acid, 50 mM EDTA) buffer. The horizontal gel was casted using an electrophoresis tray (Fisher scientific, Waltham, MA, USA). For sample preparation, 100 µl of PAFA products was centrifuged at 43 000 rpm for 1 at 4 °C h using ultracentrifuge TLX-100 (Beckman, Brea, CA, USA). The pellet was washed with 700 µl of PBS and retrieved by centrifugation at 43 000 rpm for 10 min at 4 °C. Then, the pellet was dissolved in 24 µl of SDD buffer [100 mM Tris (pH 7.5), 50 mM NaCl, 10 mM β-mercaptoethanol (Sigma-Aldrich)] and mixed with 8 µl of 4× sample loading buffer [2× TAE, 20% glycerol, 8% SDS, 0.3 M bromophenol blue (Sigma-Aldrich)]. After incubation at 37 °C for 10 min, the samples were run for 3 h at 60 V. Capillary transfer was conducted overnight using TBS [140 mM NaCl, 2 mM KCl (Sigma-Aldrich), 25 mM Tris, pH 7.4] onto a methanol (Honeywell)-activated 0.45 µm polyvinylidene difluoride (PVDF) (Merck, Piscataway, NJ, USA) membrane using the sequential piles of a sheet of TBS-wet extra thick blot filter paper (Bio-Rad, Hercules, CA, USA), 20 sheets of cellulose chromatography paper (Cytiva, Emeryville, CA, USA), and a 1 kg weight. After transfer, PVDF were subsequently blocked using 5% skim milk (BD Difco, Franklin Lakes, NJ, USA) in TBST [TBS, 0.5% Tween 20 (Sigma-Aldrich)] for 1 h. After rinsing in TBST, the membranes were incubated with anti-PrP monoclonal antibody 6D11 (Biolegend, San Diego, CA, USA) diluted to 1:30 000 in TBST for 1 h before washing in TBST for 10 min twice. Then, the membrane was further incubated with the secondary antibody (goat anti-mouse IgG HRP, Invitrogen) diluted 1:10 000 in TBST for 1 h. Following washing twice in TBST for 10 min, the membranes were developed using Amersham ECL prime (Cytiva) and visualised using a G:BOX Chemi XR5 Chemiluminescencer (Syngene, Cambrdige, UK).
Disassembling assay for pre-formed PrP aggregates
Generation of pre-formed PrP aggregates was achieved by PAFA using mouse recPrP. When the formation of PrP aggregation was reached to the equilibrium after ∼32 h-PAFA reactions, the compounds with various concentrations in 10 µl were injected to each well, and the changes of aggregate-bound ThT fluorescence were further monitored for additional 20 h in the Infinite M200 Pro Fluorescence reader (Tecan, Maennedorf, Zurich, Switzerland) as described in the PAFA section.
Real-time quaking-induced conversion
RT-QuIC was conducted according to the method described elsewhere with modificationsCitation44,Citation45. Ten µg mouse or bank vole recPrP, prion seeds (5 × 10−6 diluted prion-ill rodent or human patient brain material), and compounds for anti-prion activity tests were mixed in 100 µl of RT-QuIC buffer [0.5M NaCl, 10 µM EDTA, 10 mM phosphate buffer (pH 7.4) (Sigma-Aldrich), 10 µM ThT, 0.002% SDS]. The source of prion seeds was FVB mouse-adapted RML scrapie prions, Syrian golden hamster-adapted Hyper TME prions and sporadic CJD prions deposited in Korea CJD Diagnostic Centre (Seoul, Korea). Prion seeds were prepared from 10% (w/v) homogenate of perfused brains by serial dilution with PBS. The reaction mixture in a 96-well Black/Clear Flat Bottom Polystyrene Not Treated Microplate (Corning) sealed with a microplate adhesive film (USA Scientific) was incubated at 42 °C for 60–80 h with shaking at 700 rpm in FLUOstar Omega Fluorescence reader (BMG Labtech, Ortenberg, Germany). Fibrils formation was measured every 1 h by bottom reading of ThT fluorescence intensity using a 450 nm excitation and 480 nm emission filter.
Cytotoxicity and PrPSc assay in ScN2a cells
The cytotoxicity tests were conducted as described elsewhere using the MTT assayCitation46. Briefly, ScN2a cells were incubated with the compounds for 4 days and the level of formazan formation facilitated by cellular enzymes were measured using MTT agents. PrPSc assay was performed in ScN2a cells as described elsewhereCitation47. Briefly, ScN2a cells were incubated with the non-cytotoxic concentrations of compounds for 4 days and the proteinase K (PK)-resistant PrPSc in the cell lysate was detected by western blotting using anti-PrP monoclonal antibody 6D11 (Biolegend). Densitometry of western blots was performed using GeneTools software (Syngene). Student t-test was used for statistical analysis.
Results and discussion
Chemistry
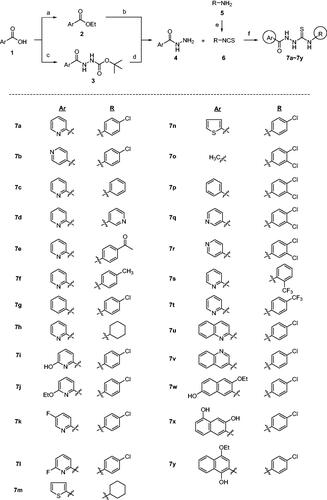
The synthesis of compounds 7 is shown in Scheme 1Citation34. Acylhydrazides 4 were prepared from acid 1 via the reaction sequence consisting of either Fischer esterification followed by hydrazine substitution or coupling with t-butyl carbazate followed by hydrolysis of t-Boc group of compound 3. Then, compounds 4 were reacted with isothiocyantes 6 to afford acylthiosemicarbazides 7 in 37–96% yields. When 6 is not commercially available, amines 5 were reacted with thiophosgene to give corresponding isothiocyanates 6. Ar group includes phenyl, thienyl, (substituted)pyridyl, quinolinyl and (substituted)naphthyl while R group covers cycloalkyl, (substituted)phenyl, and (substituted)pyridyl groups.
Scheme 1. Synthesis of 7a–7y with various Ar and R groups. Reagents and conditions: (A) H2SO4, EtOH, reflux, (B) H2NNH2H2O, EtOH, 80 °C, (C) H2N-NHt-Boc, HOAt, EDCl, NaHCO3, DMF, (D) 4 N HCl, dioxane, (E) CSCl2, Et3N, THF, 0 °C, (F) EtOH, r.t.
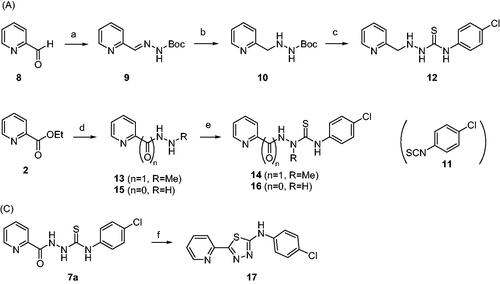
Syntheses of 12, 14, and 16 are depicted in Scheme 2. Unlike compounds 7a–7y in which Ar and R groups are modified, these compounds have modifications in thiosemicarbazide moiety of 7a that showed the highest level of anti-ER stress activityCitation34. Structure–activity relationship (SAR) of 7a could be elucidated by these analogues. Picolinaldehyde 8 was reacted with t-butyl carbazate to give compound 9 in 70% yield whose imine functional group was reduced to afford 10. Deprotection of t-Boc group of 10 followed by a reaction with 11 yielded compound 12, a reduced carbonyl version of 7a (Scheme 2(A)). Meanwhile, reaction of ethyl ester 2 with hydrazine gave 13 which was then coupled with 11 to give 14, an N-methyl analogue of 7a. A coupling reaction of 15 with 11 afforded compound 16, a decarbonyl version of 7a (Scheme 2(B)). Also, compound 17, a cyclic form of 7a, was also prepared under acidic condition to investigate the effect of structural rigidity on the activity (Scheme 2(C)).
Scheme 2. Synthesis of analogues with modifications in thiosemicarbazide moiety of 7a. Reagents and conditions: (A) NH2NHBoc, Et2O, r.t., 1 day, 70%, (B) H2, Pd/C, MeOH, (C) (i) 4 N HCl-dioxane, 0 °C, 4 h, (ii) 11, Et3N, EtOH, r.t., 4 h, 20%, (D) NH2NHCH3⋅H2SO4, Et3N, EtOH, reflux, 5 days, 46%, (E) 11, EtOH, r.t., 4 h, 36% (for compound 14), 77% (for compound 16), (F) H2SO4 or HCl, EtOH, 0 °C → r.t., 2 h, 41%.
Anti-prion aggregation activity
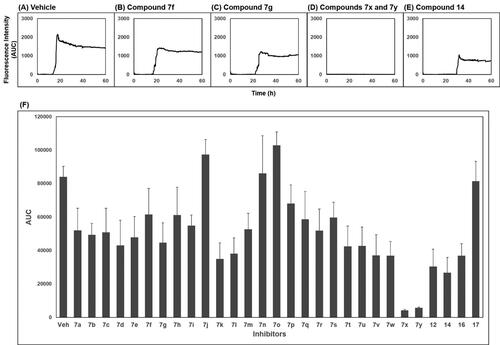
The activity of compounds 7a–7 y, 12, 14, 16, and 17 inhibiting the formation of unfolded PrP aggregates was evaluated by PAFA. This assay monitors the aggregation of unfolded PrP based on the fluorescence emitted from aggregate-bound thioflavin T (ThT)Citation39,Citation40,Citation48. Recombinant (rec) PrP, an initial component in a test tube, undergoes conformational change induced by guanidine HCl, a protein denaturant, into a partially molten form of PrP. This unfolded PrP conformer is subject to amyloid formation through self-aggregation and ThT binds to the resultant amyloids (, vehicle condition). In PAFA, the anti-prion aggregation activity of compounds can be expressed in different aspects of fluorescence curvesCitation49. As the aggregation process requires a lag period to onset, some inhibitors can prolong the lag time and reduce the degree of aggregation ()). Next, although the fluorescence level increases once the aggregation initiates at any delayed time points, other inhibitors can modulate the kinetics of aggregation or the level of plateau as each compound inhibits the aggregation at different efficacies ()). When significantly active, the inhibitors can show marginal increase in fluorescence resulting in an almost flat curve (). Taken these aspects into account, area under the curve (AUC) of fluorescence curves can be a suitable readout of PAFA to represent the overall efficiency of the inhibitor.
Figure 2. Area under the curve (AUC) of fluorescence curves obtained from PAFA. (A) vehicle condition. (B–E) Representative fluorescence curves by 7f, 7g, 7x, 7y, and 14, respectively. (F) Anti-prion activity of the compounds measured by PAFA. AUC is expressed as mean ± SEM of independent experiments (n = 9). Veh = vehicle condition where DMSO was treated instead of 100 µM compounds.
Recombinant PrP used in this study contains His tag and theoretically the activity of a certain protein should be determined in the absence of any tags that are unnatural. A study reported that the presence of His tag caused technical difficulties in protein purification due to aggregationCitation50. Hence, it was followed by some RT-QuIC studies that used recPrP without the tag. However, it is rather controversial as far as we are concerned. Our in-house experiences demonstrated the presence of His tag did not seriously affect the aggregate formation of recPrPCitation39,Citation49. This was confirmed by an independent study in which PrP aggregation reactions were conducted without the removal of the tag from PrPCitation51. Moreover, Yen et al.Citation52 showed the presence of the tag did not affect the secondary structure of PrP and PrP aggregation. At any rate, the activity of compounds that we report here is not questionable because the experiments were conducted under the identical condition using recPrP with the tag in all reactions including the vehicle control that showed a decent aggregation pattern. Then, the next concern is that whether the His tag could affect the interaction of the compounds with recPrP. Usually, the His tag shows a strong binding with divalent ions such as Ni2+ and similar sorts. Chemical structures of the active compounds clearly demonstrated that they do not contain such moieties. Thus, we can put off the verification of this issue and go forward to proving the anti-prion activity of compounds in other assays in cultured cells
The activity of the compounds displayed in shows that most compounds have weak to moderate activity against aggregation. A few analogues of 7 and 7a (7k, 7l, 7v, 7w and 12, 14, 16, respectively) have slightly better activity. Two compounds, 7x and 7y, showed particularly strong and robust anti-aggregation activity. SAR revealed that analogues with monocyclic Ar showed low activity except that fluoropyridinyl analogues, 7k and 7l, were marginally more active. Analogues of 7a (12, 14, and 16) showed better activity than the parent compound 7a. These observations indicated that the modification of Ar or thiosemicarbazide moiety can be beneficial to the activity at least to some degree when Ar group is monocyclic. The analogues with bicyclic Ar groups showed an interesting SAR. Whereas 7 u, 7v (Ar = quinoline), and 7w (Ar = hydroxynaphthalene) showed similar activity to 7k and 7l, 7x, and 7y afforded AUC of 4176 ± 565 and 5624 ± 497, respectively, that are about 15 ∼ 20-fold better activity than the vehicle condition (83 940 ± 6304). The hydroxy group on naphthalene ring of 7x and 7y can be attributed to the high activity by providing a six-membered cyclic structure with the carbonyl group via an internal hydrogen bond (). This speculation can be supported by the low activity of 7v and 7w in which such a cyclic conformation is hardly formed. Moreover, the low activity of 7w strongly demonstrated the crucial role of regiochemistry of the hydroxy group. It was also suggested that R group has very limited effect on the activity. Taken together, 7x and 7y were chosen for further study.
Figure 3. Potential cyclic structures through an internal hydrogen bond in 7x and 7y.
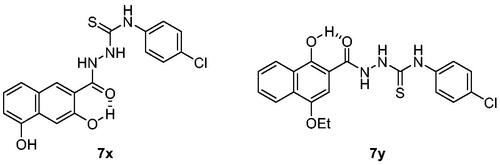
Compounds in this study were inspired by ER stress inhibitor scaffold that we have reported earlierCitation34. ER stress is triggered when abnormal level of misfolded protein is accumulated in ER, which is responsible for the correct folding process during de novo protein synthesis. Activity of the compounds (EC50) against tunicamycin-induced ER stress was measured by a GRP78-driven reporter assay systemCitation34,Citation53 and displayed in parallel with anti-prion aggregation activity (AUC) (Supplementary Figure 1). in order to investigate the correlation between two pathologies. Compounds 7a, 7c–7f, 7h, 7k, 7s, and 7t showed very strong inhibition against ER stress. SAR revealed that they all have 2-pyridyl moiety. It is speculated that pyridine N possibly form a cyclic conformation with the amide group via an internal hydrogen bond. Large differences in the inhibition between 7u vs. 7v and 7j vs. 7i can be explained in this context. Weak inhibition by 12, 14, 16, and 17 compared to the parent compound 7a demonstrated the essential role of the carbonyl group and NH of thiourea moiety in ER stress inhibition. Interestingly, in the case of the naphthalene Ar group, 7w that cannot form the cyclic structure showed better ER stress inhibition than 7x and 7y that possibly form such conformation. The strong ER stress inhibitors, however, were not among the top-tier inhibitors in PAFA assay. Although both protein aggregation and ER stress are commonly linked with protein misfolding, compounds in this study showed a considerable discrepancy between the two activities. For example, 7x and 7y, the best anti-prion compounds, only showed marginal activity against ER stress (Supplementary Figure 1). Next, the dose-responsiveness of the anti-PrP aggregation activity of 7x and 7y was evaluated (). Indeed, both compounds suppressed PrP aggregation in a dose-dependent manner. The dose–response curves based on the AUC at given concentrations determined EC50 = 4.6 µM and 5 µM for 7x and 7y, respectively.
Figure 4. Activity of compounds to inhibit PAFA and dose–response curves. (A) PAFA with different concentrations of the compounds. The multiple PAFA reactions (n = 3) were performed and the mean of each data point was plotted. (B) Dose–response curves of the compounds based on mean AUC of each concentration from independent experiments (n = 3).
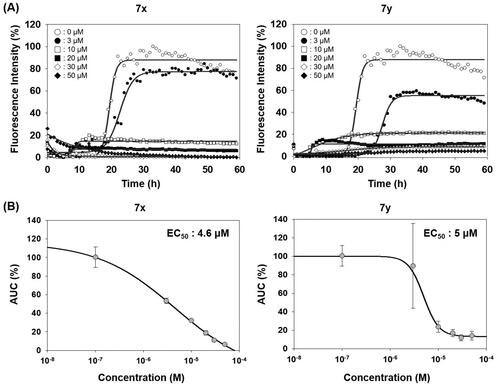
The anti-PrP aggregation activity of 7x and 7y was further compared to that of SGI-1027 (a positive control). As shown in , 7x and 7y significantly inhibited PrP aggregation in PAFA. The inhibitory activity was even better than that of the positive control, indicating the superiority of 7x and 7y as a PrP aggregation inhibitor, while 7b (a negative control) remained almost inactive. The PAFA products of 7x and 7y were further examined for the biophysical state of PrP molecules using SDD-AGE and AFM. In SDD-AGE, the migration of most PrP aggregates from PAFA with vehicle and negative control 7b was retarded, showing the massive band of very high molecular weights (). In contrast, the reaction products of PAFA with 7x and 7y showed a completely different band pattern of PrP molecules. Only PrP oligomers and monomers were observed, lacking PrP aggregates of very high molecular weights. In addition, as clearly shown in the AFM images of PAFA products, 7x and 7y significantly reduced the formation of fibrillar structure, while vehicle and 7b mostly showed fibrils (). The examination of biophysical states of PrP molecules in PAFA verified that 7x and 7y did actually inhibit aggregation to remove fibrillar aggregates observed in vehicle condition. Moreover, these results indicate that fluorescence-based detection of PrP aggregation controlled by compounds in PAFA is a reliable and robust assay to measure the efficacy of an anti-PrP aggregation inhibitor. Nevertheless, it is cautious to conclude that 7x and 7y would be beneficial to modify the course of disease in vivo because the compounds were not able to completely degrade PrP aggregates of very high molecular weights into monomers, but rather induced the emergence of a considerable level of the oligomers, which could form PrP fibrils again and, more critically, could be more harmful to neurons than PrP fibrils and monomersCitation54.
Figure 5. Biophysical states of PrP aggregates from PAFA. (A) PAFA with vehicle, positive control (SGI-1027), negative control (7b), and active compounds (7x and 7y) at 10 µM each. The multiple PAFA reactions (n = 3) were performed and the mean of each data point was plotted. (B) SDD-AGE analysis of the reaction products of PAFA shown in panel A. M, molecular weight size marker; PrP, recPrP monomer form; Veh, vehicle. (C) Micrographs of the reaction products of PAFA shown in panel A by AFM. Dotted rectangles were magnified and displayed below.
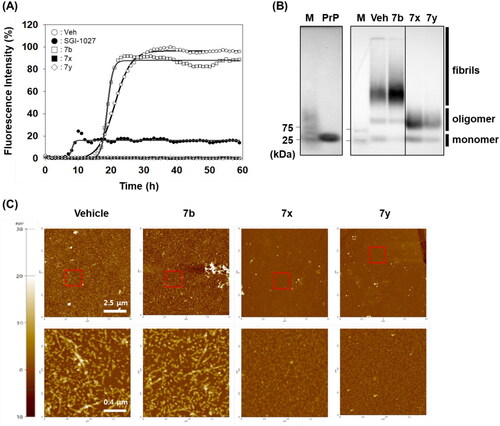
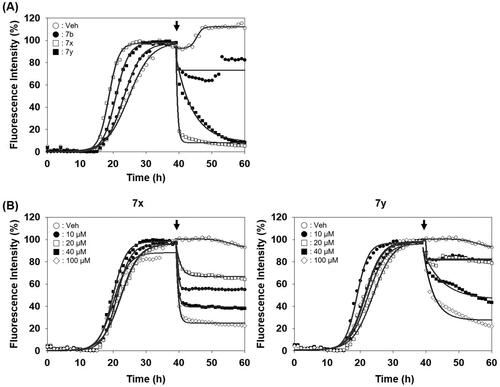
After identifying the inhibitors by PAFA that can measure the activity in a context of disease prevention, we sought to address whether they can exert the activity in a disease treatment setting as well. So, the compounds were tested if they disassemble pre-formed aggregates to smaller size aggregates or monomers. Each compound was spiked in the reaction after PrP aggregation reached the plateau in PAFA and the changes of PrP aggregation were monitored. As shown in , the fluorescence level immediately decreased upon the injection of 7x and 7y after the completion of pre-formation of PrP aggregates. It indicates that they have significant disaggregation activity against the pre-formed aggregates, while compound 7b showed no such effect. Also, the disassembling activity of the compounds was clearly confirmed in a dose-dependent manner ().
Figure 6. Activity of compounds to disassemble pre-existing aggregates. (A) Disaggregation curves of vehicle, negative control (7b), 7x, and 7y at 100 µM concentration. (B) Dose-dependent curves of 7x and 7y at different concentrations. Arrows indicate the time point of compound treatment. The multiple disaggregation reactions of PAFA products (n = 3) were conducted and the mean of each data point was plotted.
Unlike PAFA experiments in which PrP is partially unfolded by denaturants and self-aggregation of unfolded PrP occurs, the physiological observations indicate that PrPC undergoes misfolding through conformational change by PrPSc and de novo-generated PrPSc becomes aggregated. The ability of compounds 7x and 7y to inhibit the PrP conversion facilitated by PrPSc seeding was measured using RT-QuIC assay ()Citation44,Citation45,Citation49. The formation of PrP aggregates by RML PrPSc seed-induced conversion of mouse recPrP to the misfolded conformer was inhibited by 7x and 7y in a concentration-dependent fashion (). AUC curves determined the inhibitory efficacy of 7x and 7y to be EC50 = 0.9 µM and 2.8 µM, respectively (). This result suggested that 7x and 7y were able to inhibit PrPSc aggregation preceded by PrP conversion. Next, the inhibitory ability of compounds 7x and 7y in RT-QuIC was investigated with prions of various sources. RT-QuIC using bank vole recPrP substrates and seeding with RML, Hyper, and CJD prions showed robust PrP conversion and following aggregation, and compounds 7x and 7y efficiently inhibited it (). These results indicated that 7x and 7y are efficacious to a panel of prions from animals to humans.
Figure 7. Activity of compounds to inhibit RT-QuIC and dose–response curves. (A) RT-QuIC assay at different concentrations of 7x and 7y. The assay was performed using mouse recPrP and 5x1 0 −6 diluted RML-ill mouse brain homogenates. Each concentration line represents mean of multiple RT-QuIC reactions (n = 3). (B) Dose–response curves of the compounds based on AUC of each concentration from independent experiments (n = 2).
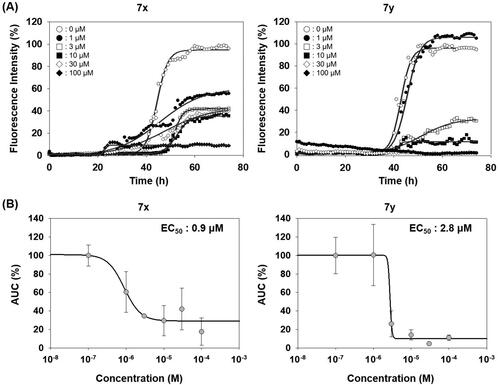
Figure 8. Activity of compounds to inhibit RT-QuIC performed with prions of various sources. Each RT-QuIC assay was performed using bank vole recPrP in the presence of 100 µM 7x and 7y. The prion seeds were 5 × 10−6 diluted brain homogenates of RML-ill mice, Hyper-ill Syrian hamsters and a CJD patient. Each concentration line represents mean of independent experiments (n = 3).
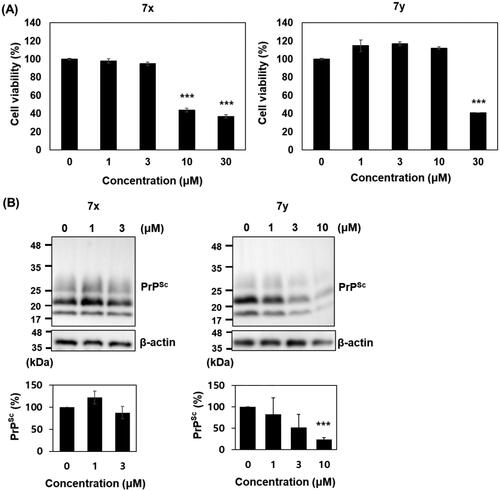
Anti-prion activity of 7x and 7y was further investigated in cultured cells with permanent prion infection. Cytotoxicity tests in ScN2a cells showed that 7x was marginally more cytotoxic than 7y: the survival of cells incubated with 7x and 7y was significantly reduced over 10 and 30 µM, respectively. The western blot detection of PK-resistant PrPSc level in ScN2a cells incubated with the non-cytotoxic concentrations of compounds showed that 10 µM 7y decreased the PrPSc level over 75% compared to vehicle control, but 7x failed ().
Figure 9. Cytotoxicity and anti-prion activity of compounds in ScN2a cells. (A) Cytotoxicity of 7x and 7y. The MTT assay were performed (n = 3) and the mean was plotted. (B) Western blots of PK-resistant PrPSc in the cells incubated with 7x and 7y. β-actin was used as a loading control. Western blotting was performed multiple times (n = 4). The PrPSc level of each blot was quantified by densitometry and the means were plotted. ***p < 0.01.
In conclusion, here, we report the synthesis of acylthiosemicarbazide compounds and their activity to inhibit PrP conversion and aggregation. PAFA indicated that compounds with monocyclic Ar groups showed low-to-medium range activity against PrP aggregation. Compounds 7x and 7y that have hydroxy-2-naphthyl Ar group were the most active inhibitors to give EC50 ≅ 5 µM in PAFA that is 15 ∼ 20-fold better activity than vehicle. Their activity can be attributed to a cyclic conformation through the internal hydrogen bond. They also showed significant activity to disassemble pre-existing PrP aggregates. Their activities were further confirmed by AFM, SDD-AGE, and RT-QuIC assay, which is relevant to the physiological condition since PrPSc from infected animal and humans is involved and therefore mimics the essence of prion propagation. Both compounds showed even better efficacy, showing EC50 = 0.9 µM and 2.8 µM, respectively in RT-QuIC assay. Furthermore, one of compounds 7y was effective to decrease the level of PrPSc in cultured cells with permanent prion infection. However, their anti-prion aggregation activity was not closely correlated with the anti-ER stress activity. Taken together, data suggested that SAR and inhibitors identified in this study can provide an excellent platform for the discovery of anti-prion therapeutics.
Supplemental Material
Download PDF (2 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95(23):13363–13383.
- Ryou C. Prions and prion diseases: fundamentals and mechanistic details. J Microbiol Biotechnol. 2007;17(7):1059–1070.
- Prusiner SB. The prion diseases. Brain Pathol. 1998;8(3):499–513.
- Ritchie DL, Ironside JW. Neuropathology of human prion diseases. Prog Mol Biol Transl Sci. 2017;150:319–339.
- Lakkaraju AKK, Frontzek K, Lemes E, Herrmann U, Losa M, Marpakwar R, Aguzzi A. Loss of PIKfyve drives the spongiform degeneration in prion diseases. EMBO Mol Med. 2021;13(9):e14714.
- Frontzek K, Bardelli M, Senatore A, Henzi A, Reimann RR, Bedir S, Marino M, Hussain R, Jurt S, Meisl G, et al. A conformational switch controlling the toxicity of the prion protein. Nat Struct Mol Biol. 2022;29(8):831–840.
- Tuite MF, Serio TR. The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol. 2010;11(12):823–833.
- Ryou C, Mays CE. Prion propagation in vitro: are we there yet? Int J Med Sci. 2008;5(6):347–353.
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144.
- Kraus A, Hoyt F, Schwartz CL, Hansen B, Artikis E, Hughson AG, Raymond GJ, Race B, Baron GS, Caughey B. High-resolution structure and strain comparison of infectious mammalian prions. Mol Cell. 2021;81(21):4540–4551.e6.
- Hoyt F, Standke HG, Artikis E, Schwartz CL, Hansen B, Li K, Hughson AG, Manca M, Thomas OR, Raymond GJ, et al. Cryo-EM structure of anchorless RML prion reveals variations in shared motifs between distinct strains. Nat Commun. 2022;13(1):4005.
- Safar J, Prusiner SB. Molecular studies of prion diseases. Prog Brain Res. 1998;117:421–434.
- Supattapone S, Nguyen HO, Cohen FE, Prusiner SB, Scott MR. Elimination of prions by branched polyamines and implications for therapeutics. Proc Natl Acad Sci USA. 1999;96(25):14529–14534.
- Solassol J, Crozet C, Perrier V, Leclaire J, Beranger F, Caminade AM, Meunier B, Dormont D, Majoral JP, Lehmann S. Cationic phosphorus-containing dendrimers reduce prion replication both in cell culture and in mice infected with scrapie. J Gen Virol. 2004;85(Pt 6):1791–1799.
- Haïk S, Marcon G, Mallet A, Tettamanti M, Welaratne A, Giaccone G, Azimi S, Pietrini V, Fabreguettes J-R, Imperiale D, et al. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomized, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(2):150–158.
- Lim YB, Mays CE, Kim Y, Titlow WB, Ryou C. The inhibition of prions through blocking prion conversion by permanently charged branched polyamines of low cytotoxicity. Biomaterials. 2010;31(8):2025–2033.
- Ryou C, Legname G, Peretz D, Craig JC, Baldwin MA, Prusiner SB. Differential inhibition of prion propagation by enantiomers of quinacrine. Lab Invest. 2003;83(6):837–843.
- Murakami-Kubo I, Doh-Ura K, Ishikawa K, Kawatake S, Sasaki K, Kira JI, Ohta S, Iwaki T. Quinoline derivatives are therapeutic candidates for transmissible spongiform encephalopathies. J Virol. 2004;78(3):1281–1288.
- Shimizu Y, Kaku-Ushiki Y, Iwamaru Y, Muramoto T, Kitamoto T, Yokoyama T, Mohri S, Tagawa Y. A novel anti-prion protein monoclonal antibody and its single-chain fragment variable derivative with ability to inhibit abnormal prion protein accumulation in cultured cells. Microbiol Immunol. 2010;54(2):112–121.
- Kim DH, Ren C, Ryou C, Li J. Direct interaction of DNMT inhibitors to PrP(C) suppresses pathogenic process of prion. Acta Pharm Sin B. 2019;9(5):952–959.
- Kuwata K, Nishida N, Matsumoto T, Kamatari YO, Hosokawa-Muto J, Kodama K, Nakamura HK, Kimura K, Kawasaki M, Takakura Y, et al. Hot spots in prion protein for pathogenic conversion. Proc Natl Acad Sci USA. 2007;104(29):11921–11926.
- Kimura T, Hosokawa-Muto J, Kamatari YO, Kuwata K. Synthesis of GN8 derivatives and evaluation of their antiprion activity in TSE-infected cells. Bioorg Med Chem Lett. 2011;21(5):1502–1507.
- Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA. 2001;98(17):9836–9841.
- Ishibashi D, Nakagaki T, Ishikawa T, Atarashi R, Watanabe K, Cruz FA, Hamada T, Nishida N. Structure-based drug discovery for prion disease using a novel binding simulation. eBiomedicine. 2016;9:238–249.
- Li JJ, Ryou C, Kim DH. Effects of SGI-1027 on formation and elimination of PrPSc in prion-infected cells. Mol Biol. 2020;54(3):412–415.
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 1995;83(1):79–90.
- Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA. 1997;94(19):10069–10074.
- Mays CE, Ryou C. Plasminogen stimulates propagation of protease-resistant prion protein in vitro. Faseb J. 2010;24(12):5102–5112.
- Baldeschi AC, Zattoni M, Vanni S, Nikolic L, Ferracin C, La Sala G, Summa M, Bertorelli R, Bertozzi SM, Giachin G, et al. Innovative non-PrP-targeted drug strategy designed to enhance prion clearance. J Med Chem. 2022;65(13):8998–9010.
- Ali T, Hannaoui S, Nemani S, Tahir W, Zemlyankina I, Cherry P, Shim SY, Sim V, Schaetzl HM, Gilch S. Oral administration of repurposed drug targeting CYP46A1 increases survival times of prion infected mice. Acta Neuropathol Commun. 2021;9(1):58.
- Ryou C, Titlow WB, Mays CE, Bae Y, Kim S. The suppression of prion propagation using poly-L-lysine by targeting plasminogen that stimulates prion protein conversion. Biomaterials. 2011;32(11):3141–3149.
- Barreca ML, Iraci N, Biggi S, Cecchetti V, Biasini E. Pharmacological agents targeting the cellular prion protein. Pathogens. 2018;7(1):27.
- Trevitt CR, Collinge J. A systemic review of prion therapeutics in experimental models. Brain. 2006;129(9):2241–2265.
- Choi H, Yun W, Lee JH, Jang S, Park SW, Kim DH, Seon KP, Hyun JM, Jeong K, Ku JM, et al. Synthesis and anti-endoplasmic reticulum stress activity of N-substituted-2-arylcarbonylhydrazinecarbothioamides. Med Chem Res. 2019;28(12):2142–2152.
- Doyle KM, Kennedy D, Gorman AM, Gupta S, Healy SJ, Samali A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J Cell Mol Med. 2011;15(10):2025–2039.
- Yoshida H. ER stress and diseases. Febs J. 2007;274(3):630–658.
- Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833(12):3460–3470.
- Jeon JH, Im S, Kim HS, Lee D, Jeong K, Ku JM, Nam TG. Chemical chaperones to inhibit endoplasmic reticulum stress: implications in diseases. DDDT. 2022;16:4385–4397.
- Kim SG, Lee HM, Ryou C. Parameters that affect macromolecular self-assembly of prion protein. Protein J. 2014;33(3):243–252.
- Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J Mol Biol. 2005;346(2):645–659.
- Hwang HG, Kim DH, Lee J, Mo Y, Lee SH, Lee Y, Hyeon JW, Lee SM, Cheon YP, Choi EK, et al. High-level production of high-purity human and murine recombinant prion proteins functionally compatible to in vitro seeding assay. J Microbiol Biotechnol. 2018;28(10):1749–1759.
- Dean DN, Das PK, Rana P, Burg F, Levites Y, Morgan SE, Ghosh P, Rangachari V. Strain-specific fibril propagation by an Abeta dodecamer. Sci Rep. 2017;7(1):40787.
- Halfmann R, Lindquist S. Screening for amyloid aggregation by semi-denaturing detergent-agarose gel electrophoresis. J Vis Exp. 2008;17:838.
- Wilham JM, Orru CD, Bessen RA, Atarashi R, Sano K, Race B, Meade-White KD, Taubner LM, Timmes A, Caughey B. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 2010;6(12):e1001217.
- Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, Onwubiko HA, Priola SA, Caughey B. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods. 2008;5(3):211–212.
- Waqas M, Lee HM, Kim J, Telling G, Kim JK, Kim DH, Ryou C. Effect of poly-L-arginine in inhibiting scrapie prion protein of cultured cells. Mol Cell Biochem. 2017;428(1–2):57–66.
- Lee S, Lee H, Kim J, Kim JH, Gao EM, Lee Y, Yoo M, Trinh THT, Kim J, Kim CY, et al. The effect of Curcuma phaeocaulis Valeton (Zingiberaceae) extract on prion propagation in cell-based and animal models. IJMS. 2022;24(1):182.
- Biancalana M, Koide S. Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim Biophys Acta. 2010;1804(7):1405–1412.
- Kang HE, Mo Y, Abd Rahim R, Lee HM, Ryou C. Prion diagnosis: application of real-time quaking-induced conversion. Biomed Res Int. 2017;2017:1–8.
- Cheng K, Sloan A, Waitt B, Vendramelli R, Godal D, Simon SLR, O'Neil J, Carpenter M, Jackson D, Eastlake J, et al. Altered rPrP substrate structures and their influence on real-time quaking induced conversion reactions. Protein Expr Purif. 2018;143:20–27.
- Abskharon R, Dang J, Elfarash A, Wang Z, Shen P, Zou LS, Hassan S, Wang F, Fujioka H, Steyaert J, et al. Soluble polymorphic bank vole prion proteins induced by co-expression of quiescin sulfhydryl oxidase in E. coli and their aggregation behaviors. Microb Cell Fact. 2017;16(1):170.
- Yen CF, Harischandra DS, Kanthasamy A, Sivasanka S. Copper-induced structural conversion templates prion protein oligomerization and neurotoxicity. Sci Adv. 2016;2(7):e1600014.
- Kim YE, Kim DH, Choi A, Jang S, Jeong K, Kim YM, Nam TG. Bi-aryl analogues of salicylic acids: design, synthesis and SAR study to ameliorate endoplasmic reticulum stress. DDDT. 2021;15:3593–3604.
- Simoneau S, Rezaei H, Salès N, Kaiser-Schulz G, Lefebvre-Roque M, Vidal C, Fournier JG, Comte J, Wopfner F, Grosclaude J, et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 2007;3(8):e125.